
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical formation and reversible base-induced cleavage of a phosphagallene†
T.
Taeufer
 a,
F.
Dankert
a,
D.
Michalik
ab,
J.
Pospech
*a,
J.
Bresien
*b and
C.
Hering-Junghans
*a
a,
F.
Dankert
a,
D.
Michalik
ab,
J.
Pospech
*a,
J.
Bresien
*b and
C.
Hering-Junghans
*a
aLeibniz Institut für Katalyse e.V. (LIKAT), A.-Einstein.-Str. 29a, 18059 Rostock, Germany. E-mail: jola.pospech@catalysis.de; christian.hering-junghans@catalysis.de; Web: https://www.catalysis.de/forschung/katalytische-funktionalisierungen Web: https://www.catalysis.de/forschung/katalyse-mit-erneuerbaren-rohstoffen/bioinspirierte-katalyse
bInstitute of Chemistry, University of Rostock, A.-Einstein.-Str. 3a, 18059 Rostock, Germany. E-mail: jonas.bresien@uni-rostock.de; Web: https://www.chemie.uni-rostock.de/arbeitsgruppen/anorganische-chemie/dr-jonas-bresien/
First published on 16th February 2023
Abstract
The reactivity of Cp*Ga (Cp* = C5Me5) towards phosphanylidenephosphoranes of the type ArTerP(PMe3) (ArTer = DipTer 2,6-(2,6-iPr2C6H3)2C6H3), TipTer 2,6-(2,4,6-iPr3C6H2)2C6H3 was investigated. While no thermal reaction was observed (in line with DFT results), irradiation at 405 nm at low temperatures resulted in the formation of phosphagallenes DipTerP = GaCp* (1a) and TipTerP = GaCp* (1b) accompanied by release of PMe3. When warming the reaction mixture to ambient temperatures without irradiation, the clean re-formation of ArTerP(PMe3) and Cp*Ga in a second-order reaction was observed. Upon removal of PMe3, 1a and 1b were isolated and fully characterized. Both derivatives were found to be labile and decomposed to the phosphafluorenes 2a and 2b, indicating generation of the transient phosphinidene ArTerP along with Cp*Ga. First reactivity studies show that CO2 and H2O cleanly reacted with 1a, affording DipTerPCO (3) and DipTerPH2 (4), respectively.
Introduction
Heavier main group element–element multiple bond systems are no longer lab curiosities and tremendous effort has been devoted to establish new bonding patterns and to utilize the transition-metal like reactivity of these systems in terms of small molecule activation and functionalization.1–11 Our group is interested in E13–E15 multiple bond systems,12 which are the iso-valent electronic heavier analogs of C–C multiple bond systems and have been proposed as intermediates in Metal–Organic Chemical Vapour Deposition (MOCVD) processes, which are based on utilizing molecular single-source E13–E15 precursors.13,14 The synthesis of E13–E15 multiple bonds is challenging due to adjacent Lewis acidic E13 and Lewis basic E15 atoms, resulting in a tendency to oligomerize.15–17 Just recently, our group isolated phospha- and arsaalumenes DipTerPn = AlCp* and TipTerP = AlCp* (DipTer = 2,6-(2,6-iPr2C6H3)2–C6H3; TipTer = 2,4,6-(2,46-iPr3C6H3)2–C6H3; Cp* = C5Me5; Pn = P (C), As; Fig. 1)18,19 by combining the pnictinidene transfer reagents ArTerPn(PMe3)20 (Ar = Dip, Tip) with (Cp*Al)4 at 80 °C.21,22 The heavier analogs of phosphaalumenes, phosphagallenes, have also been realized synthetically just recently, utilizing phosphanyl- or gallaphosphaketenes in the reaction with (DipNacnac)Ga (DipNacnac = HC[C(Me)NDip]2) facilitating CO cleavage and formation of [(S)P]–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ga(DipNacnac) (A; [(S)P] = (HnCNDip)2P; n = 1, 2)23 or (DipNacnac)GaP–Ga(Cl)(DipNacnac) (B, Fig. 1),24 respectively. Reactivity studies revealed that B reacted directly at the GaP-bond and for example the reversible insertion of two CO2 molecules, [2 + 2] cycloadditions with isocyanates and carbodiimides,25 and 1,2-oxidative additions of E–H bonds were reported.26 Interestingly, the phosphanylphosphagallene A showed mainly dipolar frustrated Lewis-pair (FLP) type 1,3-reactivity towards E–H bonds,27 and with CO2 the formation of a five-membered ring species with a P2GaCO core was described.23 We reasoned that the combination of Cp*Ga,28,29 with ArTerP(PMe3) (Ar = Dip, Tip) would afford the phosphagallenes ArTerP = GaCp* by analogy with our strategy to access phospha- and arsaalumenes. Here we show that this transformation can be achieved photochemically and that in the presence of PMe3 the reaction is fully reversible, indicating a facile and unusual ligand exchange at a phosphinidene center (Scheme 1).
