
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic pheomelanin to unravel the electronic, molecular and supramolecular structure of the natural product†
Wei
Cao
*abc,
Haochuan
Mao
ad,
Naneki C.
McCallum
a,
Xuhao
Zhou
 a,
Hao
Sun
abcg,
Christopher
Sharpe
b,
Joanna
Korpanty
a,
Ziying
Hu
a,
Qing Zhe
Ni
e,
Michael D.
Burkart
e,
Matthew D.
Shawkey
f,
Michael R.
Wasielewski
ad and
Nathan C.
Gianneschi
*abce
a,
Hao
Sun
abcg,
Christopher
Sharpe
b,
Joanna
Korpanty
a,
Ziying
Hu
a,
Qing Zhe
Ni
e,
Michael D.
Burkart
e,
Matthew D.
Shawkey
f,
Michael R.
Wasielewski
ad and
Nathan C.
Gianneschi
*abce
aDepartment of Chemistry, Northwestern University, Evanston, Illinois 60208, USA. E-mail: caowei@bnu.edu.cn; nathan.gianneschi@northwestern.edu
bDepartment of Materials Science and Engineering, Northwestern University, Evanston, Illinois 60208, USA
cDepartment of Biomedical Engineering, Department of Pharmacology, International Institute for Nanotechnology, Simpson-Querrey Institute, Chemistry of Life Processes Institute, Lurie Cancer Center, Northwestern University, Evanston, Illinois 60208, USA
dInstitute for Sustainability and Energy at Northwestern University, Evanston, Illinois 60208, USA
eDepartment of Chemistry & Biochemistry, University of California, San Diego, La Jolla, California 92093, USA
fEvolution and Optics of Nanostructures Group, Department of Biology, The University of Ghent, 9000, Ghent, Belgium
gDepartment of Chemistry and Chemical & Biomedical Engineering, Tagliatela College of Engineering, University of New Haven, West Haven, Connecticut 06516, USA
First published on 31st March 2023
Abstract
Herein, we investigate synthetic routes to a close mimic of natural pheomelanin. Three different oxidative polymerization routes were attempted to generate synthetic pheomelanin, each giving rise to structurally dissimilar materials. Among them, the route employing 5-cysteinyl-dihydroxyphenylalanine (5-CD) as a monomer was verified as a close analogue of extracted pheomelanin from humans and birds. The resulting biomimetic and natural pheomelanins were compared via various techniques, including solid-state Nuclear Magnetic Resonance (ssNMR) and Electron Paramagnetic Resonance (EPR). This synthetic pheomelanin closely mimics the structure of natural pheomelanin as determined by parallel characterization of pheomelanin extracted from multiple biological sources. With a good synthetic biomimetic material in hand, we describe cation-π interactions as an important driving force for pheomelanogenesis, further advancing our fundamental understanding of this important biological pigment.
Introduction
Melanin is an important class of polymeric biological pigment derived mainly from amino acid precursors.1 Found across different kingdoms of life, melanins have a myriad of important functions, including coloration, camouflage, thermal regulation, photoprotection, and radioprotection.2–13 Epidermal pigmentation in human beings is considered to be composed of two separate but biosynthetically related melanins: eumelanin and pheomelanin.14,15 Eumelanin is well-studied as the most important factor in protection from harmful solar UV radiation.5,16,17 While its cousin pheomelanin, which is prevalent in red hair and fair skin, is far less studied.18,19 Although pheomelanin is generally thought to be phototoxic and potentially responsible for fair skinned people being more susceptible to sunburn and skin cancer,20,21 from an evolutionary perspective, it must have played some beneficial role. In fact, several studies have shown that pheomelanin can provide better ionizing radiation protection than eumelanin.22,23 Additionally, pheomelanogenesis was suggested to be related to Parkinson's disease (PD), as red hair color is associated with a significantly higher risk for PD.24 However, other studies showed that loss of neuromelanin (a mixture of pheomelanin and eumelanin) and subsequent depigmentation of the substantia nigra is a hallmark feature of PD.25,26 Undermining our complete understanding of the material is that structural and functional studies of pheomelanin have long conflicted with one another.23,27,28 Therefore, to better understand the functions of pheomelanin, detailed structural characterization is essential.Natural pheomelanin is likely synthesized from L-dihydroxyphenylalanine (L-DOPA) and cysteine.14,25 This biosynthesis is described by the Raper–Mason pathway (RMP), in which the nucleophilic addition of cysteine to enzymatically generated DOPA quinone forms 5-cysteinyl-DOPA (5-CD), 2-cysteinyl-DOPA (2-CD), 2,5-dicysteinyl-DOPA and trace amounts of 6-cysteinyl-DOPA (6-CD) as the subunits of pheomelanin. Further oxidation leads to more hierarchical pheomelanin pigments. As with eumelanin, pheomelanin presents a significant challenge for fundamental characterization, because it is a highly crosslinked, insoluble material with no sequence-controlled structure, unlike other common biopolymers such as proteins and DNA.29 Indeed, studies have been largely hampered by ambiguity of chemical structures present in the material. One of the reasons is the inherent difficulty in isolating pure pheomelanin.14 This is not only because of the lipids and proteins found together with the pigment even after extensive attempts at purification, but also because pheomelanin covalently conjugates to and physically traps other biomolecules.30,31 Therefore, producing a high-fidelity chemical analogue of pheomelanin can play an important role in understanding the biomaterial.12 Previously, pheomelanin syntheses have been performed using a combination of L-DOPA and cysteine as comonomers, or using the 5-CD heterodimer.19,22,27,28,32–34 However, pheomelanin materials synthesized using these two main routes have not been thoroughly characterized and compared against each other. Presumably, this lack of investigation stems from the aforementioned challenges.
Furthermore, although the primary structure is caused by covalent bond forming reactions, supramolecular interactions are important for the formation of melanin pigments because aggregation of the chromophores accounts for perturbations of the π-electron systems, thereby affecting color development.29,35 The covalent pathway for pheomelanin is elucidated by the RMP,14,16 but the non-covalent pathway has been relatively underappreciated, likely due to a lack of well-established model systems. The readily-accepted interactions in pheomelanin are hydrogen bonding and π–π stacking, as demonstrated by eumelanin-based research.36–38 Recently, cation-π interactions have been shown to play a key role in the progressive assembly of polydopamine (PDA) type eumelanin.39 Since nature has utilized cation-π interactions for various important biomacromolecules,40 it is desirable to determine whether this important supramolecular interaction also exists in pheomelanin.
Here, we show the synthesis of artificial pheomelanin and directly compare it with multiple natural samples for verification and elucidation. Through comprehensive characterization, properties such as color and spectral signatures via ssNMR and EPR were identified and assigned to natural pheomelanin. This study provides a route for manipulating artificial pheomelanin synthetically, and for driving our fundamental understanding of this biomaterial both on the molecular and supramolecular level.
