
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeyond the TPP+ “gold standard”: a new generation mitochondrial delivery vector based on extended PN frameworks†
How Chee
Ong
a,
João T. S.
Coimbra
 b,
Maria J.
Ramos
b,
Bengang
Xing
a,
Pedro A.
Fernandes
*b and
Felipe
García
*cd
b,
Maria J.
Ramos
b,
Bengang
Xing
a,
Pedro A.
Fernandes
*b and
Felipe
García
*cd
aSchool of Physical and Mathematical Sciences, Division of Chemistry and Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore
bLAQV/REQUIMTE, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre 687, s/n, 4169-007 Porto, Portugal. E-mail: pafernan@fc.up.pt
cDepartamento de Química Orgánica e Inorgánica, Facultad de Química, Universidad de Oviedo, Avda Julian Claveria 8, 33006, Asturias, Spain. E-mail: garciafelipe@uniovi.es
dSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: Felipe.Garcia@monash.edu
First published on 13th February 2023
Abstract
Mitochondrial targeting represents an attractive strategy for treating metabolic, degenerative and hyperproliferative diseases, since this organelle plays key roles in essential cellular functions. Triphenylphosphonium (TPP+) moieties – the current “gold standard” – have been widely used as mitochondrial targeting vectors for a wide range of molecular cargo. Recently, further optimisation of the TPP+ platform drew considerable interest as a way to enhance mitochondrial therapies. However, although the modification of this system appears promising, the core structure of the TPP+ moiety remains largely unchanged. Thus, this study explored the use of aminophosphonium (PN+) and phosphazenylphosphonium (PPN+) main group frameworks as novel mitochondrial delivery vectors. The PPN+ moiety was found to be a highly promising platform for this purpose, owing to its unique electronic properties and high lipophilicity. This has been demonstrated by the high mitochondrial accumulation of a PPN+-conjugated fluorophore relative to its TPP+-conjugated counterpart, and has been further supported by density functional theory and molecular dynamics calculations, highlighting the PPN+ moiety's unusual electronic properties. These results demonstrate the potential of novel phosphorus-nitrogen based frameworks as highly effective mitochondrial delivery vectors over traditional TPP+ vectors.
Introduction
The ability to deliver compounds selectively to the mitochondria is rapidly growing in importance due to the increasing relevance of this organelle in disease treatment.1–4 Mitochondria-selective accumulation of compounds can be accomplished by some cationic moieties commonly known as delocalised lipophilic cations (DLCs),5–8 and this ability is frequently attributed to their delocalised cationic charge, resulting in increased lipid membrane permeability and accumulation within the negatively-charged mitochondrial matrix in accordance with the Nernst equation.7,9 While there are a broad range of mitochondrial targeting species, from rhodamines, cyanines, mitochondrial-targeting peptides to thiophene-based vectors,8,10–13 triphenylphosphonium-based vectors are arguably the most widely used and have been applied for the delivery of numerous molecular cargoes as highlighted in the literature.14 Due to the success in applying triphenylphosphonium (TPP+) systems for mitochondrial delivery, there is an increasing interest in modifying the TPP+ systems for enhanced delivery.15–23Although they are often referred to as DLCs,6,24,25 in the context of TPP+ systems, the necessity of charge delocalisation for mitochondrial accumulation is ambiguous. In contrast with DLCs such as rhodamines and cyanines, TPP+ moieties do not typically contain polyaromatic or extended conjugated moieties. While studies show that modification of the TPP+ moiety can result in improvements in biological activity, few focus on the electronic distribution or diffusing the positive charge of the TPP+ moiety.26,27 Furthermore, several studies evaluating the biological properties of tricyclohexyl-phosphonium and triphenylphosphonium vectors revealed that both aromatic and aliphatic phosphonium moieties have similar efficacies in vitro.28–30 These studies call the necessity of delocalisation into question, and additional studies are hence imperative for the design of new and unconventional mitochondrial delivery frameworks, and to understand the necessary electronic properties for an effective mitochondrial delivery vector.
