
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA convenient route to mixed cationic group 13/14/15 compounds†
Matthias T.
Ackermann
a,
Michael
Seidl
b,
Riccardo
Grande
a,
Yuqiao
Zhou
c,
Michael J.
Ferguson
 c,
Alexey Y.
Timoshkin
d,
Eric
Rivard
c and
Manfred
Scheer
*a
c,
Alexey Y.
Timoshkin
d,
Eric
Rivard
c and
Manfred
Scheer
*a
aInstitute of Inorganic Chemistry, University Regensburg, Universitätsstraße 31, 93053 Regensburg, Germany. E-mail: manfred.scheer@ur.de
bInstitute of General and Theoretical Chemistry, Leopold-Franzens Universität Innsbruck, Innrain 80-82, 6020 Innsbruck, Austria
cDepartment of Chemistry, University of Alberta, 11227 Saskatchewan Dr, Edmonton, Alberta T6G 2G2, Canada
dInstitute of Chemistry, Saint Petersburg State University, Universitetskaya emb. 7/9, 199034 St Petersburg, Russia
First published on 20th January 2023
Abstract
The formation of novel cationic mixed main group compounds is reported revealing a chain composed of different elements of group 13, 14, and 15. Reactions of different pnictogenylboranes R2EBH2·NMe3 (E = P, R = Ph, H; E = As, R = Ph, H) with the NHC-stabilized compound IDipp·GeH2BH2OTf (1) (IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene) were carried out, yielding the novel cationic, mixed group 13/14/15 compounds [IDipp·GeH2BH2ER2BH2·NMe3]+ (2a E = P; R = Ph; 2b E = As; R = Ph; 3a E = P; R = H; 3b E = As; R = H) by the nucleophilic substitution of the triflate (OTf) group. The products were analysed by NMR spectroscopy and mass spectrometry and for 2a and 2b also by X-ray structure analysis. Further reactions of 1 with H2EBH2·IDipp (E = P, As) resulted in the unprecedented parent complexes [IDipp·GeH2BH2EH2BH2·IDipp][OTf] (5a E = P; 5b E = As), which were studied by X-ray structure analysis, NMR spectroscopy and mass spectrometry. Accompanying DFT computations give insight into the stability of the formed products with respect to their decomposition.
Introduction
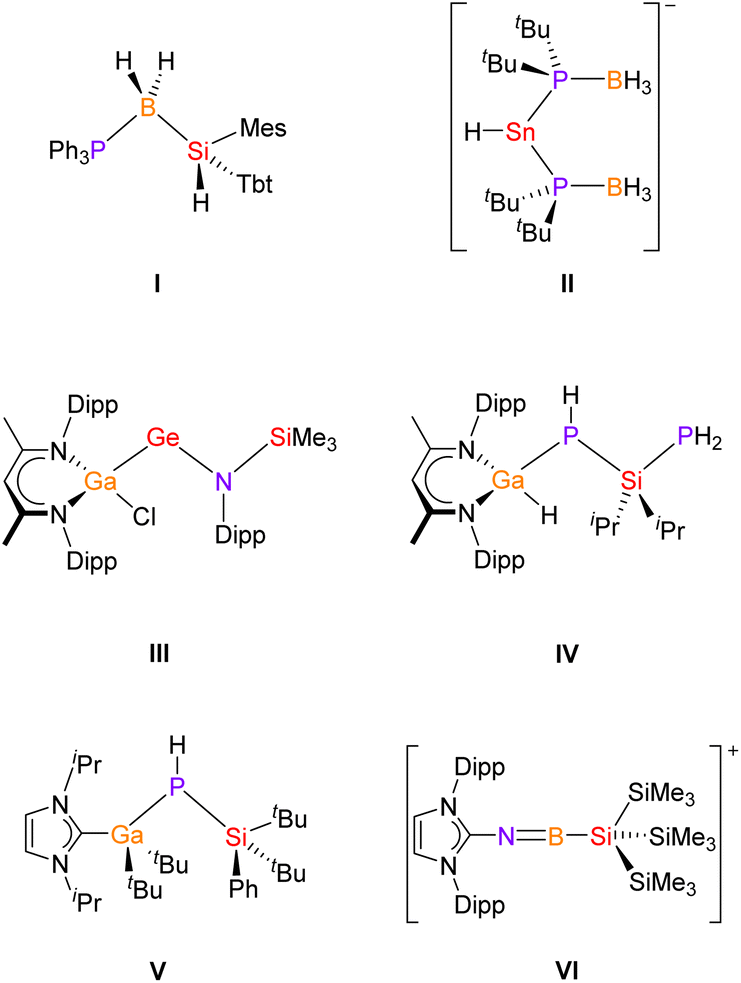
Carbon- and main group element-containing catena compounds based on C–El (e.g., El = B, Si, P) building blocks have been known for a long time and are used for various applications.1a–f For instance, 1,3-disilabutane, H3SiCH2SiH2CH3, serves as a precursor for SiC thin film deposition in several CVD (Chemical Vapor Deposition) processes.2 Similarly, inorganic chain compounds that consist of two different main group elements are well known in the literature, especially in the case of group 13/15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 and 14/154 compounds, respectively. However, comparable compounds that are assembled by three different elements of group 13, 14 and 15 are very limited, in particular, if heavier homologues are considered.5 For example, in 2004 Tokitoh and coworkers reported the neutral silylboranephosphine I, which was synthesised by reaction of a sterically crowded silylene with BH3·PPh3 (Chart 1).6 Alternatively, the anionic phosphineboranestannate II was synthesized by Wright and coworkers through the reaction of LiSn(NMe2)3 with the phosphine-borane tBu2PH·BH3.7 Examples for chain-like group 13/14/15 compounds which are stabilized by β-diketiminate ligands are the very recently reported compound III by Schulz8 and compound IV by von Hänisch and coworkers.9 Furthermore, the von Hänisch group reported unexpected reactivity of a four-membered Ga/P heterocycle towards bulky N-heterocyclic carbene (NHC) ligands, leading to the silylphosphinogallane V.10 In addition, Inoue and coworkers prepared the structurally related silylaminoborane IMe4·BH2NHSiH2SitBu3 (IMe4 = 1,3,4,5-tetra(methyl)imidazole-2-ylidene)11 and the NHC-stabilized compound VI from a salt-metathesis reaction.12