Results and discussion
Chemical synthesis of pheomelanin
Our first objective was to identify a reliable synthetic method for artificial pheomelanin. We initially employed two routes (Fig. 1). (Method 1) KMnO4 was used to oxidize a solution phase mixture of cysteine and L-DOPA in water at pH 7.32 (Method 2) Oxygen was used to oxidize cysteine and L-DOPA in phosphate-buffered saline (PBS) in the presence of tyrosinase.33 However, as shown in Fig. 1B, the aromatic region (∼100–160 ppm) of sample from methods 1 and 2 are very weak, especially compared to the aliphatic peak (55, 30 ppm) and carbonyl peak (175 ppm), while pheomelanin made of benzothiazine subunits14,25 is expected to show strong aromatic peaks, aliphatic peaks, and carboxylic acid peaks (Fig. S1†). This spectral disparity suggests strongly that the two synthetic methods do not provide mimetics for natural pheomelanin. We use ssNMR as the main characterization method because it can minimize disruptive sample preparation compared to other typical methods like chemical degradation and high-performance liquid chromatography (HPLC) separation.41,42 To verify if the possible disulfide species hinder the pheomelanin formation, KMnO4 was used to oxidize a solution phase mixture of cysteine and L-DOPA in water at pH 7, following the in situ reduction of cystine (Fig. S1†), yet this attempt resulted in a similar ssNMR spectra as method 1 and 2. The structural discrepancy of the well-established methods encouraged us to try a third method (Method 3), in which we employed 5-cysteinyl-DOPA (5-CD) as the starting monomeric material (Fig. 1A3, and see S2 and S3† for the synthesis and characterization of 5-CD).28 Here we utilized a chemoenzymatic methodology with HRP as the enzyme and hydrogen peroxide as the oxidant. Zinc sulphate was added to the reaction to aid in retaining carboxylic acid groups in the benzothiazine intermediates.28,31 As shown in Fig. 1B, only Method 3 yields a pheomelanin structure with reasonable ssNMR features. Previously, pheomelanin materials synthesized using these three methods have not been thoroughly compared. Our ssNMR data show that only the 5-CD method gives a close pheomelanin mimic, while using L-DOPA and cysteine does not.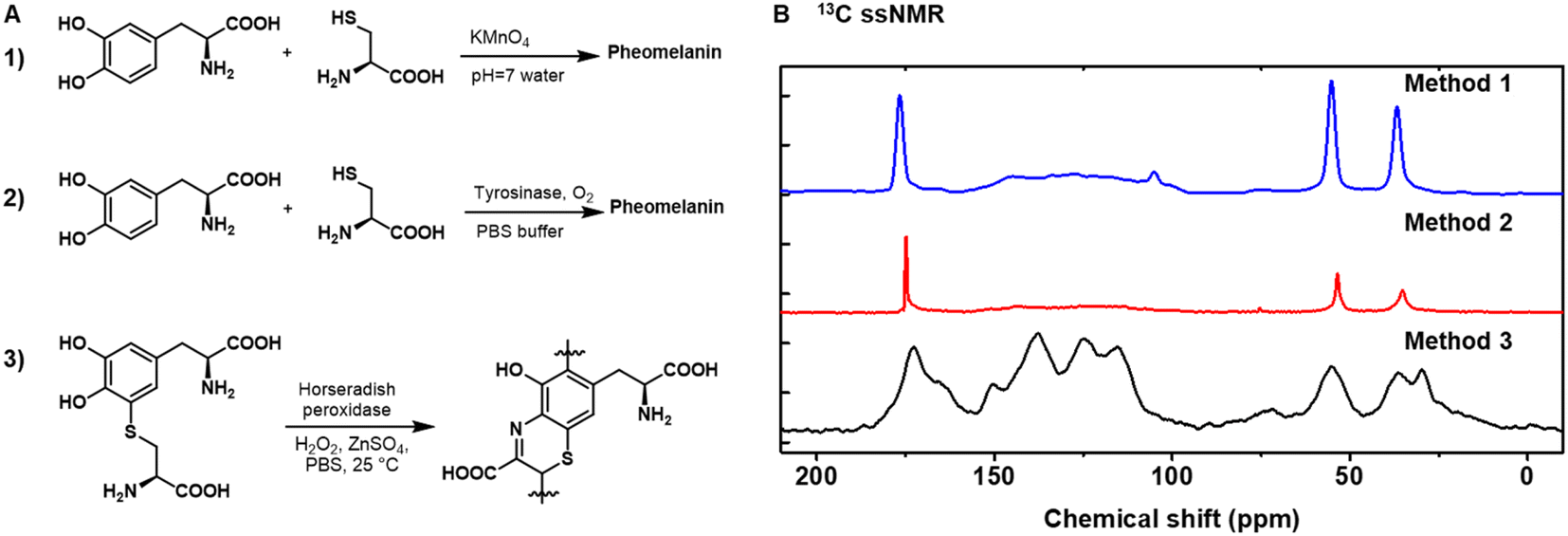 | ||
| Fig. 1 Chemical synthesis of pheomelanin using different methods. (A) (Method 1) KMnO4 was used to oxidize a solution phase mixture of cysteine and L-DOPA in water at pH 7; (Method 2) oxygen was used to oxidize cysteine and L-DOPA in phosphate buffered saline (PBS) in the presence of tyrosinase; (Method 3) 5-cysteinyl-DOPA (5-CD) was used as the starting material under chemoenzymatic oxidation with horseradish peroxidase (HRP) and H2O2. (B) Cross-polarization magic-angle spinning (CP-MAS) 13C ssNMR spectra of the pheomelanin samples synthesized from the three methods. Data for method 1 and 2 were reproduced from our previous work.23 | ||
5-CD polymerization reaction proceeds from an initially clear, light-yellow solution becoming bright yellow upon mixing the H2O2 and HRP with 5-CD (Fig. 2A). The solution changed from yellow to orange, to red, and eventually to a dark reddish brown over 24 h. The color change is distinct from the conventional PDA or poly(L-DOPA)-based eumelanin synthesis which quickly results in a yellow, to dark brown and finally to a black solution.43 As monitored by UV-vis spectroscopy (Fig. S4†), a peak around 390 nm developed during the first 5 min to 1 h period. This is followed by a more broadband absorption appearing after 2 h corresponding to an extension of the conjugated structure and more hierarchical polymers. This absorption spectroscopy change is consistent with previous observations by Napolitano.28 Absorbance at 500 nm, which can be used to quantify the total amount of melanin,18 increased over the time course of the polymerization reaction (Fig. 2B). HPLC and electrospray ionization mass spectrometry (ESI-MS) data obtained periodically from aliquots of the reaction, revealed an expected 5-CD depletion (tR = 9.3 min, [M + H]+m/z = 316.96) and its conversion to the intermediate 3-oxo-3,4-dihydrobenzothiazine (tR = 12.5 min, [M + H]+m/z= 268.88) (Fig. S4†).
