
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtroposelective brominations to access chiral biaryl scaffolds using high-valent Pd-catalysis†
Sif T.
Linde
 ,
Vasco
Corti
,
Vibeke H.
Lauridsen
,
Johannes N.
Lamhauge
,
Karl Anker
Jørgensen
and
Nomaan M.
Rezayee
*
,
Vasco
Corti
,
Vibeke H.
Lauridsen
,
Johannes N.
Lamhauge
,
Karl Anker
Jørgensen
and
Nomaan M.
Rezayee
*
Department of Chemistry, Aarhus University, DK-8000 Aarhus C, Denmark. E-mail: nmr@chem.au.dk
First published on 3rd March 2023
Abstract
Compounds featuring atropisomerism are ubiquitous in natural products, therapeutics, advanced materials, and asymmetric synthesis. However, stereoselective preparation of these compounds presents many synthetic challenges. This article introduces streamlined access to a versatile chiral biaryl template through C–H halogenation reactions employing high-valent Pd catalysis in combination with chiral transient directing groups. This methodology is highly scalable, insensitive to moisture and air, and proceeds, in select cases, with Pd-loadings as low as 1 mol%. Chiral mono-brominated, dibrominated, and bromochloro biaryls are prepared in high yield and excellent stereoselectivity. These serve as remarkable building blocks bearing orthogonal synthetic handles for a gamut of reactions. Empirical studies elucidated regioselective C–H activation to be predicated on the oxidation state of Pd and diverging site-halogenation to result from cooperative effects of Pd and oxidant.
Atropisomers are a subset of axially chiral compounds bearing restricted rotation about a σ-bond pivot—leading to isolable conformers exhibiting molecular asymmetry.1,2 Within the various classes of atropisomers, biaryls are distinguished as privileged scaffolds due to their salient contributions to the field of asymmetric catalysis found in phosphoric acid-, Brønsted base-, phase-transfer-, aldehyde-, H-bond-, Cu–H-catalysis etc.3 Consequently, significant efforts have been devoted to streamline access to novel chiral scaffolds.
Among the various methods to prepare atropisomers, the development of transition metal catalyzed asymmetric C–H functionalizations of biaryls through an enantioconvergent process has manifested into a powerful strategy.4–8 These hinge on the combined effects of directing groups/chiral auxiliaries, chiral ancillary ligands, and metal catalysts. Within this frame, elegant directing group-assisted approaches have been developed relying on either “catalyst control”9–11 or “substrate-control”12–14 for the stereoinduction. However, these methods exhibit key limitations that hinder broad application. For instance, additional steps are necessary for the stoichiometric installation and removal of optically pure (substrate-control) or achiral (catalyst-control) directing groups. These complicate syntheses and often lead to harsh conditions incompatible with structurally complex compounds.15 An attractive strategy that circumvents these limitations is the use of chiral transient directing groups (cTDGs).15–18
