
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed enantioselective diyne cyclization via C(sp2)–O bond cleavage†
Ji-Jia
Zhou‡
a,
Ya-Nan
Meng‡
a,
Li-Gao
Liu
a,
Yi-Xi
Liu
a,
Zhou
Xu
 *b,
Xin
Lu
*a,
Bo
Zhou
a and
Long-Wu
Ye
*ac
*b,
Xin
Lu
*a,
Bo
Zhou
a and
Long-Wu
Ye
*ac
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Key Laboratory of Chemical Biology of Fujian Province, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: longwuye@xmu.edu.cn; xinlu@xmu.edu.cn; xuzhou@xzhmu.edu.cn
bJiangsu Key Laboratory of New Drug Research and Clinical Pharmacy, School of Pharmacy, Xuzhou Medical University, Xuzhou 221004, China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 1st March 2023
Abstract
The functionalization of etheric C–O bonds via C–O bond cleavage is an attractive strategy for the construction of C–C and C–X bonds in organic synthesis. However, these reactions mainly involve C(sp3)–O bond cleavage, and a catalyst-controlled highly enantioselective version is extremely challenging. Here, we report a copper-catalyzed asymmetric cascade cyclization via C(sp2)–O bond cleavage, allowing the divergent and atom-economic synthesis of a range of chromeno[3,4-c]pyrroles bearing a triaryl oxa-quaternary carbon stereocenter in high yields and enantioselectivities. Importantly, this protocol not only represents the first [1,2]-Stevens-type rearrangement via C(sp2)–O bond cleavage, but also constitutes the first example of [1,2]-aryl migration reactions via vinyl cations.
Introduction
The functionalization of etheric C–O bonds via C–O bond cleavage is an attractive strategy for the construction of C–C and C–X bonds in organic synthesis as ethers are stable and readily available building blocks.1 Nevertheless, the functionalization of an etheric C–O bond is extremely challenging because of its high bond dissociation energy (BDE, 100–110 kcal mol−1, Scheme 1a).2 Currently, reactions involving C–O bond cleavage of ethers mainly focus on C–O bond activation and formal C–O bond insertion.1,3 The C–O bond activation of ethers, first realized in 1979 by E. Wenkert,4 has been demonstrated to be an effective approach for ether bond functionalization via transition metal (Ni, Pd, Ru, Fe, Cr, and Rh) catalysis,1,5 but no direct catalytic asymmetric activation of aromatic C–O bonds has been reported to the best of our knowledge.6 Formal C–O bond insertions, mainly involving [1,2]-Stevens-type rearrangement reactions of oxonium ylides, have received extensive attention in the past few decades. However, these reactions generally involve the migration of the benzyl and allyl groups via C(sp3)–O bond cleavage,3,7 and such a catalyst-controlled highly enantioselective rearrangement is also highly challenging.8 Importantly, the related aryl migration reaction via C(sp2)–O bond cleavage has not been explored yet (Scheme 1b).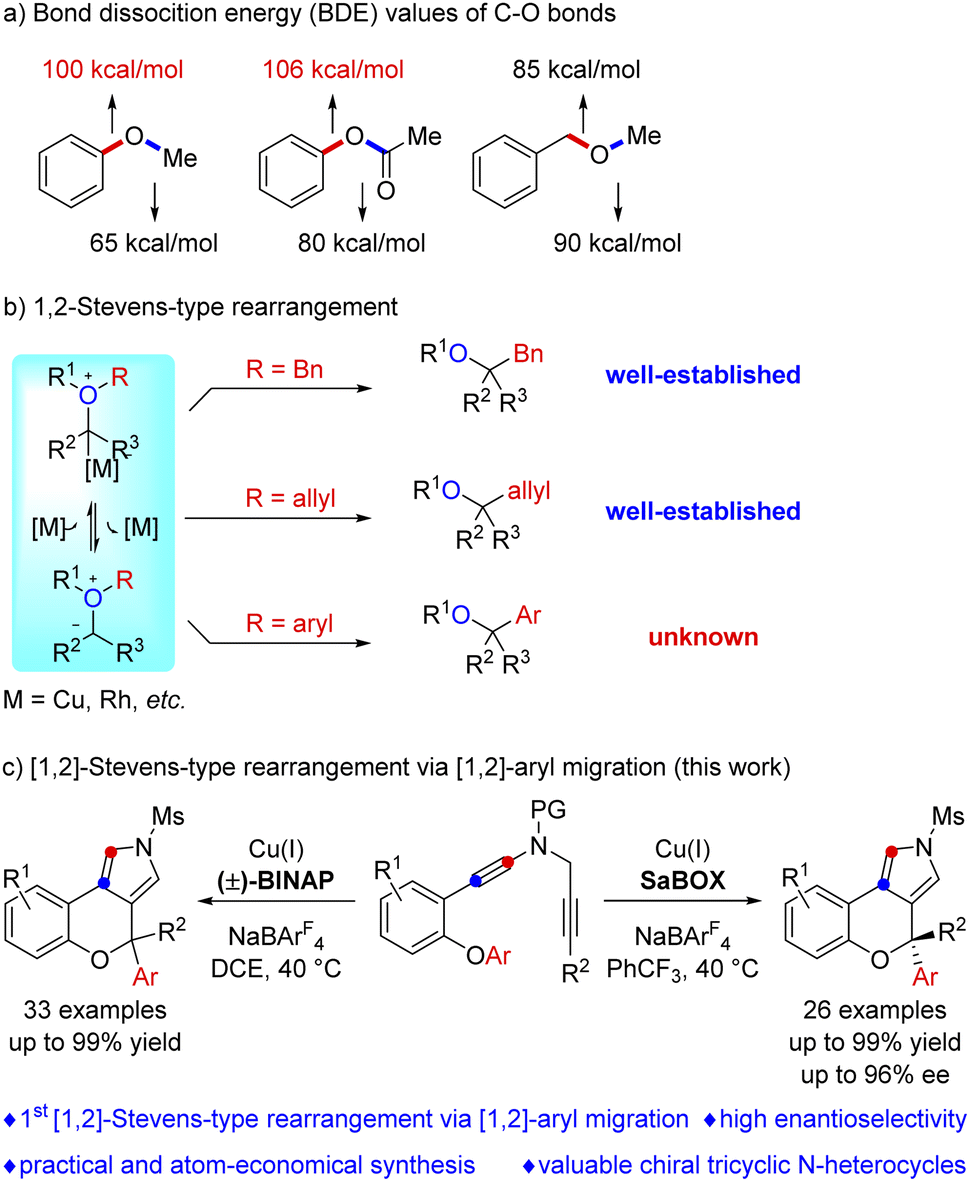 | ||
| Scheme 1 Functionalization of etheric C–O bonds involving [1,2]-Stevens-type rearrangement. | ||
Vinyl cations have proven to be versatile intermediates in organic synthesis and have attracted particular attention over the past decade due to their unique carbene-like reactivity.9 However, successful examples of asymmetric catalysis based on vinyl cation intermediates have been scarcely reported. Very recently, we have developed a facile copper-catalyzed diyne cyclization for the generation of vinyl cations,10,11 and achieved a series of asymmetric reactions by this strategy via a remote control of enantioselectivity, including intramolecular aromatic C(sp2)–H functionalization,10e vinylic C(sp2)–H functionalization,10c cyclopropanation,10e [1,2]-Stevens-type rearrangement10b and intermolecular annulations with styrenes10d and ketones.10a Inspired by the above results and by our recent study on developing ynamide chemistry for heterocycle synthesis,12,13 we envisioned that replacing the benzyl group with an aryl group may lead to C(sp2)–O bond cleavage by employing this asymmetric copper catalysis. Herein, we describe the realization of such a copper-catalyzed enantioselective cascade cyclization via C(sp2)–O bond cleavage, allowing the divergent and atom-economic synthesis of various chromeno[3,4-c]pyrroles bearing a triaryl oxa-quaternary carbon stereocenter in generally moderate to excellent yields with high enantioselectivities (Scheme 1c). Of note, structural motifs containing a triaryl oxa-quaternary carbon are widely present in natural products and drug molecules.14 To the best of our knowledge, this protocol not only represents the first [1,2]-Stevens-type rearrangement via C(sp2)–O bond cleavage, but also constitutes the first example of [1,2]-aryl migration reactions via vinyl cations.