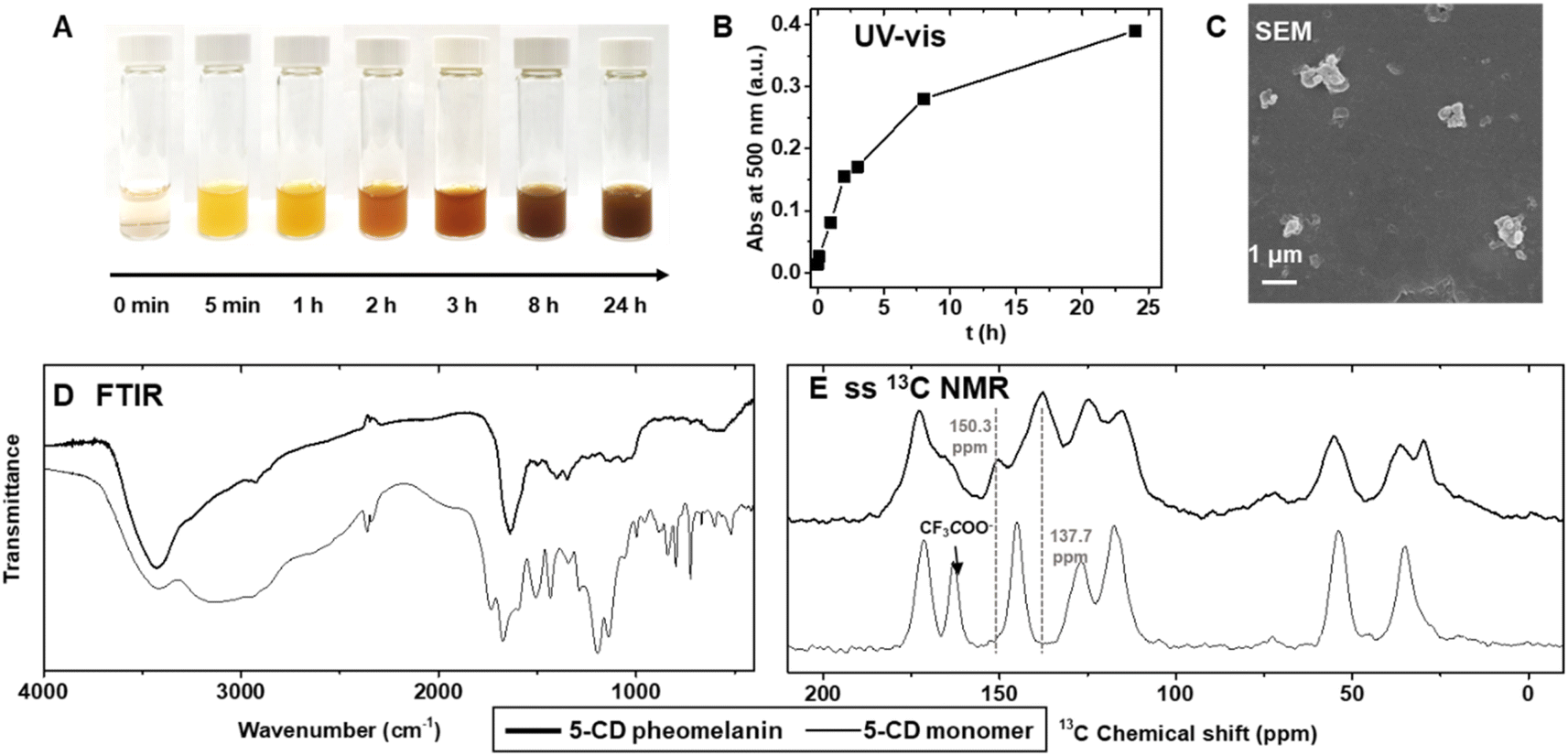 | ||
| Fig. 2 Chemical synthesis of pheomelanin using 5-cysteinyl-DOPA (5-CD) as the starting material. (A) The color change at various stages of the reaction. (B) The kinetic trace shows the continuous progress of the reaction at 500 nm. a.u., arbitrary units. (C) Representative SEM image of the synthetic pheomelanin. (D) FTIR spectra and (E) CP-MAS 13C ssNMR spectra of 5-CD pheomelanin overlaid with 5-CD monomer. | ||
The reaction yielded irregular colloidal aggregates as characterized by scanning electron microscopy (SEM) (Fig. 2C) and scanning transmission electron microscopy (STEM) (Fig. S5†). DLS showed multiple peaks with a polydispersity index of ∼0.57 (Fig. S6†). The pheomelanin particles exhibited a negative ζ-potential of −27.3 ± 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mV due to the anionic phenol and carboxylic acid groups. X-ray Photoelectron Spectroscopy (XPS) provided non-destructive information for pheomelanin (Fig. S6D–S6I†). Clear C, N, O and S signals could be identified, whereas the fluorine from the CF3COO− counter ion on the 5-CD monomer disappeared after polymerization. Energy-dispersive X-ray spectroscopy (EDS) mapping via STEM imaging verified the colocalization of sulfur and the particulates (Fig. S5†). The resulting polymer shows broad absorption bands in the Fourier Transform Infrared (FTIR) spectrum (D), which contrasts with the peak shape of the 5-CD monomer. Typical absorption bands were observed in the 5-CD pheomelanin sample at 3700–2400 cm−1 (stretching vibration of –OH, –COOH, –NH), 2920 cm−1, 2850 cm−1 (stretching vibration of aliphatic –CH, –CH2), 1640 cm−1 (bending vibrations of aromatic ring C
mV due to the anionic phenol and carboxylic acid groups. X-ray Photoelectron Spectroscopy (XPS) provided non-destructive information for pheomelanin (Fig. S6D–S6I†). Clear C, N, O and S signals could be identified, whereas the fluorine from the CF3COO− counter ion on the 5-CD monomer disappeared after polymerization. Energy-dispersive X-ray spectroscopy (EDS) mapping via STEM imaging verified the colocalization of sulfur and the particulates (Fig. S5†). The resulting polymer shows broad absorption bands in the Fourier Transform Infrared (FTIR) spectrum (D), which contrasts with the peak shape of the 5-CD monomer. Typical absorption bands were observed in the 5-CD pheomelanin sample at 3700–2400 cm−1 (stretching vibration of –OH, –COOH, –NH), 2920 cm−1, 2850 cm−1 (stretching vibration of aliphatic –CH, –CH2), 1640 cm−1 (bending vibrations of aromatic ring C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, stretching vibration of –NH2), 1400 cm−1, 1345 cm−1 (O–H bending of –COOH and phenol), and 1069 cm−1, 1040 cm−1 (aliphatic C–H deformation).
C, stretching vibration of –NH2), 1400 cm−1, 1345 cm−1 (O–H bending of –COOH and phenol), and 1069 cm−1, 1040 cm−1 (aliphatic C–H deformation).
We subsequently used 13C ssNMR to compare the chemical structure of the synthetic pheomelanin with the 5-CD monomer (Fig. 2E). The peaks at 172.7 ppm and 165.7 ppm were ascribed to the –COOH groups from 5-CD moiety (13C ssNMR of 5-CD monomer is displayed for comparison), while the decrease in the peak at 150.3 ppm corresponds to a change from the catechol structure of the monomer to the o-aminophenol structure of the benzothiazine subunit. The peak at 137.7 ppm was assigned to aromatic signals belonging to benzothiazine units as reported of polycysteinyldopamine by Ambrico.44 We note here that the spectral features in the carbonyl and aromatic regions are highly similar to that of a previously described selenomelanin, a benzoselenazine-based analogue.23 In addition, the aliphatic moieties (20–60 ppm) in the 5-CD pheomelanin spectrum have a smaller integration area relative to that of 5-CD monomer, consistent with the expected conversion of sp3 carbons. Taken together, our results support the idea that the 5-CD polymerization proceeds according to the RMP and forms benzothiazine-based pheomelanin. Therefore, method 3 should be used for future studies regarding pheomelanin function and applications.