Significant progress has been made employing cTDGs for the stereoselective preparation of atropisomers employing Pd-catalysis. A seminal contribution by the Shi group identified the chiral amino acids as a competent cTDG to reversibly condense with the carbonyl moiety of benzaldehyde derivatives (Scheme 1, precedent).19 The resulting in situ generated chiral imine directing group underwent the Fujiwara–Moritani reaction and demonstrated high proficiency for stereoinduction yielding the C–H olefinated biaryl with >99% enantiomeric excess (ee). This strategy has proven general and robust leading to C–H alkynylations,20,21 allylations,22 naphthylations,23 and alkylations24 with chiral amino acids playing an essential role in the Pd0/II cycle. Ackermann and co-workers have adapted this approach to electro-chemical setups with comparable success.25 Despite these advances, to date, the cTDG C–H functionalization reaction has been nearly exclusively limited to C–C bond formation26,27 and predominantly employs a Pd0/II cycle.6,15,28
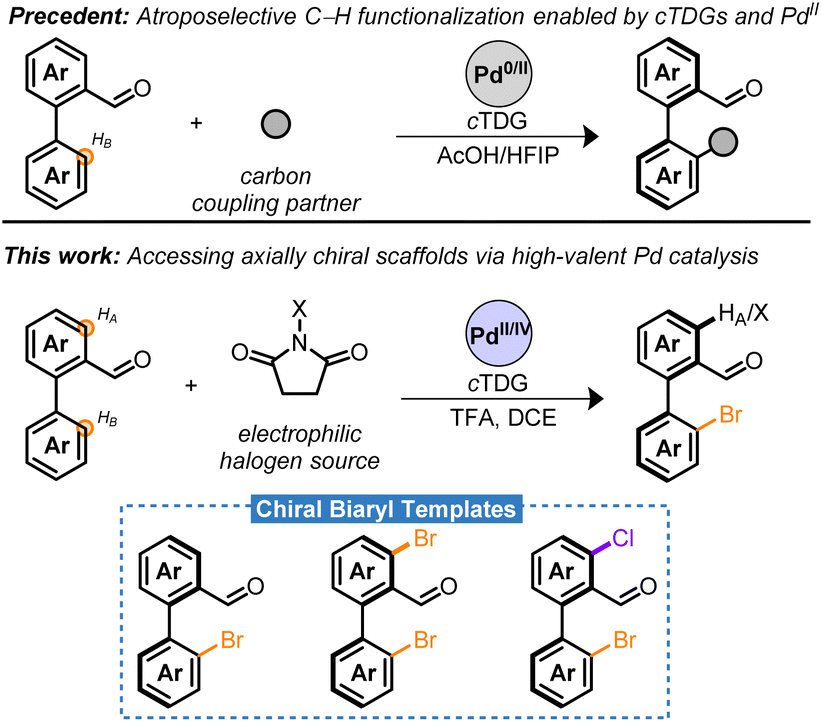 | ||
| Scheme 1 Pd-catalyzed atroposelective C–H functionalizations employing cTDGs. | ||
This article describes the atroposelective halogenation of C–H bonds by combining cTDG with high-valent Pd catalysis. Notably, reductive eliminations to forge C–X bonds are typically not feasible from PdII in a catalytic setting.29,30 Chiral dihalogenated and monohalogenated biaryls are prepared in moderate to excellent yield with, generally, excellent enantioselectivity (Scheme 1, this work). The presented method is insensitive to moisture and air, is highly scalable, and, in select cases, proceeds with Pd-loadings as low as 1 mol%. The resulting chiral bromobenzaldehyde derivatives serve as remarkable synthetic templates bearing two orthogonal handles for diversification adjacent to the axis of asymmetry. Methodologies for synthetic elaboration of aryl bromides and aldehydes are pervasive in literature. Harnessing these functional groups may bridge access to a multitude of chiral compounds that would otherwise necessitate extensive synthetic effort or the advent of new methodologies. The versatility of the chiral templates is highlighted in a series of transformations to forge C–B, C–C, C–N, C–O, C–F, and C–P bonds in a stereoretentive manner. Finally, a rare example of Pd-catalyzed oxidant-dependent regiocontrol was harnessed into telescoping reactions to furnish chiral bromochloro biaryls.
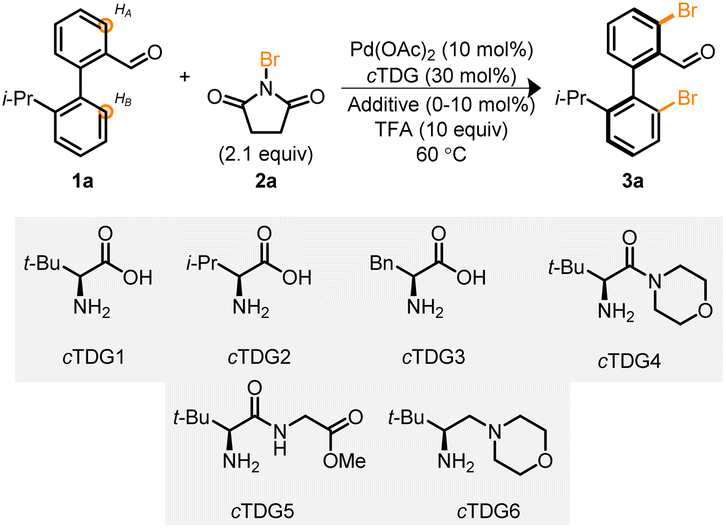
Biaryl 1a, in conjunction with cTDG1 and the halogenation reagent, NBS 2a, were selected to begin our investigation into the Pd-catalyzed reaction (Table 1). Initial attempts led to complex reaction mixtures; however, we were encouraged by the viability of the reaction and additional NBS was added. Upon the addition of 2.1 equivalents of NBS, the mixture of products coalesced into a single, di-brominated product, 3a. Investigations were undertaken to identify optimal reaction