Ga(DipNacnac) (A; [(S)P] = (HnCNDip)2P; n = 1, 2)23 or (DipNacnac)GaP–Ga(Cl)(DipNacnac) (B, Fig. 1),24 respectively. Reactivity studies revealed that B reacted directly at the GaP-bond and for example the reversible insertion of two CO2 molecules, [2 + 2] cycloadditions with isocyanates and carbodiimides,25 and 1,2-oxidative additions of E–H bonds were reported.26 Interestingly, the phosphanylphosphagallene A showed mainly dipolar frustrated Lewis-pair (FLP) type 1,3-reactivity towards E–H bonds,27 and with CO2 the formation of a five-membered ring species with a P2GaCO core was described.23 We reasoned that the combination of Cp*Ga,28,29 with ArTerP(PMe3) (Ar = Dip, Tip) would afford the phosphagallenes ArTerP = GaCp* by analogy with our strategy to access phospha- and arsaalumenes. Here we show that this transformation can be achieved photochemically and that in the presence of PMe3 the reaction is fully reversible, indicating a facile and unusual ligand exchange at a phosphinidene center (Scheme 1).

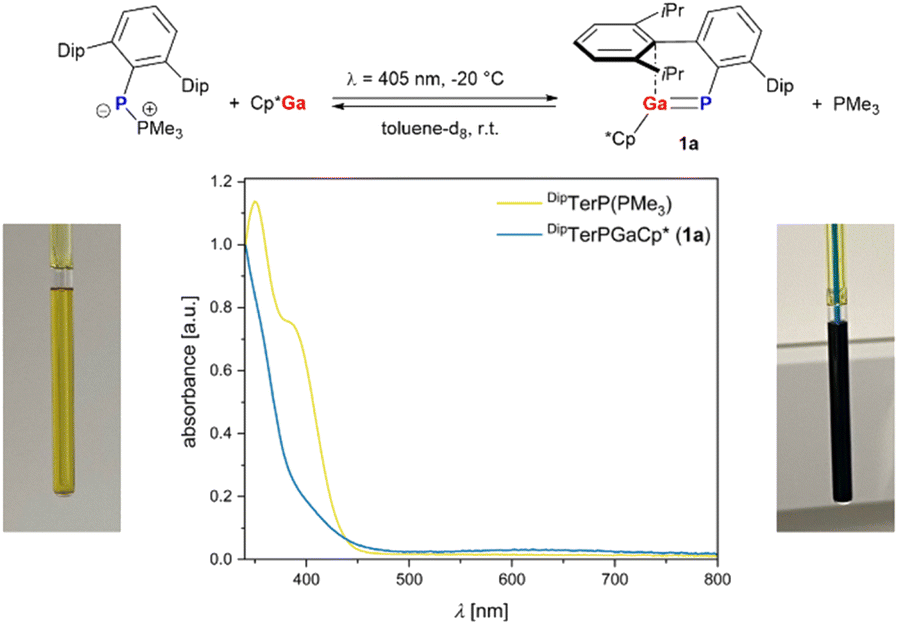 | ||
| Scheme 1 Reversible formation of DipTerPGaCp* (1a). Images of the reaction solution before (bottom left) and after irradiation (bottom right) along with the UV absorption spectra of DipTerP(PMe3) and 1a (in n-hexane, bottom middle). | ||
Results and discussion
Synthesis and kinetic studies
Our investigations started with combining Cp*Ga and DipTerP(PMe3) in toluene-d8, giving no discernible reaction at room temperature or when heated to 100 °C over a period of 72 h according to 1H and 31P NMR spectroscopy. Theoretical studies at the B3LYP-D3/def2-TZVP level of theory (cf. p. S34, ESI†) revealed an endergonic Cp*Ga for PMe3 substitution to give DipTerP = GaCp* (1a). Irradiation of the NMR sample with an LED at 396 nm, which matches the longest wave-length absorption of DipTerP(PMe3) (λmax,exp = 381 nm, λmax,calcd = 410 nm), gave a color change from yellow to green within minutes (Scheme 1). Excitation of Cp*Ga can be excluded as it shows an absorption only in the UV-region (λmax,exp = 246 nm, λmax,calcd = 241 nm cf. Table S3†). In the absence of light, the green color faded and only DipTerP(PMe3) and Cp*Ga were detected by NMR spectroscopy, indicating the formation of the desired phosphagallene 1a in reversible fashion. Recently, the reversible phosphinidene transfer in a stannaphosphene has been reported.30 We next irradiated a toluene-d8 solution containing DipTerP(PMe3) and Cp*Ga in a 1