Comparison of synthetic pheomelanin with natural pheomelanin from different sources
To perform a direct comparison of synthetic pheomelanin prepared from 5-CD with natural samples, pheomelanins were extracted from Rhode Island red rooster feathers, and human red hair from two separate individuals (Fig. 3). Enzymatic extraction was chosen over chemical extraction because the mild conditions are expected to preserve the melanin in its natural form far better than harsher conditions.45 After a series of enzymatic treatment cycles and washing steps, the sample was characterized via various techniques including SEM, DLS, and ssNMR to provide information regarding the natural chemical structure. Particles from the natural pheomelanin samples exhibit a more irregular shape than extracted eumelanin nanomaterials, as previously reported.1,21,46 The size of the resulting particles varies with rooster feathers about 330 nm in diameter by SEM microscopy (Fig. 3A) and particles from human hair measuring in the micrometer length scale (Fig. 3C), which also agree with the DLS results (Fig. 3D–F). Interestingly, we observed a size variation of the pheomelanin from two human donors likely due to the donor difference. ζ-Potential of the natural pheomelanin samples ranges from −34 mV to −44 mV (Fig. S7A†), similar to that of the synthetic sample. | ||
| Fig. 3 Natural pheomelanins extracted from bird feathers and human red hair from two individuals. (A)–(C) Optical images and SEM images of natural pheomelanin samples extracted from rooster feathers, human red hair I, and human red hair II, respectively. (D)–(F) DLS plots of the corresponding natural pheomelanins. Inset are the optical images of the pheomelanin dispersion. | ||
13C ssNMR of pheomelanin extracted from bird feathers showed a rough similarity in the aromatic region to that of the synthetic pheomelanin (Fig. 4A). ssNMR of hair pheomelanins is similar to that seen in previous literature examples (Fig. S7B and C†).30 There are some signals ascribed to proteinaceous components (172 ppm and the region 50–10 ppm). However, extra proteinase-K treatment does not significantly decrease the lipid signal intensities (Fig. S7D†), which indicates that peptides/proteins may be physically trapped within the pheomelanin, rendering them less accessible to enzyme extraction.47 In turn, three higher intensity peaks in the feather sample at ∼172 ppm, 72 ppm and 33 ppm were assigned to lipids. Similar assignments to lipids were reported by Stark in a fungal melanin sample.47 Lipid peaks were also observed in the human hair samples, which is further supported by FTIR peaks at ∼2955 cm−1 (aliphatic C–H stretch vibration, Fig. 4B). UV-vis spectra each showed broadband absorption (Fig. 4C), which is typical for melanin and melanin-like materials.29 The absorption spectra from 250 nm to 800 nm of 5-CD pheomelanin and pheomelanins from red hair showed similar features to the previous observation by Napolitano.28
These studies confirm the morphology and the chemical form by comparison of 5-CD based synthetic and natural pheomelanins. However, there are limitations to this approach, as we could not distinguish benzothiazine and benzothiazole based on the 13C ssNMR. It is known that UVA irradiation could convert benzothiazine moieties to benzothiazole units in natural pheomelanin based on HPLC analysis.31,48 We performed UV (365 nm, 7.0 mW cm−2) irradiation of our 5-CD pheomelanin for 30 h and characterized them by CP-MAS ssNMR (Fig. S7E†). Significant changes were observed in the aromatic region, which presumably results from the benzothiazine to benzothiazole conversion. Opportunities exist where isotope labeling and multi-dimensional ssNMR may reveal more structural information.
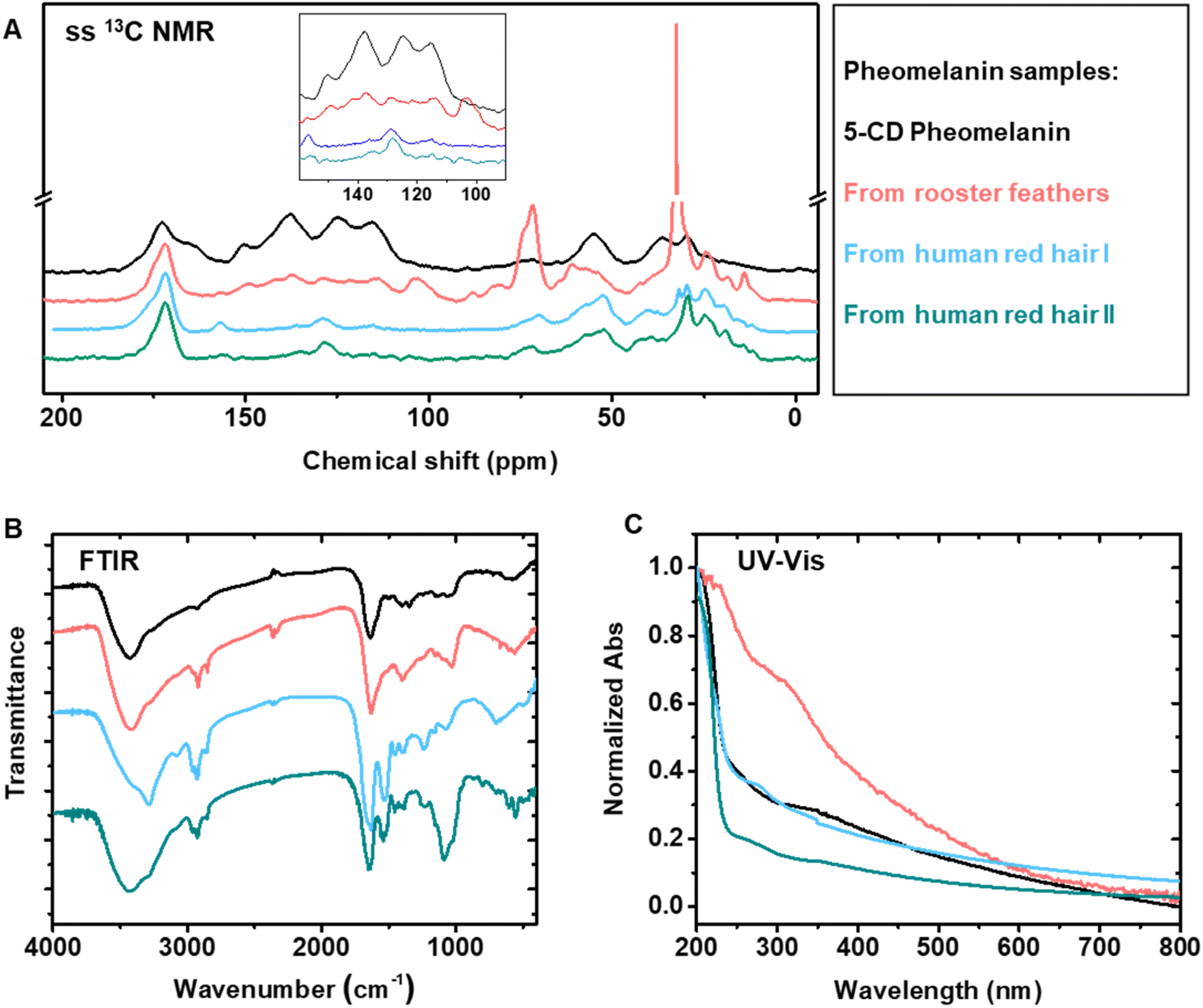 | ||
| Fig. 4 Comparison of synthetic pheomelanin with natural pheomelanin. Four samples were used in this experiment: synthetic 5-CD pheomelanin, pheomelanin extracted from rooster feathers and human red hair from two individuals. (A) CP-MAS 13C ssNMR spectra overlay of the synthetic and natural pheomelanin. Inset is the zoomed aromatic region. (B) FTIR spectra and (C) UV-vis absorption spectra of the synthetic and natural pheomelanin. | ||
Surface properties and color comparison of synthetic pheomelanin with PDA mimics of eumelanin