parameters. To our surprise 1,1,1,3,3,3-hexafluoro-propan-2-ol (HFIP),19,24,31,32 the typical solvent used with cTDGs, did not lead to marked improvement (entry 1). Instead, a survey (see ESI†) of solvents identified 1,2-dichloroethane (DCE) to provide the desired product in 48% yield as determined by NMR spectroscopy with >99% ee (entry 2) when employing cTDG1 and 10 equivalents of trifluoracetic acid (TFA).33 A suite of other amino acids and tert-leucine derived compounds (cTDGs 2–6) led to diminished yields and enantioselectivity (entries 2–7)—securing the use of commercially available L-tert-leucine (cTDG1). Notably, the addition of TFA was essential for the reaction to proceed (entry 8). Finally, a brief evaluation of additives found Ag2CO3 to improve yields to 84% (entries 9 and 10).34
|
|
|||||
|---|---|---|---|---|---|
| Entry | cTDG | Solvent | Additive | Yield (%) | ee (%) |
| a Conditions: 0.1 mmol 1a, 1.0 mL solvent, 0.1 equiv. of additive, 16 h. Yield determined by 1H NMR, ee determined by UPC2. b TFA omitted. | |||||
| 1 | cTDG1 | HFIP | — | 0 | — |
| 2 | cTDG1 | DCE | — | 48 | >99 |
| 3 | cTDG2 | DCE | — | 22 | 94 |
| 4 | cTDG3 | DCE | — | 4 | nd |
| 5 | cTDG4 | DCE | — | 42 | 85 |
| 6 | cTDG5 | DCE | — | 8 | nd |
| 7 | cTDG6 | DCE | — | 0 | — |
| 8b | cTDG1 | DCE | — | 0 | — |
| 9 | cTDG1 | DCE | AgTFA | 64 | >99 |
| 10 | cTDG1 | DCE | Ag2CO3 | 84 | >99 |
With the optimized conditions in hand, we next focused our attention on the Pd-catalyzed bromination of other biaryl compounds. The resulting enantioconvergent strategy proved universal—accommodating diverse steric profiles on the bottom ring (3b–m), top ring (3c–d, and 3h–p) and electronic parameters (3e–g, 3l, and 3p), in moderate to excellent yield and excellent enantioselectivity (Table 2). In the context of compounds 3g and 3h, poor mass balance was observed. Notably, the atroposelective halogenation of the electron-rich biaryl 1l led to the tribrominated product 3l in high yield and enantioselectivity. Previous works in the C–H halogenation of electron-rich arenes have revealed an uncatalyzed, background pathway consistent with the regioselectivity observed in 3l.35
| a Conditions: 0.1 mmol 1, 1.0 mL DCE, 0.1 equiv. Ag2CO3, 16 h. Yield are isolated yields, ee determined by UPC2. b ee determined by 1H NMR employing (R)-2-methyl-2-propanesulfinamide. c 3.1 equiv. of 2a. d 1.1 equiv. of 2a. e 2.0 mmol scale, 20 mL DCE. f 1.0 mol% Pd(OAc)2, 1.8 equiv. 2a, and Ag2CO3 omitted. |
|---|
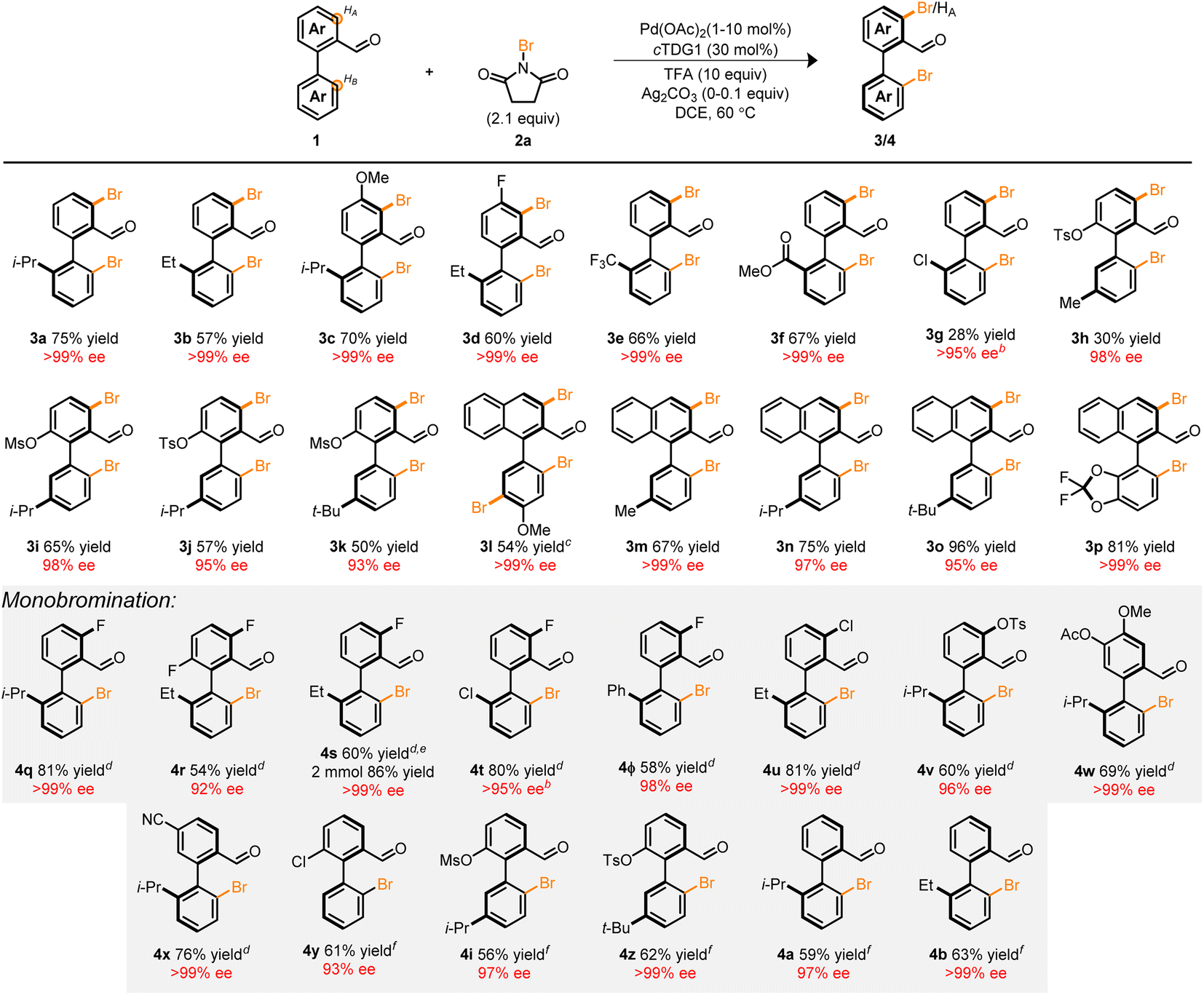
|
These products serve as versatile, catalytically-relevant scaffolds typified in 3p which resembles a hybrid among the core structures of BINAP and SEGPHOS. Notably, the installation of the Br-functional group is compatible with other synthetic handles such as Cl (3g) and pseudohalides (3h–k) directly adjacent to the axis of chirality. This allows for a myriad of orthogonal synthetic manipulations with three distinct sites for synthetic diversification flanking a pre-set stereogenic axis.