to give DipTerP = GaCp* (1a). Irradiation of the NMR sample with an LED at 396 nm, which matches the longest wave-length absorption of DipTerP(PMe3) (λmax,exp = 381 nm, λmax,calcd = 410 nm), gave a color change from yellow to green within minutes (Scheme 1). Excitation of Cp*Ga can be excluded as it shows an absorption only in the UV-region (λmax,exp = 246 nm, λmax,calcd = 241 nm cf. Table S3†). In the absence of light, the green color faded and only DipTerP(PMe3) and Cp*Ga were detected by NMR spectroscopy, indicating the formation of the desired phosphagallene 1a in reversible fashion. Recently, the reversible phosphinidene transfer in a stannaphosphene has been reported.30 We next irradiated a toluene-d8 solution containing DipTerP(PMe3) and Cp*Ga in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio inside the NMR spectrometer using a laser diode (λ = 405 nm, 140 mW).
:1 ratio inside the NMR spectrometer using a laser diode (λ = 405 nm, 140 mW).
The formation of 1a was traced by 1H and 31P NMR spectroscopy at room temperature, showing two new resonances in the 31P NMR spectrum at −62.9 ppm (PMe3) and −104.8 ppm for 1a, which corresponds well with the theoretically predicted value for 1a (δ(31P)calcd = −105.2 ppm). After irradiation for 2 h a dynamic steady state between 1a, PMe3 and DipTerP(PMe3), Cp*Ga was reached. When switching off the laser the concentration of 1a and PMe3 decreased, and the starting materials were re-formed. Irradiation at −20 °C gave 1a nearly quantitatively, effectively suppressing the thermal back reaction. The thermal reverse reaction was then studied at 10, 15, 20 and 25 °C using the same sample repeatedly, indicating a fully reversible system. Using the concentrations derived from the relative integrals of 1a, PMe3, Cp*Ga and DipTerP(PMe3) in the 1H NMR data, the reaction kinetics of the thermal reverse reaction were investigated. The reaction follows 2nd order kinetics. However, the concentration profiles could be described reasonably well using two different models, one including an associative (nucleophilic attack of PMe3 at 1a in a SN2-type reaction), the other a dissociative mechanism (with the dissociation reaction 1a ⇌ DipTerP + Cp*Ga as its first step and reaction with PMe3 in a subsequent step; cf. p. S30†).Thus, the experimental data alone do not allow a definitive statement regarding the mechanism of the reaction. Yet, since the dissociative mechanism would include free phosphinidenes, albeit in very low concentrations, it is certainly less likely. Nonetheless, both models gave similar activation barriers (ΔG‡ ≈ 90 kJ mol−1, Table S6†) when analyzing the temperature dependence of the rate constants using transition state theory (TST).