In this context, phosphazenylphosphonium (PPN+) cations are novel species with unusual charge distributions, which has never been explored a priori for mitochondria targeting (Fig. 1). They are isoelectronic with carbodiphosphoranes, and can be described with multiple canonical structures.31,32 The general structure of a PPN+ cation is best described as a dication bridged by a negatively-charged nitrogen bridge, stabilised by negative hyperconjugation of the nitrogen lone pair into the P–C σ* orbitals (Fig. 2A).33–36 Most notably, these species have found applications as weakly-coordinating cations, which are especially useful for stabilising reactive anions or increasing the solubility of inorganic anions in organic solvents.37,38 The latter is indicative of high lipophilicity, which can, in turn, increase lipid membrane permeability. Furthermore, PPN+ cations are among the largest weakly-coordinating cations, making them particularly promising as a mitochondrial delivery vector, as molecular volume was shown to be a useful parameter for this purpose.18,20,37
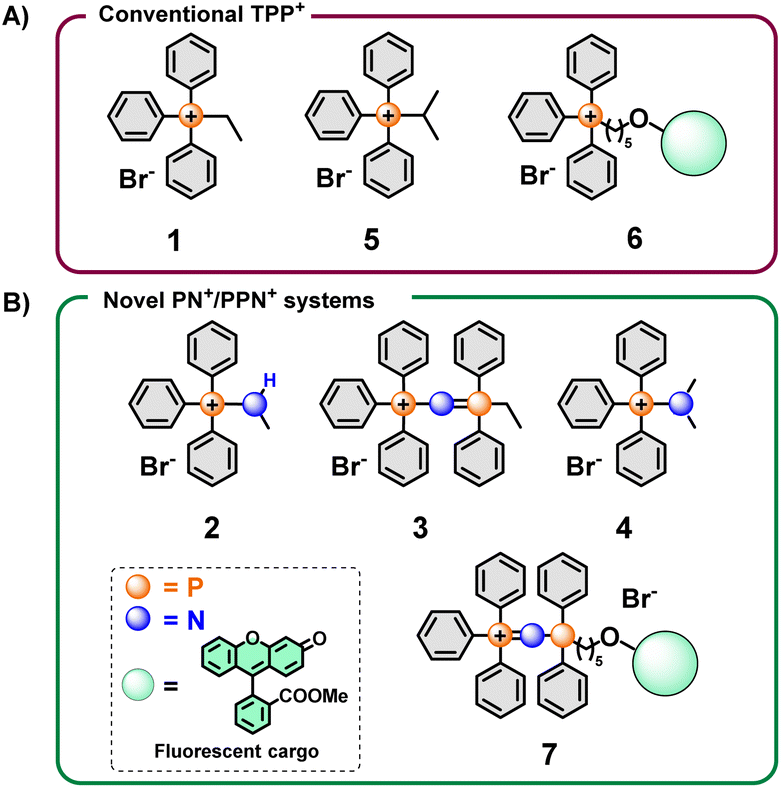 | ||
| Fig. 1 Design of conventional and non-conventional mitochondrial delivery vectors based on main group elements. (A) Conventional vector tested in our study. (B) Novel mitochondrial vectors designed and evaluated. | ||
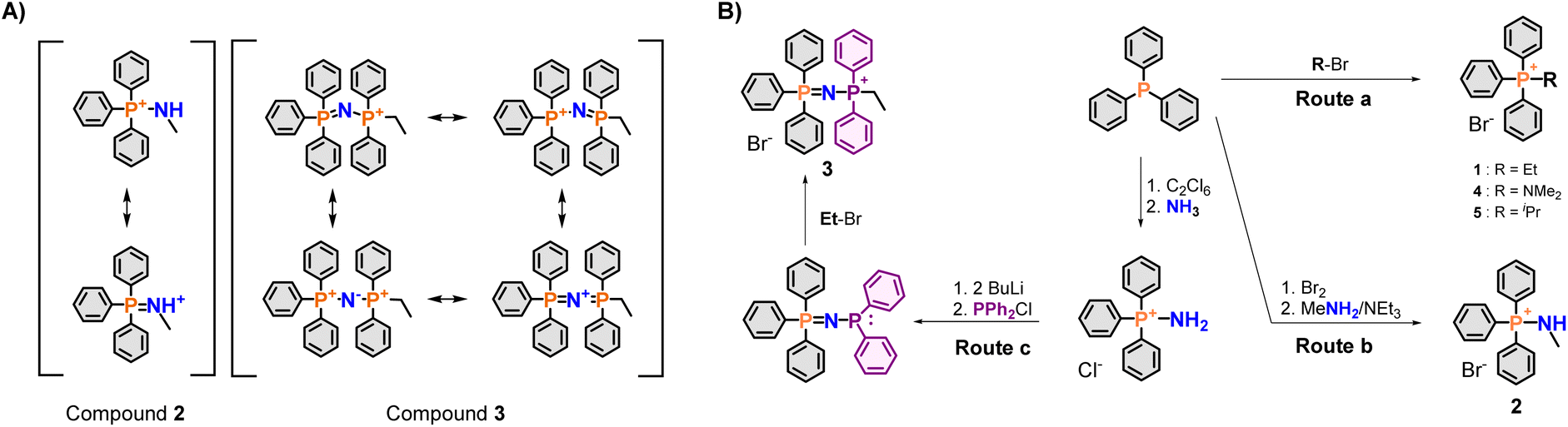 | ||
| Fig. 2 Electronic properties and synthesis of PPN+ mitochondrial delivery vectors. (A) Canonical structures of aminophosphonium (PN+) (2) and phosphazenyl phosphonium (PPN+) (3) cations, left and right, respectively. (B) Synthesis of compounds 1–5. | ||
Similarly, aminophosphonium salts (PN+) (Fig. 1 and 2A), which could be seen as a nitrogen analogue of the traditional alkyl TPP+ (or truncated PPN+) salts, are potentially viable alternative mitochondria-targeting agents due to their increased charge delocalisation from hyperconjugation.39 Furthermore, comparing these species with PPN+ moieties could reveal further insights into both the electronic and steric effects resulting from extending the number of heteroatoms present.
Overall, considering that the basic framework of the TPP+ vector for mitochondrial targeting has remained essentially unchanged since inception, and that modifications of the TPP+ vector has produced improved biological properties, there is considerable value in exploring a wider range of novel alternatives. Herein we present the synthesis and characterisation of PN+ and PPN+ salts, and their evaluation as improved novel mitochondrial delivery vectors. The enhanced ability of the PPN+ moiety to deliver a fluorescent cargo selectively to the mitochondria was also demonstrated and was shown to be superior to the “gold standard” TPP+ moiety. The main reason for this behaviour is attributed to the more extensive charge delocalisation of the PPN+ salts, as demonstrated here through the calculation of electrostatic potential surface (ESP) maps for the studied compounds. This work seeks to provide insights into the effects of charge delocalisation on the physical and biological properties of these compounds, as well as demonstrate the efficacy of the PPN+ framework as a powerful tool for mitochondrial delivery.
Results and discussion
Synthesis and characterisation
Initial screening of the uptake was conducted using the new phosphonium vectors with a short alkyl chain as a model compound (Fig. 1, compounds 1–3). The synthetic pathway is summarised in Fig. 2B (routes a–c). The synthesis of the TPP+ control (1) was achieved by reacting triphenylphosphine with ethyl bromide (route a).20 The aminotriphenyl-phosphonium vector (2) was synthesised through the bromination of triphenylphosphine, followed by treatment with methylamine in the presence of triethylamine (route b).40 The PPN+ vector (3) was synthesised via a two-step process (route c): (1) bromination of triphenylphosphine followed by bubbling ammonia through the mixture to obtain aminotriphenyl-phosphonium bromide, (2) double deprotonation of the amino protons using n-butyllithium, treatment with diphenylchloro-phosphine and ethyl bromide.41 All three compounds were characterised via NMR spectroscopy (1H, 13C, 31P{1H}), HRMS and single-crystal XRD studies. The NMR and HRMS spectra were consistent with the proposed structures (see ESI†).The X-ray solid-state structures obtained present a shortened P–N bond length in both 2 and 3 – 1.625 and 1.582 Å, respectively – which are shorter than the typical phosphazane P–N.39 Notably, the bond angle of the P–N–C fragment in 2 was 119.22° and closely matched the expected sp2 geometry. Compound 3 showed a bent P–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P geometry, with an increased bond angle of 139.70°, consistent with known compounds with a P–NP backbone. Interestingly, despite the co-crystallization of compound 3 with a water molecule, there were no short contacts observed between the water molecule and the P–NP backbone, highlighting the weakly-coordinating nature of the cation.
