DOI:
10.1039/D2SC06442A
(Edge Article)
Chem. Sci., 2023,
14, 3247-3256
Mechanistic study of homoleptic trisamidolanthanide-catalyzed aldehyde and ketone hydroboration. Chemically non-innocent ligand participation†
Received
22nd November 2022
, Accepted 23rd February 2023
First published on 24th February 2023
Abstract
Carbonyl bond hydroboration is a valuable synthetic route to functionalized alcohols but relies on sometimes unselective and sluggish reagents. While rapid and selective aldehyde and ketone hydroboration mediated by trisamidolanthanide catalysts is known, the origin of the selectivity is not well-understood and is the subject of this contribution. Here the aldehyde and ketone HBpin hydroboration reaction mechanisms catalyzed by La[N(SiMe3)2]3 are investigated both experimentally and theoretically. The results support initial carbonyl oxygen coordination to the acidic La center, followed by intramolecular ligand-assisted hydroboration of the carbonyl moiety by bound HBpin. Interestingly, ketone hydroboration has a higher energetic barrier than that of aldehydes due to the increased steric encumbrance and decreased electrophilicity. Utilizing NMR spectroscopy and X-ray diffraction, a bidentate acylamino lanthanide complex associated with the aldehyde hydroboration is isolated and characterized, consistent with the relative reaction rates. Furthermore, an aminomonoboronate–lanthanide complex produced when the La catalyst is exposed to excess HBpin is isolated and characterized by X-ray diffraction, illuminating unusual aminomonoboronate coordination. These results shed new light on the origin of the catalytic activity patterns, reveal a unique ligand-assisted hydroboration pathway, and uncover previously unknown catalyst deactivation pathways.
Introduction
The selective reduction of unsaturated bonds in complex organic molecules is an essential tool for natural-product, pharmaceutical, and fine chemical synthesis.1–5 Furthermore, functional group tolerance and chemoselectivity are vital in reducing complex organic targets. Metal hydrides based on boron have become commonplace in organic synthesis due to their chemoselectivity and availability; however, they suffer from a limited substrate scope and sluggish reaction rates with sterically hindered substrates. To access a broader variety of substrates, more aggressive reducing agents, such as aluminum hydrides, can be employed but typically reduce various functional groups with minimal discrimination and incur significant safety hazards.6,7 The application of precious transition-metal catalysts has aided in the growth of hydroelementation research, but these often require complex ligands, hence high catalyst costs, limiting their applications.8–13 Therefore, the availability of milder, more selective, and earth-abundant catalytic reduction methodologies would greatly accelerate the pace of complex molecular synthesis and discovery with fewer safety concerns.14,15
Earth-abundant and readily available homogeneous catalysts have demonstrated exceptional competency for the hydroboration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bonds (X = C, N, O) while introducing minimally toxic metal ions.16–22 In this regard, lanthanides are promising as they are relatively non-toxic, inexpensive, earth-abundant, and due to the f orbital contraction, have large and flexible coordination geometries as well as substitutional lability while retaining a highly Lewis acidic character.23–26 Furthermore, lanthanide-organic catalysts have demonstrated significant advances in the hydroelementation of olefins, allenes, and other unsaturated functionalities.27–36 Regarding specific hydroelementation strategies, hydroboration has proven to be a valuable transformation, enabling the introduction of boron-based functional groups, which offer synthetic handles for subsequent modifications with an extensive scope.37 Lanthanide-organic catalysts are efficient and selective hydroboration catalysts and, regarding specific unsaturations, have proven proficient in the hydroboration of carbonyl functionalities.38–44 They lend themselves well to this transformation due to the oxophilic nature of the lanthanide ion, which aids in carbonyl coordination/activation. While the use of rare earth catalysts for the hydroboration of CX bonds has been on the rise, there has been limited mechanistic information which would provide valuable insight into reaction pathways and their potential manipulation in this emerging field.45–49 Quantum chemical studies of these catalytic reaction mechanisms would provide a fundamental understanding of the structures and energetics in question,50–54 and offer a predictive model to accelerate catalyst design while forecasting reaction outcomes and potential deactivation pathways.55,56
X bonds (X = C, N, O) while introducing minimally toxic metal ions.16–22 In this regard, lanthanides are promising as they are relatively non-toxic, inexpensive, earth-abundant, and due to the f orbital contraction, have large and flexible coordination geometries as well as substitutional lability while retaining a highly Lewis acidic character.23–26 Furthermore, lanthanide-organic catalysts have demonstrated significant advances in the hydroelementation of olefins, allenes, and other unsaturated functionalities.27–36 Regarding specific hydroelementation strategies, hydroboration has proven to be a valuable transformation, enabling the introduction of boron-based functional groups, which offer synthetic handles for subsequent modifications with an extensive scope.37 Lanthanide-organic catalysts are efficient and selective hydroboration catalysts and, regarding specific unsaturations, have proven proficient in the hydroboration of carbonyl functionalities.38–44 They lend themselves well to this transformation due to the oxophilic nature of the lanthanide ion, which aids in carbonyl coordination/activation. While the use of rare earth catalysts for the hydroboration of CX bonds has been on the rise, there has been limited mechanistic information which would provide valuable insight into reaction pathways and their potential manipulation in this emerging field.45–49 Quantum chemical studies of these catalytic reaction mechanisms would provide a fundamental understanding of the structures and energetics in question,50–54 and offer a predictive model to accelerate catalyst design while forecasting reaction outcomes and potential deactivation pathways.55,56
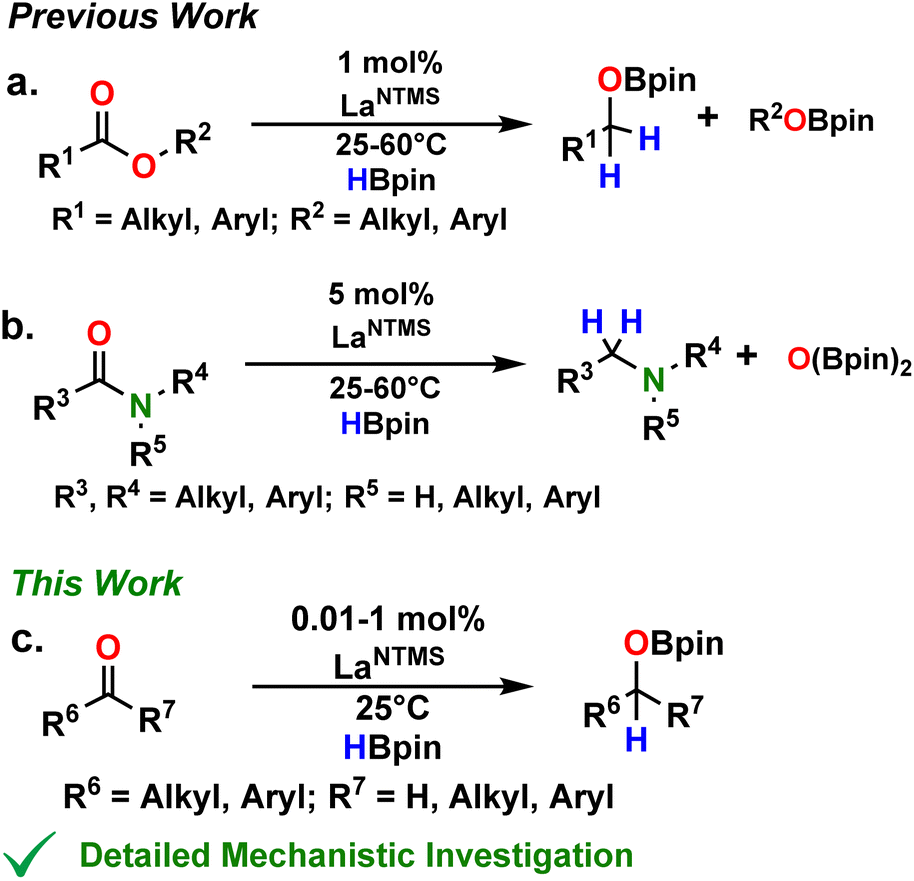
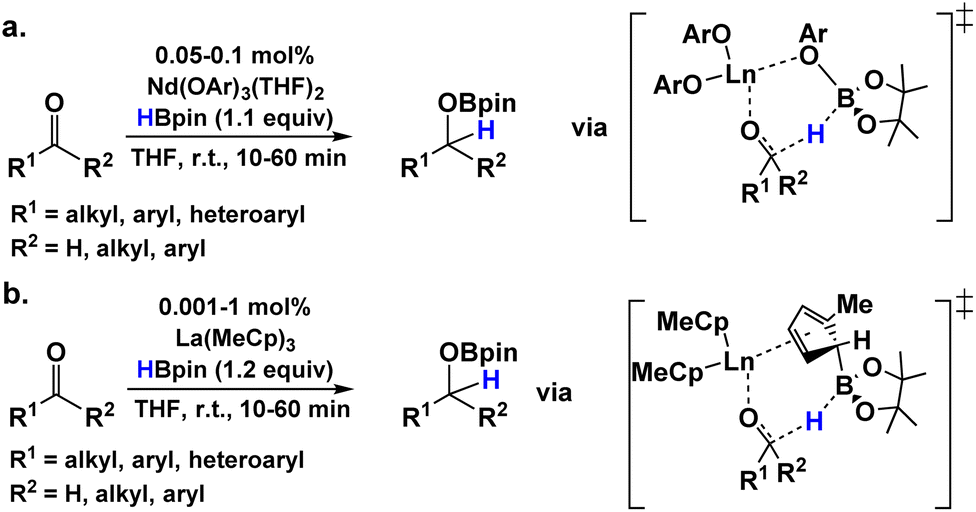
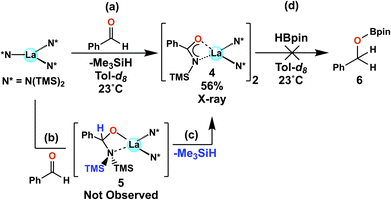
In recent reports from this Laboratory, the rapid and selective hydroboration of aldehydes, ketones, esters, and amides was achieved using homoleptic lanthanide trisamido precatalysts Ln[N(SiMe3)2]3 (LnNTMS) (TMS = SiMe3) and pinacolborane (HBpin) as the reducing agent (Fig. 1).42–44 In the case of esters and amides, kinetic, isotopic labeling, and preliminary theoretical studies were carried out to interrogate the reaction mechanisms (Fig. 1a and b). The DFT-derived mechanisms were in good agreement with the experimental observations. Interestingly, some of the key intermediate structures in the proposed mechanisms featured bidentate chelation of esters or amides with the lanthanide center. Note that such structural modes for aldehydes and ketones are rare.57 In the case of aldehydes and ketones, the room temperature hydroboration is significantly faster than for esters and amides, with TOFs as high as 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 with catalyst loadings in the range of 0.01–1 mol%. In aldehyde/ketone competition studies, aldehydes invariably exhibit higher activity toward hydroboration.