Results and discussion
We started our investigations by using OPh-substituted N-propargyl ynamide 1a as the model substrate, and selected results are listed in Table 1. To our delight, the desired chromeno[3,4-c]pyrrole 2a bearing an oxo-quaternary carbon stereocenter could be obtained in 62% yield in the presence of Cu(CH3CN)4PF6 (10 mol%) as the catalyst according to our designed [1,2]-aryl migration, albeit together with a significant amount of seven-membered product 2a′via a Cu-catalyzed 7-endo-dig cyclization (Table 1, entry 1). Further studies revealed that the use of NaBArF4 (12 mol%)15 as the additive completely prohibited the formation of byproduct 2a′, and the expected 2a was delivered in 87% yield (Table 1, entry 2). Of note, NaBArF4 has been widely used as an additive in transition-metal catalysis to enhance the acidity and/or solubility of metal catalysts, thus leading to significantly improved reactivity and selectivity.10d Next, various typical solvents including DCM, toluene, PhCF3, Et2O and THF were screened, but failed to give better results (Table 1, entries 3–7). Subsequently, several copper catalysts were also evaluated, and it was found that the use of Cu(CH3CN)4BF4, CuOTf or CuBr as the catalyst led to a slightly decreased yield (Table 1, entries 8, 9, and 11), whereas CuI could not catalyze this cascade cyclization (Table 1, entry 12). When Cu(OTf)2 was used as the catalyst, 2a′ was obtained as the main product in 40% yield (Table 1, entry 10). Gratifyingly, the desired 2a could be formed in nearly quantitative yield by employing 12 mol% (±)-BINAP as the ligand (Table 1, entry 13). Finally, decreasing the reaction temperature to 30 °C or 20 °C led to slightly decreased yields (Table 1, entries 14 and 15). Notably, no background benzofuran formation was observed via a direct 5-endo-dig cyclization in all cases under these Cu-catalyzed conditions.|
|
|||
|---|---|---|---|
| Entry | [Cu] | Reaction conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.05 mmol), [Cu] (0.005 mmol), NaBArF4 (0.006 mmol), solvent (1 mL), 20–40 °C, 2–40 h, in vials. b Measured by 1H NMR using 1,3,5-trimethoxybenzene as the internal standard. c Without NaBArF4. d Yield of 2a′. e With (±)-BINAP (0.006 mmol). Ms = methanesulfonyl, PMP = 4-methoxyphenyl, NaBArF4 = sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate, DCE = 1,2-dichloroethane. | |||
| 1c | Cu(CH3CN)4PF6 | DCE, 40 °C, 2 h | 62(15d) |
| 2 | Cu(CH3CN)4PF6 | DCE, 40 °C, 2 h | 87 |
| 3 | Cu(CH3CN)4PF6 | DCM, 40 °C, 2 h | 86 |
| 4 | Cu(CH3CN)4PF6 | Toluene, 40 °C, 2 h | 72 |
| 5 | Cu(CH3CN)4PF6 | PhCF3, 40 °C, 2 h | 78 |
| 6 | Cu(CH3CN)4PF6 | Et2O, 40 °C, 2 h | <10 |
| 7 | Cu(CH3CN)4PF6 | THF, 40 °C, 2 h | <10 |
| 8 | Cu(CH3CN)4BF4 | DCE, 40 °C, 2 h | 86 |
| 9 | CuOTf | DCE, 40 °C, 2 h | 81 |
| 10 | Cu(OTf)2 | DCE, 40 °C, 2 h | 32(40d) |
| 11 | CuBr | DCE, 40 °C, 2 h | 85 |
| 12 | CuI | DCE, 40 °C, 2 h | <10 |
| 13 | Cu(CH 3 CN) 4 PF 6 | DCE, 40 °C, 2 h | 99 |
| 14e | Cu(CH3CN)4PF6 | DCE, 30 °C, 12 h | 90 |
| 15e | Cu(CH3CN)4PF6 | DCE, 20 °C, 40 h | 84 |
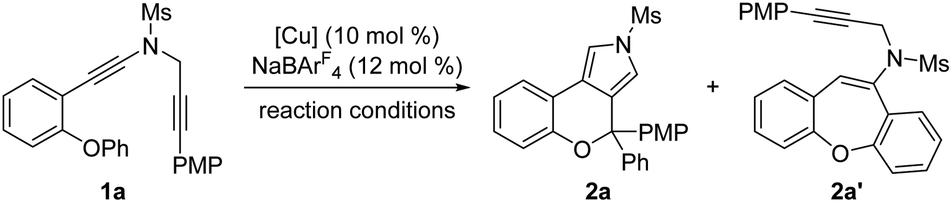
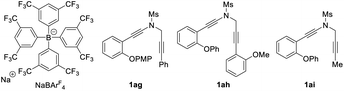
Having established the optimal reaction conditions (Table 1, entry 13), we next sought to explore the scope of this cascade reaction. As shown in Table 2, we initially investigated the scope of different N-protecting groups of the N-propargyl ynamides. Apart from the Ms-protected ynamide, the reaction proceeded smoothly with other alkyl sulfonyl groups, providing the corresponding chromeno[3,4-c]pyrroles 2b and 2c bearing an oxo-quaternary carbon stereocenter in 82–85% yields. Then, diynes with a diverse array of aryl sulfonyl groups, such as Bs (4-bromobenzenesulfonyl), Ts, and MBS were suitable substrates for this cascade cyclization, affording the desired products 2d–2h in generally good to excellent yields. The role of the electronic properties of the