3 and 14/154 compounds, respectively. However, comparable compounds that are assembled by three different elements of group 13, 14 and 15 are very limited, in particular, if heavier homologues are considered.5 For example, in 2004 Tokitoh and coworkers reported the neutral silylboranephosphine I, which was synthesised by reaction of a sterically crowded silylene with BH3·PPh3 (Chart 1).6 Alternatively, the anionic phosphineboranestannate II was synthesized by Wright and coworkers through the reaction of LiSn(NMe2)3 with the phosphine-borane tBu2PH·BH3.7 Examples for chain-like group 13/14/15 compounds which are stabilized by β-diketiminate ligands are the very recently reported compound III by Schulz8 and compound IV by von Hänisch and coworkers.9 Furthermore, the von Hänisch group reported unexpected reactivity of a four-membered Ga/P heterocycle towards bulky N-heterocyclic carbene (NHC) ligands, leading to the silylphosphinogallane V.10 In addition, Inoue and coworkers prepared the structurally related silylaminoborane IMe4·BH2NHSiH2SitBu3 (IMe4 = 1,3,4,5-tetra(methyl)imidazole-2-ylidene)11 and the NHC-stabilized compound VI from a salt-metathesis reaction.12
 | ||
| Chart 1 Selected examples for mixed group 13/14/15 chain compounds. | ||
The few existing examples of linear group 13/14/15 compounds are usually short (3-membered) chains and possess organic groups to stabilize them. Thus, the only examples of parent group 13/14/15 chains containing only hydrogen substituents, including the triple mixed hydrides H3TetPH2BH3 (Tet = Si, Ge) and our recently reported compound [IDipp·GeH2BH2PH3][OTf] (IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene), could not be isolated or show very limited stability, which prevented their full characterization.13 Thus, it is still very desirable to develop efficient synthetic routes for longer parent chain compounds, as they might represent suitable single-source precursors to bulk and nanodimensional ternary solids of tuneable composition and function, which can serve as small bandgap semiconductors for optoelectronics.14
Our group has a special interest in hydride-substituted (parent) group 13/15 compounds, and donor/acceptor stabilization is an elegant method for the development of several group 13/15 parent compounds of the type LA·H2EE‘H2·LB (E = group 15 element, E’ = group 13 element, LA = Lewis acid, LB = Lewis base).15 This concept was successfully refined to the LB-only stabilization of pnictogenyltriels H2EE‘H2·LB.16 Besides neutral compounds, the NMe3-stabilized pnictogenylboranes H2EBH2·NMe3 (E = P, As) could also be applied to build up cationic chain complexes.3c,d Hence, we wondered if it is possible to obtain long mixed group 13/14/15 chain compounds (group 14 element ≠ C) by the reaction of LB-stabilized pnictogenylboranes with the mixed group 13/14 starting material IDipp·GeH2BH2OTf (1) and if so, this approach could enable access to the longest cationic group 13/14/15 chains bearing only hydrogen substituents.