P geometry, with an increased bond angle of 139.70°, consistent with known compounds with a P–NP backbone. Interestingly, despite the co-crystallization of compound 3 with a water molecule, there were no short contacts observed between the water molecule and the P–NP backbone, highlighting the weakly-coordinating nature of the cation.
The DFT optimised structures of compounds 2 and 3 for the subsequent computational studies, showed that the P–N bond length was of 1.65 Å and 1.60 Å, respectively. In addition, the P–N–C and P–NP angles were 126.21° and 132.88°. Furthermore, an NBO-charge analysis on compound 3, supported a dication bridged by a negatively charged nitrogen – with P and N atomic charges of 1.862 a.u. and −1.462 a.u., respectively. These results were thus in line with the experimental characterisation of compounds 2 and 3.
Cytotoxicity and lipophilicity
Due to the high accumulation of lipophilic cations within the mitochondria, DLCs induce mitochondrial membrane depolarisation at high concentrations – which causes cell death.7 While not a rigorous metric, cytotoxicity assays can serve as a surrogate to compare the relative in vitro mitochondrial accumulation of the mitochondrial vectors.18 Subsequent sections will thus be focused on the more promising mitochondrial vectors (vide infra).In this study, cytotoxicity assays using HeLa cells were carried out to obtain the 72 h IC50 values. The results obtained are summarised in Table 1. Relative to the TPP+ control, compound 2 showed an increase in the IC50 values from 16.15 μM to 46.36 μM, indicating poorer performance. On the other hand, compound 3 has a drastically lowered IC50 at 0.77 μM. To rationalise these findings, the water–octanol partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), was experimentally obtained via an HPLC method reported in the literature and summarised in Table 1.42 LogP, frequently used as a measure of lipophilicity, is a critical parameter in biological systems Quantitative Structure–Activity Relationship (QSAR) models for mitochondrial uptake and has been found to be particularly relevant in TPP+ systems.43–47
P), was experimentally obtained via an HPLC method reported in the literature and summarised in Table 1.42 LogP, frequently used as a measure of lipophilicity, is a critical parameter in biological systems Quantitative Structure–Activity Relationship (QSAR) models for mitochondrial uptake and has been found to be particularly relevant in TPP+ systems.43–47
| Compound | IC50 (±95% CI)/μM | logP (±SD) |
logPmem (±SD) |
V s,max/kcal mol−1 | SASA/nm2 | Volume/nm3 |
|---|---|---|---|---|---|---|
| 1 | 16.15 ± 1.29 | −1.36 ± 0.09 | −0.73 ± 0.01 | 96.15 | 5.381 ± 0.189 | 0.891 ± 0.026 |
| 2 | 46.36 ± 5.43 | −1.28 ± 0.01 | −0.70 ± 0.06 | 117.36 | 5.340 ± 0.196 | 0.877 ± 0.027 |
| 3 | 0.77 ± 0.27 | 0.507 ± 0.03 | −0.53 ± 0.20 | 80.20 | 7.312 ± 0.219 | 1.323 ± 0.033 |
| 4 | 6.90 ± 1.94 | −1.29 ± 0.194 | −0.74 ± 0.02 | 88.86 | 5.451 ± 0.185 | 0.912 ± 0.027 |
| 5 | 8.98 ± 1.52 | −1.28 ± 0.01 | −0.69 ± 0.07 | 94.79 | 5.475 ± 0.190 | 0.920 ± 0.027 |
According to QSAR models developed by Horobin et al., logP is required to fall between 0 to 5 for optimal mitochondrial uptake.48 It has been previously established that an increase in logP is generally well correlated with an increase in mitochondrial accumulation.15,18,20 Relative to compound 1 (logP = −1.36), – the current “gold-standard” – compound 2 had a marginally higher logP of −1.28, but displayed a much higher IC50, which goes against the expected trends. We attributed this discrepancy to the increased ion-pairing tendency of this compound (vide infra). Compound 3, on the other hand, had a logP of 0.51, which is within QSAR the range for optimal mitochondrial accumulation.48 This is consistent with the lowered IC50 observed, indicating that the PPN+ compounds are potentially highly efficient mitochondrial vectors.
The apparent discrepancy between IC50 and logP for compound 2 was attributed to the increased formation of an ion-pair for compound 2 due to the introduction of the solvent-exposed N–H hydrogen bond donor (HBD). logP measurements conducted in phosphate-buffered saline for 1 and 2 revealed an apparent increase in lipophilicity for both compounds (from −1.36 to −0.429 and −1.28 to 0.527, respectively, see Table S1†). The higher logP in a solution with increased salt concentration is expected due to the increased counterion concentration. However, the relative increase in lipophilicity was significantly larger in 2, indicating a stronger influence from the high salt concentrations. The increased tendency to form ion-pairs due to the presence of the solvent-exposed HBD moiety renders compound 2 as an inefficient mitochondrial delivery vector since the ion-pair is electrically neutral.
To eliminate the increased ion pairing potential from the N–H bond, two additional compounds were synthesised – an aminophosphonium salt with a tertiary amino group, together with the alkyl substituted counterpart as a control (i.e., –NMe2 and –CHMe2, compounds 4 and 5, respectively – see Fig. 1).49 The cytotoxicity assays revealed that the IC50 values for these two compounds are 6.90 μM and 8.98 μM, respectively, in line with our hypothesis that the presence of the –NH moiety in compound 2 is the key contributor to the anomalous logP/IC50 result – see Table 1. This is further supported by the single-crystal structures, with the presence of short contacts between the P/P–N moiety and the bromide counterion only observed in compound 2 (NH⋯Br− = 2.445 Å) and absent in all other compounds studied (compounds 1 and 3–5) – see Fig. 3 and ESI.† Although a slight improvement of IC50 is observed for compounds 4 and 5 with respect to 2, the differences are not sufficiently significant for subsequent biological studies. Therefore, further studies on PN+ system were not pursued.