aromatic ring was next studied, and it was found that substitutions including both electron-donating and -withdrawing groups on the aromatic ring occurred efficiently, delivering the corresponding products 2i–2m with yields ranging from 88% to 93%. Substitutions with various electronic properties at the different positions of the phenyl ring had little effect on this protocol, and the expected chromeno[3,4-c]pyrroles 2n–2s were obtained in 54–92% yields. In addition, different aryl-substituted diynes on N-propargyl moieties (Ar2) proved to be applicable substrates for this reaction, thus leading to anticipated products 2t–2w in high yields. Ynamides bearing disubstituents on the aryl ring could participate in this transformation as well to give rise to 2x–2z in 83–95% yields. Furthermore, the reaction was also extended to naphthyl- and thienyl-substituted diynes, enabling an approach to the expected products 2aa in 87% yield and 2ab in 71% yield, respectively. Finally, we examined the effect of the migratory aryl groups, and found that the reaction of 4-Me, 4-OMe, 4-Ph and 4-Cl substituted diynes was also well compatible with the established conditions, thus providing the desired chromeno[3,4-c]pyrroles 2ac–2af in 52–90% yields. Of note, the synthesis of product 2a could also be achieved in 77% yield from ynamide substrate 1ag.15 Attempts to extend the reaction to ynamide 1ah bearing a substituent at the ortho position of the aryl group of Ar2 and ynamide 1ai with an alkyl group only gave a complex mixture of products.15
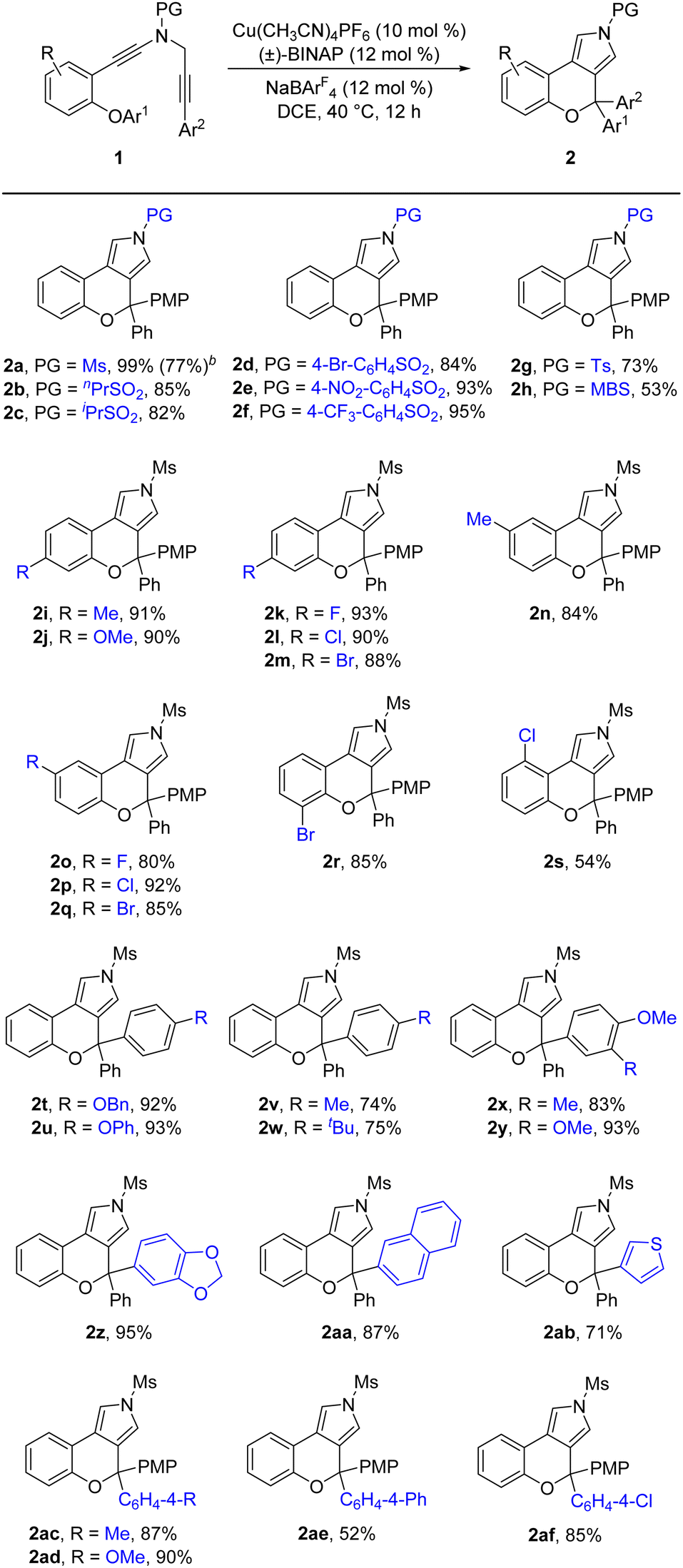
After accomplishing the above copper-catalyzed cascade cyclization/[1,2]-aryl migration reaction of N-propargyl ynamides, we attempted to establish the catalytic asymmetric version of this reaction (for more details see ESI Tables 1 and 2).† As summarized in Table 3, we were pleased to find that the expected chiral chromeno[3,4-c]pyrrole (+)-2a bearing a oxo-quaternary carbon stereocenter could be furnished in 75% yield with 75% ee by the use of phenyl-substituted BOX (bisoxazoline) ligand L1 (Table 3, entry 1). Encouraged by this preliminary result, we next investigated assorted types of chiral BOX ligands. However, employing several typical chiral BOX ligands L2–L6 led to diminished yields and enantioselectivities (Table 3, entries 2–6). In view of the fact that BOX ligands could be readily modified with the side-arm strategy established by Tang's group,16 we attempted to alter the dibenzyl group of the BOX ligand. Gratifyingly, the enantioselectivity was significantly improved by employing 3,5-diethyoxyl benzyl-substituted BOX ligand L14 after a large amount of explorations (Table 3, entries 7–17), and the desired chiral chromeno[3,4-c]pyrrole (+)-2a was delivered in 82% yield with 92% ee (Table 3, entry 14). Among various typical organic solvents screened for this cascade cyclization (Table 3, entries 18–21), PhCF3 was found to be optimal, providing the anticipated (+)-2a in 92% yield with 94% ee (Table 3, entry 19).
|
|
||||
|---|---|---|---|---|
| Entry | L | Reaction conditions | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.05 mmol), Cu(CH3CN)4PF6 (0.005 mmol), L (0.006 mmol), NaBArF4 (0.006 mmol), solvent (1 mL), in vials. b Measured by 1H NMR using 1,3,5-trimethoxybenzene as the internal standard. c Determined by HPLC analysis. | ||||
| 1 | L1 | DCE, 40 °C, 12 h | 75 | 75 (+) |
| 2 | L2 | DCE, 40 °C, 18 h | <10 | — |
| 3 | L3 | DCE, 40 °C, 24 h | 42 | 10 (+) |
| 4 | L4 | DCE, 40 °C, 12 h | 71 | 58 (+) |
| 5 | L5 | DCE, 40 °C, 12 h | 74 | 74 (+) |
| 6 | L6 | DCE, 40 °C, 12 h | 72 | 69 (+) |
| 7 | L7 | DCE, 40 °C, 12 h | 74 | 75 (+) |
| 8 | L8 | DCE, 40 °C, 12 h | 73 | 72 (+) |
| 9 | L9 | DCE, 40 °C, 12 h | 71 | 72 (+) |
| 10 | L10 | DCE, 40 °C, 12 h | 71 | 70 (+) |
| 11 | L11 | DCE, 40 °C, 18 h | 72 | 58 (+) |
| 12 | L12 | DCE, 40 °C, 12 h | 75 | 77 (+) |