Results and discussion
Reactions of IDipp·GeH2BH2OTf (1) with the LB-stabilized diphenyl substituted pnictogenylboranes Ph2EBH2·NMe3 (E = P, As) led to the formation of the unprecedented mixed cationic group 13/14/15 chains as OTf− salts of [IDipp·GeH2BH2EPh2BH2·NMe3]+ (2a E = P; 2b E = As) (Scheme 1) and represent, to our knowledge, the longest cationic, mixed element 13/14/15 (group 14 element ≠ C) chain compounds so far. After stirring the reaction mixture overnight, the Ge–B–E–B–N chains 2a and 2b could be isolated in high yields of 94 and 93%, respectively. The reactions were performed in Et2O solutions, as the cationic complexes precipitate out of the reaction mixture upon formation and can then be isolated as pure white powders. Compounds 2a and 2b are well soluble in more polar solvents like CH2Cl2 or THF and are stable in solution and as solids, at ambient temperatures under an inert atmosphere. After the successful formation of the organo-substituted cationic group 13/14/15 chains in 2a and 2b, we wondered if the all hydrogen-substituted Ge-B-E-B chains [IDipp·GeH2BH2EH2BH2·NMe3][OTf] (3a E = P; 3b E = As) could be synthesized analogously. Reactions were performed under the same conditions as for 2a and 2b by using IDipp·GeH2BH2OTf (1) and the parent pnictogenylboranes H2EBH2·NMe3 (E = P, As) as starting materials. Similar to the diphenyl-substituted compounds, the formed ionic products 3a and 3b precipitate out of the reaction mixture and could be isolated as white powders, however, in slightly lower yields of 68 and 74%, respectively (Scheme 1). While both products are stable as solids at ambient temperatures in an inert atmosphere, they slowly decompose within several days in solutions of CH2Cl2 or THF.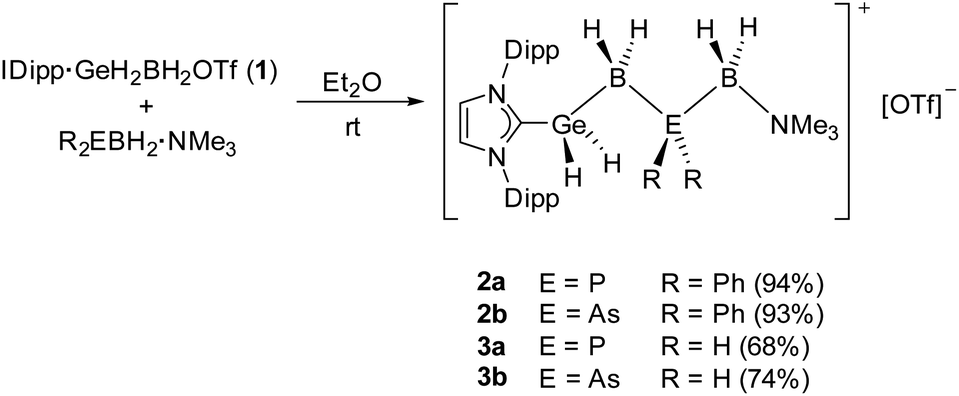 | ||
| Scheme 1 Synthesis of the group 13/14/15 chain compounds 2a, 2b, 3a and 3b. Yields are given in parentheses. | ||
While single crystals of the diphenyl-substituted compounds 2a and 2b were successfully obtained (Fig. 1) by layering a THF solution of the corresponding compounds with n-hexane, numerous attempts to crystalize the parent compounds 3a and 3b failed due to their rather fast decomposition in solution, leading only to the crystallization of [IDippH][OTf].