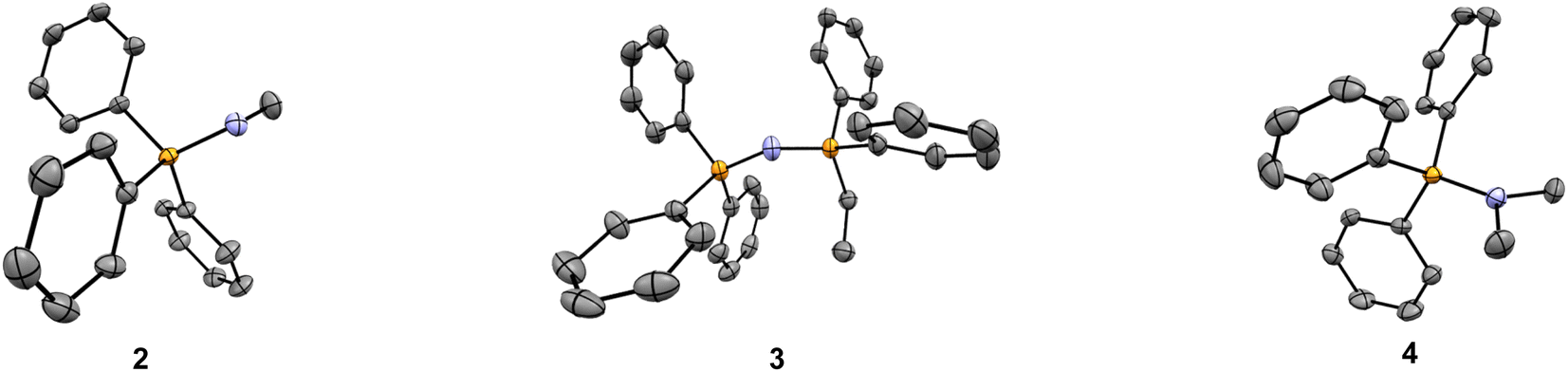 | ||
| Fig. 3 Molecular structures of the new main group molecular vectors developed. Solid-state structure of compounds 2–4. Hydrogen atoms, solvent molecules, and bromide counterions have been omitted for clarity. | ||
The evidence obtained during our quest for non-conventional mitochondrial delivery vectors highlights the limitations of lipophilicity as a parameter for mitochondrial accumulation when applied to DLCs. This limitation is especially pronounced when a large difference in hydrogen bonding ability is present between the series compounds studies, as logP measures the lipophilicity of the ion pair, while mitochondrial targeting ability depends largely on the cationic moiety.
Structural and computational studies
To further understand the influence of molecular modifications on electronic properties, charge distribution, and lipophilicity, we performed a molecular dynamics study and quantum mechanical calculations for compounds 1–5. In line with previous works, the molecular volume (Vol), solvent-accessible surface area (SASA), electrostatic surface potential (Vs), and membrane translocation free energy profiles were calculated.One of the most promising parameters for the studies series was the maximum of the molecular electrostatic potential surface of the cations, Vs,max – represented in Fig. 4. More positive Vs,max values have been related to stronger halogen/hydrogen bonding-anion intermolecular interactions.50 The charge distribution at the molecular surface (delocalisation and shielding) has also been related to the ability of DLCs to cross hydrophobic membranes.26 However, to the best of our knowledge, this parameter was never used to rationalise the mitochondrial uptake of these species.
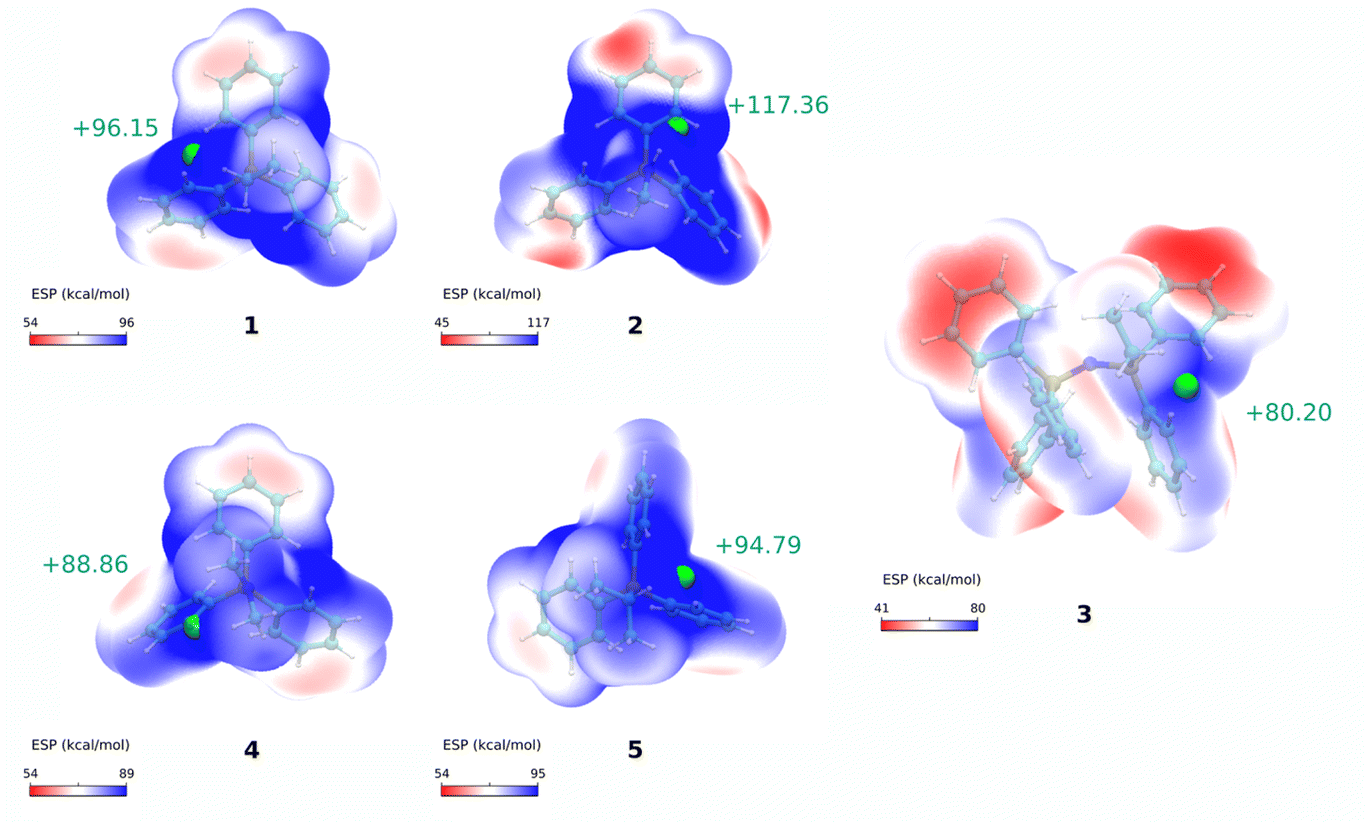 | ||
| Fig. 4 Electrostatic potential maps. Calculated electrostatic potential surfaces for cations 1–5, calculated at the ωB97XD/6-311++G(d,p) level of theory. Maximum ESP values, Vs,max, are shown as green spheres and their numerical value are reported next to them. | ||
When correlating Vs,max values with the IC50 results for compounds 1–5, a very significant linear correlation was observed (r2 = 0.95, see ESI, Fig. S3†). This could indicate that Vs,max might be used as a parameter of mitochondrial vector ability. We have then increased the compound dataset to include previously tested cations (including dications) from our previous reports (compounds 8–23 in Fig. S4 and Table S4†).15,18,20 This enlarged dataset made up a total of 21 cations (16 additional compounds) with a series of experimental correlations shown in Fig. S5.†
The IC50 and Vs,max results still display significative correlation, even though r2 was lower (r2 = 0.46, and r2 = 0.55 excluding the dications in Fig. S5a†). Still, the overall trend for the cations with similar molecular features seemed to indicate that those that presented lower IC50 had lower Vs,max. The lower, but significant r2 values, might result from several factors: (1) some of the compounds might be approaching the upper limit for the lipophilicity-linked toxicity, with negligible changes in IC50 over an order of magnitude of [TPP+];15 (2) the lack of data for cations with IC50 within the 50–150 μM range, and (3) by using a more diverse set of compounds, the correlation may become non-linear.