Several tactics can be taken to attenuate the extent of bromination in these systems to achieve monohalogenation (Table 2, monobromination). For instance: (1) the incorporation of a substituent at the HA-position (4q–v), (2) sterically occlude the undesired site (4w), (3) electronically deactivate the reactive site (4x), and (4) decrease the Pd loading to 1 mol% (4y, 4i, 4z, 4a, and 4b). Of particular significance is compound 4ϕ which provides a vector for the preparation of libraries of chiral terphenyls with high enantiopurity. Among the strategies, the latter approach necessitated delicate refinement and reoptimization (see ESI†). Nonetheless, we were pleased to find that 1 mol% of Pd(OAc)2 was sufficient to promote the reaction in moderate to good yield and high enantioselectivity. The influence of palladium loading on iteroselectivity may potentially arise from competitive hydrolysis, condensation, and ligand substitution of unfunctionalized starting material prior to a second C–H functionalization event (see ESI†). Furthermore, a two mmol scale-up reaction of 4s demonstrated the scalability of the transformation leading to an improved yield of 86% and >99% ee. The fidelity of the stereogenic axis for a variety of halogenated biaryls was interrogated experimentally and computationally with half-lives ranging from 92.3–114.9 h at 180 °C (see ESI†).
The brominated benzaldehyde derivatives behave as chiral templates for the construction of synthetically valuable biaryls through functionalization of either the aryl halide or aldehyde motif. Together, the orthogonal reactivity of the synthetic handles allows for the introduction of an array of functional groups. Summarized in Scheme 2 is a tableau of reactions to derivatize compounds 4q, 4s, or 4s′. The acetal protected 4s′ was amenable to a variety of C–C bond forming reactions such as carboxylation via lithiation, the Suzuki–Miyuara coupling, and Pd-catalyzed cyanation in a stereoretentive manner (Scheme 2a–c). In addition, heteroatoms can also be installed through established cross-coupling methodologies to construct, C–B, C–N, and C–P36 bonds adjacent to the stereogenic axis (Scheme 2d–f). Atroposelective methods to form these bonds through other means have remained elusive. Functionalization of the aldehyde moiety introduces a complementary avenue for diversification without erosion of the optical purity through reductive amination, deoxyfluorination, Baeyer–Villiger oxidation, and oxazoline forming reactions (Scheme 2g–j). An attractive prospect of the stereospecific Baeyer–Villiger reaction is the access to phenols which may be conveniently converted into a pseudohalide. In addition, the dibrominated benzaldehyde products exhibited preference for regioselective Suzuki–Miyuara couplings at the HA-site, allowing for potential iterative cross-couplings (see ESI†).