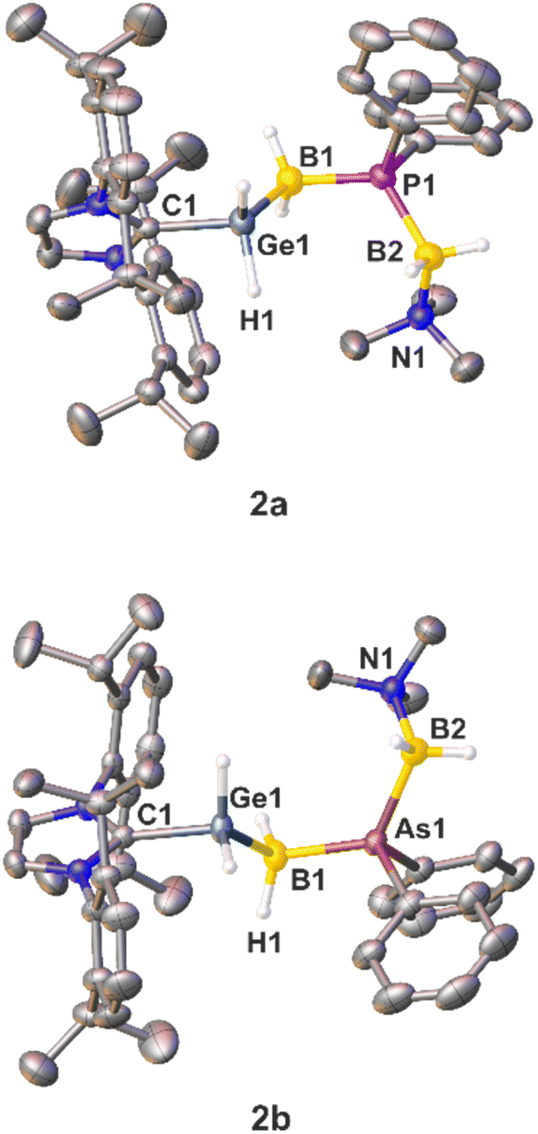 | ||
| Fig. 1 Molecular structures of the cations in 2a (top) and 2b (bottom) in the solid state with thermal ellipsoids at a 50% probability level. Hydrogen atoms bound to carbon atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: 2a: C1–Ge1 2.009(2), Ge1–B2 2.051(3), B1–P1 1.937(3), P1–B2 1.969(3), B2–N1 1.609(3); C1–Ge1–B1 111.86(10), Ge1–B1–P1 112.17(14), P1–B2–N1 116.50(18). 2b: C1–Ge1 2.0075(18), Ge1–B1 2.054(2), B1–As1 2.047(2), As1–B2 2.072(2), B2–N1 1.599(3); C1–Ge1–B1 112.01(8), Ge1–B1–As1 111.19(10), As1–B2–N1 114.43(13). | ||
In order to perform X-ray structure analysis on salts of the parent chains 3a/b, attempts to exchange the [OTf]− anions with the larger weakly coordinating anions (WCAs) [BArF20]− (BArF20 = [B(C6F5)4]) and [TEF]− (TEF = [Al{OC(CF3)3}4]) were carried out, as the bigger anions might promote a more favorable packing of the ion pairs within the solid state. However, those attempts failed to yield crystals. Thus, experiments to exchange the rather small LB NMe3 with the sterically more demanding NHCs were performed. Out of the NHCs used IMe4, IMes (IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene) and IDipp, only the latter was suitable for the crystallization of the desired products (vide infra). Whereas only decomposition was observed for the reaction of 1 and H2PBH2·IMe4, the parent compound [IDipp·GeH2BH2PH2BH2·IMes][OTf] (4) could be isolated as white powder in 68% yield from the reaction of 1 and H2PBH2·IMes. While 4 was verified by NMR spectroscopy, all crystallization attempts failed and only decomposition was observed. Similarly to H2PBH2·IMes, reactions of 1 with H2EBH2·IDipp (E = P, As) in Et2O result in the formation of the desired parent complexes [IDipp·GeH2BH2PH2BH2·IDipp][OTf] (5a E = P; 5b E = As) in high yields for 5a (91%) and good yields for 5b (69%) (Scheme 2).
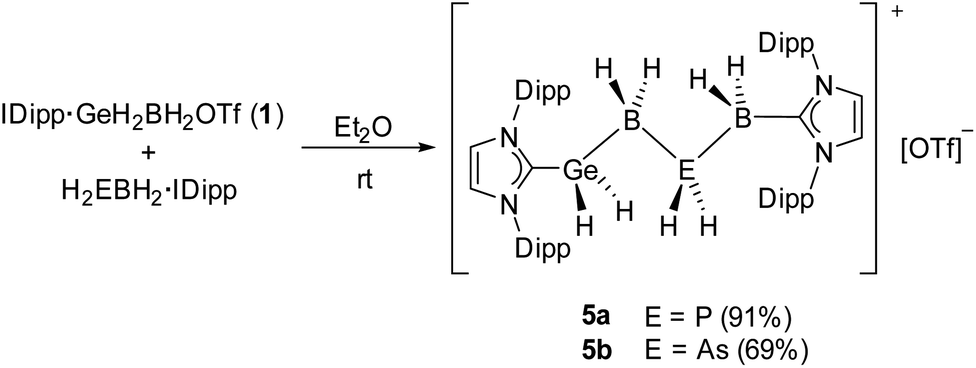 | ||
| Scheme 2 Synthesis of the parent compounds 5a and 5b. Yields are given in parentheses. | ||
The 1H NMR spectra in CD2Cl2 of all isolated group 13/14/15 chain compounds show signals for the GeH2-moieties in the expected range of δ = 3.20–3.57 ppm. Furthermore, the 31P NMR spectrum of 2a contains a broad singlet at δ = −24.9 ppm due to the coupling with the 11B nuclei and shows a downfield shift compared to the starting material Ph2PBH2·NMe3 (δ = −39.5 ppm).17 Analogously, the hydrogen-substituted compounds 3a, 4 and 5a each display a broad 31P NMR triplet resonance with almost identical chemical shifts for the PH2 moieties (δ = −112.1 to −116.6 ppm; 1JP,H = 334–340 Hz). The 11B NMR spectra of 2a – 3b reveal two broad signals for the BH2 moieties in the range of δ = −6.4 to −10.8 ppm and δ = −37.2 to −43.5 ppm. In contrast to the NMe3-stabilized compounds 2a – 3b, the 11B NMR spectra of 