To investigate the possible mechanism of the thermal reverse reaction further, we performed a series of semi-empirical GFN2-xTB computations31 as well as DFT calculations at the B3LYP-D3/def2-TZVP level of theory (cf. p. S46†).32–34 We could in fact identify both an associative as well as a dissociative reaction pathway. The associative SN2-type mechanism involves an activation barrier of ΔG‡ = 80.1 kJ mol−1 ( , TS2, Scheme 2), in good agreement with the experimental data. A second potential associative pathway, involving a weak donor–acceptor complex between PMe3 and 1a was also identified, however this path is associated with a significantly higher barrier (ΔG‡ = 121.4 kJ mol−1, TS1, Scheme 2) and is therefore not in line with the experimental values. The free dissociation energy of 1a into PMe3 and singlet DipTerP, on the other hand, was estimated at +107.2 kJ mol−1 (note a rather high uncertainty for this value due to the singlet biradical character of DipTerP, which is not well represented within DFT), which puts this first reaction step of the dissociative pathway in a reasonable energetic range. Assuming a low activation barrier for this dissociative pathway, it lies in a similar energy window as the SN2-type substitution with PMe3, and thus could be a (minor) contributing factor to the thermal reverse reaction. Note that singlet DipTerP would only be formed in situ, as it is predicted to possess a triplet ground state (ΔES–T = 45.5 kJ mol−1). Singlet phosphinidenes have previously been shown to be electrophilic and irreversible ligand exchange reactions were described.35–37
, TS2, Scheme 2), in good agreement with the experimental data. A second potential associative pathway, involving a weak donor–acceptor complex between PMe3 and 1a was also identified, however this path is associated with a significantly higher barrier (ΔG‡ = 121.4 kJ mol−1, TS1, Scheme 2) and is therefore not in line with the experimental values. The free dissociation energy of 1a into PMe3 and singlet DipTerP, on the other hand, was estimated at +107.2 kJ mol−1 (note a rather high uncertainty for this value due to the singlet biradical character of DipTerP, which is not well represented within DFT), which puts this first reaction step of the dissociative pathway in a reasonable energetic range. Assuming a low activation barrier for this dissociative pathway, it lies in a similar energy window as the SN2-type substitution with PMe3, and thus could be a (minor) contributing factor to the thermal reverse reaction. Note that singlet DipTerP would only be formed in situ, as it is predicted to possess a triplet ground state (ΔES–T = 45.5 kJ mol−1). Singlet phosphinidenes have previously been shown to be electrophilic and irreversible ligand exchange reactions were described.35–37
 | ||
| Scheme 2 Computed reaction pathway for the thermal reverse reaction of 1a with PMe3 (B3LYP-D3/def2-TZVP, c° = 1 mol L−1). | ||
Characterization
Next, we sought to isolate 1a rather than generating it in situ.A flask containing a 1:1 mixture of DipTerP(PMe3) and Cp*Ga in toluene was connected to a receiving flask via a U-shaped glass tube and the headspace was evacuated (Scheme 3, top). After irradiating the mixture for 4 h (λLED = 396 nm) a color change to deep green was observed and the receiving flask was then cooled to ca. −70 °C to slowly evaporate PMe3 and toluene into the receiving flask, while the product crystallized in the reaction flask. After washing with n-heptane at −78 °C, 1a was isolated as a dark turquoise solid in good yield (77%). X-ray quality crystals of 1a were grown from a saturated n-hexane solution at −30 °C.
 | ||
| Scheme 3 Isolation of 1a and 1b, its thermal decomposition and reactivity of 1a towards CO2 and H2O. | ||
The 1H NMR data of 1a are similar to those of DipTerPAlCp* (C) with three characteristic signals for the Dip-groups in the alkyl region and a singlet resonance for the Cp*-substituent, indicating η5-coordination, or at least a fast sigmatropic rearrangement in solution. The 31P NMR signal of 1a (δ(31P)(C6D6) = −104.8 ppm; δ(31P)(C7D8) = −105.5 ppm) is significantly deshielded compared to C (cf. δ(31P) = −203.9 ppm). The Ga–P stretching vibration in 1a at 448 cm−1 (calcd 439 cm−1) is red-shifted compared to the Al–P stretch in C at 558 cm−1 in the IR spectrum, while the lowest energy absorption in the UV-vis (λmax,exp = 620 nm, λmax,calcd = 635 nm) is similar to that in DipTerAsAlCp* (λmax = 590 nm).5
1a crystallizes in the monoclinic space group P21/c and is isomorphous with C with a P–Ga distance of 2.2104(5) Å (cf.C 2.2113(6); ∑rcov(PGa) = 2.19 Å)38 and a C1–P1–Ga1 angle of 105.02(5)°(Fig. 2, left), minimally narrower than in C, resulting in a close contact between Ga1 and one of the flanking Dip-groups of the DipTer-moiety (d(Ga1–C19) = 2.8869(15) Å).17 The Ga–CCp* distances range from 2.1526(18) to 2.5202(17), with two short and three rather long contacts, indicating that in the solid state the Cp* substituent is not perfectly η5-coordinated.
 | ||
| Fig. 2 Molecular structures of 1a (left), 1b (middle) and 3 (right). ORTEPs drawn at 50% probability. Selected bond lengths (Å) and angles (°) of 1a: P1–Ga1 2.2104(5), Ga1–C19 2.8869(15); C1–P1–Ga1 105.02(5); 1b: P1–Ga1 2.2176(5), Ga1–C7 2.8555(16); C1–P1–Ga1 104.44(6); 3: P1–C31 1.6833(12), C311–O1 1.1559(14); C1–P1–C311 105.17(5), P1–C31–O1 162.45(10). | ||
Starting from TipTerP(PMe3), TipTerPGaCp* (1b, 49%), was synthesized in similar fashion and X-ray quality crystals of 1b were grown from a saturated n-heptane solution at −30 °C, revealing similar structural parameters compared to 1a (Fig. 2, middle), with a short Ga1–P1 atomic distance of 2.2176(5) Å and a similar short Ga1⋯CTip contact (d(Ga1–C7) = 2.8555(16) Å) and a C1–P1–Ga1 angle of 104.44(6)°. This shows that the terphenyl moiety has minimal influence on the structure of 1. 1b showed a singlet signal in the 31P NMR spectrum at −109.8 ppm in C6D6 (calcd −107.9 ppm) and in the 1H NMR spectrum two characteristic resonances in a 2:1 ratio were detected for the methine protons of the TipTer-substituent, along with three doublet signals for the iPr-groups and one singlet for the Cp* substituent at Ga.