Nevertheless, it is evident that the Vs,max property is a valuable descriptor to differentiate compounds when charge delocalisation is significant (i.e., PN vs. PC systems). When delocalisation is not pronounced, or when approaching the lipophilicity-linked toxicity upper limit, accumulation properties may be better described by parameters based on molecular ion charge-to-volume ratios (i.e., charge density ratios, Z/Vol or Vol/Z).15,18
Thus, we have calculated a non-linear relationship between Vol/Z and IC50, which worked similarly to the logP vs. IC50 correlation (see Fig. S5b†). This similarity is consistent with the good linear correlation observed between logP and Vol/Z (r2 = 0.86, Fig. S5c†).
When Z/Vol is used instead, a good logarithmic correlation is observed with IC50. Furthermore, if our compound series (excluding the dications) is divided into two regions (IC50 < 3 μM and IC50 > 3 μM), it can be observed that for the first region the linear correlation is better for the Z/Vol parameter than for Vs,max (r2 = 0.71 vs. r2 = 0.47, respectively, see Fig. S5e†). However, when moving to the region of IC50 > 3 μM, the linear correlation is better with Vs,max (r2 = 0.92) than with Z/Vol (r2 = 0.31) – as shown in Fig. S5f.†
In addition, we studied the partition of cations 1–5 at a biological membrane model (100% POPC) using molecular dynamics simulations, to assess if in a more realistic water:membrane system than water:octanol, small differences in the lipophilicity and translocation of the cations could be captured, which could better correlate with the toxicity IC50 results. Fig. S1† presents the translocation free energy profiles of all five cations (1–5) across a water:POPC bilayer system. These profiles were then used to compute the membrane partition coefficients (logPmem) by calculating the standard binding free energy of the cations to the membrane (see ESI for the full details†).
The logPmem values gave similar trends to the experimental logP results (see Table 1). More specifically, we obtained a very similar membrane partition for the TPP+ (1) and PN+ (2) cations (−0.73 ± 0.01 and −0.70 ± 0.06, respectively), and a slightly higher membrane partition for the PPN+ (3) cation (−0.53 ± 0.20). As for the other two cations, 4 and 5, we have obtained very similar logPmem results to 1 and 2 (−0.74 ± 0.02 and −0.69 ± 0.07, respectively). We also analysed other parameters taken from the free energy profiles, such as the free energy barrier at the centre of the bilayer (ΔGB). However, none of these correlated linearly with the observed IC50 results (see ESI, Table S2 and Fig. S2†). These results corroborate the limitations of lipophilicity as a parameter for predicting mitochondrial toxicity when applied to our sets of TPP+ molecules.
We then evaluated electronic and molecular properties of the cations that have been related to aqueous solubility, more specifically the SASA, Vol, HOMO–LUMO orbitals, isotropic polarizability, and dipole moment.51 The electronic properties were calculated at the DFT level using the software Gaussian 09 (see ESI for the full details†). These properties were correlated with the lipophilicity and toxicity of the tested molecules, but again, they could not explain the toxicity IC50 results of the five cations. However, they correlated well with the experimental logP values available (compounds 1–5). These results are presented in Table S3 (see ESI†).
Finally, using a polarisable continuum solvent framework, we have assessed the propensity for ion-pair formation of two of the cations in the dataset (cations 1 and 2). In this regard, we have calculated the solvation free energy of the cations and the respective ion-pairs (with Cl−) in water and hexane (a model lipophilic liquid representing the inner part of the bilayer). – see Table S5 in the ESI.†
The calculations show that for the ion-pairs comprising compounds cations 1 and 2, the energy of transfer from water to n-hexane is 64 kJ mol−1 and 38 kJ mol−1, respectively. In contrast, the transfer free energy between water and n-hexane for both cations was approximately the same (49 kJ mol−1). Thus, ion pairing leads to better charge neutralisation, and less zwitterionic character in compound 2.
An additional analysis of the partial charges on the ion-pair was performed to examine the charge delocalisation over the cation–anion complex for compounds 1 and 2. It was also observed that the partial charge on the chloride anion was less negative for the cation 2·Cl− complex when compared with the 1·Cl− ion pair (see Fig. S6†). This supports a higher charge delocalisation of the DLC for the 2·Cl− ion-pair, which could decrease the energy of the ion-pair in the hydrophobic phase. A similar effect has previously been proposed for anion–triazole ion pair complexes.50 In addition, these results are in accordance with the previous previously discussed logP and IC50 experimental results observed for cation 2 (vide supra).