000 h−1 with catalyst loadings in the range of 0.01–1 mol%. In aldehyde/ketone competition studies, aldehydes invariably exhibit higher activity toward hydroboration.
 |
| | Fig. 1 Status of LaNTMS + HBpin catalytic hydroboration mechanistic understanding for the indicated carbonyl substrates.42–44 | |
Note that mechanistic studies were not carried out on the aldehyde and ketone hydroborations, and the proposed mechanism(s) were necessarily speculative. Understanding the reaction pathways of these effective methodologies should provide valuable information for designing future lanthanide catalysts with enhanced activity toward even more challenging substrates. Furthermore, proposed transition states in the ester43 and amide44 hydroborations featured a ligand-assisted HBpin-centered hydride transfer which raised the question of whether similar ligand involvement was necessary in the aldehyde and ketone mechanisms. While this type of interaction is well-documented in heteroleptic organometallic catalysis with redox non-innocent ligands, the concept of chemically non-innocent ligands is just emerging within the homogeneous homoleptic catalytic research community.58–60
In regard to the catalytic cycle(s) for LaNTMS-mediated aldehyde and ketone hydroboration with HBpin, here we report the observation of multiple significant intermediates and deactivated products which are characterized by multinuclear NMR spectroscopy and X-ray diffraction. Along with the results of relevant stoichiometric test reactions, these new insights expand the mechanistic understanding. Interestingly, one of the isolated solid complexes argues for the chemically non-innocent nature of the LnNTMS ligands. In addition, the DFT computed activation energetics for LaNTMS-mediated aldehyde, and ketone hydroborations are found to be in good agreement with the experimental kinetic data. Due to the lack of an additional substrate heteroatom to engage in bidentate La(III) chelation, it will be seen that these mechanisms differ from those previously identified in the deoxygenative reduction of amides and esters.43,44
In situ experimental mechanistic studies
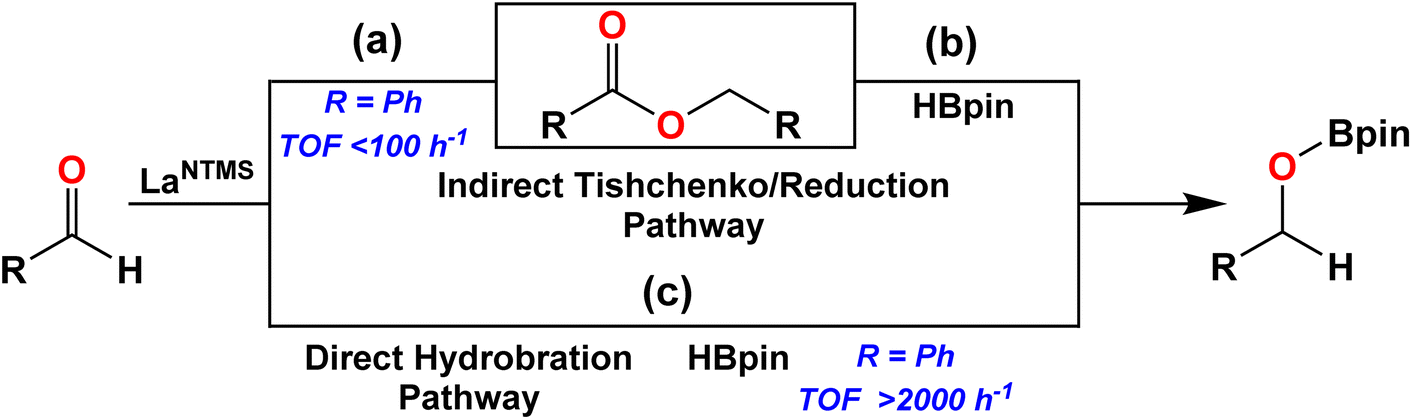
In pioneering work, LaNTMS, the solid-state structure of which was recently published,61 was previously reported to catalyze the Tishchenko reaction, producing esters from the cross-coupling of aldehydes at TOFs < 100 h−1 (Scheme 1, path (a)),62,63 and as noted above, LaNTMS is also an efficient ester hydroboration catalyst (Fig. 1c).43 Thus, a possible reaction pathway, specific for aldehydes, might include initial catalytic coupling mediated by LaNTMS (Scheme 1, path (a)), and then, in the presence of HBpin, LaNTMS would mediate ester reduction to yield the hydroborated aldehyde (Scheme 1, step (b)). To test this hypothesis, benzaldehyde was first reacted with 1 mol% of LaNTMS for 4 h at 25 °C, producing the benzyl benzoate Tishchenko product in 7.4% yield. In contrast, when 1 equiv. of HBpin is added to this reaction mixture (Scheme 1, step (b)) full conversion to the hydroborated product occurs within 1 h at 25 °C, while the Tishchenko product (benzyl benzoate) is still present (Fig. S37†). Note that efficient reduction of the benzyl benzoate requires a temperature of 60 °C.43 Due to the rapidity of the aldehyde hydroboration (TOF ∼2000 h−1),42 slow formation of the intermediate ester and subsequent HBpin reduction must therefore be a minor pathway. With this information in hand, a more in-depth NMR and crystallographic study of the actual hydroboration pathway (Scheme 1c) was next undertaken.
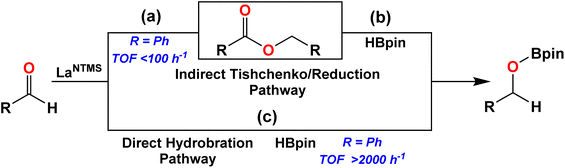 |
| | Scheme 1 Comparison of hydroboration and Tishchenko/reduction pathways. TOFs from ref. 41 and 62. | |
Stoichiometric 1H, 13C, 11B NMR, and crystallographic studies
To shed light on the present ketone and aldehyde hydroboration pathway(s), stoichiometric reactions of LaNTMS were first carried out with representative substrates to probe the catalyst–substrate interactions by in situ NMR spectroscopy and then by X-ray crystallography. In order to isolate and crystallize relevant structures and due to the very rapid reaction rates, we omitted some substrates from study (vide infra).
LaNTMS reactions with benzophenone.