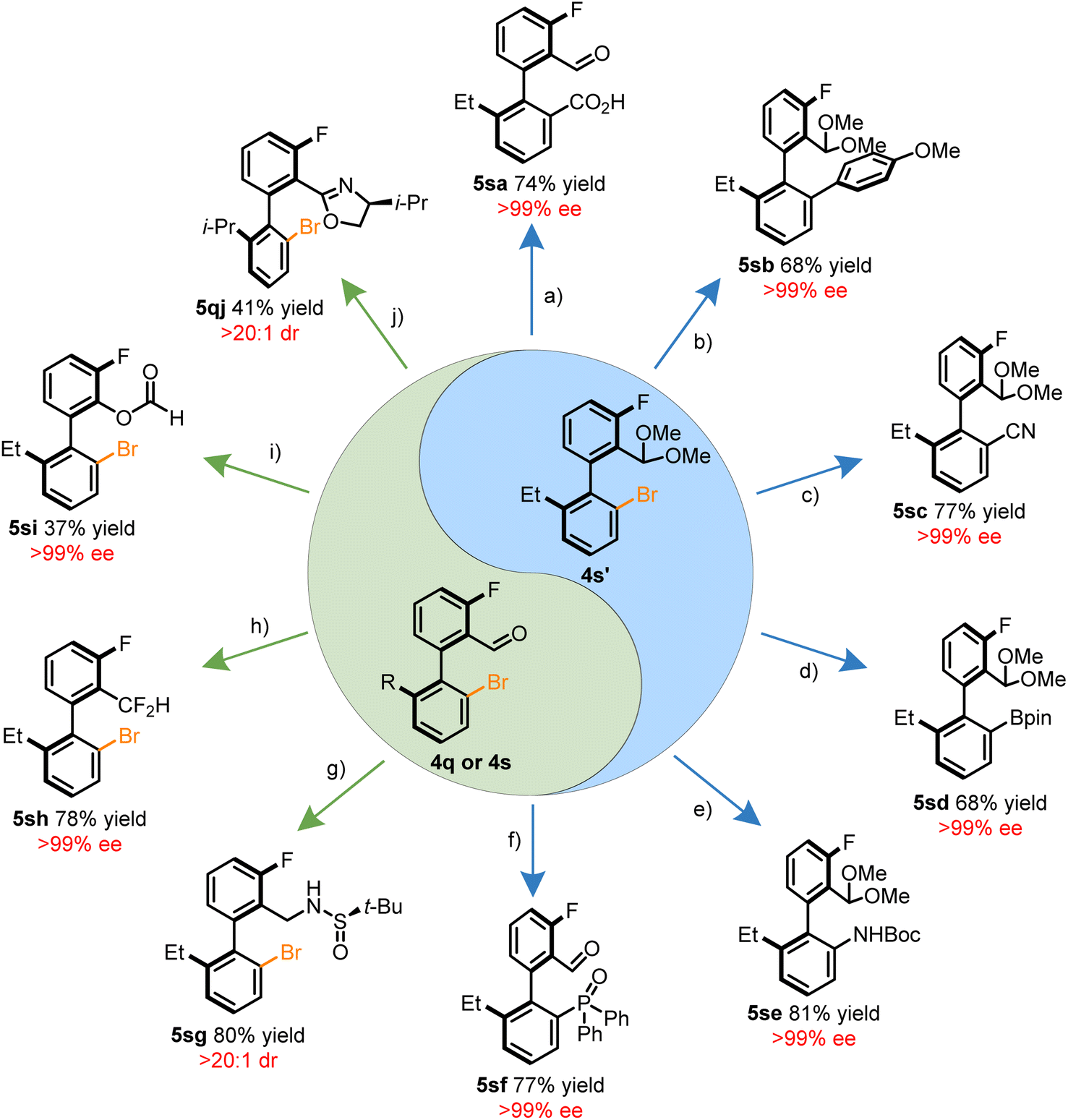 | ||
| Scheme 2 Application of chiral biaryl templates to access novel chiral scaffolds. (a) tert-BuLi (2.0 equiv.), then CO2, −90 °C. (b) p-Methoxyphenylboronic acid (1.5 equiv.), Pd(XPhos)-G4 (2 mol%), K3PO4 (2 equiv.). rt. (c) Na4[Fe(CN)6]·10H2O (0.5 equiv.), Pd2(dba)3 (2.5 mol%), XPhos (20 mol%), KOAc (12.5 mol%), 100 °C. (d) B2Pin2 (4.0 equiv.), Pd(dppf)Cl2 (10 mol%), KOAc (5.0 equiv.), 90 °C. (e) BocNH2 (2.0 equiv.), Pd(OAc)2 (10 mol%), XPhos (30 mol%), Cs2CO3 (1.4 equiv.), 100 °C. (f) Ph2P(O)H (2.0 equiv.), Pd(OAc)2 (20 mol%), dppb (30 mol%), DIPEA (4.8 equiv.), 120 °C. (g) (R)-2-Methyl-2-propanesulfinamide (1.2 equiv.), Ti(i-PrO)4 (4.0 equiv.), 40 °C then NaBH4 (4.0 equiv.), rt. (h) DAST (3.5 equiv.), rt. (i) mCPBA (3.3 equiv.) 0 °C to rt. (j) L-Valinol (1.0 equiv.), then NBS (1.0 equiv.), rt. | ||
Attempts to extend this methodology to other halogens proved unfruitful37–39 despite precedent for atroposelective iodinations employing chiral auxiliaries13 or chiral ancillary ligands.10 However, through our studies, a rare example of Pd-mediated oxidant dependent regiocontrol was identified. Unlike NBS, NCS 2b exclusively functionalized HA. This feature was recruited into telescoping reactions depicted in Table 3. At the onset, Pd, cTDG1, in conjunction with NCS, selectively furnished the mono, ortho-chlorinated product despite the vast excess in NCS. Subsequent addition of NBS installed Br at the HB-position, providing a chiral, mixed halogenated biaryl template with complete stereo- and regiocontrol. Remarkably, no reaction took place in the absence of Pd and cTDG1; indicating a cooperative effect among oxidant and Pd. Products arising from mixed chloro- and bromo-halogens provide synthetically valuable platforms allowing for chemodivergent cross-coupling reactions.40 This allows for facile initial tailoring at HB and subsequent refinement at HA. Notably, the resulting mixed halogen products deliver a complementary pathway for divergent iterative cross-couplings to the dibrominated products 4 (see ESI†). While a host of substrates are amenable to these telescoping reactions, Table 3 provides a brief survey.