Confocal colocalization studies
To investigate the mitochondrial targeting ability of the PPN+ moiety, it was conjugated to a fluorescein derivative previously reported in the literature.15,52 However, to control regioselectivity and eliminate the protonophoric site, a methyl ester of fluorescein was employed to avoid mixtures of products. An analogous TPP+ variant was also synthesised as a control.The structures of the synthesised conjugates are shown in Fig. 1 (i.e., compounds 6 and 7). The logP of the dye conjugates were also measured experimentally (Table 2). As expected, the conjugation of the highly lipophilic PPN+ moiety resulted in a dye conjugate with a higher logP of 2.55, as compared to 0.417 in the TPP+ conjugate.
| Compound | Fluo/MT (±SD) | logP (±SD) |
Pearson's coefficient | M1 | M2 |
|---|---|---|---|---|---|
| 6 | 0.065 ± 0.05 | 0.417 ± 0.04 | 0.568 ± 0.108 | 0.954 ± 0.023 | 0.874 ± 0.013 |
| 7 | 0.666 ± 0.04 | 2.55 ± 0.10 | 0.887 ± 0.007 | 0.961 ± 0.011 | 0.971 ± 0.011 |
These fluorescein dyes were subsequently used together with MitoTracker DeepRed FM in colocalisation studies to verify the ability of the new vector to target the mitochondria, as well as to quantify the relative uptake of the new PPN+ vector. HeLa cells were treated the MitoTracker together with compound 6/7, and the images were examined under a confocal laser microscope (see ESI for Experimental details†).
The fluorescence intensity ratio between the two fluorophores (Fluo/MT) are calculated to compare the relative accumulation of the two compounds. The confocal microscopy images and Fluo/MT values are presented in Fig. 5 and Table 2 respectively. The images and Fluo/MT values evidently revealed that compound 7 had a much higher rate of accumulation. A high degree of colocalization was observed as well, indicating that 7 had localised within the mitochondria, as supported by the Van Steensel's cross correlation function, high Pearson's coefficient and Mander's coefficients (see Table 2 and ESI†). On the other hand, compound 6 had a low uptake with poorer colocalisation. This observation is consistent with other studies, where it was noted that an increase in the alkyl linker to 10 carbons was necessary for higher uptake for a similar fluorophore.52 The Fluo/MT was approximately 10-fold higher in 7, showing drastically improved performance of the PPN+ moiety in mitochondrial targeting compared to the traditional TPP+.
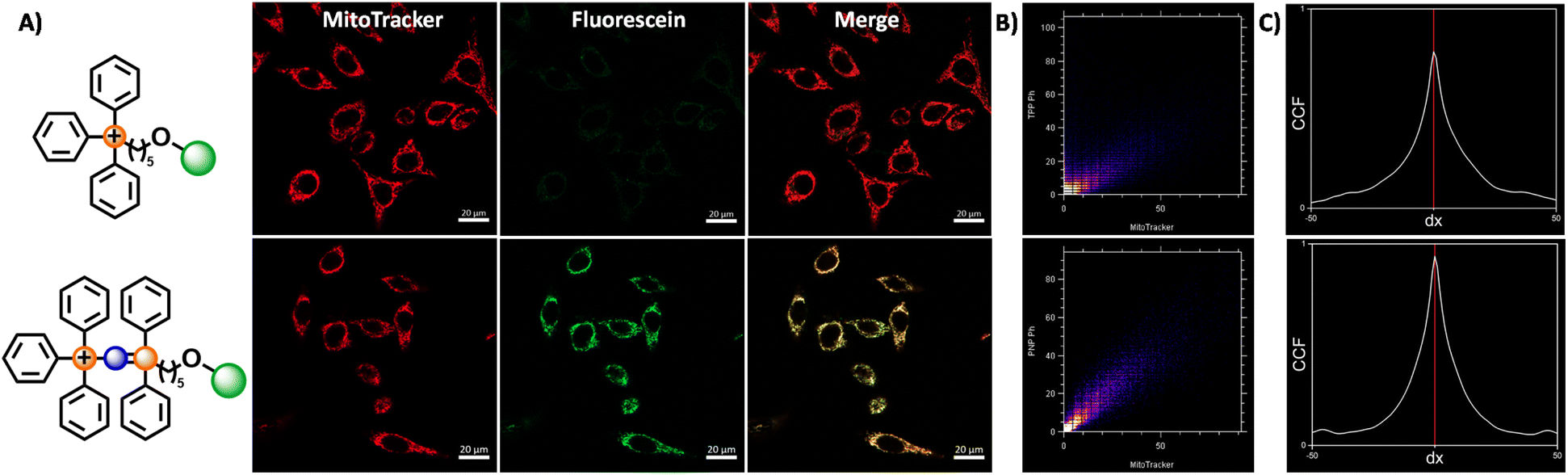 | ||
| Fig. 5 Fluorescence microscopy images of HeLa cells stained with MitoTracker and compound 6 and 7. (A) Confocal fluorescence microscopy images of HeLa cells treated with 6/7 (100 nM) and MitoTracker Deep Red FM (50 nM) for 60 minutes, upon excitation with 488 nm and 644 nm for fluorescein and Mitotracker channels respectively, with a total exposure time of 14.90 seconds. (B) Cytofluorogram for compound 6 (top) and 7 (bottom), with the fluorescence intensity of the MitoTracker and fluorescein channels represented by the x and y-axis respectively. (C) Selected Van Steensel's curves for compound 6 (top) and 7 (bottom). The cross-correlation function is maximal for a shift of dx = 0 for all images obtained (n = 5) for both compounds 6 and 7. | ||
Conclusions
In summary, we designed and synthesised aminophosphonium and phosphazenylphosphonium main group frameworks as enhanced mitochondrial targeting vectors. We have also demonstrated the first application of PPN+ compounds as viable mitochondrial delivery vectors through confocal imaging. These species were shown to be superior molecular vectors compared to the current ‘gold standard’, with a 10-fold increase in mitochondrial accumulation while maintaining high mitochondrial selectivity.Efforts were made to find new molecular parameters to adequately explain trends observed in cytotoxicity and lipophilicity, with the maximum of the molecular electrostatic potential surface of the cations, Vs,max showing good correlation for the newly species synthesised. This new parameter demonstrates the positive effect of charge delocalisation on mitochondrial accumulation.