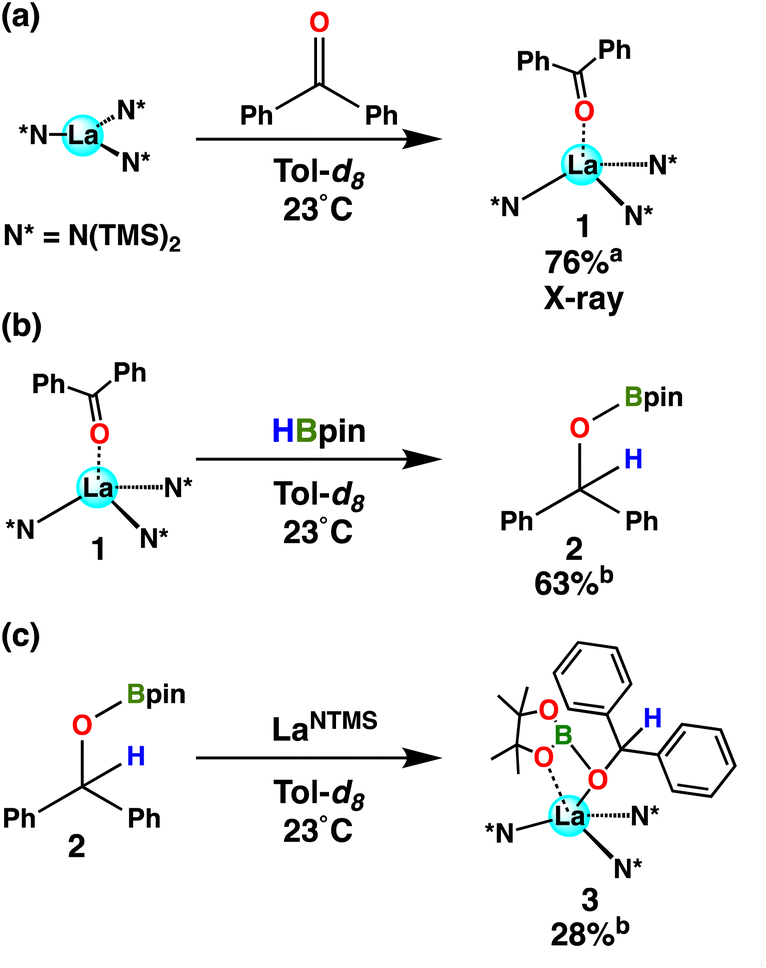
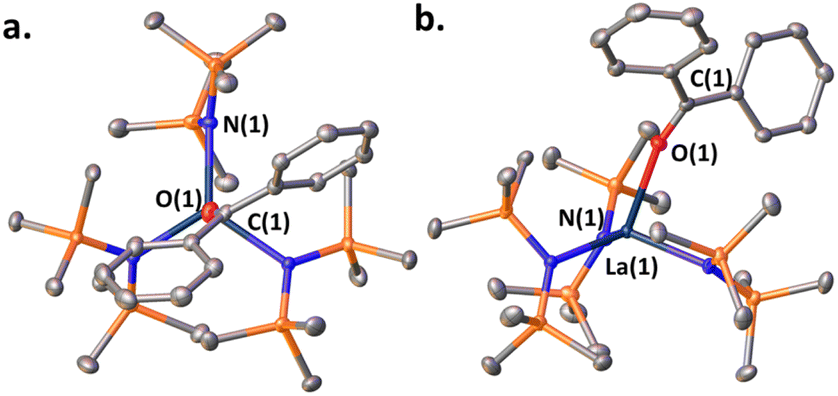
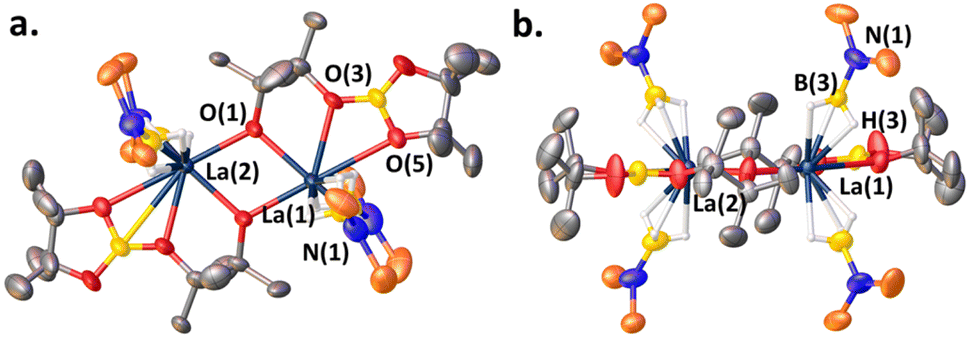
When a ketone (benzophenone is used for ease of crystallization) is exposed to LaNTMS (Scheme 2, eqn (a)) the carbonyl oxygen coordinates to the La center and forms complex 1, which was characterized by NMR spectroscopy, elemental analysis, and X-ray diffraction (Fig. 2). As expected, the C(1)–O(1)–La(1) angle is slightly bent (163.79°) (Fig. 2, top-view), and the La atom is located 0.63 Å above the centroid of the plane containing the N atoms of the three N(TMS)2 ligands with a torsion angle (N(3)–N(1)–N(2)–La(1)) of 25.58° (Fig. 2, side-view).
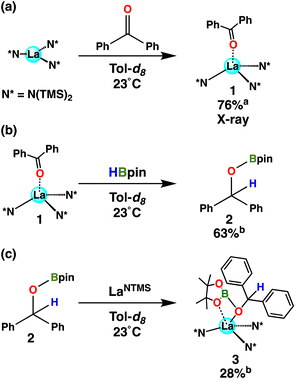 |
| | Scheme 2 Stoichiometric reaction between LaNTMS and benzophenone (eqn (a)), with the subsequent reaction of product 1 with HBpin to yield 2 (eqn (b)) and 3 (eqn (c)). a Isolated yield. b NMR yield determined using mesitylene as an internal standard. | |
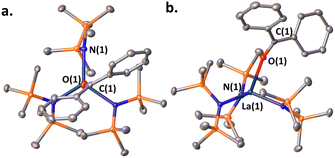 |
| | Fig. 2 (a) Top-view of the molecular structure of complex 1. (b) Side-view. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (deg): La(1)–O(1), 2.503(1); O(1)–C(1), 1.241(2); La(1)–N(1), 2.393(1); C(1)–O(1)–La(1), 163.79(12); N(3)–N(1)–N(2)–La(1), 25.58. | |
When 1 equiv. of HBpin is added to complex 1, a 63% yield of hydroborated product 2 is observed (Scheme 2, eqn (b)), supporting the contention that 1 is an intermediate in the catalytic cycle. Interestingly, according to NMR spectroscopy, the subsequent reaction of LaNTMS with 2 yields a complex formulate as structure 3 in 28% yield (Scheme 2, eqn (c)). The 11B NMR spectrum of 3 reveals a 25.7 ppm signal downfield of that in hydroborated product 2 (δ 22.9 ppm) by 2.86 ppm, presumably reflecting the product coordinating to electrophilic La center. Furthermore, the proton on the ketyl carbon in 3 is shifted upfield by 0.34 ppm to 5.98 ppm, presumably reflecting the β-oxygen coordination to the Lewis acidic La center. These observations were augmented by 2D NMR experiments (Fig. S19 and S20†), which reveal magnetically distinct proton signals from complexes 2 and 3 bound to the oxy-carbon atoms of their respective structures. The same effect is observed when comparing the aryl protons of benzophenone vs. complex 1 (see Fig. S1†). Compound 3 is proposed to be an intermediate in the hydroboration of ketones (vide infra).
LaNTMS reactions with benzaldehyde.
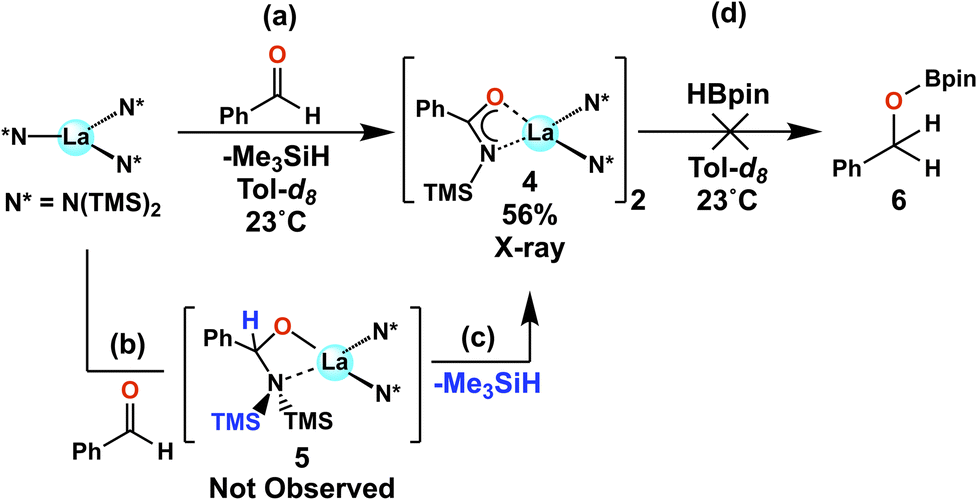
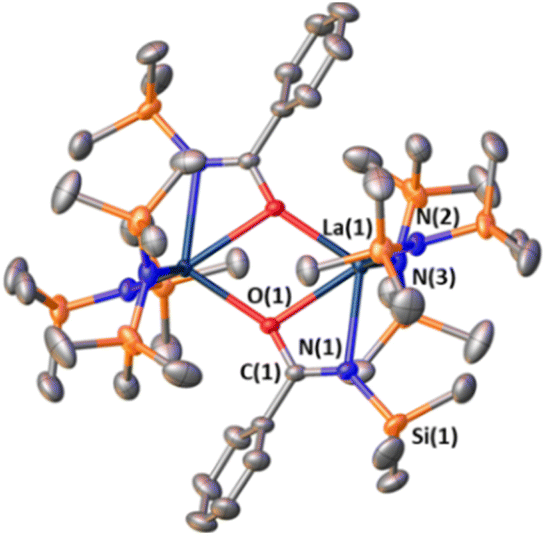
In an attempt to ascertain whether intermediates analogous to complexes 1 and/or 3 are involved in the aldehyde hydroborations, stoichiometric experiments were conducted with LaNTMS and benzaldehyde. Benzaldehyde was used for ease of crystallization. Surprisingly, instead of an expected aldehyde–La coordination complex, bidentate acylaminato–La complex 4 is observed by NMR together with Me3SiH formation (Scheme 3, step (a)). The structure of 4 is supported by 1D/2D NMR spectroscopy, elemental analysis, and X-ray crystallography. In the solid state, 4 crystallizes as a dimer (Fig. 3). The C(1)–N(1) = 1.290(5) Å, significantly shorted than C–N single bonds (∼1.47 Å)64 and slightly longer than CN double bonds (∼1.25 Å).64 The plane containing O(1), C(1), N(1), and La(1) has a dihedral angle of only 8.69°. The La(1)–N(1) distance is 2.591(3) Å which is significantly longer than that of La(1)–N(2), which is 2.370(3) Å, suggesting N(1) has adopted more of an imino character compared to that of the amido N(2). Complex 4 is best described as a bidentate acylaminolate anion bound to La, and the structure is similar to several previously reported amido–La complexes.65–67
 |
| | Scheme 3 Stoichiometric NMR scale reaction between LaNTMS and benzaldehyde, and between complex 4 and HBpin. Isolated yield reported for 4. | |
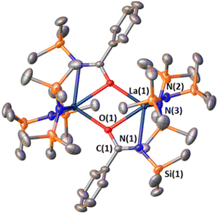 |
| | Fig. 3 Solid-state structure of dimeric complex 4. Hydrogens have been omitted for clarity. Selected bond distances (Å) and angles (deg): La(1)–N(1), 2.591(3); La(1)–O(1), 2.627(2), O(1)–C(1), 1.327(4); N(1)–C(1), 1.290(5); La(1)–N(2), 2.370(3); O(1)–La(1)–N(1), 51.06(8); N(1)–C(1)–O(1), 118.5(3); C(1)–N(1)–La(1), 95.503; O(1)–C(1)–N(1)–La(1), 8.688; N(3)–N(1)–N(2)–La(1), 30.852. | |
Complex 4 is presumably formed by the initial insertion of the aldehyde CO bond into the LaNTMS N–La bond to produce Intermediate 5 (Scheme 3, path (b)), which then undergoes rapid silane elimination to yield 4 (Scheme 3, step (c)). When HBpin is added to 4 (Scheme 3, step (d)), a complex mixture is obtained, which does not include the benzaldehyde (6) hydroboration product. Complex 4 is proposed to be a deactivated product produced when aldehydes but not HBpin are present, and this scenario is supported by DFT calculations (vide infra). Note that higher catalyst loadings were reported to be necessary for some aldehydes,42 and the formation of this deactivated product could be the cause. An interesting observation from the chemistry shown in Scheme 3 is that the N(TMS)2 ligand is clearly non-innocent and is capable of interacting with the substrate while remaining coordinated to the La center. This will be a key feature in the proposed reaction mechanism (vide infra).