| 13 | L13 | DCE, 40 °C, 12 h | 80 | 91 (+) |
| 14 | L14 | DCE, 40 °C, 12 h | 82 | 92 (+) |
| 15 | L15 | DCE, 40 °C, 16 h | 80 | 92 (+) |
| 16 | L16 | DCE, 40 °C, 16 h | 79 | 92 (+) |
| 17 | L17 | DCE, 40 °C, 30 h | 65 | <10 (+) |
| 18 | L14 | Toluene, 40 °C, 10 h | 82 | 93 (+) |
| 19 | L14 | PhCF3, 40 °C, 12 h | 92 | 94 (+) |
| 20 | L14 | PhF, 40 °C, 10 h | 85 | 92 (+) |
| 21 | L14 | PhCl, 40 °C, 10 h | 84 | 92 (+) |
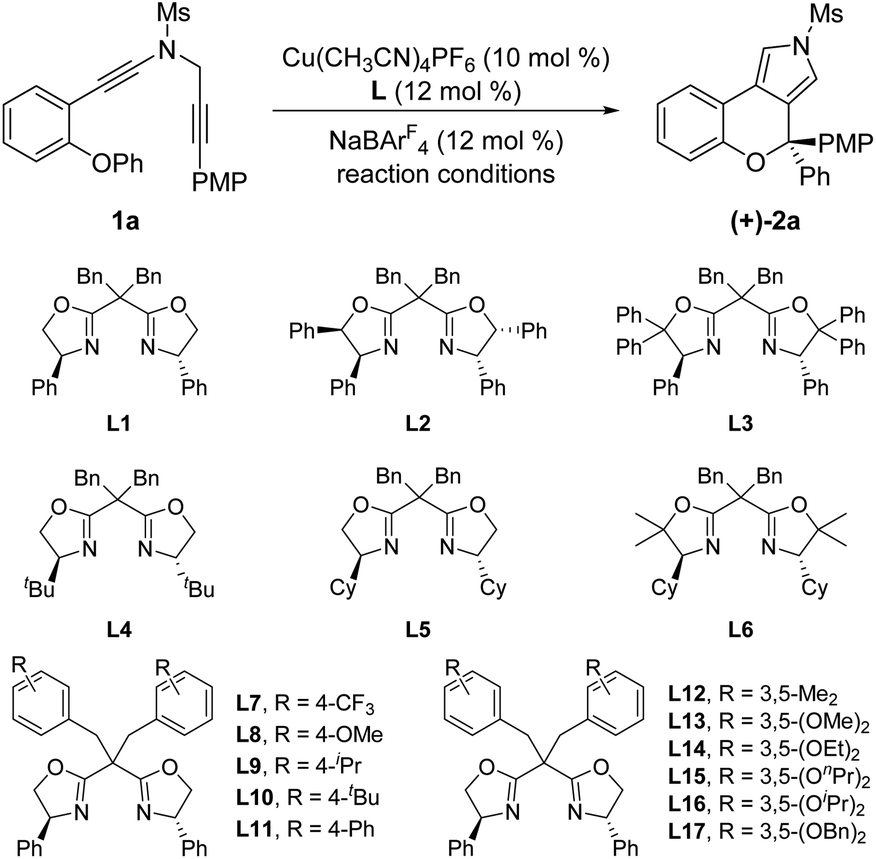
Under the optimized conditions for the asymmetric version of this cascade cyclization (Table 3, entry 19), the substrate scope for synthesis of enantioenriched chromeno[3,4-c]pyrroles was then examined. As depicted in Table 4, except for the Ms-protected ynamide 1a, this enantioselective cyclization is remarkably tolerant of various N-protected ynamides, including nPrSO2-, iPrSO2-, Bs-, 4-NO2-C6H4SO2-, 4-CF3-C6H4SO2-, Ts- and MBS-protected N-propargyl ynamides, thus affording the corresponding chiral chromeno[3,4-c]pyrroles (+)-2b–(+)-2h in 60–99% yields and 78–94% ees. Furthermore, this asymmetric process readily accommodated substitution at the 3- or 4-position of the phenyl ring with electron-deficient and -rich substituents comprising Me, OMe, F, Cl, and Br, enabling the assembly of the expected enantioenriched chromeno[3,4-c]pyrroles (+)-2i–(+)-2n in generally excellent yields with excellent enantioselectivities. Additionally, switching the PMP-substituent of diyne to other electron-rich substituents such as OBn-, OPh-, Me-, and tBu-substituted diynes led to the asymmetric rearrangement process to furnish the desired products (+)-2t–(+)-2w in high yields and enantiopurities. Of note, the reaction time was considerably prolonged on decreasing the electronic density of the phenyl groups, such as Me- and tBu-substituted ynamides 1v and 1w, which is similar to our previous reports.10 What's more, diynes bearing different disubstituents on the phenyl ring were accommodated to provide the expected products (+)-2x–(+)-2z in 95–99% yields and 93–96% ees. Next, naphthyl- and thienyl-substituted N-propargyl ynamides performed well in this asymmetric tandem reaction, and the corresponding products (+)-2aa and (+)-2ab were isolated in 86% yield with 93% ee and 93% yield with 95% ee, respectively. Further modifications on the substituents of the migratory aryl group such as 4-Me, 4-Ph and 4-Cl substituents were also accomplished to furnish the desired products (+)-2ac, (+)-2ae and (+)-2af in excellent yields with remarkably high enantioselectivities. Finally, the reaction occurred smoothly by employing a chiral catalyst with the opposite configuration, and delivered the desired (−)-2a with the opposite configuration in 96% yield with 94% ee. The absolute configuration of (+)-2a was confirmed by X-ray diffraction analysis (Fig. 1)17
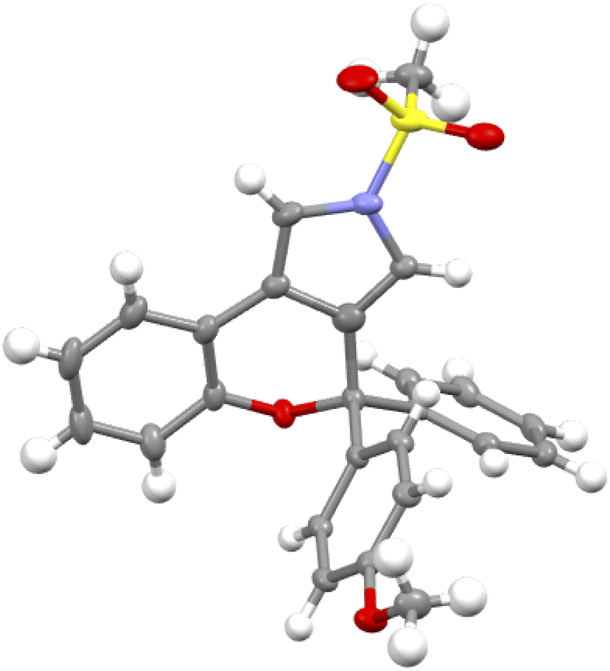 | ||
| Fig. 1 Structure of compound (+)-2a in its crystal form. | ||
Gram-scale reactions and further synthetic applications of the as-synthesized tricyclic heterocycles were then explored (Scheme 2). First, the Ms protecting group in chromeno[3,4-c]pyrrole 2a, which could be synthesized on the gram scale in 73% yield under standard conditions, was readily removed by treatment with KOH, followed by protection with the Boc group, site-selective bromination by NBS (N-bromosuccinimide) and oxidation by the UHP (urea-hydrogen peroxide complex) to lead to the corresponding products 3a (90%, 2 steps), 4a (65%, 2 steps) and 5a (60%, 2 steps), respectively (Scheme 2a). Moreover, the asymmetric cyclization reaction of 1a on the gram scale under standard conditions was also performed, and the desired (+)-2a was formed in 99% yield and 94% ee, which could be reduced into the pyrrolidine-fused product (−)-6a containing three contiguous stereocenters in 63% yield with 94% ee and 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr (Scheme 2b).
:1 dr (Scheme 2b).