Surprisingly, even isolated 1a and 1b are thermally labile in solution. Particularly for 1b decomposition was already observed after 10 min in C6D6 solution, with new signals at 1.91 and at 0.9 ppm in the 1H NMR spectrum indicating the formation of Cp*Ga and a phosphafluorene (2b, Scheme 3), previously observed when TipTerP(PMe3) was irradiated at λ = 365 nm.39 Albeit slower, 1a showed a similar behavior in solution, decomposing to give Cp*Ga and the related phosphafluorene 2a. The thermal decomposition in C7D8 solution was then studied by 1H NMR spectroscopy over time, which revealed a 1st order decay of 1a and 1b with half lives of 47.8 and 7.8 h, respectively. This compares well with the theoretically determined free dissociation energy of 1a into DipTerP and Cp*Ga (ΔGdiss,calcd = 107.2 kJ mol−1, 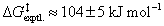 ; cf. Section in the ESI†) and corroborates the formation of 2a as decomposition product of DipTerP. Moreover, the faster decomposition of 1b can be understood in terms of electrophilic attack of the phosphinidene on one of the flanking aryl groups, which should be faster for the more electron-rich Tip-substituent present in 1b.
; cf. Section in the ESI†) and corroborates the formation of 2a as decomposition product of DipTerP. Moreover, the faster decomposition of 1b can be understood in terms of electrophilic attack of the phosphinidene on one of the flanking aryl groups, which should be faster for the more electron-rich Tip-substituent present in 1b.
Electronic structure
The electronic structure of 1a was probed using NBO,40,41 AIM,42,43 and ELF analyses.44 The P–Ga bonding is best described as a polarized double bond (Scheme 4): The σ bond, which is formed by the 3px(P) and 4s(Ga) orbitals, is evenly distributed between both atoms. The π bond, on the other hand, is strongly polarized towards the P atom [∼87% 3pz(P), ∼13% 4pz(Ga); resonances type II and III], which agrees with the NBO data for known variants A and B.23,24 The polarization of the P–Ga double bond is also reflected in the natural charges of P (−0.28e) and Ga (+1.11e) as well as in the Wiberg bond index of 1.31. In structure II-a, there are two formal lone valences at the Ga atom (4px and 4py orbitals), which are stabilized by donor–acceptor interactions with the s-type LP at the P atom (resonance I) as well as the Cp* ligand (resonances of type b), respectively (see also Scheme S2†). Note that resonance I, to which we attribute only a small weight, should be understood in terms of a non-classical multiple bond, i.e. the LP(P) → 4px(Ga) interaction is not a π bond (in-depth description of the electronic structure, p. S40ff†). Inspection of the Electron Localization Function (ELF, Fig. 3) supported the results from NBO analysis, with a LP on the P atom and strongly polarized π-electron density towards the P atom perpendicular to the Ga–P–CTer plane, in line with resonance between Lewis structures II and III (Scheme 3).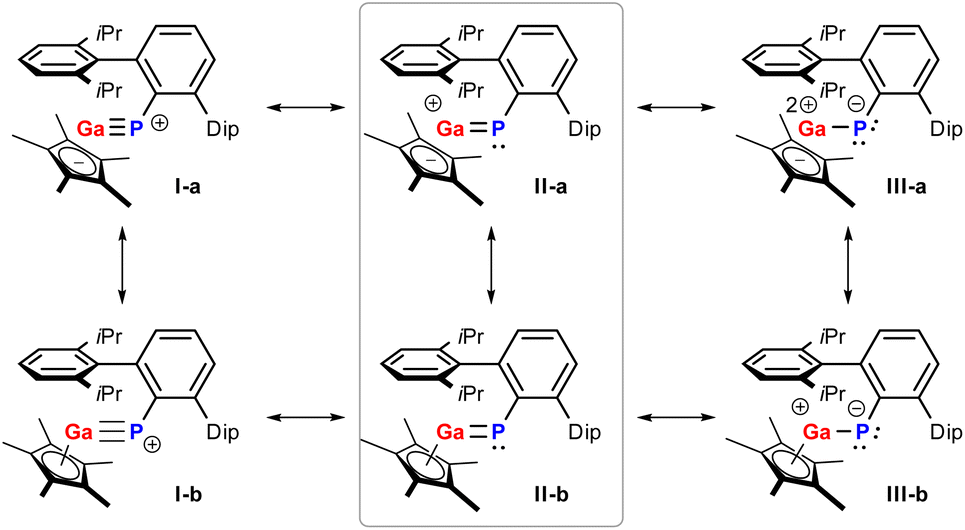 | ||