LaNTMS reactions with HBpin.
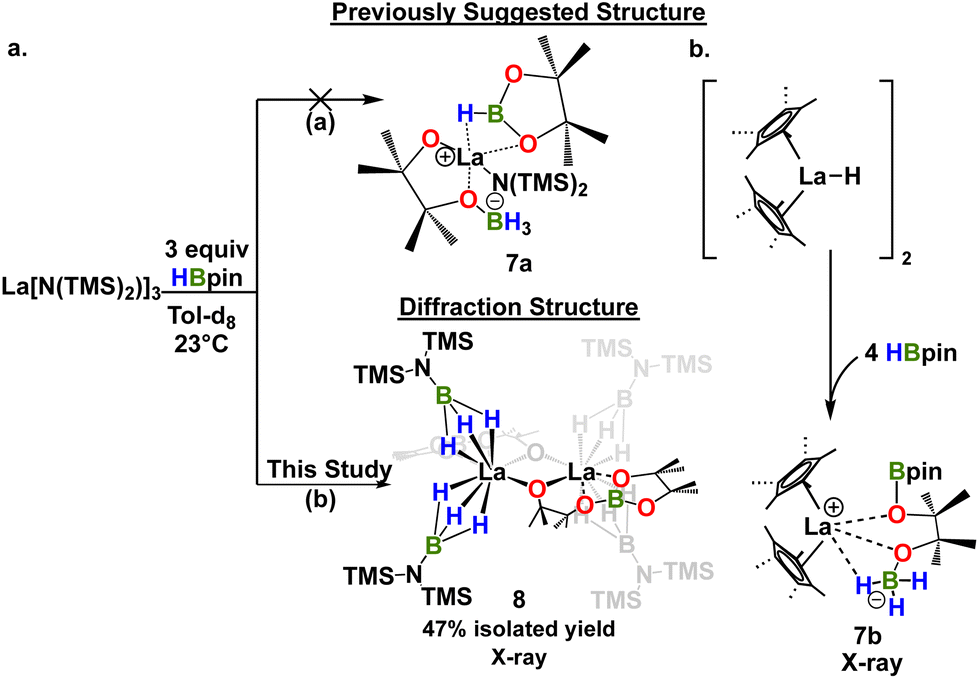
We next investigated the interaction of LaNTMS with HBpin. In initial work the NMR data for stoichiometric reactions of LaNTMS with excess HBpin suggested the possible formation of a zwitterionic deactivated product such as 7a (Scheme 4a, path (a)) by analogy to a similar Cp*2La-deactivated product characterized by NMR and X-ray diffraction23 (7b) (Scheme 4b).43 Here we isolated the LaNTMS + HBpin product and characterized it by multinuclear NMR and X-ray diffraction and suggest a plausible mechanism for its formation (Fig. S39†). The new data require reassigning the tentative structure to that of dimeric complex 8 (Scheme 4a, path (b)). The new NMR data are quite similar to those initially reported43 and those of the Cp*2La-23complex. Note that 3 equiv. of HBpin are required to fully convert LaNTMS (Fig. S14†). The solid-state structure of complex 8 can be described as a dimer with two La fragments bridged by alkoxide-like ligands. Each La ion is bound to a boronated pinacol alkoxy ligand, and two aminomonoboronate ligands coordinate to each La center in a κ3-H fashion. The aminomonoboronate coordination is essentially linear as La(1)–B(3)–N(1) is 177.85°, similar to an analogous actinide structure.68
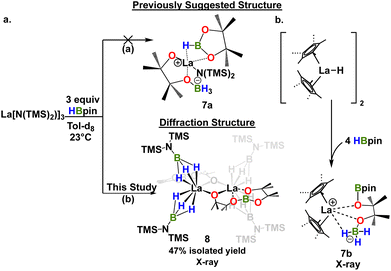 |
| | Scheme 4 (a) Proposed structure of deactivated product 8 based on NMR (ref. 42) and analogy to a diffraction-derived deactivated product in ref. 23 (a) and revised, diffraction-derived structure from this work ((b) and Fig. 4). (b) Synthesis and structure of relevant deactivated product lanthanide complex from ref. 23. | |
Note that the pinacolato oxygen in the Bpin fragment of 8 is coordinated to the La ion. This is the second known structure featuring this type of coordination between Bpin and a lanthanide; the other structure is a polyoxometalate La catalyst synthesized from stoichiometric studies similar to those reported here.69 In addition, this coordination motif has been proposed as an important interaction in other catalytic processes70–73 involving homoleptic lanthanide complexes,43,44,74,75 and is related to the reaction mechanism proposed here (vide infra). The second pinacolato oxygen of 8 bridges the two La centers in an approximately equatorial plane, having a torsion angle of only 4.34°. Ligand insertion has been observed with similar rare earth silylamide complexes.76,77
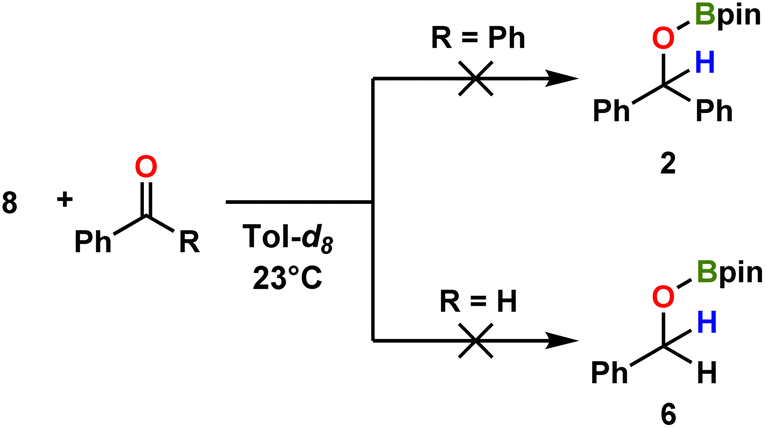
Regarding aminoboronate ligand utility, note that their f-element complexes have gained relevance as potential chemical vapor deposition and atomic layer deposition precursors, as ligands for the chemical separation of lanthanides,78 as volatile carriers for the use in brachytherapy,79 and for H2 storage.80 To the best of our knowledge, this is the first isolated structure of an amino-mono-boronate lanthanide complex. When 8 is reacted with benzophenone or benzaldehyde, the corresponding hydroborated products 2 or 6 are not obtained (Scheme 5), suggesting that 8 is not involved in the catalysis but is rather a deactivated side product.