5a and 5b show a significantly upfield-shifted signal for the terminal boron atom within the Ge–B–E–B chain (5aδ = −34.8 ppm; 5bδ = −33.3 ppm) due to the exchange of NMe3 with IDipp. This is also observed for the substitution with IMes in compound 4 (δ = −35.3 ppm). Similar to the NMe3-stabilized compounds, a broad signal for the second boron atom is found in the range of δ = −39.6 to −42.8 ppm. Moreover, all molecular ion peaks of the products 2a–5b (except 4) are detected in the ESI-MS spectra (see ESI† for details).
The solid state structures of 2a and 2b reveal similar C–Ge bond lengths [2a: 2.009(2) Å; 2b: 2.0075(18) Å] (Fig. 1). In addition, the Ge−B distances are almost identical [2a: 2.051(3) Å; 2b: 2.054(2) Å]. The P–B bond lengths of 2a [1.937(3) and 1.969(3) Å] and the As–B bond lengths of 2b [2.047(2) and 2.072(2) Å] are in the expected range for single bonds. The B–P–B angle measures 119.27(13)°, while the corresponding B–As–B angle is slightly wider [122.45(9)°]. Both B–N distances [2a: 1.609(3) Å; 2b: 1.599(3) Å] are within the expected range of single bonds.18 In the Ge–B–E–B chain (2a: E = P, 2b: E = As) all substituents adopt a staggered conformation about the Ge−B and both E–B bond axes. Specifically, an antiperiplanar conformation is present about the Ge1–B1 axis, whilst a synperiplanar arrangement is found about the B1–E1-axis, and a synclinal arrangement exists about the E1–B2-axis. Similar to 2a and 2b, single crystals of 5a and 5b could be obtained by layering of a CH2Cl2 solution of the products with n-hexane at room temperature. Both compounds 5a and 5b crystallize with two independent molecules in the asymmetric unit and show a very disordered Ge–B–E–B (E = P, As) chain (Fig. 2; see the ESI† for details). In the solid state both compounds reveal Ge−B single bond distances. The P–B bond lengths of compound 5a and the As–B bond lengths of 5b are in the expected range for single bonds.18
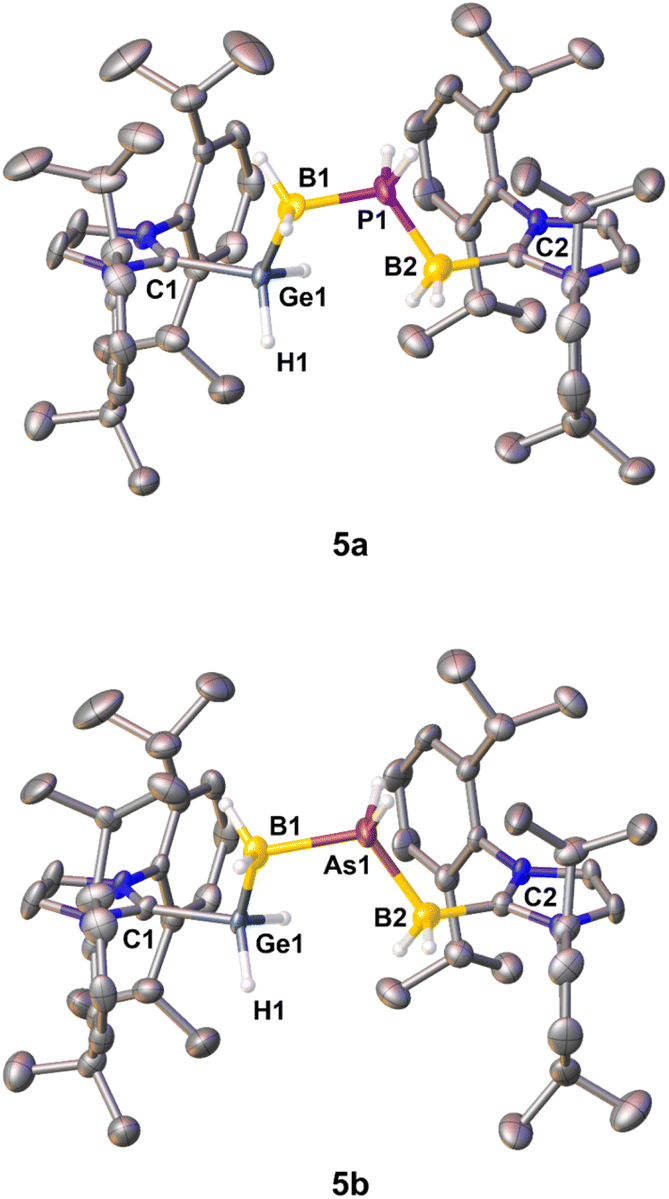 | ||
| Fig. 2 Major parts of the disordered molecular structures of the cations in 5a (top) and 5b (bottom) in the solid state with thermal ellipsoids at a 50% probability level. Only one cation of the asymmetric unit is depicted, respectively. Hydrogen atoms bound to carbon atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]:5a: C1–Ge1 1.955(3), Ge1–B1 2.014(7), B1–P1 1.934(6), P1–B2 1.922(13), B2–C2 1.665(11); C1–Ge1–B1 111.3(2), Ge1–B1–P1 105.7(3), B1–P1–B2 114.8(4), P1–B2–C2 116.1(8). 5b: C1–Ge1 1.994(3), Ge1–B1 2.055(8), B1–As1 2.076(6), As1–B2 2.034(10), B2–C2 1.595(9); C1–Ge1–B1 112.1(2), Ge1–B1–As1 103.4(3), B1–As1–B2 119.0(4), As1–B2–C2 112.2(7). | ||