As the polydopamine-type eumelanin chemistry on the surface has aroused broad interest,49 we next set out to investigate whether synthetic pheomelanin led to different surface properties compared to PDA. Here we choose PDA as a synthetic eumelanin mimic because PDA is the most commonly-used and well-studied standard for eumelanin in literature.12,49 In contrast to PDA-based film studies, pheomelanin films from close mimics of natural pheomelanin have not been studied.44,50 Pheomelanin films were prepared by submerging a glass slide in the polymerization reaction solution (Fig. 5). PDA films were prepared similarly via the oxidative polymerization of dopamine under alkaline conditions in the presence of a glass slide.51 The water contact angle of pheomelanin was 20.7° (Fig. 5A), smaller than that of PDA (34.8°, Fig. 5B) and blank glass (25.5°, Fig. 5C), indicative of a higher hydrophilicity. Our data showed that a continuous pheomelanin film can also be prepared (Fig. 5D, see PDA control 5E and blank glass 5F). The synthetic film showed the typical broadband absorption seen in aqueous dispersion (Fig. S8†). Other substrates like polystyrene and gold substrates could also be coated (Fig. 5G). Additionally, the pheomelanin could also be deposited onto a 3D-printed poly-methacrylate object (Fig. 5H), showing the versatility of surface modification with these materials. Furthermore, synthetic pheomelanin appears reddish brown in color compared with the darker, black PDA at identical particle concentration by mass (1 mg mL−1) (Fig. 5I). Reflectance spectra of the films made of synthetic and natural pheomelanin showed a peak around 670 nm, corresponding to the visual red color of pheomelanin (Fig. S8†). Considering the long-lasting enthusiasm of polydopamine chemistry on surfaces for numerous applications,37,49,52 these pheomelanin films should be of significant interest as a surface modification and photochemical responsive material.44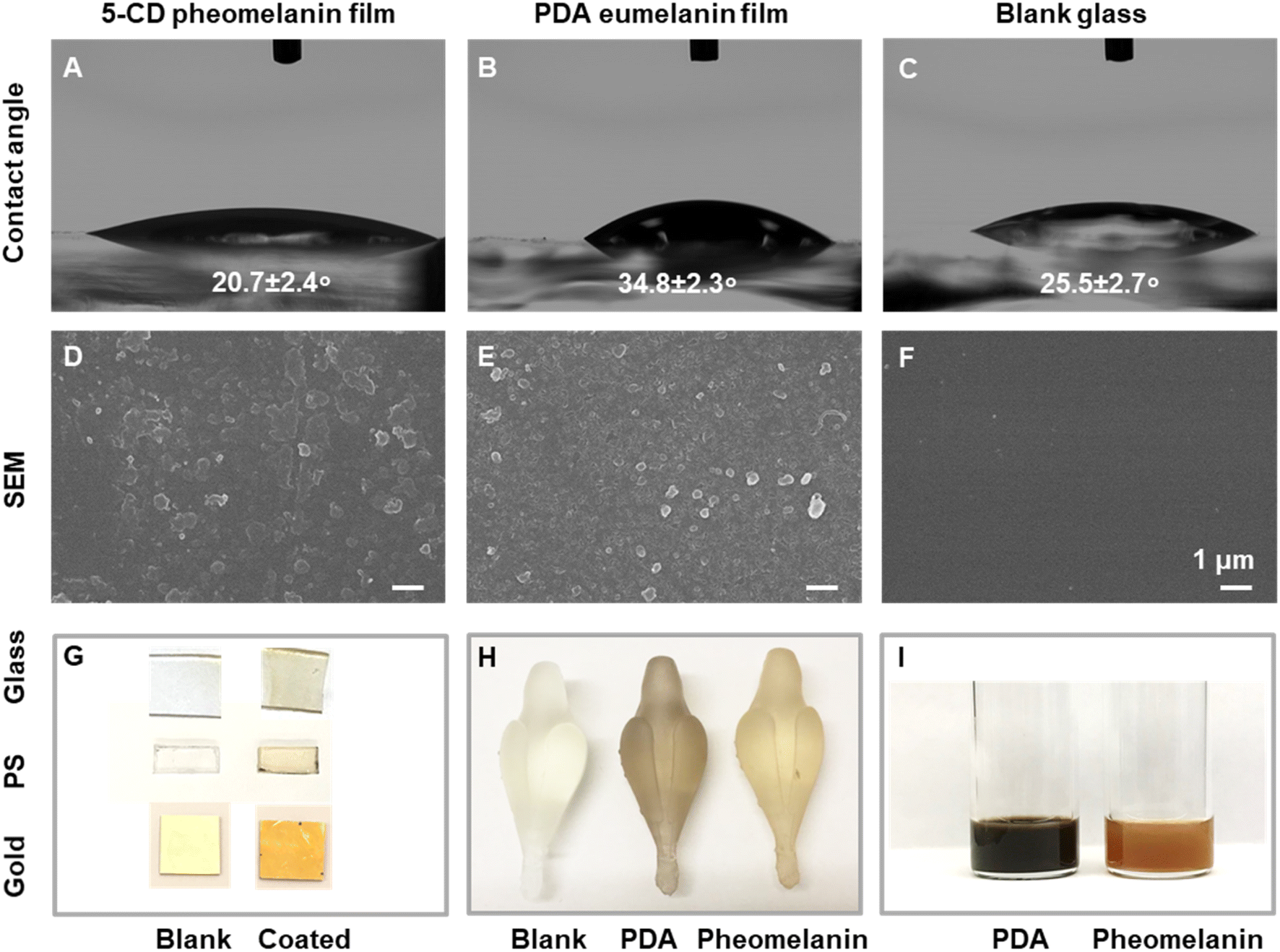 | ||
| Fig. 5 Surface properties and color comparison of synthetic pheomelanin with PDA mimics of eumelanin. The static water contact angle images for (A) pheomelanin film, (B) PDA film and (C) blank glass. Surface morphology by SEM for (D) pheomelanin film, (E) PDA film and (F) blank glass. (G) Optical photos show the color changes before and after pheomelanin coating on planar substrates, including glass, polystyrene and gold substrates. (H) 3D-printed objects before and after the coating with synthetic eumelanin and pheomelanin. (I) The color of synthetic eumelanin and pheomelanin dispersion at 1 mg mL−1 concentration. | ||
Electronic structure comparison
Electron paramagnetic resonance (EPR) reveals that stable free radicals exist in pheomelanin (Fig. 6). The anisotropic nitrogen hyperfine splitting (Fig. 6A) is similar to that of natural pheomelanin extracted from rooster feathers, indicating a similar chemical structure. Despite the inhomogeneous broadening of the spectra, one of the principal values of the nitrogen hyperfine tensor, Azz,53 can be read out directly from the peak-to-peak linewidth (Fig. 6A). The Azz values extracted from the EPR spectra of 5-CD pheomelanin and feather pheomelanin are virtually the same, and are significantly lower than nitroxide radicals,54 suggesting the existence of a semiquinoneimine radical at an o-aminophenol site, a signature for natural pheomelanin.55 By contrast, PDA eumelanin spectra of the 5-CD pheomelanin and feather pheomelanin as aqueous dispersions (Fig. S9†) are similar to those of the powder form (Fig. 6A). In addition, quantitative EPR in dispersion elucidated that the spin concentration of the pheomelanin is ∼1.7 times higher than that of PDA-type eumelanin (Fig. S9†), consistent with previous studies.22 | ||
| Fig. 6 Paramagnetic properties comparison of synthetic pheomelanin with PDA mimics of eumelanin and natural pheomelanin. (A) EPR spectra under attenuation powers of 30 dB. (B) Power saturation curves for the three samples. (C) The T1 relaxation time and fitted plot for the three samples. Open circles are the pulse EPR experimental values, and the solid lines are the fitted curves. (D) Table of the summarized values of spin concentration, P1/2 and T1 relaxation time. | ||
To further study the difference between pheomelanin and eumelanin, a continuous-wave EPR power saturation curve was measured (Fig. 6B). In these plots, the signal amplitude of an EPR spectrum was plotted against the square root of the incident microwave power P. The signal amplitude increased linearly with the square root of P until it began to saturate, and then decreased in intensity. The 5-CD pheomelanin (Fig. 6B) behaved similarly to the pheomelanin from rooster feathers, whereas the slower saturation of PDA melanin (Fig. 6B) with increasing power compared with pheomelanin suggests a longer spin-lattice relaxation time caused by the different chemical structures. For PDA eumelanin, the P1/2, which is the power at which the signal amplitude is half-saturated, is much smaller than the pheomelanin samples (Fig. 6D). To further quantify the spin-lattice relaxation time T1, pulse EPR measurements were utilized. Pulse experiments can measure relaxation time more directly than the continuous-wave saturation experiment. The T1 is 18.8 μs for synthetic pheomelanin (Fig. 6C), which agrees reasonably well with the feather pheomelanin (15.4 μs) and is much shorter than PDA eumelanin (55.5 μs, Fig. 6D).