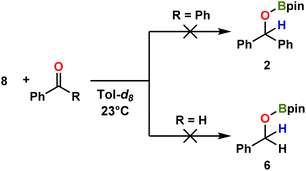 |
| | Scheme 5 Investigated catalytic reactions of benzophenone and benzaldehyde with complex 8, indicating negligible catalytic turnover. | |
DFT mechanistic analysis
With an improved understanding of how each substrate interacts with the LaNTMS catalyst, DFT analysis was next performed to better define the LaNTMS-catalyzed mechanisms of ketone and aldehyde hydroborations. The DFT results will be compared with the experimentally derived kinetic rate law and activation parameters, which for di-cyclohexyl ketone are rate = k[LaNTMS]1[HBpin]1[ketone]1, ΔH‡ = 17(1) kcal mol−1, and ΔS‡ = −15(2) e.u., and for cyclohexanecarboxaldehyde rate = k[LaNTMS]1[HBpin]0[aldehyde]0, ΔH‡ = 12(2) kcal mol−1, and ΔS‡ = −33(7) e.u.42 Calculations were carried out using the MO6/6-31G** (LANL2DZ) for the La level of theory. To validate the selected methodology, the structures of 1 and 4 were first computed and compared to the experimental X-ray structures. In both cases, the computed data are in excellent agreement with the experiment (Tables S4 and S5†).
Mechanism of catalytic ketone hydroboration
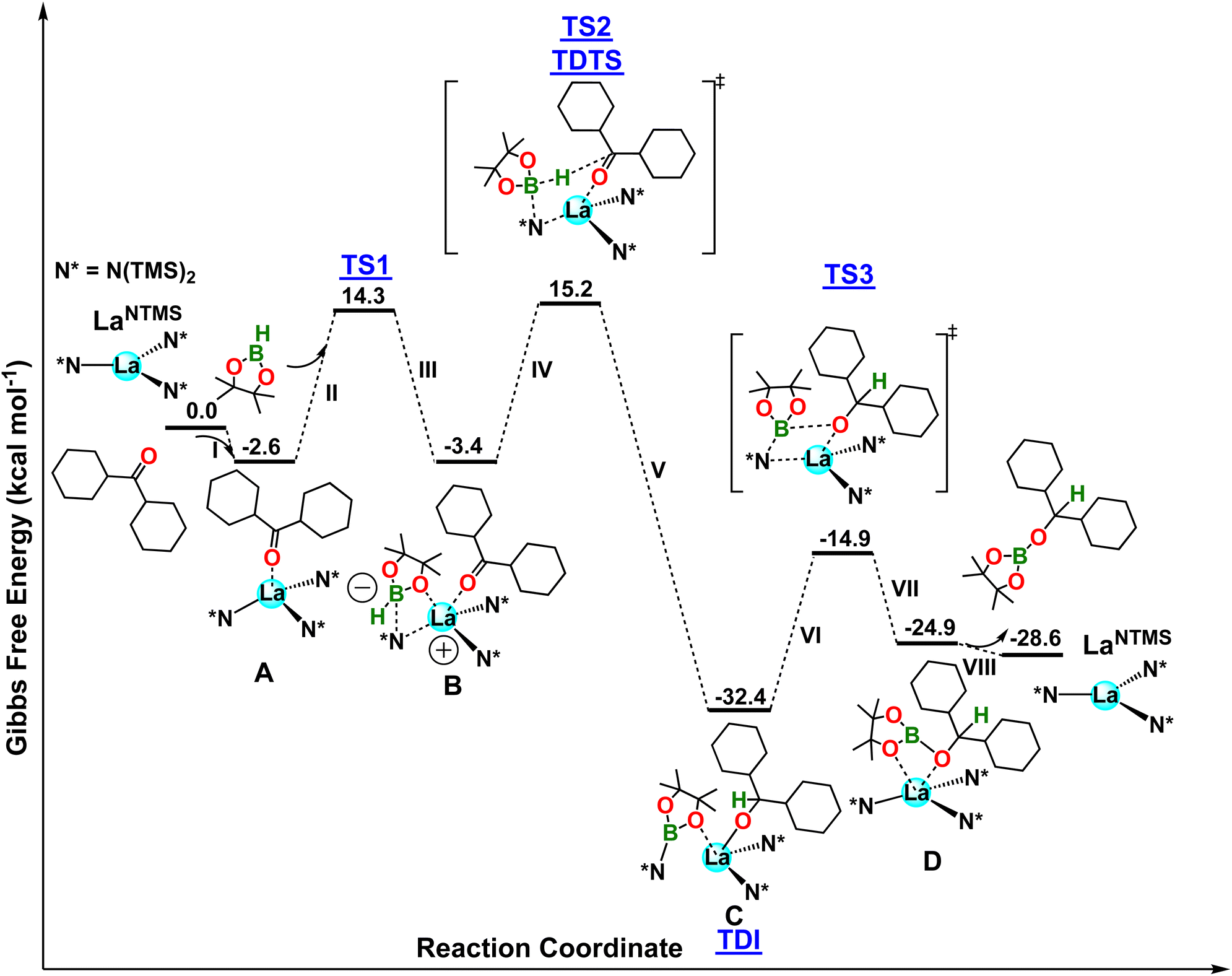
In the first step, the ketone carbonyl oxygen coordinates to the La center to form complex A in a slightly exergonic step (ΔG = −2.6 kcal mol−1; step I in Fig. 5). The formation of A is supported by solution phase NMR spectroscopy and the solid-state structure of complex 1 (Fig. 2). In addition, complex 1 releases the hydroborated product when HBpin is present, further supporting the involvement in the hydroboration pathway. Next, HBpin interacts with the La–N bond, yielding complex B (steps II and III). This step has a barrier of 16.9 kcal mol−1 (TS1) and is isoergonic by ΔG = −0.8 kcal mol−1. Note that the HBpin pinacolato oxygen coordinates to the acidic La center in a fashion similar to that in complex 8 (Fig. 4). Note also that the boron atom in B adopts a tetrahedral geometry with an additional N(TMS)2 ligand coordination. This yields a lanthanum center with a formal positive charge and boron with a formal negative charge. Next, the B–H hydride atom migrates to the electrophilic carbonyl carbon, yielding species C (steps IV and V), and reestablishing a neutral charge environment around the La as it loses coordination to an N(TMS)2 ligand (La–N distance = 4.38 Å). This latter step has a barrier of ΔG‡ = 18.6 kcal mol−1 (TS2) and is highly exergonic (ΔG = −29.0 kcal mol−1). While still coordinated to the La center, the bound pinB-N(TMS)2 moiety approaches the dicyclohexylmethoxide ligand, and an insertion into the La–O bond follows, yielding intermediate D with new B–O and La–N bonds. This step has a barrier of 17.5 kcal mol−1 (TS3) and is endergonic (ΔG = 7.5 kcal mol−1) (steps VI and VII in Fig. 5).43,44 In the final step, the hydroborated ketone is released from the catalyst (ΔG = −3.7 kcal mol−1), restoring the active LaNTMS catalyst (step VIII). The overall energetic span is 19.0 kcal mol−1 when considering C as the TDI species and TS2 as the TDTS species, in good agreement with the experiment. Moreover, a first order in both HBpin and ketone is expected from the location of TDI and TDTS, in good agreement with the experiments.