| a Conditions: 0.1 mmol 1, 1.0 mL DCE, 48 h then 16 h. |
|---|
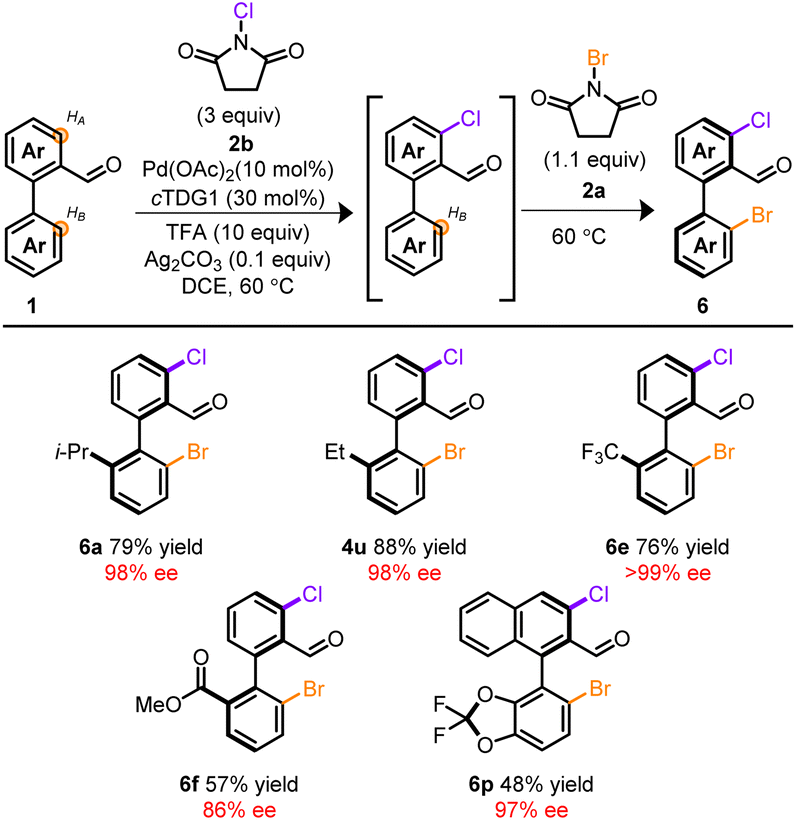
|
Based on the atypical circumstance of functionalization proceeding at chemically distinct sites,41,42 we were interested in gaining insight into the origin of the regiodivergent C–H activation. Previous atroposelective strategies employing TDGs in a Pd0/II manifold afforded exclusive functionalization of HB with no indications of competitive HA- or di-functionalization despite employing the same cTDG and Pd source.19–24,31,32 However, a distinguishing facet from prior works is the implementation of a high-valent Pd manifold.29,30,35,37 To interrogate whether the oxidation state of Pd was the root of regiocontrol, empirical studies were undertaken. Depicted in Scheme 3 is a pair of deuterium labeling experiments evaluating the reversible C–H functionalization(s) under contrasting reaction conditions. In Scheme 3a, conditions suitable to access high-valent Pd catalysis are achieved employing NCS and TFA-d. The resulting monochlorinated biaryl product 7a–d, exhibited selective chlorination at HA and 67% deuterium incorporation at HB. These results indicate that under oxidative reaction conditions, both sites undergo C–H activation, however, when employing NCS, chlorination exclusively occurs vicinal to the aldehyde. Conversely, Scheme 3b reveals the reversible C–H activation, in the absence of exogeneous oxidant, to solely proceed at HB—consistent with previous atroposelective reports at PdII.19–24,31,32 Taken together, Scheme 3 suggests a variance in site selectivity for C–H activation to be predicated on the oxidation state of Pd.43 Empirical distinctions between NCS and NBS suggest a divergence in site halogenation. NCS initially functionalizes HA (Scheme 3a) whereas NBS exclusively halogenates HB (Scheme 3c). The origin of this divergence remains unclear. Nevertheless, this marks a rare example of Pd-catalyzed oxidant-dependent regiocontrol.