Cation-π interactions elucidated by controlled disassembly
Although the RMP provides a molecular pathway for the synthesis of pheomelanin, the concomitant non-covalent pathway for pheomelanin remains elusive. Nature is known to use cation-π interactions to bind important small molecules, like acetylcholine.40,56 In biomacromolecules like proteins, cation-π interactions make significant energetic contributions to protein stability.57 In the Protein Data Bank, 1 out of 77 amino acid residues has cation-π interactions.40 Melanin is a biomacromolecule that has conjugated π systems and cationic groups, making cation-π interactions readily accessible. For instance, it was reported that cation-π interactions were the primary mechanism for progressive assembly in PDA mimics of eumelanin.39 Based on the chemical structure of pheomelanin, we hypothesize that cation-π interactions could exist in pheomelanin, along with the more commonly accepted hydrogen bonding and π–π interactions. Compared with the well-studied PDA, the supramolecular interactions of the pheomelanin are more difficult to decipher due to the lack of a well-established model system. Here, we utilized our synthetic pheomelanin films as a model system to study the supramolecular interactions in pheomelanin.We speculated that deprotonation of the cationic ammonium might result in disassembly of the entire pheomelanin thin film if cation-π bonding is the dominant driving force for pheomelanin supramolecular formation (Fig. 7). First, we found that KOH aqueous solution (pH 10) could quickly trigger the disassembly of the pheomelanin film (Fig. 7A and B),58 whereas the film is stable in deionized water, pH 7.4 buffer and 1% acetic acid solution (Fig. S8†), suggesting the high stability at neutral or acidic environment. The pKa of phenol hydroxyl group in tyrosine is 10.10. The pKa of –COOH is 1.91 and 2.18 for cystine and tyrosine. Therefore, the phenol remains protonated and –COOH remains deprotonated at the experimental conditions: pH 7 and pH 10.0. The disassembly of pheomelanin triggered by pH change from 7 to 10.0 indicates that the deprotonation of ammonium is the dominant factor for this change. Secondly, the solution salinity strongly impacts the disassembly process under identical pH conditions. The addition of KCl (0.5 M) provides a strong cation-π interaction, and therefore can compensate for the deprotonation of cationic amine (Fig. 7B). Furthermore, lowering potassium concentrations leads to a smaller compensation effect (Fig. S10†). The observed KCl salt compensation could be caused by salt bridge interactions or cation-π interactions. However, typical cation-π bonding between a cation and a hydrophobic aromatic group is known to be stronger than salt bridges between oppositely charged species in aqueous solution because of its low desolvation penalty.59 Therefore, we speculate that the loss of cation- π bonding is the main reason for pheomelanin film disassembly. We note here that salt bridge interaction should also exist in the synthetic pheomelanin film. To test this, another cation, Na+ was used, because Na+ has a lower binding affinity to the electron-rich π system than K+ based on previous experimental and simulation studies.60 In our experiment, NaCl (0.5 M) showed only a very small compensation effect for disassembly, suggesting that the cation- π interaction is the main driving force for the stabilization of pheomelanin disassembly. UV-vis spectra were recorded of the resulting film (Fig. 7C), and the statistical test showed that 0.5 M KCl treated film has the same absorbance at 400 nm with the pristine one (P = 0.41, Fig. 7D), whereas the 0.5 M NaCl and pH 10 solution led to much lower absorbance values (P = 0.0002, 0.0005 with reference to 0.5 M KCl treated film). After the salt treatment, SEM micrographs (Fig. 7E) confirmed that the film was mostly preserved in KCl solution, slightly preserved in NaCl solution, and only blank glass substrate could be observed under SEM after treatment with a pH 10 KOH solution. Additionally, to decouple this cation compensation effect from the type of anion, we then screened different counterions including Br− and SO42− (Fig. S10†). Similarly colored films were observed for alternated anions as compared to treatment with the KCl solution, and absorption mapping at 400 nm of the film supported a similar trend (Fig. S10†), indicating that the cationic species is the dominating factor in the pheomelanin disassembly process. The results collectively suggest the cation-π interaction contributes in a dominant fashion to the assembly of the synthetic pheomelanin pigment. Although we mainly highlight the intermolecular cation-π interaction, it is anticipated that intramolecular cation-π61,62 should also exist in pheomelanin biomacromolecule. Understanding the supramolecular pathway of pheomelaningenesis could lead to novel routes for manipulating artificial pheomelanin synthetically. This high-fidelity pheomelanin model also could enable the elucidation of other enigmatic properties and biological functions of pheomelanin.
 | ||
| Fig. 7 Cation-π interactions in the synthetic pheomelanin. (A) A schematic description for cation-π interaction and alkali-triggered disassembly of the pheomelanin film. (B) The optical images showcase the color change of the pristine pheomelanin film, and films after treatment with pH 10 solution, NaCl (0.5 M), and KCl (0.5 M) solution, respectively. (C) UV-vis spectra of each pheomelanin film sample. a.u., arbitrary units. (D) Absorbance comparison at 400 nm and statistical test. Error bars represent the standard deviation of >4 measurements in a single experiment. NS means no statistical difference P >0.1, ***: P <0.001. (E) Representative SEM micrographs of each pheomelanin film. Scale bar is 1 μm and applies to all images in the set. | ||
Conclusions
Pheomelanin is an important part of the diverse melanin family. Producing a high-fidelity chemical analogue of pheomelanin can play an important role in understanding the biomaterial. Biosynthetic and synthetic attempts at pheomelanin have been investigated multiple times over several decades by different groups,19,27,28 with most efforts focused on reaction intermediate studies and degradation product identification.28,63 Yet thorough characterization of the resulting polymeric materials with reference to the natural product has been lacking. Here we reiterate the importance of parallel comparison and full characterization of different synthetic routes and natural products. This work identified the false methods and therefore, is an important guidance and benchmark for the future synthesis and utilization of pheomelanin biomaterials. We demonstrated that the chemoenzymatic oxidation of 5-CD could yield benzothiazine-based pheomelanin, but using cysteine and L-DOPA failed, possibly because of the slow redox step to give cysteinyldopaquinone.25 Although the 5-CD synthetic method is only one step further in the RMP, it altered the polymerization pathway significantly,27 likely due to the suppression of the addition of cysteine to the quickly growing polymer chains.19 Moreover, these approaches could also help to elucidate the more enigmatic neuromelanin, which is believed to have structural features of both eumelanin and pheomelanin.In nature, pheomelanin is found as a mixture of melanins according to the previously proposed “casing” model (pheomelanin cores encased by eumelanin surfaces).14,26 This makes it a formidable challenge to study the properties of the pheomelanin polymer from natural sources. Synthetic methods, such as the use of 5-CD monomers, provide a route to circumvent the casing model since no eumelanin monomer is involved. As eumelanin typically has a well-defined shape, the absence of the casing eumelanin may account for the irregular morphology of 5-CD synthetic pheomelanin. To be clear, we note that despite structural similarity at the molecular level, morphologically the chemoenzymatic approach to synthetic pheomelanin yields irregular structures as opposed to the largely oval shapes of natural, isolated pheomelanin. Presumably, this is because biological processes guide the in vivo pheomelanogenesis utilizing confinement (liposomal, or casing induced) and genetically programmed progression of the melanosome. To truly mimic the shape and chemistry of the pheomelanosomes, synthetic templation or self-assembled nanoarchitecture approaches should be developed. Insight derived from our study highlighting the importance of cation-π interactions may give direction to such an approach.