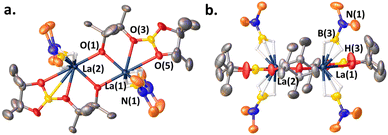 |
| | Fig. 4 (a) Top-view of the solid-state structure of complex 8. (b) Side-view. Hydrogens and carbons bound to Si have been omitted for clarity. See ESI† for the full structure. Selected bond distances (Å) and angles (deg): N(1)–B(3), 1.51(2); La(1)–B(3), 2.669; B(3)–H(3), 1.003; La(1)–H(3), 2.36(3); La(1)–O(5), 2.898(8); La(1)–O(3), 2.555(9); La(1)–O(1), 2.407(8); La(1)–B(3)–N(1), 177.9; La(1)–O(1)–La(2), 110.3(3); La(2)–La(1)–O(2), 35.47; La(1)–O(2)–O(1), 55.25; La(2)–La(1)–O(2)–O(1), 4.344. | |
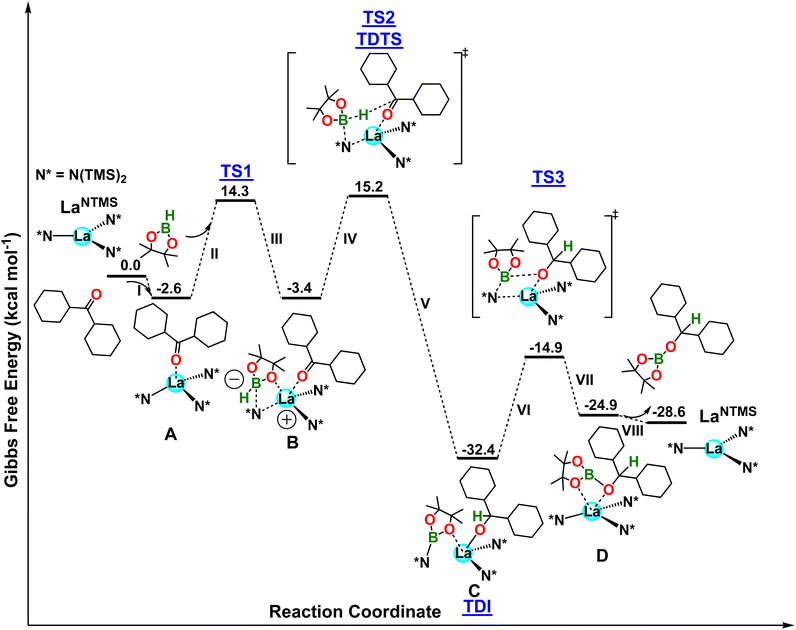 |
| | Fig. 5 DFT-derived Gibbs free energy profile for the LaNTMS-catalyzed hydroboration of a dicyclohexylketone with HBpin (kcal mol−1). | |
Mechanism of catalytic aldehyde hydroboration
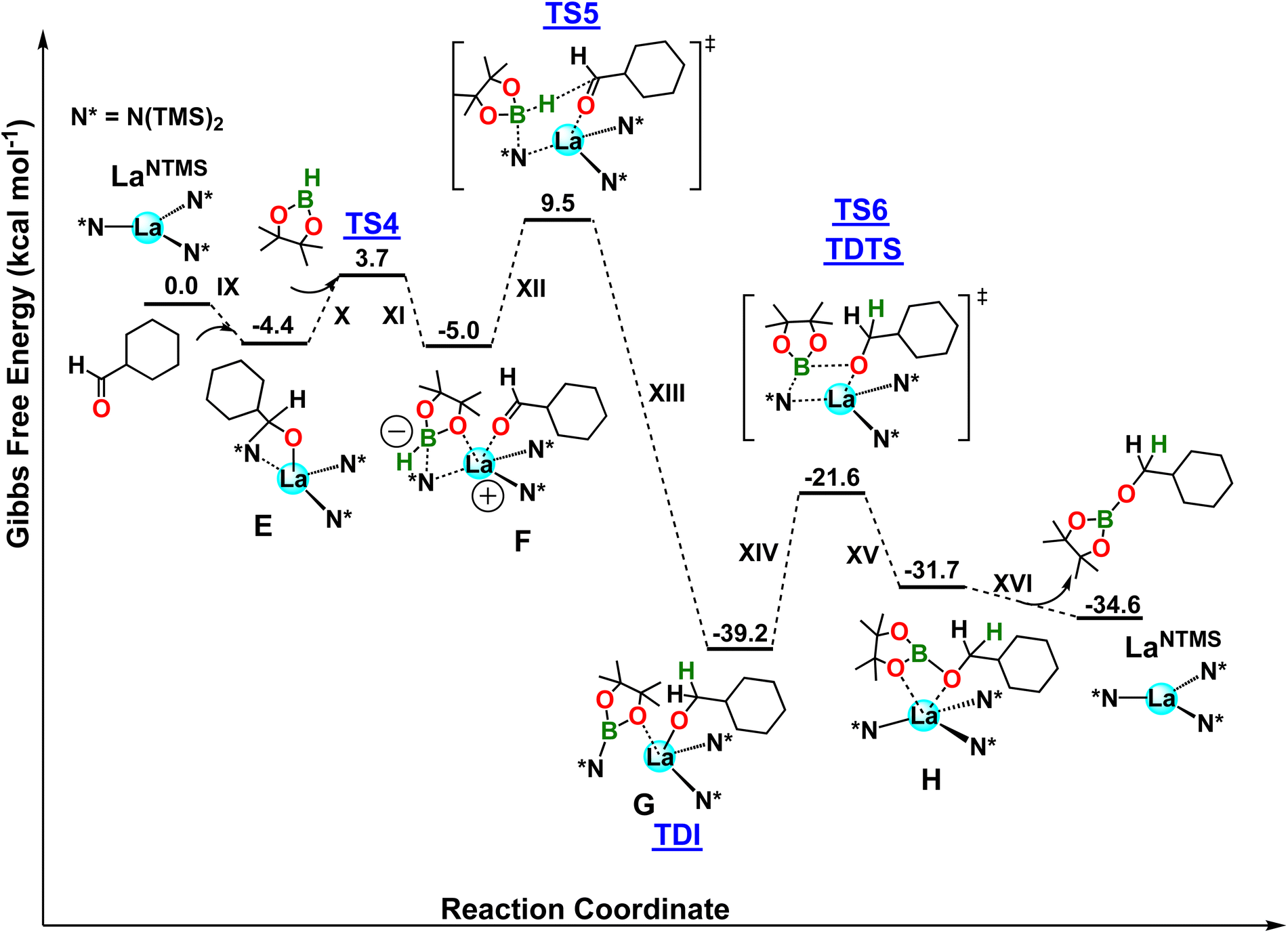
Next, the aldehyde hydroboration mechanism was investigated (Fig. 6). In the first step, the approach of aldehyde to the acidic LaNTMS center leads to complex E in a exergonic step (ΔG = −4.4 kcal mol−1; step IX, Fig. 6). This complex is reminiscent of species 5 hypothesized in Scheme 3 and it requires an energy barrier of 11.9 kcal mol−1 (not shown in Fig. 6). Once the substrate is coordinated to/activated by the La center, in a process similar to that in the ketone example, HBpin interacts with the La–N bond, forming structure F with a barrier of ΔG‡ = 8.1 kcal mol−1 (TS4; steps X and XI). Similar to the ketone mechanism, note the coordination of the pinacolato oxygen to the electrophilic La center, as seen in complex 8. The boron adopts a tetrahedral geometry with one N(TMS)2 ligand disassociating from the La center. Similar to the ketone pathway, the N(TMS)2 ligand is not innocent and creates a zwitterionic species with a formal positive charge on La and a formal negative charge on the boron atom. This process is isoergonic with a ΔG = −0.6 kcal mol−1. Next, the hydridic B⋯H− in F is delivered to the electrophilic carbonyl carbon yielding complex G which is the TDI of the proposed mechanism. This step has a barrier of 14.5 kcal mol−1 (TS5) and is highly exergonic by ΔG = −34.2 kcal mol−1 (G). Next, a metathesis-like step involves the pinB-N(TMS)2 group, which is not coordinated to the metal center (La–N distance = 4.37 Å), approaching the cyclohexanemethoxy ligand, forming complex H with a B–O bond, and subsequently dissociating the B–N bond. Complex H formation (steps XIV and XV) is endergonic (ΔG = 7.5 kcal mol−1) and is the turnover-determining step of the cycle with a ΔG‡ = 17.6 kcal mol−1 (TS6, TDTS). Finally, the hydroborated aldehyde is released from the catalyst (ΔG = −2.9 kcal mol−1), restoring the active LaNTMS catalyst (step XVI). The overall energetic span is 17.6 kcal mol−1 and is in favorable agreement with the experiment. Moreover, a zero order in both HBpin and ketone is expected from the location of TDI and TDTS, in good agreement with the experiments.
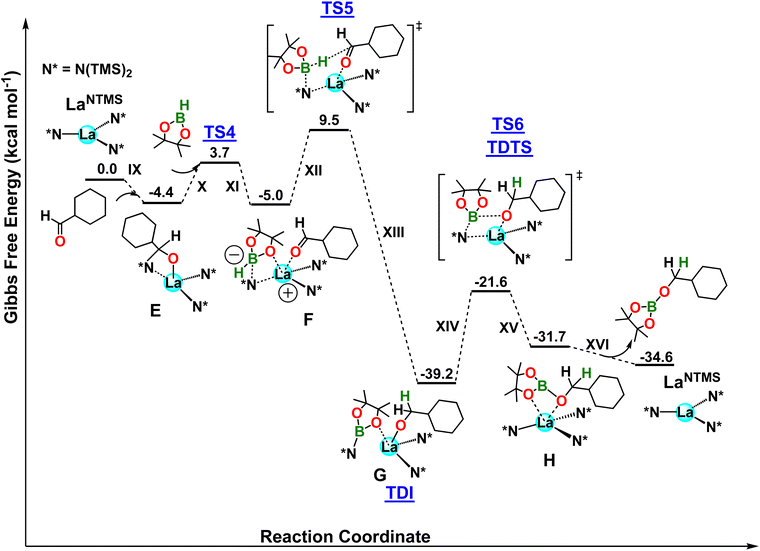 |
| | Fig. 6 DFT-derived Gibbs free energy profile for the LaNTMS-catalyzed hydroboration of cyclohexanecarboxaldehyde with HBpin (kcal mol−1). | |
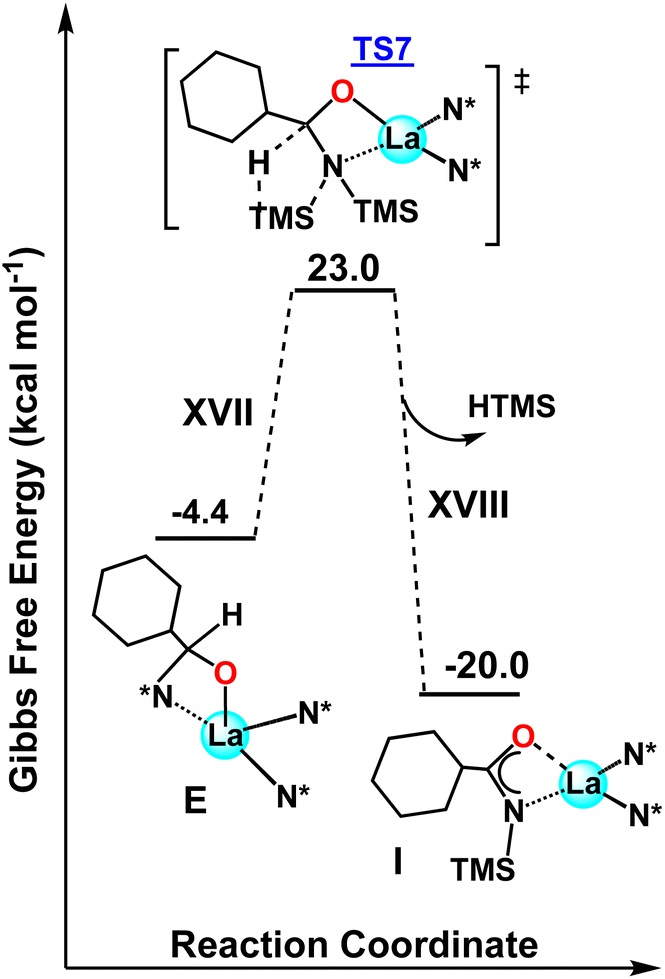
Next, the possible deactivation of the catalyst in the presence of aldehydes without HBpin was investigated. After initial aldehyde coordination to the La center (E), the H transfer between the aldehyde and a vicinal TMS group can occur, forming complex I (analogous to complex 4 in Scheme 3) with a computed barrier of ΔG‡ = 27.4 kcal mol−1 (Fig. 7). This step is exergonic (ΔG = −15.6 kcal mol−1). Note that this barrier is higher by 9.8 kcal mol−1 than when HBpin is present (Fig. 6, step XIV) and explains why, only when HBpin is absent or in not sufficient quantity, the catalysis could form complex 4. Despite the higher energetic barriers than when HBpin is present, this process of forming complex 4 from E could be the cause for the higher catalyst loadings required for some aldehydes.42 That being said, the hydroboration reaction stills dominate the product outcome based on both the theoretical and experimental data presented.