| Scheme 4 Lewis resonance scheme of compound 1a. | ||
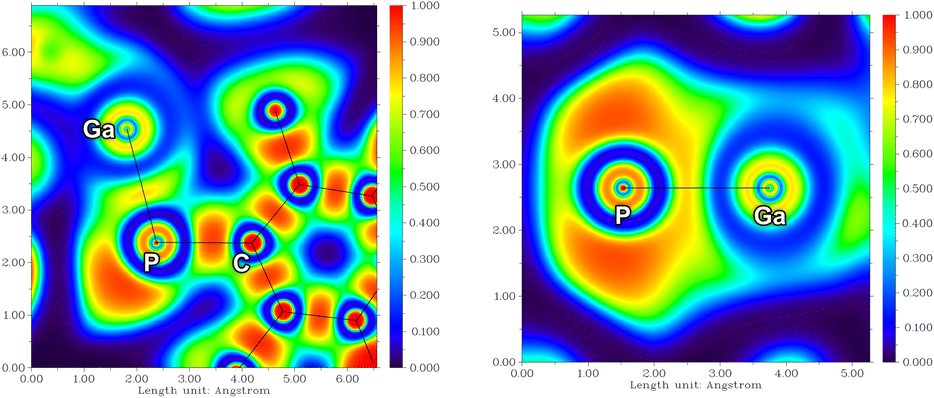 | ||
| Fig. 3 ELF of 1a in the Ga–P–C plane (left) and perpendicular to that plane (right). | ||
Inspection of the Laplacian of the electron density ∇2ρ corroborates charge accumulation near the P atom in the π bonding system, in agreement with the NBO and ELF results. The ellipticity ε at the bond critical point (BCP) of 0.31 indicates double bond character (cf. ∼0.3 for ethylene, ∼0.2 for benzene; Table S15†).
Reactivity studies
Next, we tested the reactivity of 1a towards CO2. Phosphagellenes were previously shown to react with CO2 to give five-membered ring species23 or to reversibly insert two CO2 molecules into the Ga–P bond.24 When a green toluene-d8 solution of 1a was exposed to an atmosphere of CO2 a color change to orange was observed after 20 min at room temperature.A resonance in the 31P{1H} NMR spectrum at −201.3 ppm, along with a doublet in the 13C NMR spectrum at 199.9 ppm (1JPC = 102.4) indicated the formation of the arylphosphaketene DipTerPCO (3), which was verified by SC-XRD experiments on crystals grown from n-hexane at −30 °C. Additionally, a broad feature between 1.65–2.01 ppm in the 1H NMR spectrum with an integral value of ca. 15 indicated the formation of various Cp*-containing species. The PCO unit in 3 is in-plane with the central aryl ring of the DipTer-substituent, and deviates from linearity (⦠(P1–C31–CO) = 162.45(10)°) with short P1–C31 (1.6833(12) Å) and C31–O1 (1.1559(14) Å) distances (Fig. 2, right). Surprisingly, 3 represents the first structurally characterized arylphosphaketene. Phosphaketene, tBu–PCO, was first isolated by Appel and Paulen in 1983 through treatment of tBuP(SiMe3)2 with phosgene at −70 °C, with a characteristic 31P NMR shift of −180 ppm. In the same year Mes*PCO (Mes* = 2,4,6-tBu3C6H2) was isolated as stable orange crystalline solid,45 and has been utilized as phosphinidene transfer reagent.46,47 Recently, the chemistry of phosphaketenes has seen a resurgance based on Na(dioxane)xPCO as an easily accessible form of the phosphaethynolate anion [PCO]−,48 allowing to access heteroatom-substituted E–PCO systems through salt metathesis reactions.23,24,35–37,49–51 The deoxygenation of CO2 to access 3 is reminiscent of the bora-phospha-Wittig reaction, in which phosphaborenes are utilized to make phosphaalkenes.52 Due to the presence of Cp*-containing impurities it was not possible to isolate 3 in pure form, however, it can be generated in situ in clean fashion, which will allow further reactivity studies in the future.