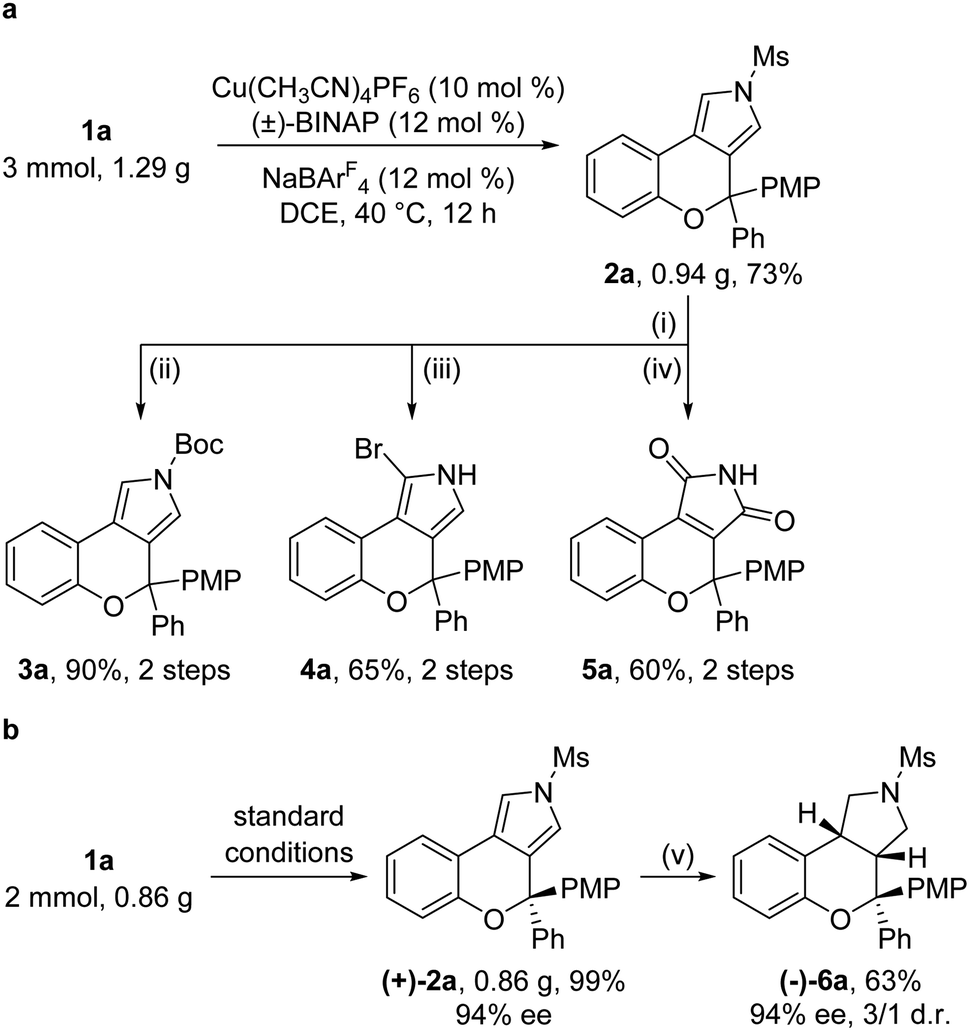 | ||
| Scheme 2 Scale-up reaction and product elaboration. Reagents and conditions: (i) KOH (5 equiv.), THF:MeOH/1:1, 50 °C, 2 h. (ii) 4-DMAP (20 mol%), (Boc)2O (3 equiv.), Et3N (4 equiv.), DCM, rt, 2 h. (iii) NBS (1.05 equiv.), THF, −78 °C, 1 h. (iv) UHP (5 equiv.), HFIP (hexafluoroisopropanol), 45 °C, 24 h. (v) H2 (7 Mpa), Pd/C (20 mol%), EtOH:AcOH/10:1, 100 °C, 3.5 h. | ||
Based on the above experimental observations, our previously published results10 and present density functional theory (DFT) calculations (for more details see the ESI†), a plausible mechanism involving vinyl cation intermediates11 for the formation of chiral chromeno[3,4-c]pyrrole (+)-2a from diyne 1a is displayed in Scheme 3. The CuI catalyst first preferentially coordinates to the electron-richer amide-tethered C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond of 1a to form precursor A, followed by intramolecular trapping by the N-propargyl moiety, affording the vinyl cation intermediate B. Subsequent intramolecular trapping of intermediate B by the phenoxy group leads to the chiral oxonium intermediate C with a free energy barrier of 51.6 kJ mol−1. Next, a [1,2]-Stevens-type rearrangement within the oxonium cation C affords the copper carbene intermediate Dvia a stereospecific one-step [1,2]-aryl migration pathway with a free energy barrier of 111.0 kJ mol−1. Finally, the same as our previous findings,10 a formal [1,4]-H migration assisted by trace H2O in the reaction system via two consecutive steps of proton transfer and demetallation occurs to deliver the final chiral product (+)-2a, with a free energy barrier of 120.3 kJ mol−1. As alluded above, the reaction can smoothly occur under the experimental conditions, with the H2O-assisted 1,4-H migration being the rate-determining step.
C bond of 1a to form precursor A, followed by intramolecular trapping by the N-propargyl moiety, affording the vinyl cation intermediate B. Subsequent intramolecular trapping of intermediate B by the phenoxy group leads to the chiral oxonium intermediate C with a free energy barrier of 51.6 kJ mol−1. Next, a [1,2]-Stevens-type rearrangement within the oxonium cation C affords the copper carbene intermediate Dvia a stereospecific one-step [1,2]-aryl migration pathway with a free energy barrier of 111.0 kJ mol−1. Finally, the same as our previous findings,10 a formal [1,4]-H migration assisted by trace H2O in the reaction system via two consecutive steps of proton transfer and demetallation occurs to deliver the final chiral product (+)-2a, with a free energy barrier of 120.3 kJ mol−1. As alluded above, the reaction can smoothly occur under the experimental conditions, with the H2O-assisted 1,4-H migration being the rate-determining step.