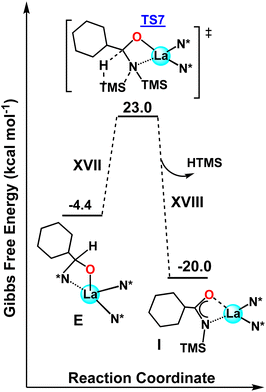 |
| | Fig. 7 DFT-derived energetic profile for the hydroboration process of aldehydes when HBpin is absent. | |
Discussion of hydroboration mechanisms
The hydroboration mechanisms for both ketones and aldehydes are found to be similar; however, they differ at the substrate coordination/activation step, which is more exothermic for ketones, while the turnover-determining step for ketones requires 1.4 kcal mol−1 more energy than the turnover-determining step for aldehydes. Thus, the overall energetic profile for dicyclohexyl ketone hydroboration lies at higher energy than that of the aldehydes likely due to the greater steric hindrance and decreased electrophilicity of the carbonyl associated with the ketone. The calculated rate-determining step is a H− transfer between HBpin and carbonyl group for the ketone and a σ-bond metathesis step for aldehyde. In addition, the differences between the hydroboration of ketones and aldehydes to the reaction patterns of the LaNTMS catalyzed reductions of esters43 and amides44 can be summarized as: (1) the LaNTMS catalyst does not lose an NTMS ligand during the hydroboration of ketones and aldehydes, while it does during the more demanding amide and ester reductions. (2) While both classes of reduction involve a ligand-assisted mechanism, the former class involves a NTMS ligand already bound to the initial catalyst, while the amide reduction invokes participation of HBpin, a substrate molecule, in the ligand assisting step of the proposed mechanism. (3) Amides and esters can chelate to the lanthanide in a bidentate fashion, as in the proposed amide deoxygenation process, whereas ketones and aldehydes rarely engage in bidentate coordination and are not involved in their reduction process. Overall, the energetic span of the hydroboration of ketones and aldehydes is far lower compared to the energy of the LaNTMS-catalyzed reductions of esters43 and amides.44 Moreover, other homoleptic lanthanide catalytic processes have been theorized to involve a ligand-assisted mechanism in the hydroboration/reduction of aldehydes (Fig. 8).39,74,75 In Bao et al.'s DFT-computed mechanisms for the hydroboration of aldehydes using either a lanthanide-alkoxide or MeCp catalyst, the ligand-assisted pathway was found to be significantly lower in energy than HBpin-Ln-coordinated carbonyl metathesis. Likewise, the experimental and theoretical data reported here support the chemical non-innocence of the amido ligands in the hydroboration of aldehydes and ketones.
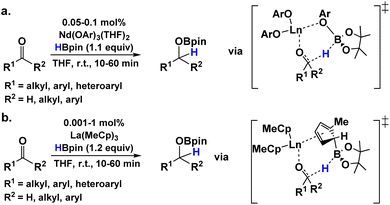 |
| | Fig. 8 Previously proposed ligand-assisted hydroboration mechanisms associated with homoleptic lanthanide-organic catalysts.74,75 | |
Conclusions
This study examined plausible reaction mechanisms for the rapid and selective LaNTMS-catalyzed hydroboration of ketones and aldehydes with HBpin. Several key intermediates and deactivated product structures were characterized via multinuclear NMR spectroscopy, elemental analysis, and X-ray crystallography, and thereby provide detailed comparison and contrast of the ketone and aldehyde catalytic cycles. It is found vital that the carbonyl oxygen of the ketones and aldehydes binds to the Lewis acidic La center to initiate both pathways, and the structure of the ketone-coordinated La complex 1 supports this claim. Moreover, identifying the amido ligand interaction with the substrate in the bidentate acylaminato–La complex 4 supports the catalytically important and chemically non-innocent nature of these –N(TMS)2 ligands as they interact with the substrate while remaining coordinated to the catalytic metal center. Furthermore, the formation of complex 4 may be the origin of the higher required catalyst loadings and lower reaction rates observed for the hydroboration of aldehydes versus that of ketones. In addition, the first aminomonoboronate bound to a lanthanide ion is reported (complex 8), which can alter hydridoborate chemistry in comparison to BH3-lanthanide complexes. This structure should inform the design of future aminomonoboronates and their potential applications. Finally, the DFT calculations identify ligand-assisted hydroboration mechanisms that are in excellent agreement with experiment. These results further support the growing interest in lanthanide-organic amido catalysts and the chemical non-innocence of these important ligands.
Data availability
Full experimental and computational details are provided as part of the ESI.†
Author contributions
T. J. M. and Y. K. supervised the study. J. O. R. carried out the synthetic work. A. M. carried out the computational studies. All authors analyzed the results and prepared the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the NSF CAT program for support under grant CHE-1856619 (J. O. R, T. J. M.). This work made use of the IMSERC NMR facility at Northwestern U., which received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), Int. Institute of Nanotechnology, the State of Illinois, and Northwestern U. Computational resources were provided by the Northwestern U. Quest High Performance Computing Cluster and CINECA award no. HP10CC5WSY 2020 under the ISCRA initiative. Financial support by the Office of Basic Energy Sciences, U.S. Department of Energy (DE-FG02-03ER15457) to the Institute for Catalysis in Energy Processes (ICEP) at Northwestern University (Y. K.). J. O. R. thanks Northwestern U. for an Academy Graduate Fellowship, Drs C. Barger, T. Lohr, and V. Weidner for helpful discussions, and C. Stern for crystallographic service.
References
- S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673–686 CrossRef CAS PubMed.
- M. Magre, M. Szewczyk and M. Rueping, Chem. Rev., 2022, 122, 8261–8312 CrossRef CAS PubMed.
- L. Mao and S. K. Bose, Adv. Synth. Catal., 2020, 362, 4174–4188 CrossRef CAS.
- H. S. Ban and H. Nakamura, Chem. Rec., 2015, 15, 616–635 CrossRef CAS PubMed.
- S. J. Baker, Y.-K. Zhang, T. Akama, A. Lau, H. Zhou, V. Hernandez, W. Mao, M. R. K. Alley, V. Sanders and J. J. Plattner, J. Med. Chem., 2006, 49, 4447–4450 CrossRef CAS PubMed.
- C. A. Merlic, C. J. Ferber and I. Schröder, ACS Chem. Health Saf., 2022, 29(4), 362–365 CrossRef CAS.
- A. K. Jaladi, W. K. Shin and D. K. An, RSC Adv., 2019, 9, 26483–26486 RSC.
- D. Männig and H. Nöth, Angew. Chem., Int. Ed., 1985, 24, 878–879 CrossRef.
- S. Rej, A. Das and T. K. Panda, Adv. Synth. Catal., 2021, 363, 4818–4840 CrossRef CAS.
- L. Zhang, Z. Zuo, X. Leng and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 2696–2700 CrossRef CAS PubMed.
- K. Utimoto, Pure Appl. Chem., 1983, 55, 1845–1852 CrossRef CAS.
- Z. Zhang, C. Liu, R. E. Kinder, X. Han, H. Qian and R. A. Widenhoefer, J. Am. Chem. Soc., 2006, 128, 9066–9073 CrossRef CAS PubMed.
- X. Li, A. R. Chianese, T. Vogel and R. H. Crabtree, Org. Lett., 2005, 7, 5437–5440 CrossRef CAS PubMed.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed.
- T. Ghatak, K. Makarov, N. Fridman and M. S. Eisen, Chem. Commun., 2018, 54, 11001–11004 RSC.
- M. L. Shegavi and S. K. Bose, Catal. Sci. Technol., 2019, 9, 3307–3336 RSC.
- C. Li, S. Song, Y. Li, C. Xu, Q. Luo, Y. Guo and X. Wang, Nat. Commun., 2021, 12, 3813 CrossRef CAS PubMed.
- F. E. McDonald and C. C. Schultz, J. Am. Chem. Soc., 1994, 116, 9363–9364 CrossRef CAS.
- Y. K. Jang, M. Magre and M. Rueping, Org. Lett., 2019, 21, 8349–8352 CrossRef CAS PubMed.