Computational studies of the gas phase reactions of IDipp·GeH2BH2OTf with R2EBH2·LB leading to the ion pairs reveal that those processes are exothermic for all cases (Table 1). The favorability increases in the order 3b < 2b < 3a < 2a < 5b < 5a and thus, confirms the experimental findings well. In detail, the formation of the phosphorus derivatives is more exothermic (by 18–35 kJ mol−1) than for the corresponding arsenic compounds. Furthermore, the formal substitution of NMe3 by IDipp is exothermic by 75–80 kJ mol−1 and therefore significantly increases the stability of the respective hydride-only-substituted products.
| Process | ΔE0° | ΔH298° | ΔS298° | ΔG298° |
|---|---|---|---|---|
| 1 + Ph2PBH2·NMe3 = 2a | −77.9 | −69.0 | −183.6 | −14.3 |
| 1 + Ph2AsBH2·NMe3 = 2b | −51.3 | −43.8 | −168.9 | 6.6 |
| 1 + H2PBH2·NMe3 = 3a | −67.5 | −58.1 | −173.9 | −6.2 |
| 1 + H2AsBH2·NMe3 = 3b | −39.9 | −31.8 | −168.5 | 18.4 |
| 1 + H2PBH2·IDipp = 5a | −122.8 | −113.8 | −151.0 | −68.7 |
| 1 + H2AsBH2·IDipp = 5b | −88.1 | −80.0 | −146.7 | −36.3 |
| 3a + IDipp = 5a + NMe3 | −79.4 | −82.0 | −33.7 | −72.0 |
| 3b + IDipp = 5b + NMe3 | −75.8 | −78.2 | −35.4 | −67.6 |
Conclusions
The results show that the synthetic concept to generate cationic linear chain compounds by using IDipp·GeH2BH2OTf (1) in the reactions with different pnictogen sources could be successfully applied to obtain the longer, parent, mixed group 13/14/15 chain compounds containing a Ge–B–E–B (E = P, As) sequence. As a result, the diphenyl-substituted compounds 2a and 2b were synthesized in very good yields and were fully characterized. In addition, the cationic chain compounds of only hydrogen-substituted parent group 13/14/15 elements [IDipp·GeH2BH2EH2BH2·NMe3][OTf] (3a E = P; 3b E = As) could be isolated successfully. Subsequent substitution of NMe3 with IDipp for the group 13/14/15 chains containing only hydrogen substituents allowed X-ray structure analysis on the unprecedented parent compounds 5a and 5b. Further investigations are directed to generate anionic or neutral chain compounds of mixed group 13/14/15 elements.Data availability
All experimental procedures, spectroscopic data, information on the theoretical calculations and crystallographic data can be found in the ESI.†Author contributions
M. T. A. and R. C. performed the experimental work. M. T. A wrote the original draft. A. Y. T. performed the DFT calculations. M. Seidl, Y. Z. and M. J. F. performed single crystal X-ray experiments and interpreted the structural data. E. R. and M. Scheer supervised and acquired funding for the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by joint DFG-RSF grant (RSF project 21-43-04404 and DFG project Sche 384/41-1). The use of computational resources of the research center «Computing Center» of the research park of St. Petersburg State University is acknowledged. M. T. A. thanks the Deutscher Akademischer Austauschdienst (DAAD) for travel support. E. R. thanks NSERC of Canada for funding.References