In summary, 5-cysteinyl-DOPA oxidation under enzymatic conditions yields a close structural mimic to natural pheomelanin. Notably, synthetic pheomelanin was found to be more hydrophilic, exhibits a lower T1 relaxation time than polydopamine-type eumelanin and is a reddish-brown color at the same mass concentration. Alongside the extensive structural characterization of pheomelanin, with this synthetic tool in hand, we found that cation-π interactions exist in synthetic pheomelanin. This insight may provide a deeper understanding of the melanogenesis of one of the most important melanin subfamilies in nature. Further research will focus on controlling monomer composition, introducing other functionalities, and controlling nanoscale/microscale morphology. Moreover, the pursuit of better synthetic mimics for melanin classes, including for eumelanin, where it is known that PDA is a common but incomplete analogue, promises to shed much needed light on function and molecular structure of melanins more generally.12
Data availability
All experimental procedures and characterization data are available in the ESI.†Author contributions
W. C. and N. C. G. conceived the project and designed the experiments. W. C. conducted most experiments. H. M. and M. R. W. performed EPR. N. C. M. collected the STEM and SEM images. X. Z. and H. S. obtained HPLC and MS spectra analysis. C. S. printed the 3D objects. J. K. did the analysis of the film mapping. Z. H. did the contact angle analysis. Q. Z. N. and M. D. B. interpreted solid-state 13C NMR. M. D. S. measured the reflectance spectra. W. C. and N. C. G. co-wrote the manuscript with contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Air Force Office of Scientific Research through a MURI Grant (FA9550-18-1-0142), and MURI supplemental grant (AFOSR FA9550-18-1-0477). EPR spectroscopy was supported by the National Science Foundation under Award No CHE-1900422 (M. R. W.). We thank Dr Yuyang Wu and Dr Yongbo Zhang in the IMSERC core facility at Northwestern University for assisting with ssNMR. We thank James Seale and Tirzah Abbott from Northwestern University for red hair donation.References
- L. D'Alba and M. D. Shawkey, Physiol. Rev., 2019, 99, 1–19 CrossRef PubMed.
- M. Xiao, Z. Hu, Z. Wang, Y. Li, A. D. Tormo, N. Le Thomas, B. Wang, N. C. Gianneschi, M. D. Shawkey and A. Dhinojwala, Sci. Adv., 2017, 3, e1701151 CrossRef PubMed.
- A. Lampel, S. A. McPhee, H.-A. Park, G. G. Scott, S. Humagain, D. R. Hekstra, B. Yoo, P. W. J. M. Frederix, T.-D. Li, R. R. Abzalimov, S. G. Greenbaum, T. Tuttle, C. Hu, C. J. Bettinger and R. V. Ulijn, Science, 2017, 356, 1064–1068 CrossRef CAS PubMed.
- A. B. Mostert, S. B. Rienecker, C. Noble, G. R. Hanson and P. Meredith, Sci. Adv., 2018, 4, eaaq1293 CrossRef PubMed.
- Y. Huang, Y. Li, Z. Hu, X. Yue, M. T. Proetto, Y. Jones and N. C. Gianneschi, ACS Cent. Sci., 2017, 3, 564–569 CrossRef CAS PubMed.
- X. Zhou, N. C. McCallum, Z. Hu, W. Cao, K. Gnanasekaran, Y. Feng, J. F. Stoddart, Z. Wang and N. C. Gianneschi, ACS Nano, 2019, 13, 10980–10990 CrossRef CAS PubMed.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS PubMed.
- Z. Wang, T. Tschirhart, Z. Schultzhaus, E. E. Kelly, A. Chen, E. Oh, O. Nag, E. R. Glaser, E. Kim, P. F. Lloyd, P. T. Charles, W. Li, D. Leary, J. Compton, D. A. Phillips, A. Dhinojwala, G. F. Payne and G. J. Vora, Appl. Environ. Microbiol., 2019, 86, e02749 CrossRef PubMed.
- Q. Li, K.-Q. Gao, Q. Meng, J. A. Clarke, M. D. Shawkey, L. D'Alba, R. Pei, M. Ellison, M. A. Norell and J. Vinther, Science, 2012, 335, 1215–1219 CrossRef CAS PubMed.
- J. Lindgren, P. Sjovall, R. M. Carney, P. Uvdal, J. A. Gren, G. Dyke, B. P. Schultz, M. D. Shawkey, K. R. Barnes and M. J. Polcyn, Nature, 2014, 506, 484–488 CrossRef CAS PubMed.
- H. Liu, Y. Yang, Y. Liu, J. Pan, J. Wang, F. Man, W. Zhang and G. Liu, Adv. Sci., 2020, 1903129 CrossRef CAS PubMed.
- W. Cao, X. Zhou, N. C. McCallum, Z. Hu, Q. Z. Ni, U. Kapoor, C. M. Heil, K. S. Cay, T. Zand, A. J. Mantanona, A. Jayaraman, A. Dhinojwala, D. D. Deheyn, M. D. Shawkey, M. D. Burkart, J. D. Rinehart and N. C. Gianneschi, J. Am. Chem. Soc., 2021, 143, 2622–2637 CrossRef CAS PubMed.
- X. Zhou, X. Gong, W. Cao, C. J. Forman, J. Oktawiec, L. D’ Alba, H. Sun, M. P. Thompson, Z. Hu, U. Kapoor, N. C. McCallum, C. D. Malliakas, O. K. Farha, A. Jayaraman, M. D. Shawkey and N. C. Gianneschi, Angew. Chem., Int. Ed., 2021, 60, 17464–17471 CrossRef CAS PubMed.
- J. D. Simon and D. N. Peles, Acc. Chem. Res., 2010, 43, 1452–1460 CrossRef CAS PubMed.
- S. Ito and K. Fujita, Anal. Biochem., 1985, 144, 527–536 CrossRef CAS PubMed.
- Q. Ni, B. Sierra, J. J. LaClair and M. Burkart, Chem. Sci., 2020, 11, 7836–7841 RSC.
- J. Wang and L. Blancafort, Angew. Chem., Int. Ed., 2021, 60, 18800–18809 CrossRef CAS PubMed.
- I. Galván, A. Jorge, F. Solano and K. Wakamatsu, Spectrochim. Acta, Part A, 2013, 110, 55–59 CrossRef PubMed.
- R. B. Deibel and M. R. Chedekel, J. Am. Chem. Soc., 1982, 104, 7306–7309 CrossRef CAS.
- F. Solano, Polym. Int., 2016, 65, 1276–1287 CrossRef CAS.
- T. H. Nasti and L. Timares, Photochem. Photobiol., 2015, 91, 188–200 CrossRef CAS PubMed.