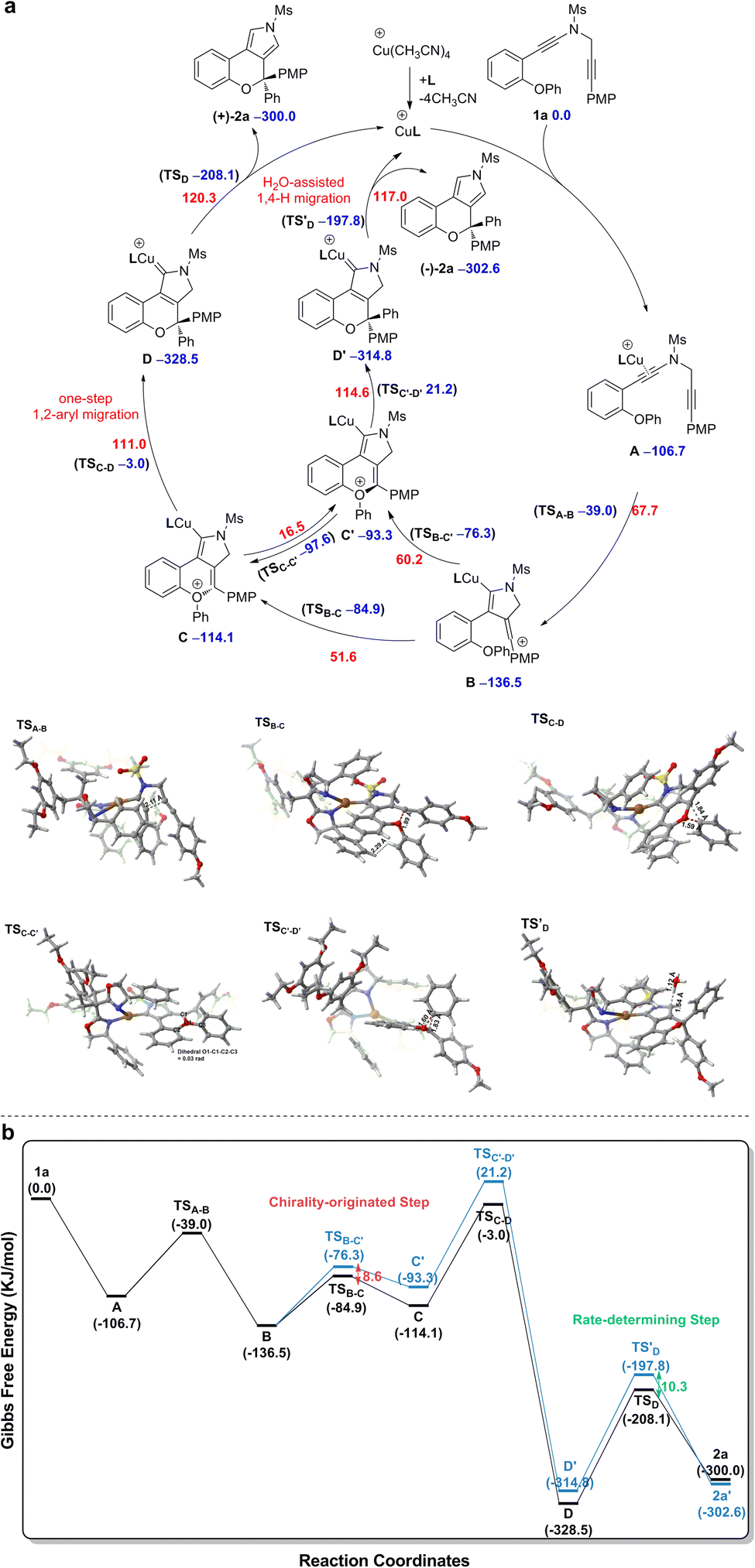 | ||
| Scheme 3 (a) Mechanism for the synthesis of chromeno[3,4-c]pyrrole (+)-2a from diyne substrate 1a. (b) Free energy profile for the synthesis of chromeno[3,4-c]pyrrole (+)-2a from diyne substrate 1a. Relative free energies (ΔG, in kJ mol−1) of all the transition states and intermediates were computed at the SMD(PhCF3)-M06/6-31G(d,p)&LANL2DZ level of theory, with the electronic energy of all the transition states and intermediates recomputed at the SMD(PhCF3)-M06-D3/def2-TZVPP level of theory. Color code: red = O; white = H; gray = C; yellow = S; blue = N; brown = Cu. | ||
The origin of enantioselective synthesis of chiral (+)-2a is also theoretically unraveled with the chiral L14 ligated to the CuI center in the process of intramolecular trapping of the vinyl cation by the phenoxy group (B → C), as the O atom orientation of the phenoxy group separates the configuration of the final chiral product in the process of intramolecular cyclization. More importantly, the difference in barrier heights of the subsequent rate-determining step, i.e. the H2O-assisted 1,4-H migration process helps maintain the enantioselectivity in a kinetic control manner, although it is unraveled by DFT computations that the chiral oxonium cation generated in the aforementioned chirality-originated step easily undergoes an inversion in the reaction.
The free energy of the transition state [CuL14]-R TSB-C is predicted to be 8.6 kJ mol−1 lower than that of [CuL14]-S TSB-C, which is well in line with the experimental ee value of 94%. Also, the difference in free energies (10.3 kJ mol−1) of the two transition states in the rate-determining step, i.e. the H2O-assisted H-migration process (TSD and TS′D) again supports the experimental ee value of 94%, which shows that there is no loss of enantioselectivity in the process from C to 2a attributed to the kinetic control of the subsequent rate-determining step as well. Further observation on their stereogenic configurations indicates that the steric repulsion between the bulky chiral ligand L14 and the branched groups in the substrate is critical to the generation of enantioselectivity in a remote control manner (Scheme 4).
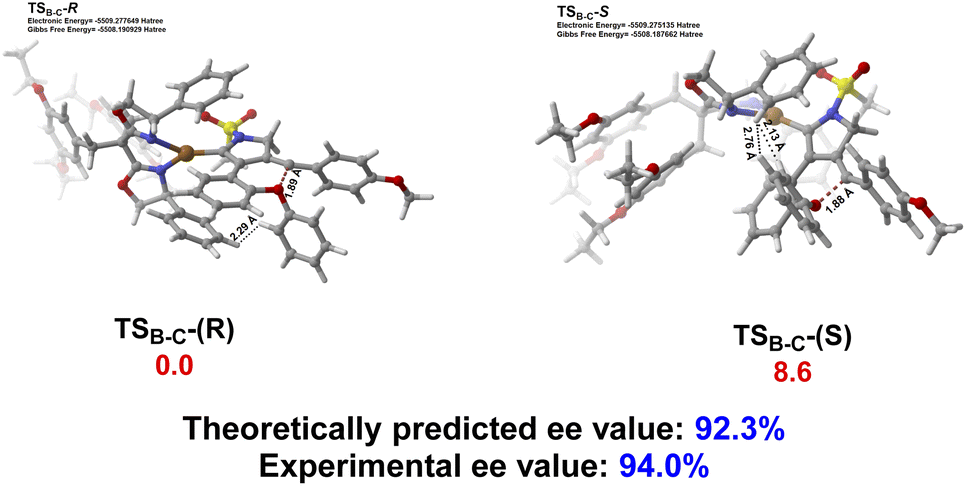 | ||
| Scheme 4 The geometries and relative free energies (ΔG, in kJ mol−1) of the transition state [CuL14]-R TSB-C and [CuL14]-S TSB-C with the chiral ligand. Relative free energies (ΔG, in kJ mol−1) of all the transition states and intermediates were computed at the SMD(PhCF3)-M06/6-31G(d,p)&LANL2DZ level of theory, with the electronic energy of all the transition states and intermediates recomputed at the SMD(PhCF3)-M06-D3/def2-TZVPP level of theory. Color code: red = O; white = H; gray = C; yellow = S; blue = N; brown = Cu. | ||
Conclusions
In summary, we have developed a copper-catalyzed asymmetric cascade cyclization via C(sp2)–O bond cleavage, which not only represents the first [1,2]-Stevens-type rearrangement via C(sp2)–O bond cleavage, but also constitutes the first example of [1,2]-aryl migration reactions via vinyl cations. This method enables the divergent, practical and atom-economic synthesis of a range of chromeno[3,4-c]pyrroles bearing a triaryl oxa-quaternary carbon stereocenter in high yields and enantioselectivities (up to 96% ee) under mild conditions. Moreover, theoretical calculations provide further evidence for this vinyl cation involved cyclization/rearrangement and the origin of enantioselectivity. We envision that the above findings will open up new horizons in the field of catalytic asymmetric reactions based on etheric C–O bond functionalization, [1,2]-Stevens-type rearrangement and vinyl cations.Data availability
Data for the crystal structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition numbers CCDC 2192857 ((+)-2a). All other data supporting the findings of this study, including experimental procedures and compound characterization, are available within the paper and its ESI files, or from the corresponding authors on request.†Author contributions
J.-J. Z., Y.-N. M., Y.-X. L., Z. X., and B. Z. performed the experiments. L.-G. L. and X. L. performed the DFT calculations. L.-W. Y. conceived and directed the project and wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from MOST (2021YFC2100100), the National Natural Science Foundation of China (22125108, 22121001 and 92056104), the Natural Science Foundation of Jiangsu Province (BK20211059), the President Research Funds from Xiamen University (20720210002), and NFFTBS (J1310024). We thank Mr Zanbin Wei from Xiamen University for assistance with X-ray crystallographic analysis.Notes and references
- For recent selected reviews, see: (a) J. Becica and D. C. Leitch, Synlett, 2021, 32, 641 CrossRef CAS; (b) B. Zhao, T. Rogge, L. Ackermann and Z. Shi, Chem. Soc. Rev., 2021, 50, 8903 RSC; (c) Z. Qiu and C.-J. Li, Chem. Rev., 2020, 120, 10454 CrossRef CAS PubMed; (d) T. B. Boit, A. S. Bulger, J. E. Dander and N. K. Garg, ACS Catal., 2020, 10, 12109 CrossRef CAS PubMed.