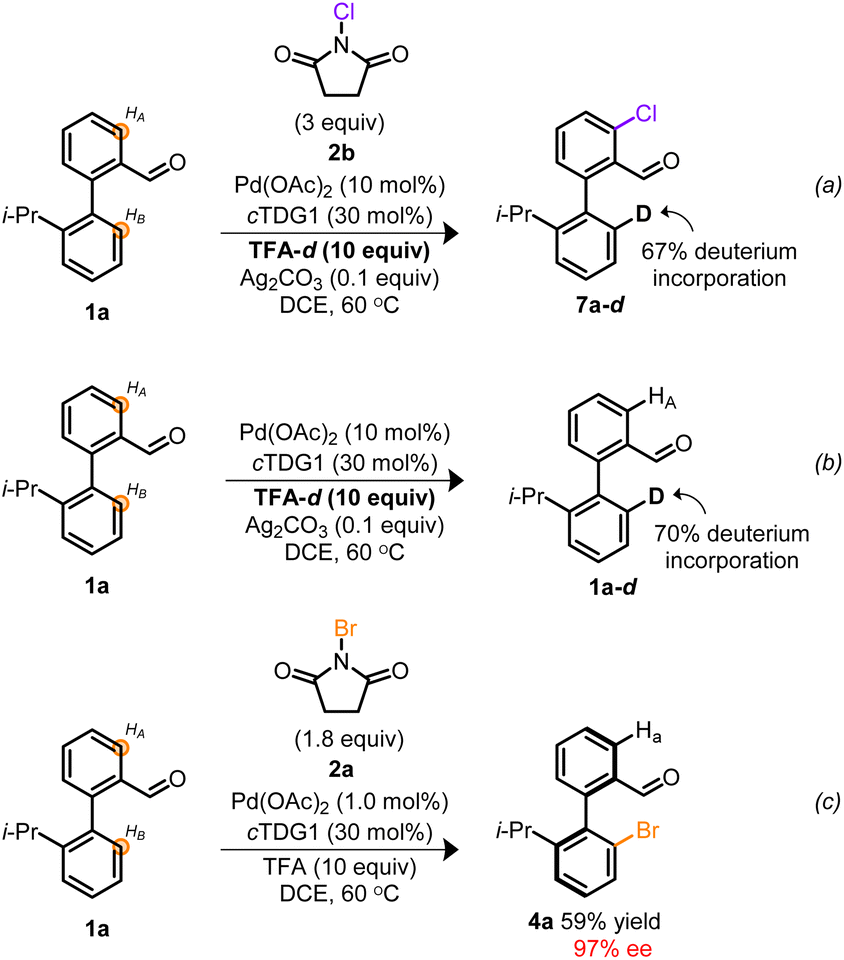 | ||
| Scheme 3 (a) Deuterium labeling experiment under oxidative conditions employing NCS. (b) Deuterium labeling experiment at PdII and in the absence of oxidant. (c) Empirical result from Table 24a. | ||
In summary, this article describes the first example of an atroposelective methodology to form C–X bonds using high-valent Pd-catalysis in conjunction with cTDGs. These transformations were achieved with Pd loadings ranging from 10 mol% to as low as 1 mol%. The resulting mono- and dihalogenated chiral biaryls were formed with excellent stereoselectivity and proved to be outstanding building blocks to undergo a gamut of stereoretentive transformations. Remarkably, empirical C–H activation studies reveal variances in regioselectivity based on the oxidation state of Pd and diverging sites of halogenation based on the oxidant. We anticipate this methodology to spur developments into novel asymmetric ligands and catalysts.
Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.† Crystallographic data for compounds 3c and 6p have been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC 2169123 (3c) and 2168153 (6p).Author contributions
N. M. R. conceived and supervised the work. S. T. L. optimized and identified the initial reaction conditions. S. T. L., V. C., and N. M. R. designed and executed the experiments. V. H. L. obtained the initial reaction result. J. N. L. performed experimental edits requested in the revision process. N. M. R., S. T. L., V. C., and K. A. J. contributed to writing and editing the manuscript. All authors agreed on the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. A. J. thanks Villum Investigator grant (no. 25867), the Carlsberg Foundation “Semper Ardens”, FNU and Aarhus University.Notes and references
- E. V. D. Anslyn and A. Dennis, Modern Physical Organic Chemistry, University Science Books, Herndon, VA, 2005 Search PubMed.
- G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef PubMed.
- J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed.
- Q. Wang, Z.-J. Cai, C.-X. Liu, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2019, 141, 9504–9510 CrossRef CAS PubMed.
- Q. Wang, W.-W. Zhang, H. Song, J. Wang, C. Zheng, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2020, 142, 15678–15685 CrossRef CAS PubMed.
- C.-X. Liu, W.-W. Zhang, S.-Y. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 14025–14040 CrossRef CAS PubMed.
- J. Frey, X. Hou and L. Ackermann, Chem. Sci., 2022, 13, 2729–2734 RSC.
- Y. Li, Y.-C. Liou, X. Chen and L. Ackermann, Chem. Sci., 2022, 13, 4088–4094 RSC.