With H2O 1a reacted to give DipTerPH2 (4) quantitatively,53 clearly underlining the potential of 1a to act as an deoxygenation reagent, while also clearly deviating from the reactivity of DipTerP(PMe3) towards H2O, which afforded the primary phosphine oxide DipTerP(O)H2 instead.54
Conclusion
Phosphagallenes have previously been obtained by reacting phosphaketenes with the Ga(I) source DipNacnacGa as thermally stable entities. Here we show that DipTerPGaCp* (1a) is only formed upon irradiation of DipTerP(PMe3) in the presence of Cp*Ga. Under thermal conditions 1a reacted with PMe3 back to the starting materials in a 2nd-order reaction, indicating a facile and reversible PMe3 for Cp*Ga exchange at a phosphinidene. When removing PMe3 while irradiating the reaction mixture, 1a and 1b were isolated. However, in solution the isolated phosphagallenes dissociate into the phosphinidene ArTerP and Cp*Ga. ArTerP is highly reactive and formed the phosphafluorenes 2a and 2b. The bonding in 1a was investigated by combined theoretical means (NBO, ELF, AIM), clearly showing a strongly polarized (towards P) P–Ga bond with significant double bond character. Considering the longer half-life of 1a, its reactivity towards CO2 and H2O was probed, giving phosphaketene 3 and phosphine 4, respectively. Further reactivity studies to harness the reversible formation of 1a and its lability with respect to phosphinidene release are currently underway.Data availability
Crystallographic data for 1a, 1b and 3 has been deposited at the CCDC under 2216822–2216824. The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
J. P., J. B. and C. H.-J. conceptualized the project and designed the experiments. T. T., J. B. and C. H.-J. carried out most of the experimental work. T. T., F. D., J. B., J. P. and C. H.-J. characterized the compounds and analyzed the data. D. M., J. B. and C. H.-J. performed the in situ NMR studies and analyzed the data. J. B. carried out the computational studies. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. D. and C. H.-J. thank the Leibniz Association for funding within the scope of the Leibniz ScienceCampus Phosphorus Research Rostock (https://www.sciencecampus-rostock.de). We wish to thank the ITMZ at the University of Rostock for access to the cluster computer, and especially Malte Willert for his assistance with the queuing system and software installations.Notes and references
- L. Weber, Chem. Rev., 1992, 92, 1839–1906 CrossRef CAS
.
- P. P. Power, Chem. Rev., 1999, 99, 3463–3504 CrossRef CAS PubMed
- Y. Wang and G. H. Robinson, Chem. Commun., 2009, 5201–5213 RSC
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed
- P. P. Power, Acc. Chem. Res., 2011, 44, 627–637 CrossRef CAS PubMed
- P. Bag, C. Weetman and S. Inoue, Angew. Chem., Int. Ed., 2018, 57, 14394–14413 CrossRef CAS PubMed
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed
- J.-D. Guo and T. Sasamori, Chem.–Asian J., 2018, 13, 3800–3817 CrossRef CAS PubMed
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS
- R. L. Melen, Science, 2019, 363, 479–484 CrossRef CAS PubMed
- R. Borthakur and V. Chandrasekhar, Coord. Chem. Rev., 2021, 429, 213647 CrossRef CAS
- F. Dankert and C. Hering-Junghans, Chem. Commun., 2022, 58, 1242–1262 RSC
-
S. Schulz, in Advances in Organometallic Chemistry, Academic Press, 2003, vol. 49, pp. 225–317 Search PubMed
- M. A. Malik, M. Afzaal and P. O'Brien, Chem. Rev., 2010, 110, 4417–4446 CrossRef CAS PubMed
- M. Kapitein and C. von Hänisch, Eur. J. Inorg. Chem., 2015, 2015, 837–844 CrossRef CAS
- M. Kapitein, M. Balmer, L. Niemeier and C. von Hänisch, Dalton Trans., 2016, 45, 6275–6281 RSC
- S. Nees, F. Fantuzzi, T. Wellnitz, M. Fischer, J.-E. Siewert, J. T. Goettel, A. Hofmann, M. Härterich, H. Braunschweig and C. Hering-Junghans, Angew. Chem., Int. Ed., 2021, 60, 24318–24325 CrossRef CAS PubMed