- A. D. Schweitzer, R. C. Howell, Z. Jiang, R. A. Bryan, G. Gerfen, C. C. Chen, D. Mah, S. Cahill, A. Casadevall and E. Dadachova, PLoS One, 2009, 4, e7229 CrossRef PubMed.
- W. Cao, N. C. McCallum, Q. Z. Ni, W. Li, H. Boyce, H. Mao, X. Zhou, H. Sun, M. P. Thompson, C. Battistella, M. R. Wasielewski, A. Dhinojwala, M. D. Shawkey, M. D. Burkart, Z. Wang and N. C. Gianneschi, J. Am. Chem. Soc., 2020, 142, 12802–12810 CrossRef CAS PubMed.
- X. Chen, D. Feng, M. A. Schwarzschild and X. Gao, Ann. Clin. Transl. Neurol., 2017, 4, 212–216 CrossRef CAS PubMed.
- S. Ito, Anal. Biochem., 2003, 16, 230–236 CAS.
- W. D. Bush, J. Garguilo, F. A. Zucca, A. Albertini, L. Zecca, G. S. Edwards, R. J. Nemanich and J. D. Simon, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14785–14789 CrossRef CAS PubMed.
- R. M. B. Deibel and M. R. Chedekel, J. Am. Chem. Soc., 1984, 106, 5884–5888 CrossRef CAS.
- A. Napolitano, M. De Lucia, L. Panzella and M. d'Ischia, Photochem. Photobiol., 2008, 84, 593–599 CrossRef CAS PubMed.
- M. d'Ischia, A. Napolitano, A. Pezzella, P. Meredith and M. Buehler, Angew. Chem., Int. Ed., 2020, 59, 11196–11205 CrossRef PubMed.
- P. Thureau, F. Ziarelli, A. Thevand, R. W. Martin, P. J. Farmer, S. Viel and G. Mollica, Chem. – Eur. J., 2012, 18, 10689–10700 CrossRef CAS PubMed.
- A. Napolitano, L. Panzella, G. Monfrecola and M. d'Ischia, Pigm. Cell Melanoma Res., 2014, 27, 721–733 CrossRef CAS PubMed.
- J. Pyo, K. Y. Ju and J. K. Lee, J. Photochem. Photobiol., B, 2016, 160, 330–335 CrossRef CAS PubMed.
- S. Ito, Pigm. Cell Res., 1989, 2, 53–56 CrossRef CAS PubMed.
- R. C. Sealy, J. S. Hyde, C. C. Felix, I. A. Menon and G. Prota, Science, 1982, 217, 545–547 CrossRef CAS PubMed.
- A. Büngeler, B. Hämisch and O. I. Strube, Int. J. Mol. Sci., 2017, 18, 1901 CrossRef PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- M. d'Ischia, A. Napolitano, V. Ball, C. T. Chen and M. J. Buehler, Acc. Chem. Res., 2014, 47, 3541–3550 CrossRef PubMed.
- L. Panzella, G. Gentile, G. D'Errico, N. F. Della Vecchia, M. E. Errico, A. Napolitano, C. Carfagna and M. d'Ischia, Angew. Chem., Int. Ed., 2013, 52, 12684–12687 CrossRef CAS PubMed.
- S. Hong, Y. Wang, S. Y. Park and H. Lee, Sci. Adv., 2018, 4, eaat7457 CrossRef PubMed.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed.
- S. Tian, J. Garcia-Rivera, B. Yan, A. Casadevall and R. E. Stark, Biochemistry, 2003, 42, 8105–8109 CrossRef CAS PubMed.
- W. Li, Z. Wang, M. Xiao, T. Miyoshi, X. Yang, Z. Hu, C. Liu, S. S. C. Chuang, M. D. Shawkey, N. C. Gianneschi and A. Dhinojwala, Biomacromolecules, 2019, 20, 4593–4601 CrossRef CAS PubMed.
- K.-Y. Ju, Y. Lee, S. Lee, S. B. Park and J.-K. Lee, Biomacromolecules, 2011, 12, 625–632 CrossRef CAS PubMed.
- M. Ambrico, N. F. Della Vecchia, P. F. Ambrico, A. Cardone, S. R. Cicco, T. Ligonzo, R. Avolio, A. Napolitano and M. d'Ischia, Adv. Funct. Mater., 2014, 24, 7161–7172 CAS.
- Y. Liu, V. R. Kempf, J. Brian Nofsinger, E. E. Weinert, M. Rudnicki, K. Wakamatsu, S. Ito and J. D. Simon, Pigm. Cell Res., 2003, 16, 355–365 CrossRef CAS PubMed.
- Y. Liu, L. Hong, K. Wakamatsu, S. Ito, B. Adhyaru, C. Y. Cheng, C. R. Bowers and J. D. Simon, Photochem. Photobiol., 2005, 81, 135–144 CrossRef CAS PubMed.
- S. Chatterjee, R. Prados-Rosales, S. Tan, V. C. Phan, C. Chrissian, B. Itin, H. Wang, A. Khajo, R. S. Magliozzo, A. Casadevall and R. E. Stark, J. Biol. Chem., 2018, 293, 20157–20168 CrossRef CAS PubMed.
- K. Wakamatsu, Y. Nakanishi, N. Miyazaki, L. Kolbe and S. Ito, Pigm. Cell Melanoma Res., 2012, 25, 434–445 CrossRef CAS PubMed.
- J. H. Ryu, P. B. Messersmith and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 7523–7540 CrossRef CAS PubMed.
- N. F. Della Vecchia, R. Marega, M. Ambrico, M. Iacomino, R. Micillo, A. Napolitano, D. Bonifazi and M. d'Ischia, J. Mater. Chem. C, 2015, 3, 6525–6531 RSC.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- P. Yang, F. Zhu, Z. Zhang, Y. Cheng, Z. Wang and Y. Li, Chem. Soc. Rev., 2021, 50, 8319–8343 RSC.
- E. Bordignon, eMagRes, 2017, 6, 235–254 CAS.
- O. H. Griffith, D. W. Cornell and H. M. McConnell, J. Chem. Phys., 1965, 43, 2909–2910 CrossRef CAS.
- R. C. Sealy, J. S. Hyde, C. C. Felix, I. A. Menon, G. Prota, H. M. Swartz, S. Persad and H. F. Haberman, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 2885–2889 CrossRef CAS PubMed.
- W. Zhong, J. P. Gallivan, Y. Zhang, L. Li, H. A. Lester and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12088–12093 CrossRef CAS PubMed.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS PubMed.
- R. D. Lillie, J. Histochem. Cytochem., 1955, 3, 453–454 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- Q. Lu, D. X. Oh, Y. Lee, Y. Jho, D. S. Hwang and H. Zeng, Angew. Chem., Int. Ed., 2013, 52, 3944–3948 CrossRef CAS PubMed.
- J. Sivaguru, R. B. Sunoj, T. Wada, Y. Origane, Y. Inoue and V. Ramamurthy, J. Org. Chem., 2004, 69, 6533–6547 CrossRef CAS PubMed.
- L. S. Kaanumalle, J. Sivaguru, R. B. Sunoj, P. H. Lakshminarasimhan, J. Chandrasekhar and V. Ramamurthy, J. Org. Chem., 2002, 67, 8711–8720 CrossRef CAS PubMed.
- A. Napolitano, S. Memoli and G. Prota, J. Org. Chem., 1999, 64, 3009–3011 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06418a |
| This journal is © The Royal Society of Chemistry 2023 |