Benchmarking against a wider range of molecular vectors in the future, may further broaden the applicability of the parameters discussed to other subsets of compounds. Thus, there is a need to study and expand the scope of modified TPP+ compounds as well as alternative delivery vectors – such as the ones described herein in the search for a universal mitochondrial accumulation predictive tool.
This work expands the scope of mitochondrial delivery vectors based on main group frameworks and underscores the need to explore non-conventional delivery vectors beyond the traditional TPP+ systems toward future enhanced mitochondrial therapies.
Author contributions
HCO: conceptualization, formal analysis, investigation, visualization, methodology, writing – original draft. writing – review & editing; JTSC: conceptualization, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing; MJR: funding acquisition, resources, supervision, writing – review & editing; BX: funding acquisition, resources; FG: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – original draft, writing – review & editing; PAF: conceptualization, funding acquisition, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. G. would like to thank A*STAR AME IRG (A1783c0003 and A2083c0050), NTU for a start-up grant (M4080552) and MOE Tier 1 grants (RG 11/15 and RG 113/16) for financial support. F. G. also thanks the support of Fundación para el Fomento en Asturias de la Investigación Científica Aplicada y la Tecnología (FICYT) through the Margarita Salas Senior Program (AYUD/2021/59709) and the Ministerio de Ciencia e Innovación through the project PID2021-127407NB-I00. F. G. would like to thank Monash University for affiliate position. We would like to also thank Nanyang Technological University for technical support. H. C. O. would like to thank NTU for NPGS scholarship. J. T. S. C., M. J. R. and P. A. F. acknowledge financial support from the Associate Laboratory for Green Chemistry Unit – LAQV@REQUIMTE, which received financial support from PT national funds (FCT/MCTES, Fundação para a Ciência e Tecnologia and Ministério da Ciência, Tecnologia e Ensino Superior) through the project UIDB/50006/2020 | UIDP/50006/2020. J. T. S. C. thanks FCT for funding through the Individual Call to Scientific Employment Stimulus (CEECIND/01374/2018).Notes and references
- M. P. Murphy and R. A. Smith, Adv. Drug Delivery Rev., 2000, 41, 235–250 CrossRef CAS PubMed.
- M. C. Frantz and P. Wipf, Environ. Mol. Mutagen., 2010, 51, 462–475 CAS.
- M. P. Murphy and R. C. Hartley, Nat. Rev. Drug Discovery, 2018, 17, 865–886 CrossRef CAS PubMed.
- B. Kalyanaraman, G. Cheng, M. Hardy, O. Ouari, M. Lopez, J. Joseph, J. Zielonka and M. B. Dwinell, Redox Biol., 2018, 14, 316–327 CrossRef CAS PubMed.
- V. R. Fantin, M. J. Berardi, L. Scorrano, S. J. Korsmeyer and P. Leder, Cancer Cells, 2002, 2, 29–42 CrossRef CAS PubMed.
- J. A. Jara, V. Castro-Castillo, J. Saavedra-Olavarría, L. Peredo, M. Pavanni, F. Jaña, M. E. Letelier, E. Parra, M. I. Becker, A. Morello, U. Kemmerling, J. D. Maya and J. Ferreira, J. Med. Chem., 2014, 57, 2440–2454 CrossRef CAS PubMed.
- J. S. Modica-Napolitano and J. R. Aprille, Adv. Drug Delivery Rev., 2001, 49, 63–70 CrossRef CAS PubMed.
- B. A. D. Neto, J. R. Corrêa and R. G. Silva, RSC Adv., 2013, 3, 5291–5301 RSC.
- M. P. Murphy and R. A. Smith, Annu. Rev. Pharmacol. Toxicol., 2007, 47, 629–656 CrossRef CAS PubMed.
- C. Ripoll, P. Herrero-Foncubierta, V. Puente-Muñoz, M. C. Gonzalez-Garcia, D. Miguel, S. Resa, J. M. Paredes, M. J. Ruedas-Rama, E. Garcia-Fernandez, M. Roldan, S. Rocha, H. D. Keersmaecker, J. Hofkens, M. Martin, J. M. Cuerva and A. Orte, Pharmaceutics, 2021, 13, 254 CrossRef CAS PubMed.
- A. Cossarizza, M. Baccaranicontri, G. Kalashnikova and C. Franceschi, Biochem. Biophys. Res. Commun., 1993, 197, 40–45 CrossRef CAS PubMed.
- L. V. Johnson, M. L. Walsh and L. B. Chen, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 990–994 CrossRef CAS PubMed.
- H. H. Szeto, AAPS J., 2006, 8, E277–E283 CrossRef CAS PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- H. C. Ong, J. T. S. Coimbra, G. Kwek, M. J. Ramos, B. Xing, P. A. Fernandes and F. García, RSC Chem. Biol., 2021, 2, 1643–1650 RSC.
- G. Schanne, L. Henry, H. C. Ong, A. Somogyi, K. Medjoubi, N. Delsuc, C. Policar, F. Garcia and H. C. Bertrand, Inorg. Chem. Front., 2021, 8, 3905–3915 RSC.
- A. J. Smith, B. E. Osborne, G. P. Keeling, P. J. Blower, R. Southworth and N. J. Long, Dalton Trans., 2020, 49, 1097–1106 RSC.
- H. C. Ong, Z. Hu, J. T. S. Coimbra, M. J. Ramos, O. L. Kon, B. Xing, E. K. L. Yeow, P. A. Fernandes and F. Garcia, Inorg. Chem., 2019, 58, 8293–8299 CrossRef CAS PubMed.
- N. Lalwani, D. W. Allen, P. N. Horton, S. J. Coles, N. A. Cross and N. Bricklebank, Polyhedron, 2019, 158, 515–523 CrossRef CAS.
- Z. Hu, Y. Sim, O. L. Kon, W. H. Ng, A. J. Ribeiro, M. J. Ramos, P. A. Fernandes, R. Ganguly, B. Xing, F. Garcia and E. K. Yeow, Bioconjugate Chem., 2017, 28, 590–599 CrossRef CAS PubMed.
- A. Haslop, L. Wells, A. Gee, C. Plisson and N. Long, Mol. Pharm., 2014, 11, 3818–3822 CrossRef CAS PubMed.