- M. Fischer, S. Nees, T. Kupfer, J. T. Goettel, H. Braunschweig and C. Hering-Junghans, J. Am. Chem. Soc., 2021, 143, 4106–4111 CrossRef CAS PubMed
- S. Nees, T. Wellnitz, F. Dankert, M. Härterich, S. Dotzauer, M. Feldt, H. Braunschweig and C. Hering-Junghans, Angew. Chem., Int. Ed., 2023, e202215838 CAS
- P. Gupta, J.-E. Siewert, T. Wellnitz, M. Fischer, W. Baumann, T. Beweries and C. Hering-Junghans, Dalton Trans., 2021, 50, 1838–1844 RSC
- C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, Angew. Chem., Int. Ed., 1991, 30, 564–565 CrossRef
- H. Sitzmann, M. F. Lappert, C. Dohmeier, C. Üffing and H. Schnöckel, J. Organomet. Chem., 1998, 561, 203–208 CrossRef CAS
- D. W. N. Wilson, J. Feld and J. M. Goicoechea, Angew. Chem., Int. Ed., 2020, 59, 20914–20918 CrossRef CAS PubMed
- M. K. Sharma, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 6784–6790 CrossRef CAS PubMed
- M. K. Sharma, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 21784–21788 CrossRef CAS PubMed
- M. K. Sharma, C. Wölper and S. Schulz, Dalton Trans., 2022, 51, 1612–1616 RSC
- J. Feld, D. W. N. Wilson and J. M. Goicoechea, Angew. Chem., Int. Ed., 2021, 60, 22057–22061 CrossRef CAS PubMed
- D. Loos and H. Schnöckel, J. Organomet. Chem., 1993, 463, 37–40 CrossRef CAS
- P. Jutzi and L. O. Schebaum, J. Organomet. Chem., 2002, 654, 176–179 CrossRef CAS
- M. Fischer, M. M. D. Roy, L. L. Wales, M. A. Ellwanger, A. Heilmann and S. Aldridge, J. Am. Chem. Soc., 2022, 144, 8908–8913 CrossRef CAS PubMed
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1493 CAS
-
H. Jonsson, G. Mills and K. W. Jacobsen, in Classical and Quantum Dynamics in Condensed Phase Simulations, 1998, pp. 385–404 Search PubMed
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed
- M. M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 15885–15888 CrossRef CAS PubMed
- M. M. Hansmann, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 8356–8359 CrossRef CAS PubMed
- L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CAS
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed
- S. Shah, M. C. Simpson, R. C. Smith and J. D. Protasiewicz, J. Am. Chem. Soc., 2001, 123, 6925–6926 CrossRef CAS PubMed
-
E. D. Glendening, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013 Search PubMed
- F. Weinhold, C. R. Landis and E. D. Glendening, Int. Rev. Phys. Chem., 2016, 35, 399–440 Search PubMed
-
R. F. W. Bader, Atoms in Molecules: A Quantum Theory,Oxford University Press, 1994 Search PubMed
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS
- R. Appel and W. Paulen, Angew. Chem., Int. Ed. Engl., 1983, 22, 785–786 Search PubMed
- A. H. Cowley, B. Pellerin, J. L. Atwood and S. G. Bott, J. Am. Chem. Soc., 1990, 112, 6734–6735 CrossRef CAS
- A. H. Cowley, F. Gabbai, R. Schluter and D. Atwood, J. Am. Chem. Soc., 1992, 114, 3142–3144 CrossRef CAS
- J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS PubMed
- A. R. Jupp and J. M. Goicoechea, J. Am. Chem. Soc., 2013, 135, 19131–19134 CrossRef CAS PubMed
- D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 5920–5928 RSC
- K. M. Szkop, A. R. Jupp, H. Razumkov and D. W. Stephan, Dalton Trans., 2020, 49, 885–890 RSC
- A. M. Borys, E. F. Rice, G. S. Nichol and M. J. Cowley, J. Am. Chem. Soc., 2021, 143, 14065–14070 CrossRef CAS PubMed
- W. A. Merrill, E. Rivard, J. S. DeRopp, X. Wang, B. D. Ellis, J. C. Fettinger, B. Wrackmeyer and P. P. Power, Inorg. Chem., 2010, 49, 8481–8486 CrossRef CAS PubMed
- F. Dankert, M. Fischer and C. Hering-Junghans, Dalton Trans., 2022, 51, 11267–11276 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, NMR spectra, crystallographic, and computational details. CCDC 2216822–2216824. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06292e |
| This journal is © The Royal Society of Chemistry 2023 |