- F. Kakiuchi, P. Le Gendre, A. Yamada, H. Ohtaki and S. Murai, Tetrahedron: Asymmetry, 2000, 11, 2647–2651 CrossRef CAS.
- D.-W. Gao, Q. Gu and S.-L. You, ACS Catal., 2014, 4, 2741–2745 CrossRef CAS.
- Y.-N. Ma, H.-Y. Zhang and S.-D. Yang, Org. Lett., 2015, 17, 2034–2037 CrossRef CAS PubMed.
- T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139–2144 CrossRef CAS.
- C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871–13875 CrossRef CAS PubMed.
- Q. Dherbassy, J.-P. Djukic, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2018, 57, 4668–4672 CrossRef CAS PubMed.
- G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 CrossRef CAS PubMed.
- M. I. Lapuh, S. Mazeh and T. Besset, ACS Catal., 2020, 10, 12898–12919 CrossRef CAS.
- C. Jacob, B. U. W. Maes and G. Evano, Chem. - Eur. J., 2021, 27, 13899–13952 CrossRef CAS PubMed.
- P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CAS.
- Q. J. Yao, S. Zhang, B. B. Zhan and B. F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617–6621 CrossRef CAS PubMed.
- S. Zhang, Q.-J. Yao, G. Liao, X. Li, H. Li, H.-M. Chen, X. Hong and B.-F. Shi, ACS Catal., 2019, 9, 1956–1961 CrossRef CAS.
- G. Liao, Q.-J. Yao, Z.-Z. Zhang, Y.-J. Wu, D.-Y. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 3661–3665 CrossRef CAS PubMed.
- H.-M. Chen, S. Zhang, G. Liao, Q.-J. Yao, X.-T. Xu, K. Zhang and B.-F. Shi, Organometallics, 2019, 38, 4022–4028 CrossRef CAS.
- G. Liao, H.-M. Chen, Y.-N. Xia, B. Li, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 11464–11468 CrossRef CAS PubMed.
- H.-M. Chen, G. Liao, C.-K. Xu, Q.-J. Yao, S. Zhang and B.-F. Shi, CCS Chem., 2021, 3, 455–465 CrossRef CAS.
- U. Dhawa, C. Tian, T. Wdowik, J. C. A. Oliveira, J. Hao and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 13451–13457 CrossRef CAS PubMed.
- Q. Gao, C. Wu, S. Deng, L. Li, Z.-S. Liu, Y. Hua, J. Ye, C. Liu, H.-G. Cheng, H. Cong, Y. Jiao and Q. Zhou, J. Am. Chem. Soc., 2021, 143, 7253–7260 CrossRef CAS PubMed.
- Z.-S. Liu, Y. Hua, Q. Gao, Y. Ma, H. Tang, Y. Shang, H.-G. Cheng and Q. Zhou, Nat. Catal., 2020, 3, 727–733 CrossRef CAS.
- J. Zhang, J. Fan, Y. Wu, Z. Guo, J. Wu and M. Xie, Org. Lett., 2022, 24, 5143–5148 CrossRef CAS PubMed.
- A. H. Roy and J. F. Hartwig, Organometallics, 2004, 23, 1533–1541 CrossRef CAS.
- A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177–185 CrossRef CAS PubMed.
- J. Zhang, Q. Xu, J. Wu, J. Fan and M. Xie, Org. Lett., 2019, 21, 6361–6365 CrossRef CAS PubMed.
- J. Fan, Q.-J. Yao, Y.-H. Liu, G. Liao, S. Zhang and B.-F. Shi, Org. Lett., 2019, 21, 3352–3356 CrossRef CAS PubMed.
- X.-H. Liu, H. Park, J.-H. Hu, Y. Hu, Q.-L. Zhang, B.-L. Wang, B. Sun, K.-S. Yeung, F.-L. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 888–896 CrossRef CAS PubMed.
- T. Bhattacharya, S. Dutta and D. Maiti, ACS Catal., 2021, 11, 9702–9714 CrossRef CAS.
- D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Tetrahedron, 2006, 62, 11483–11498 CrossRef CAS.
- Q.-Y. Zhao and M. Shi, Tetrahedron, 2011, 67, 3724–3732 CrossRef CAS.
- D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Org. Lett., 2006, 8, 2523–2526 CrossRef CAS PubMed.
- D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302–309 CrossRef CAS PubMed.
- D. C. Powers, D. Y. Xiao, M. A. L. Geibel and T. Ritter, J. Am. Chem. Soc., 2010, 132, 14530–14536 CrossRef CAS PubMed.
- E. K. Reeves, E. D. Entz and S. R. Neufeldt, Chem. - Eur. J., 2021, 27, 6161–6177 CrossRef PubMed.
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed.
- G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS PubMed.
- J. M. Racowski, N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18022–18025 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2169123 and 2168153. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06131g |
| This journal is © The Royal Society of Chemistry 2023 |