- L. Wells, A. Haslop, C. Coello, N. Keat, A. Gee, J. Passchier, N. Long and C. Plisson, J. Nucl. Med., 2014, 55, 121 CrossRef PubMed.
- D. E. Morrison, J. B. Aitken, M. D. de Jonge, J. A. Ioppolo, H. H. Harris and L. M. Rendina, Chem. Commun., 2014, 50, 2252–2254 RSC.
- M. Kardashinsky, N. Lengkeek and L. M. Rendina, J. Labelled Compd. Radiopharm., 2017, 60, 4–11 CrossRef CAS PubMed.
- C. Reily, T. Mitchell, B. K. Chacko, G. Benavides, M. P. Murphy and V. Darley-Usmar, Redox Biol., 2013, 1, 86–93 CrossRef CAS PubMed.
- E. L. Robb, J. M. Gawel, D. Aksentijevic, H. M. Cocheme, T. S. Stewart, M. M. Shchepinova, H. Qiang, T. A. Prime, T. P. Bright, A. M. James, M. J. Shattock, H. M. Senn, R. C. Hartley and M. P. Murphy, Free Radical Biol. Med., 2015, 89, 883–894 CrossRef CAS PubMed.
- M. F. Ross, T. D. Ros, F. H. Blaikie, T. A. Prime, C. M. Porteous, I. Severina, V. P. Skulachev, H. G. Kjaergaard, R. A. Smith and M. P. Murphy, Biochem. J., 2006, 400, 199–208 CrossRef CAS PubMed.
- T. I. Rokitskaya, E. A. Kotova, V. B. Luzhkov, R. S. Kirsanov, E. V. Aleksandrova, G. A. Korshunova, V. N. Tashlitsky and Y. N. Antonenko, Biochim. Biophys. Acta, Biomembr., 2021, 1863, 183483 CrossRef CAS PubMed.
- J. R. Luque-Ortega, P. Reuther, L. Rivas and C. Dardonville, J. Med. Chem., 2010, 53, 1788–1798 CrossRef CAS PubMed.
- P.-H. Lou, B. S. Hansen, P. H. Olsen, S. Tullin, M. P. Murphy and M. D. Brand, Biochem. J., 2007, 407, 129–140 CrossRef CAS PubMed.
- M. Gruber, W. Bauer, H. Maid, K. Schöll and R. R. Tykwinski, Inorg. Chim. Acta, 2017, 468, 152–158 CrossRef CAS.
- R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2006, 45, 8038–8042 CrossRef CAS PubMed.
- A. Widelöv, B. Folkesson and C. Andersson, Spectrosc. Lett., 1989, 22, 1101–1110 CrossRef.
- A. Bauzá, A. Frontera, T. J. Mooibroek and J. Reedijk, Crystengcomm, 2015, 17, 3768–3771 RSC.
- J. A. Dobado, H. Martínez-García, J. Molina and M. R. Sundberg, J. Am. Chem. Soc., 2000, 122, 1144–1149 CrossRef CAS.
- D. G. Gilheany, Chem. Rev., 1994, 94, 1339–1374 CrossRef CAS PubMed.
- L. Mann, E. Hornberger, S. Steinhauer and S. Riedel, Chem.–Eur. J., 2018, 24, 3902–3908 CrossRef CAS PubMed.
- F. J. Lalor and S. Chaona, J. Organomet. Chem., 1988, 344, 163–165 CrossRef CAS.
- L. Chiche and H. Christol, J. Chem. Soc., Perkin Trans. 2, 1984, 753–755 RSC.
- H. Zimmer and G. Singh, J. Org. Chem., 1963, 28, 483–486 CrossRef CAS.
- N. J. Rahier, J.-N. Volle, M. A. Lacour and M. Taillefer, Tetrahedron, 2008, 64, 6645–6650 CrossRef CAS.
- A. Andres, M. Roses, C. Rafols, E. Bosch, S. Espinosa, V. Segarra and J. M. Huerta, Eur. J. Pharm. Sci., 2015, 76, 181–191 CrossRef CAS PubMed.
- F. Sodano, B. Rolando, F. Spyrakis, M. Failla, L. Lazzarato, E. Gazzano, C. Riganti, R. Fruttero, A. Gasco and S. Sortino, ChemMedChem, 2018, 13, 1238–1245 CrossRef CAS PubMed.
- A. J. Smith, P. J. Gawne, M. T. Ma, P. J. Blower, R. Southworth and N. J. Long, Dalton Trans., 2018, 47, 15448–15457 RSC.
- S. P. Oldfield, M. D. Hall and J. A. Platts, J. Med. Chem., 2007, 50, 5227–5237 CrossRef CAS PubMed.
- R. W. Horobin, S. Trapp and V. Weissig, J. Controlled Release, 2007, 121, 125–136 CrossRef CAS PubMed.
- M. J. McKeage, S. J. Berners-Price, P. Galettis, R. J. Bowen, W. Brouwer, L. Ding, L. Zhuang and B. C. Baguley, Cancer Chemother. Pharmacol., 2000, 46, 343–350 CrossRef CAS PubMed.
- R. W. Horobin, Methods Mol. Biol., 2015, 1265, 13–23 CrossRef CAS PubMed.
- K. Fukui and R. Sudo, Bull. Chem. Soc. Jpn., 1970, 43, 1160–1163 CrossRef CAS.
- L. E. Bickerton, A. J. Sterling, P. D. Beer, F. Duarte and M. J. Langton, Chem. Sci., 2020, 11, 4722–4729 RSC.
- S. Md. A. Rauf, P. I. Arvidsson, F. Albericio, T. Govender, G. E. M. Maguire, H. G. Kruger and B. Honarparvar, Org. Biomol. Chem., 2015, 13, 9993–10006 RSC.
- Y. N. Antonenko, S. S. Denisov, D. N. Silachev, L. S. Khailova, S. S. Jankauskas, T. I. Rokitskaya, T. I. Danilina, E. A. Kotova, G. A. Korshunova, E. Y. Plotnikov and D. B. Zorov, Biochim. Biophys. Acta, 2016, 1860, 2463–2473 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, lipophilicity and cell viability studies, theoretical methods, and compound characterisations (i.e., NMR and mass spectra, and single crystal studies). CCDC 2084067, 2084071, 2084073 and 2084074. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06508h |
| This journal is © The Royal Society of Chemistry 2023 |