- (a) H. I. Schlesinger, N. W. Flodin and A. B. Burg, J. Am. Chem. Soc., 1939, 61, 1078–1083 CrossRef CAS; (b) F. Gol, G. Hasselkuss, P. C. Knüppel and O. Stelzer, Z. Naturforsch., B: J. Chem. Sci., 1988, 43, 31–44 CrossRef CAS; (c) I. Kovacs and G. Fritz, Z. Anorg. Allg. Chem., 1993, 619, 1491–1493 CrossRef CAS; (d) A. Wörsching and G. Fritz, Z. Anorg. Allg. Chem., 1984, 512, 131–163 CrossRef; (e) G. Fritz, Angew. Chem., Int. Ed. Engl., 1987, 26, 1111–1132 CrossRef; (f) D. P. Gates, in New Aspects in Phosphorus Chemistry V, Springer, Berlin, Heidelberg, 2005, vol. 250, pp. 107–126 Search PubMed.
- (a) J.-H. Boo, K.-S. Yu, Y. Kim, S. H. Yeon and I. N. Jung, Chem. Mater., 1995, 7, 694–698 CrossRef CAS; (b) K.-W. Lee, K.-S. Yu and Y. Kim, J. Cryst. Growth, 1997, 179, 153–160 CrossRef CAS; (c) C. R. Stoldt, C. Carraro, W. R. Ashurst, D. Gao, R. T. Howe and R. Maboudian, Sens. Actuators, A, 2002, 97–98, 410–415 CrossRef CAS.
- (a) C. Marquardt, C. Thoms, A. Stauber, G. Balázs, M. Bodensteiner and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 3727–3730 CrossRef CAS PubMed; (b) C. Marquardt, T. Kahoun, A. Stauber, G. Balázs, M. Bodensteiner, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2016, 55, 14828–14832 CrossRef CAS PubMed; (c) C. Marquardt, G. Balázs, J. Baumann, A. V. Virovets and M. Scheer, Chem. - Eur. J., 2017, 23, 11423–11429 CrossRef CAS PubMed; (d) O. Hegen, C. Marquardt, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2017, 56, 12783–12787 CrossRef CAS PubMed; (e) L. Tuscher, C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 10657–10661 CrossRef CAS PubMed; (f) M. Köster, A. Kreher and C. von Hänisch, Dalton Trans., 2018, 47, 7875–7878 RSC.
- (a) K. Hassler and U. Katzenbeisser, J. Organomet. Chem., 1990, 399, C18–C20 CrossRef CAS; (b) K. Hassler and S. Seidl, Monatsh. Chem., 1988, 119, 1241–1244 CrossRef CAS; (c) M. S. Balakrishna, P. Chandrasekaran and P. P. George, Coord. Chem. Rev., 2003, 241, 87–117 CrossRef CAS; (d) M. Driess, S. Block, M. Brym and M. T. Gamer, Angew. Chem., Int. Ed., 2006, 45, 2293–2296 CrossRef CAS PubMed; (e) M. Balmer and C. von Hänisch, Z. Anorg. Allg. Chem., 2018, 644, 1143–1148 CrossRef CAS; (f) A. R. Dahl, A. D. Norman, H. Shenav and R. Schaeffer, J. Am. Chem. Soc., 1975, 97, 6364–6370 CrossRef CAS.
- (a) M. Fan, R. T. Paine, E. N. Duesler and H. Nöth, Z. Anorg. Allg. Chem., 2006, 632, 2443–2446 CrossRef CAS; (b) S. Inoue and K. Leszczyńska, Angew. Chem., Int. Ed., 2012, 51, 8589–8593 CrossRef CAS PubMed; (c) R. J. Wilson, J. R. Jones and M. V. Bennett, Chem. Commun., 2013, 49, 5049–5051 RSC; (d) M. Kapitein, M. Balmer and C. von Hänisch, Phosphorus, Sulfur, Silicon Relat. Elem., 2016, 191, 641–644 CrossRef CAS; (e) D. Wendel, A. Porzelt, F. A. D. Herz, D. Sarkar, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 8134–8137 CrossRef CAS PubMed.