- K. Chen, X.-S. Zhang and Z.-J. Shi, Pure Appl. Chem., 2014, 86, 361 CrossRef CAS.
- For recent selected reviews, see: (a) V. N. Nair and U. K. Tambar, Org. Biomol. Chem., 2022, 20, 3427 RSC; (b) C. Empel, S. Jana and R. M. Koenigs, Synthesis, 2021, 53, 4567 CrossRef CAS; (c) S. Jana, Y. Guo and R. M. Koenigs, Chem.–Eur. J., 2021, 27, 1270 CrossRef CAS PubMed; (d) G. K. Murphy, C. Stewart and F. G. West, Tetrahedron, 2013, 69, 2667 CrossRef CAS; (e) D. M. Hodgson, F. Y. T. M. Pierard and P. A. Stupple, Chem. Soc. Rev., 2001, 30, 50 RSC.
- E. Wenkert, E. L. Michelotti and C. S. Swindell, J. Am. Chem. Soc., 1979, 101, 2246 CrossRef CAS.
- For selected reviews, see: (a) Y.-Y. Huang, C. Cai, X. Yang, Z.-C. Lv and U. Schneider, ACS Catal., 2016, 6, 5747 CrossRef CAS; (b) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886 CrossRef CAS PubMed; (c) D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486 CrossRef CAS PubMed.
- For a recent example on the atroposelective cross-coupling of an inert aromatic C–O bond, see: J. Zhang, T. Sun, Z. Zhang, H. Cao, Z. Bai and Z.-C. Cao, J. Am. Chem. Soc., 2021, 143, 18380 CrossRef CAS PubMed.
- For selected examples, see: (a) V. N. Nair, V. Kojasoy, C. J. Laconsay, W. Y. Kong, D. J. Tantillo and U. K. Tambar, J. Am. Chem. Soc., 2021, 143, 9016 CrossRef CAS PubMed; (b) N. Guranova, D. Dar’in, G. Kantin and M. Krasavin, Eur. J. Org. Chem., 2021, 3411 CrossRef CAS; (c) D. Dar’in, G. Kantin, O. Bakulina, A. Inyutina, E. Chupakhin and M. Krasavin, J. Org. Chem., 2020, 85, 15586 CrossRef PubMed; (d) S. Jana, Z. Yang, C. Pei, X. Xu and R. M. Koenigs, Chem. Sci., 2019, 10, 10129 RSC; (e) J. Zhang, Z. Liao, L. Chen and S. Zhu, Chem.–Eur. J., 2019, 25, 9405 CrossRef CAS PubMed; (f) M. Kitamura, M. Kisanuki, K. Kanemura and T. Okauchi, Org. Lett., 2014, 16, 1554 CrossRef CAS PubMed; (g) D. M. Jaber, R. N. Burgin, M. Helper, P. Y. Zavalij and M. P. Doyle, Org. Lett., 2012, 14, 1676 CrossRef CAS PubMed; (h) D. J. Mack, L. A. Batory and J. T. Njardarson, Org. Lett., 2012, 14, 378 CrossRef CAS PubMed; (i) D. M. Jaber, R. N. Burgin, M. Hepler, P. Zavalij and M. P. Doyle, Chem. Commun., 2011, 47, 7623 RSC; (j) J. Wang, in Comprehensive Organometallic Chemistry III, ed. D. M. P. Mingos and R. H. Crabtree, Elsevier, Oxford, 2007, vol. 11, p. 151 Search PubMed; (k) G. K. Murphy and F. G. West, Org. Lett., 2006, 8, 4359 CrossRef CAS PubMed; (l) G. K. Murphy and F. G. West, Org. Lett., 2005, 7, 1801 CrossRef CAS PubMed; (m) F. P. Marmsäter, G. K. Murphy and F. G. West, J. Am. Chem. Soc., 2003, 125, 14724 CrossRef PubMed; (n) N. P. Karche, S. M. Jachak and D. D. Dhavale, J. Org. Chem., 2001, 66, 6323 CrossRef CAS PubMed; (o) F. G. West, B. N. Naidu and R. W. Tester, J. Org. Chem., 1994, 59, 6892 CrossRef CAS; (p) F. G. West, T. H. Eberlein and R. W. Tester, J. Chem. Soc., Perkin Trans. 1, 1993, 1, 2857 RSC; (q) T. H. Eberlein, F. G. West and R. W. Tester, J. Org. Chem., 1992, 57, 3479 CrossRef CAS; (r) E. J. Roskamp and C. R. Johnson, J. Am. Chem. Soc., 1986, 108, 6062 CrossRef CAS PubMed; (s) J. B. Brogan and C. K. Zercher, Tetrahedron Lett., 1998, 39, 1691 CrossRef CAS; (t) J. B. Brogan, C. K. Zercher, C. B. Bauer and R. D. Rogers, J. Org. Chem., 1997, 62, 3902 CrossRef CAS; (u) K. Friedrich, U. Jansen and W. Kirmse, Tetrahedron Lett., 1985, 26, 193 CrossRef CAS; (v) H. Nozaki, H. Takaya and R. Noyori, Tetrahedron, 1966, 22, 3393 CrossRef CAS; (w) C. D. Gutsche and M. Hillman, J. Am. Chem. Soc., 1954, 76, 2236 CrossRef CAS.
- For recent selected examples, see: (a) M. M.-C. Lo and G. C. Fu, Tetrahedron, 2001, 57, 2621 CrossRef CAS; (b) S. Kitagaki, Y. Yanamoto, H. Tsutsui, M. Anada, M. Nakajima and S. Hashimoto, Tetrahedron Lett., 2001, 42, 6361 CrossRef CAS; (c) J. S. Clark, M. Fretwell, G. A. Whitlock, C. J. Burns and D. N. A. Fox, Tetrahedron Lett., 1998, 39, 97 CrossRef CAS; (d) M. P. Doyle, D. G. Ene, D. C. Forbes and J. S. Tedrow, Tetrahedron Lett., 1997, 38, 4367 CrossRef CAS; (e) K. Ito, M. Yoshitake and T. Katsuki, Tetrahedron, 1996, 52, 3905 CrossRef CAS; (f) H. Nozaki, H. Takaya, S. Moriuti and R. Noyori, Tetrahedron, 1968, 24, 3655 CrossRef CAS.