- S. R. Tamang and M. Findlater, J. Org. Chem., 2017, 82, 12857–12862 CrossRef CAS PubMed.
- N. Eedugurala, Z. Wang, U. Chaudhary, N. Nelson, K. Kandel, T. Kobayashi, I. I. Slowing, M. Pruski and A. D. Sadow, ACS Catal., 2015, 5, 7399–7414 CrossRef CAS.
- S. Bagherzadeh and N. P. Mankad, Chem. Commun., 2016, 52, 3844–3846 RSC.
- A. S. Dudnik, V. L. Weidner, A. Motta, M. Delferro and T. J. Marks, Nat. Chem., 2014, 6, 1100–1107 CrossRef CAS PubMed.
- P. Nuss and M. J. Eckelman, PLoS One, 2014, 9, e101298 CrossRef PubMed.
- K. K. Turekian and K. H. Wedepohl, Geol. Soc. Am. Bull., 1961, 72, 175–192 CrossRef CAS.
- J. O. Rothbaum, A. Motta, Y. Kratish and T. J. Marks, J. Am. Chem. Soc., 2022, 144, 17086–17096 CrossRef CAS PubMed.
- B. D. Stubbert and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 6149–6167 CrossRef CAS PubMed.
- Y. Horino and T. Livinghouse, Organometallics, 2004, 23, 12–14 CrossRef CAS.
- Y. Horino, T. Livinghouse and M. Stan, Synlett, 2004, 2004, 2639–2641 CrossRef.
- C. J. Weiss and T. J. Marks, Dalton Trans., 2010, 39, 6576–6588 RSC.
- X. Yu, S. Seo and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 7244–7245 CrossRef CAS PubMed.
- S. Seo, X. Yu and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 263–276 CrossRef CAS PubMed.
- A. M. Kawaoka, M. R. Douglass and T. J. Marks, Organometallics, 2003, 22, 4630–4632 CrossRef CAS.
- A. M. Kawaoka and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 6311–6324 CrossRef CAS PubMed.
- K. Komeyama, T. Kawabata, K. Takehira and K. Takaki, J. Org. Chem., 2005, 70, 7260–7266 CrossRef CAS PubMed.
- S. Seo and T. J. Marks, Chem. – Eur. J., 2010, 16, 5148–5162 CrossRef CAS PubMed.
- G. Zweifel and H. C. Brown, Org. React., 2004, 13, 1–54 Search PubMed.
- S. Patnaik and A. D. Sadow, Angew. Chem., Int. Ed., 2019, 58, 2505–2509 CrossRef CAS PubMed.
- R. D. Dicken, A. Motta and T. J. Marks, ACS Catal., 2021, 11, 2715–2734 CrossRef CAS.
- K. Nie, Y. Han, C. Wang and X. Cheng, Appl. Organomet. Chem., 2022, 36, e6570 CrossRef CAS.
- H. Liu and M. S. Eisen, Synthesis, 2020, 52, 629–644 CrossRef CAS.
- V. L. Weidner, C. J. Barger, M. Delferro, T. L. Lohr and T. J. Marks, ACS Catal., 2017, 7, 1244–1247 CrossRef CAS.
- C. J. Barger, A. Motta, V. L. Weidner, T. L. Lohr and T. J. Marks, ACS Catal., 2019, 9, 9015–9024 CrossRef CAS.
- C. J. Barger, R. D. Dicken, V. L. Weidner, A. Motta, T. L. Lohr and T. J. Marks, J. Am. Chem. Soc., 2020, 142, 8019–8028 CrossRef CAS PubMed.
- W. Wang, X. Shen, F. Zhao, H. Jiang, W. Yao, S. A. Pullarkat, L. Xu and M. Ma, J. Org. Chem., 2018, 83, 69–74 CrossRef CAS PubMed.
- X. Xu, Z. Kang, D. Yan and M. Xue, Chin. J. Chem., 2019, 37, 1142–1146 CrossRef CAS.
- G. Zhang, H. Zeng, J. Wu, Z. Yin, S. Zheng and J. C. Fettinger, Angew. Chem., Int. Ed., 2016, 128, 14581–14584 CrossRef.
- D. Mukherjee, S. Shirase, T. P. Spaniol, K. Mashima and J. Okuda, Chem. Commun., 2016, 52, 13155–13158 RSC.
- S. Bagherzadeh and N. P. Mankad, Chem. Commun., 2016, 52, 3844–3846 RSC.
- E. Nieto-Sepulveda, A. D. Bage, L. A. Evans, T. A. Hunt, A. G. Leach, S. P. Thomas and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2019, 141, 18600–18611 CrossRef CAS PubMed.
- S. De, S. Dutta and D. Koley, Organometallics, 2022, 41(15), 2132–2145 CrossRef CAS.
- A. Motta, I. L. Fragalà and T. J. Marks, Organometallics, 2010, 29, 2004–2012 CrossRef CAS.
- S. Tobisch, Chem. – Eur. J., 2010, 16, 4995–4998 CrossRef CAS PubMed.
- K. Chang, I. del Rosal, X. Zheng, L. Maron and X. Xu, Dalton Trans., 2021, 50, 7804–7809 RSC.
- A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, ACS Catal., 2020, 10, 13479–13486 CrossRef CAS.
- T. L. Lohr, Z. Li and T. J. Marks, Acc. Chem. Res., 2016, 49, 824–834 CrossRef CAS PubMed.
- S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2794 CrossRef CAS PubMed.
- R. G. Belli, V. C. Tafuri, M. V. Joannou and C. C. Roberts, ACS Catal., 2022, 12, 3094–3099 CrossRef CAS.
- R. G. Belli, V. C. Tafuri and C. C. Roberts, ACS Catal., 2022, 9430–9436, DOI:10.1021/acscatal.2c02785.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- L. Wursthorn, K. Beckett, J. O. Rothbaum, R. M. Cywar, C. Lincoln, Y. Kratish and T. J. Marks, Angew. Chem., Int. Ed., 2023, 62, e202212543 CrossRef CAS PubMed.
- M. R. Bürgstein, H. Berberich and P. W. Roesky, Chem. – Eur. J., 2001, 7, 3078–3085 CrossRef.
- H. Berberich and P. W. Roesky, Angew. Chem., Int. Ed., 1998, 37, 1569–1571 CrossRef PubMed.
-
W. M. Haynes, D. R. Lide and T. J. Bruno, CRC handbook of chemistry and physics, CRC press, 2016 Search PubMed.
- Y.-L. Wang, Y.-X. Zhou, L.-Q. Deng, Q.-S. Hu, X. Tao and Y.-Z. Shen, J. Organomet. Chem., 2016, 805, 77–86 CrossRef CAS.
- P. Zheng, J. Hong, R. Liu, Z. Zhang, Z. Pang, L. Weng and X. Zhou, Organometallics, 2010, 29, 1284–1289 CrossRef CAS.
- H. Ding, C. Lu, X. Hu, B. Zhao, B. Wu and Y. Yao, Synlett, 2013, 24, 1269–1274 CrossRef CAS.
- H. Braunschweig, A. Gackstatter, T. Kupfer, K. Radacki, S. Franke, K. Meyer, K. Fucke and M. H. Lemée-Cailleau, Inorg. Chem., 2015, 54, 8022–8028 CrossRef CAS PubMed.
- S. R. Tamang, A. Singh, D. Bedi, A. R. Bazkiaei, A. A. Warner, K. Glogau, C. McDonald, D. K. Unruh and M. Findlater, Nat. Catal., 2020, 3, 154–162 CrossRef CAS.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808–12814 CrossRef CAS PubMed.
- Z. Li, L. Zhang, M. Nishiura, G. Luo, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2020, 142, 1966–1974 CrossRef CAS PubMed.
- S. Krautwald, M. J. Bezdek and P. J. Chirik, J. Am. Chem. Soc., 2017, 139, 3868–3875 CrossRef CAS PubMed.
- N. G. Léonard, M. J. Bezdek and P. J. Chirik, Organometallics, 2017, 36, 142–150 CrossRef.
- D. Yan, P. Dai, S. Chen, M. Xue, Y. Yao, Q. Shen and X. Bao, Org. Biomol. Chem., 2018, 16, 2787–2791 RSC.
- Z. Zhu, P. Dai, Z. Wu, M. Xue, Y. Yao, Q. Shen and X. Bao, Catal. Commun., 2018, 112, 26–30 CrossRef CAS.
- H. Yin, P. J. Carroll and E. J. Schelter, Chem. Commun., 2016, 52, 9813–9816 RSC.
- Y. Li and Y. Gong, Chem. Commun., 2022, 58(90), 12552–12555 RSC.
- S. R. Daly, D. Y. Kim, Y. Yang, J. R. Abelson and G. S. Girolami, J. Am. Chem. Soc., 2010, 132, 2106–2107 CrossRef CAS PubMed.
- S. R. Daly, B. J. Bellott, D. R. McAlister, E. P. Horwitz and G. S. Girolami, Inorg. Chem., 2022, 61, 7217–7221 CrossRef CAS PubMed.
- K. A. Erickson and J. L. Kiplinger, ACS Catal., 2017, 7, 4276–4280 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, X-ray crystallographic data, and computational details. CCDC 2196153, 2196157, and 2196158. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06442a. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Alessandro
Motta
a,
Alessandro
Motta