- T. Kajiwara, N. Takeda, T. Sasamori and N. Tokitoh, Organometallics, 2004, 23, 4723–4734 CrossRef CAS.
- M. Fernández-Millán, L. K. Allen, R. García-Rodríguez, A. D. Bond, M. E. G. Mosquera and D. S. Wright, Chem. Commun., 2016, 52, 5993–5996 RSC.
- A. Bücker, C. Wölper, G. Haberhauer and S. Schulz, Chem. Commun., 2022, 58, 9758–9761 RSC.
- S. Schneider and C. von Hänisch, Eur. J. Inorg. Chem., 2021, 4655–4660 CrossRef CAS.
- M. Balmer, M. Kapitein and C. von Hänisch, Dalton Trans., 2017, 46, 7074–7081 RSC.
- G. Dübek, D. Franz, C. Eisenhut, P. J. Altmann and S. Inoue, Dalton Trans., 2019, 48, 5756–5765 RSC.
- D. Franz, T. Szilvási, A. Pöthig and S. Inoue, Chem. - Eur. J., 2019, 25, 11036–11041 CrossRef CAS PubMed.
- (a) A. B. Burg and E. S. Kuljian, J. Am. Chem. Soc., 1950, 72, 3103–3107 CrossRef CAS; (b) J. E. Drake and J. Simpson, Inorg. Chem., 1967, 6, 1984–1986 CrossRef CAS; (c) J. E. Drake and J. Simpson, Chem. Commun., 1967, 249–250 RSC; (d) J. E. Drake and C. Riddle, J. Chem. Soc. A, 1968, 7, 1675–1678 RSC; (e) N. Goddard and J. E. Drake, J. Chem. Soc. A, 1969, 662–665 Search PubMed; (f) M. T. Ackermann, M. Seidl, F. Wen, M. J. Ferguson, A. Y. Timoshkin, E. Rivard and M. Scheer, Chem. – Eur. J., 2022, 28, e202103780 CrossRef CAS PubMed.
- (a) A. G. Norman, J. M. Olson, J. F. Geisz, H. R. Moutinho, A. Mason, M. M. Al-Jassim and S. M. Vernon, Appl. Phys. Lett., 1999, 74, 1382–1384 CrossRef CAS; (b) T. Watkins, A. V. G. Chizmeshya, L. Jiang, D. J. Smith, R. T. Beeler, G. Grzybowski, C. D. Poweleit, J. Menéndez and J. Kouvetakis, J. Am. Chem. Soc., 2011, 133, 16212–16218 CrossRef CAS PubMed; (c) R. Jia, T. Zhu, V. Bulović and E. A. Fitzgerald, J. Appl. Phys., 2018, 123, 175101 CrossRef; (d) T. K. Purkait, A. K. Swarnakar, G. B. de Los Reyes, F. A. Hegmann, E. Rivard and J. G. C. Veinot, Nanoscale, 2015, 7, 2241–2244 RSC; (e) A. A. Omaña, R. K. Green, R. Kobayashi, Y. He, E. R. Antoniuk, M. J. Ferguson, Y. Zhou, J. G. C. Veinot, T. Iwamoto, A. Brown and E. Rivard, Angew. Chem., Int. Ed., 2021, 60, 228–231 CrossRef PubMed; (f) M. M. D. Roy, A. A. Omaña, A. S. S. Wilson, M. S. Hill, S. Aldridge and E. Rivard, Chem. Rev., 2021, 121, 12784–12965 CrossRef CAS PubMed.
- (a) U. Vogel, A. Y. Timoshkin and M. Scheer, Angew. Chem., Int. Ed., 2001, 40, 4409–4412 CrossRef CAS; (b) U. Vogel, P. Hoemensch, K.-C. Schwan, A. Y. Timoshkin and M. Scheer, Chem. – Eur. J., 2003, 9, 515–519 CrossRef CAS PubMed.
- (a) K.-C. Schwan, A. Y. Timoskin, M. Zabel and M. Scheer, Chem. - Eur. J., 2006, 12, 4900–4908 CrossRef CAS PubMed; (b) C. Marquardt, A. Adolf, A. Stauber, M. Bodensteiner, A. V. Virovets, A. Y. Timoshkin and M. Scheer, Chem. – Eur. J., 2013, 19, 11887–11891 CrossRef CAS PubMed.
- C. Marquardt, T. Jurca, K.-C. Schwan, A. Stauber, A. V. Virovets, G. R. Whittell, I. Manners and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 13782–13786 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2219857, 2219858, 2219859 and 2219860. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06594k |
| This journal is © The Royal Society of Chemistry 2023 |