- For recent reviews, see: (a) X.-J. Liu, Y. Xu, C. Tang, P.-C. Qian and L.-W. Ye, Sci. China: Chem., 2022, 65, 20 CrossRef CAS; (b) M. Niggemann and S. Gao, Angew. Chem., Int. Ed., 2018, 57, 16942 CrossRef CAS PubMed ; for recent selected examples, see: ; (c) T. Ikeuchi, S. Inuki, S. Oishi and H. Ohno, Angew. Chem., Int. Ed., 2019, 58, 7792 CrossRef CAS PubMed; (d) B. Wigman, S. Popov, A. L. Bagdasarian, B. Shao, T. R. Benton, C. G. Williams, S. P. Fisher, V. Lavallo, K. N. Houk and H. M. Nelson, J. Am. Chem. Soc., 2019, 141, 9140 CrossRef CAS PubMed; (e) S. Popov, B. Shao, A. L. Bagdasarian, T. R. Benton, L. Zou, Z. Yang, K. N. Houk and H. M. Nelson, Science, 2018, 361, 381 CrossRef CAS PubMed; (f) S. E. Cleary, M. J. Hensinger and M. Brewer, Chem. Sci., 2017, 8, 6810 RSC.
- For recent selected examples, see: (a) L.-J. Qi, C.-T. Li, Z.-Q. Huang, J.-T. Jiang, X.-Q. Zhu, X. Lu and L.-W. Ye, Angew. Chem., Int. Ed., 2022, 61, e202210637 CAS; (b) F.-L. Hong, C.-Y. Shi, P. Hong, T.-Y. Zhai, X.-Q. Zhu, X. Lu and L.-W. Ye, Angew. Chem., Int. Ed., 2022, 61, e202115554 CAS; (c) X.-Q. Zhu, P. Hong, Y.-X. Zheng, Y.-Y. Zhen, F.-L. Hong, X. Lu and L.-W. Ye, Chem. Sci., 2021, 12, 9466 RSC; (d) F.-L. Hong, Y.-B. Chen, S.-H. Ye, G.-Y. Zhu, X.-Q. Zhu, X. Lu, R.-S. Liu and L.-W. Ye, J. Am. Chem. Soc., 2020, 142, 7618 CrossRef CAS PubMed; (e) F.-L. Hong, Z.-S. Wang, D.-D. Wei, T.-Y. Zhai, G.-C. Deng, X. Lu, R.-S. Liu and L.-W. Ye, J. Am. Chem. Soc., 2019, 141, 16961 CrossRef CAS PubMed.
- For the related gold-catalyzed generation of vinyl cations from diynes, see: (a) A. Ahrens, J. Schwarz, D. M. Lustosa, R. Pourkaveh, M. Hoffmann, F. Rominger, M. Rudolph, A. Dreuw and A. S. K. Hashmi, Chem.–Eur. J., 2020, 26, 5280 CrossRef CAS PubMed; (b) S. Tavakkolifard, K. Sekine, L. Reichert, M. Ebrahimi, K. Museridz, E. Michel, F. Rominger, R. Babaahmadi, A. Ariafard, B. F. Yates, M. Rudolph and A. S. K. Hashmi, Chem.–Eur. J., 2019, 25, 12180 CrossRef CAS PubMed; (c) T. Wurm, J. Bucher, S. B. Duckworth, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2017, 56, 3364 CrossRef CAS PubMed.
- For recent reviews on ynamide reactivity, see: (a) Y.-C. Hu, Y. Zhao, B. Wan and Q.-A. Chen, Chem. Soc. Rev., 2021, 50, 2582 RSC; (b) C. C. Lynch, A. Sripada and C. Wolf, Chem. Soc. Rev., 2020, 49, 8543 RSC; (c) Y.-B. Chen, P.-C. Qian and L.-W. Ye, Chem. Soc. Rev., 2020, 49, 889 Search PubMed; (d) F.-L. Hong and L.-W. Ye, Acc. Chem. Res., 2020, 53, 2003 CrossRef CAS PubMed; (e) J. Luo, G.-S. Chen, S.-J. Chen, J.-S. Yu, Z.-D. Li and Y.-L. Liu, ACS Catal., 2020, 10, 13978 CrossRef CAS; (f) B. Zhou, T.-D. Tan, X.-Q. Zhu, M. Shang and L.-W. Ye, ACS Catal., 2019, 9, 6393 CrossRef CAS; (g) G. Evano, C. Theunissen and M. Lecomte, Aldrichimica Acta, 2015, 48, 59 CAS; (h) X.-N. Wang, H.-S. Yeom, L.-C. Fang, S. He, Z.-X. Ma, B. L. Kedrowski and R. P. Hsung, Acc. Chem. Res., 2014, 47, 560 CrossRef CAS PubMed.
- For recent selected examples by our group, see: (a) G.-Y. Zhu, J.-J. Zhou, L.-G. Liu, X. Li, X.-Q. Zhu, X. Lu, J.-M. Zhou and L.-W. Ye, Angew. Chem., Int. Ed., 2022, 61, e202204603 CAS; (b) Z.-S. Wang, L.-J. Zhu, C.-T. Li, B.-Y. Liu, X. Hong and L.-W. Ye, Angew. Chem., Int. Ed., 2022, 61, e202201436 CAS; (c) Y.-Q. Zhang, Y.-B. Chen, J.-R. Liu, S.-Q. Wu, X.-Y. Fan, Z.-X. Zhang, X. Hong and L.-W. Ye, Nat. Chem., 2021, 13, 1093 CrossRef CAS PubMed; (d) P.-F. Chen, B. Zhou, P. Wu, B. Wang and L.-W. Ye, Angew. Chem., Int. Ed., 2021, 60, 27164 CrossRef CAS PubMed; (e) Y.-Q. Zhang, Y.-P. Zhang, Y.-X. Zheng, Z.-Y. Li and L.-W. Ye, Cell Rep. Phys. Sci., 2021, 2, 100448 CrossRef CAS; (f) Z.-S. Wang, Y.-B. Chen, H.-W. Zhang, Z. Sun, C. Zhu and L.-W. Ye, J. Am. Chem. Soc., 2020, 142, 3636 CrossRef CAS PubMed; (g) X. Liu, Z.-S. Wang, T.-Y. Zhai, C. Luo, Y.-P. Zhang, Y.-B. Chen, C. Deng, R.-S. Liu and L.-W. Ye, Angew. Chem., Int. Ed., 2020, 59, 17984 CrossRef CAS PubMed.
- For recent selected examples, see: (a) G. Shen, Z. Wang, X. Huang, M. Hong, S. Fan and X. Lv, Org. Lett., 2020, 22, 8860 CrossRef CAS PubMed; (b) S. DeBonis, D. A. Skoufias, R.-L. Indorato, F. Liger, B. Marquet, C. Laggner, B. Joseph and F. Kozielski, J. Med. Chem., 2008, 51, 1115 CrossRef CAS PubMed; (c) L. Alain and G. Frederic, EP 197232, 2008; (d) B. Hellrung and H. Balli, Helv. Chim. Acta, 1980, 63, 1284 CrossRef CAS.
- For the structures of NaBArF4 and ynamides 1ag–1ai, see below:
 .
. - (a) S. Liao, X.-L. Sun and Y. Tang, Acc. Chem. Res., 2014, 47, 2260 CrossRef CAS PubMed; (b) L. Wang and Y. Tang, Tetrahedron, 2022, 129, 133121 CrossRef CAS.
- CCDC 2192857 ((+)-2a).†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2192857. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06152j |
| ‡ J.-J. Z. and Y.-N. M. contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |