
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot cascade construction of nonsubstituted quinoline-bridged covalent organic frameworks†
Huaji
Pang
ab,
Dekang
Huang
 a,
Yanqiu
Zhu
ab,
Xiaodong
Zhao
ab and
Yonggang
Xiang
*ab
a,
Yanqiu
Zhu
ab,
Xiaodong
Zhao
ab and
Yonggang
Xiang
*ab
aDepartment of Chemistry, College of Science, Huazhong Agricultural University, Wuhan, Hubei 430070, China. E-mail: ygxiang@mail.hzau.edu.cn
bCollege of Resources and Environment, Huazhong Agricultural University, 430070, Wuhan, P. R. China
First published on 6th January 2023
Abstract
Irreversible locking of imine linkages into stable linkages represents a promising strategy to improve the robustness and functionality of covalent organic frameworks (COFs). We report, for the first time, a multi-component one-pot reaction (OPR) for imine annulation to construct highly stable nonsubstituted quinoline-bridged COFs (NQ-COFs), and that equilibrium regulation of reversible/irreversible cascade reactions by addition of MgSO4 desiccant is crucial to achieve high conversion efficiency and crystallinity. The higher long-range order and surface area of NQ-COFs synthesized by this OPR than those of the reported two-step post-synthetic modification (PSM) facilitate charge carrier transfer and photogeneration ability of superoxide radicals (O2˙−), which makes these NQ-COFs more efficient photocatalysts for O2˙− mediated synthesis of 2-benzimidazole derivatives. The general applicability of this synthetic strategy is demonstrated by fabricating 12 other crystalline NQ-COFs with a diversity of topologies and functional groups.
Introduction
Covalent organic frameworks (COFs) are a unique class of polymer materials that are fabricated by atomically connecting building blocks with one another through covalent bonds.1–3 With a periodic structure, high accessible surface area, well-defined porosity, and designable construction, COFs are burgeoning as a new research frontier in energy storage,4,5 separation,6,7 sensing,8–10 and catalysis.11–15 It is well recognized that the reversibility of the covalent linkages serves as the major driving force for the formation of long-range ordered COFs. This dynamic nature of the bonds, however, has a negative impact on the chemical stability of COFs, thus giving rise to an unavoidable trade-off between crystallinity and robustness.16 To address this intractable issue, the past few years have witnessed growing interest in the exploitation of efficient methodologies to improve the stability of COFs.17–23One of the methodologies to construct ultrastable COFs is to chemically convert reversible covalent linkages into irreversible robust ones by post synthetic modification (PSM).24,25 For instance, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds in imine-linked COFs could be transformed to more robust amide linkages under an oxidizing condition with high conversion efficiency and crystallinity.26–29 Similarly, locking of the imine linkage by other organic reactions could afford stable benzothiazole,30,31 quinoline,32–35 and benzoxazole linkages,36 and C–N bonds.37,38 Following these pioneering works, we recently reported a novel oxidant-free PSM method to construct ultrastable nonsubstituted quinoline-linked COFs (labelled as NQ-COFs) via rhodium-catalyzed [4 + 2] annulation.34 However, these PSM strategies, including ours, still suffer from some drawbacks, such as collapse of the framework and decreased crystallinity during the solid-state transformation, which might hamper the separation and transfer of photoinduced electron–hole pairs when these PSM derived COFs are applied in photocatalytic processes. Therefore, although the NQ-COFs synthesized in our work have demonstrated their first application in photoredox organic transformation, there is still much room to boost their photocatalytic activities. One-pot reaction (OPR) is considered a possible solution to the above dilemma, because such a methodology allows the building of ordered oligomeric structures from the very beginning of the reaction and thus is expected to produce more highly crystalline frameworks.39–45 However, examples of producing ultrastable COFs through OPR are rare, because balancing the cascade reaction conditions between reversible imine formation and subsequent irreversible locking is extremely challenging.
N bonds in imine-linked COFs could be transformed to more robust amide linkages under an oxidizing condition with high conversion efficiency and crystallinity.26–29 Similarly, locking of the imine linkage by other organic reactions could afford stable benzothiazole,30,31 quinoline,32–35 and benzoxazole linkages,36 and C–N bonds.37,38 Following these pioneering works, we recently reported a novel oxidant-free PSM method to construct ultrastable nonsubstituted quinoline-linked COFs (labelled as NQ-COFs) via rhodium-catalyzed [4 + 2] annulation.34 However, these PSM strategies, including ours, still suffer from some drawbacks, such as collapse of the framework and decreased crystallinity during the solid-state transformation, which might hamper the separation and transfer of photoinduced electron–hole pairs when these PSM derived COFs are applied in photocatalytic processes. Therefore, although the NQ-COFs synthesized in our work have demonstrated their first application in photoredox organic transformation, there is still much room to boost their photocatalytic activities. One-pot reaction (OPR) is considered a possible solution to the above dilemma, because such a methodology allows the building of ordered oligomeric structures from the very beginning of the reaction and thus is expected to produce more highly crystalline frameworks.39–45 However, examples of producing ultrastable COFs through OPR are rare, because balancing the cascade reaction conditions between reversible imine formation and subsequent irreversible locking is extremely challenging.
In this contribution, we successfully realized the one-pot synthesis of NQ-COFs after extensive screening of reaction conditions. It was found that the addition of MgSO4 was crucial to complete imine locking and form high crystallinity NQ-COFs. Detailed characterization indicated that a more ordered framework and higher surface area were obtained for the NQ-COFs synthesized by OPR than the ones synthesized by PSM, and these properties were beneficial to the exposure of active sites and charge carrier transfer. As a result, the NQ-COF obtained by OPR displayed superior photocatalytic performance for driving the O2˙− mediated synthesis of 2-benzimidazole derivatives. By means of this newly developed strategy, 12 other NQ-COFs were also obtained, substantiating its general applicability.
Results and discussion
Given that OPR has obvious advantages over PSM for preparation of COFs, particularly those containing structural diversity and complexity, we made a rapid attempt to synthesize NQ-COFA1via OPR under the reported PSM reaction conditions. However, it failed with only amorphous polymer obtained (Scheme 1b). Such reaction conditions were also not suitable for the smooth operation of the model reaction, as only a 61% yield of 2-phenylquinoline was achieved (Scheme 1a). To drive the multicomponent reaction, extensive screening of reaction conditions was conducted (supplementary Table 1†). It was found that the presence of MgSO4 was essential in constructing highly crystalline frameworks, and all experiments aiming to crystallize NQ-COFA1 were proven to be unsuccessful if MgSO4 was absent. Next, the amount of MgSO4 was screened, whereby MgSO4 (1.0, 2.0, 4.0, 6.0 and 10.0 equiv. per imine) was added in the process of synthesizing NQ-COFA1-OPR. It turned out that a low conversion efficiency was observed with a great amount of unreacted imine and aldehyde in the presence of 1.0 and 2.0 equivalents of MgSO4 according to Fourier transform infrared (FT-IR) measurements, and a high conversion efficiency was achieved when 4.0, 6.0 and 10.0 equivalents of MgSO4 were added (supplementary Fig. 2a†). However, it was found that addition of 4.0 equivalents of MgSO4 resulted in the strongest powder X-ray diffraction (PXRD) intensity (supplementary Fig. 2b†). Inspired by this result, another typical desiccant, CaCl2 (4.0 equiv. per imine), was also used instead of MgSO4, however a low conversion efficiency and complete loss of crystallinity were observed via FT-IR and PXRD measurements (supplementary Fig. 3†). The optimal reaction conditions to obtain NQ-COFA1 were determined to be as follows: vinylene carbonate (3.0 equiv. per imine), [Cp*RhCl2]2 (1.5 mol% per imine), MgSO4 (4.0 equiv. per imine), HOAc (50 μL), reaction temperature of 120 °C, reaction time of 3 days, and a mixed solvent system of mesitylene/dioxane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v). Under these reaction conditions, the yield of model compound 2-phenylquinoline was also greatly increased to 95%, further verifying the significant role of MgSO4. It is known that the mechanism for the construction of nonsubstituted quinoline linkages starts with the formation of imine by condensation of aldehyde and amine, followed by Rh-catalyzed [4 + 2] annulation between the in situ generated imine and vinylene carbonate. To form an ordered structure, the reversible Schiff-base reaction must be fast enough to promote error correction before locking by the subsequent irreversible annulation reaction. The drying agent MgSO4 was regarded as crucial in the first step of reversible Schiff-base reaction, as the in situ generated water could be removed to accelerate the formation rate of imine intermediates. The water content with or without addition of MgSO4 in the reaction solution was determined by Karl Fisher titration, and the results indicated that the content of water in the solution for preparing COFA1 was 4.0 times that for preparing NQ-COFA1-OPR (supplementary Tables 4–6†). Therefore, the role of MgSO4 is speculated to adjust the balance between reversible Schiff-base reaction and irreversible annulation reaction. It is noted that, for simplicity in illustration, the NQ-COFA1 samples prepared by OPR and PSM are labelled as NQ-COFA1-OPR and NQ-COFA1-PSM in this work, respectively.
:2, v/v). Under these reaction conditions, the yield of model compound 2-phenylquinoline was also greatly increased to 95%, further verifying the significant role of MgSO4. It is known that the mechanism for the construction of nonsubstituted quinoline linkages starts with the formation of imine by condensation of aldehyde and amine, followed by Rh-catalyzed [4 + 2] annulation between the in situ generated imine and vinylene carbonate. To form an ordered structure, the reversible Schiff-base reaction must be fast enough to promote error correction before locking by the subsequent irreversible annulation reaction. The drying agent MgSO4 was regarded as crucial in the first step of reversible Schiff-base reaction, as the in situ generated water could be removed to accelerate the formation rate of imine intermediates. The water content with or without addition of MgSO4 in the reaction solution was determined by Karl Fisher titration, and the results indicated that the content of water in the solution for preparing COFA1 was 4.0 times that for preparing NQ-COFA1-OPR (supplementary Tables 4–6†). Therefore, the role of MgSO4 is speculated to adjust the balance between reversible Schiff-base reaction and irreversible annulation reaction. It is noted that, for simplicity in illustration, the NQ-COFA1 samples prepared by OPR and PSM are labelled as NQ-COFA1-OPR and NQ-COFA1-PSM in this work, respectively.
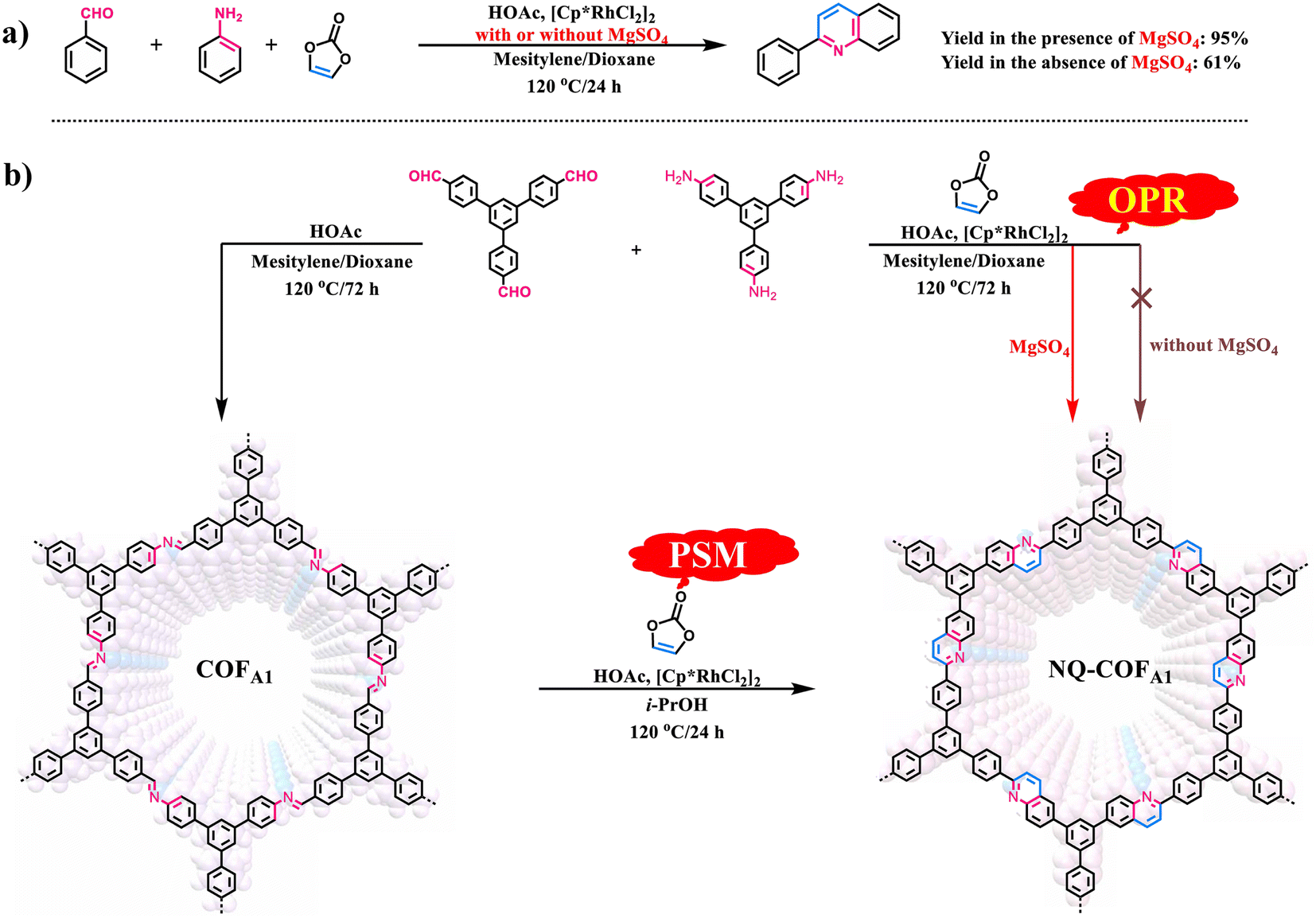 | ||
| Scheme 1 (a) Synthesis of model compound 2-phenlyquinoline via multicomponent reactions with or without addition of MgSO4. (b) Construction of NQ-COFA1via OPR and PSM methods. | ||
The long-range ordered structure of NQ-COFA1-OPR was verified using PXRD measurement. As shown in Fig. 1a, the PXRD pattern of NQ-COFA1-OPR shows six prominent diffraction peaks at 4.0°, 7.0°, 8.0°, 10.6°, 14.5°, and 25.6°, which are assigned to the (100), (110), (020), (210), (400), and (001) facets, respectively. Using the optimized monolayer structure, the simulated eclipsed (AA) stacking in the space group P1 was found to fit very well with the experimental PXRD pattern, and Pawley refinement of the crystal model afforded the unit cell of α = β = 90°, γ = 120°, a = 25.47 Å, b = 26.22 Å, c = 3.59 Å with satisfactory R-factors (Rp = 1.1%, Rwp = 1.49%). In comparison, NQ-COFA1-PSM shows a similar PXRD pattern to NQ-COFA1-OPR but an obviously lower intensity of the peaks. Meanwhile, the (100) peak of NQ-COFA1-OPR has a narrower full-width at half-maximum (FWHM) value of 0.47° than that of NQ-COFA1-PSM (0.49°), also suggesting a higher crystallinity of NQ-COFA1-OPR. It can be seen that an amorphous polymer was obtained with no obvious PXRD peaks in the absence of MgSO4. The more ordered crystal structure of NQ-COFA1-OPR over NQ-COFA1-PSM was also revealed by N2 adsorption–desorption measurements at 77 K (Fig. 1b). The adsorption profiles of all three COFs display reversible type IV isotherms, which are typical of mesoporous materials, and the narrow pore size distributions of COFA1, NQ-COFA1-OPR, and NQ-COFA1-PSM were determined by the non-local density flooding theory (NLDFT) fitted curves to be centered at 2.8, 2.6, and 2.6 nm, respectively, in good agreement with their simulated structures (supplementary Fig. 4†). Significantly, the Brunauer–Emmett–Teller (BET) specific surface areas of COFA1, NQ-COFA1-OPR, and NQ-COFA1-PSM were determined to be 1377, 1211, and 964 m2 g−1, respectively, which are 68%, 68%, and 54% of the theoretical values of COFA1 (2028 m2 g−1) and NQ-COFA1 (1779 m2 g−1). This result clearly indicates that the PSM strategy decreases the surface area of COFA1 as a result of some unavoidable factors. The OPR strategy can endow NQ-COFA1 with a higher surface area because NQ-COFA1-OPR has higher crystallinity and less amorphous monomer in the framework pores.
 | ||
| Fig. 1 (a) Experimental PXRD patterns of COFA1, NQ-COFA1-OPR, and NQ-COFA1-PSM. The simulated PXRD pattern was generated using NQ-COFA1-OPR. (b) N2 sorption isotherm curves of COFA1, NQ-COFA1-OPR, and NQ-COFA1-PSM. (c) FT-IR spectra of COFA1, NQ-COFA1-OPR, and NQ-COFA1-PSM. (d) Solid-state isotopically enriched 13C CP/MAS NMR spectra. (e) SEM images of COFA1, NQ-COFA1-OPR, and NQ-COFA1-PSM. (f) HRTEM image of NQ-COFA1-OPR. Inset: fast Fourier transform (FFT) pattern. | ||
Considering that PSM and OPR afforded NQ-COFA1 with a similar conversion efficiency, the more ordered periodic structure of NQ-COFA1-OPR may arise from the different mechanism of crystallization. The morphologies were then studied using field emission scanning electron microscopy (SEM). As shown in Fig. 1e, COFA1 was obtained in the form of layer-like blocks with a smooth surface, and the imine locking by the heterogeneous PSM has no significant impact on the morphology. In sharp contrast, NQ-COFA1-OPR is composed of irregular coral-like lamellae decorated on the outer ring of nanosheets with a relatively rough surface, which may contribute to more exposure of surface area. Based on the above result, we assumed that the more rigid skeleton of the quinoline-based oligomers than imine-based counterparts formed from the very beginning may enhance the interlayer π–π stacking, which can act as an impetus to drive the formation of a more ordered periodic structure with high crystallinity. This was further clearly visualized by high-resolution transmission electron microscopy (HRTEM) (Fig. 1f), and a well-ordered arrangement of continuous honeycomb-like pore channels was observed to extend through the whole crystal domain. The hexagonal shape with a pore size of 26 Å is in good consistence with the simulated AA-stacking mode of NQ-COFA1-OPR. Meanwhile, the ordered layered π–π stacking structure along the (001) direction can also be observed with an interlayer distance of 3.59 Å (supplementary Fig. 5†).
As it was previously reported that Rh-catalyzed annulation of imine by the PSM strategy could achieve quantitative conversion, we first compared the FT-IR spectra of NQ-COFA1-OPR and NQ-COFA1-PSM. The complete disappearance of the characteristic imine stretching vibration at 1623 cm−1 for NQ-COFA1-PSM verifies the high conversion efficiency (Fig. 1c). Satisfactorily, NQ-COFA1-OPR shows an almost identical spectrum to that of NQ-COFA1-PSM. On the other hand, there is still an obvious residue of CN bonds in NQ-COFA1 without MgSO4 addition, confirming that OPR can only smoothly operate in the presence of MgSO4. Subsequently, isotopically enriched 13C-labeled aldehyde was synthesized. After the structure was verified using FT-IR spectra (supplementary Fig. 6†), the 13C-labeled COFA1 and NQ-COFA1-OPR were afforded under the same conditions for collecting the solid-state 13C cross-polarization magic angle rotation (CP-MAS) NMR spectra. As shown in Fig. 1d, the imine carbon peak in 13C-labeled NQ-COFA1-OPR at 157 ppm disappears completely while a new peak appears at 155 ppm for the quinoline carbon of 13C-labeled NQ-COFA1-OPR. The formation of the quinoline-linked COF was further supported by high-resolution X-ray photoelectron spectroscopy (XPS) analysis as the binding energy of N 1s in CN fragments shifts up from ∼398.9 eV to ∼399.2 eV (supplementary Fig. 7†). The above measurements revealed that both the formation of reversible imine and subsequent Rh-catalyzed annulation in the multi-component OPR collaborated very well in the presence of MgSO4.
The chemical stability of the NQ-COFs was evaluated by subjecting COFA1 and NQ-COFA1-OPR to several harsh chemical conditions for 24 hours, including a strong acid (12 M HCl at 60 °C, 98% trifluoroacetic acid at 60 °C), strong base (14M NaOH in H2O/MeOH at 60 °C), and strong reducing agent (NaBH4 in MeOH at 60 °C). It was found that the crystallinity of NQ-COFA1-OPR was retained well according to PXRD measurements whereas COFA1 became amorphous, proving the robust nature of the frameworks of NQ-COFA1-OPR. Both COFs were also treated with n-hexylamine solution at 120 °C for 24 hours (supplementary Fig. 8†). Aminolysis of the imine bridges in COFA1 converted the suspension into a transparent state; however, the quinoline linkages were not affected. In addition, the thermal stability of the COFs was studied using thermogravimetric analysis (TGA), and the thermal decomposition temperature of NQ-COFA1-OPR was found to be higher than that of NQ-COFA1-PSM (supplementary Fig. 9†).
To demonstrate the generality of OPR for COF synthesis, a range of building blocks with different skeletons and geometries was utilized to construct NQ-COFs following the topology design principle (Fig. 2). After extensive optimization of polycondensation conditions, the combination of [3 + 2] and [3 + 3] can afford six hexagonal NQ-COFs (NQ-COFB1, NQ-COFB2, NQ-COFC1, NQ-COFC2, NQ-COFF1, and NQ-COFF2) with mesoporous or microporous structures in yields of 89–95%. All of them display high crystallinity according to PXRD measurements, and the diffraction patterns were found to be in good agreement with the eclipsed AA-stacking model. On the other hand, NQ-COFD4, NQ-COFD5, and NQ-COFF3 with rhombic pores were also constructed with high crystallinity and yields using the [4 + 2] combination. Moreover, we successfully synthesized three functional NQ-COFs (NQ-COFE4, NQ-COFE5, and NQ-COFG1) starting from substituted aromatic building blocks, which might provide more opportunities for NQ-COFs to be applied in wide areas in the future. 12 of the corresponding imine-linked COFs were also synthesized to confirm the high locking efficiency using FT-IR spectra and XPS analysis (supplementary Fig. 10 and 11†). Meanwhile, all the NQ-COFs displayed smaller BET specific surface areas and narrower pore size distributions than the corresponding imine-linked COFs, and this trend is also consistent with the comparison between COFA1 and NQ-COFA1-OPR (supplementary Fig. 12 and 13†). The robust skeletons of these NQ-COFs were confirmed using PXRD and FT-IR measurements before and after aminolysis tests (supplementary Fig. 14 and 15†). SEM observation revealed that the NQ-COFs displayed different morphologies from the corresponding imine-linked COFs, which might arise from the different crystallization process (supplementary Fig. 16†). Furthermore, all these 12 NQ-COFs were also characterized using HRTEM, and their high crystallinities were also demonstrated (supplementary Fig. 17†).
 | ||
| Fig. 2 XRD patterns of 12 different NQ-COFs (a-l). Green dot: experimental data, green line: Pawley refined profiles, orange line: simulated patterns of eclipsed (AA) stacking, blue bar: Bragg positions, and claret line: refinement differences. | ||
As we previously reported that enhanced π-electron delocalization of NQ-COFs favors visible-light absorption and carrier transfer in comparison to imine-linked COFs, it is particularly important to further clarify the effects of crystallinity and surface area differences of NQ-COFA1 synthesized by the PSM and OPR strategies on the photophysical properties. Ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) measurements revealed that quinoline formation contributes to the redshifted absorption, but NQ-COFA1-OPR displays a longer absorption onset than NQ-COFA1-PSM. The optical energy bandgap (Eg) of COFA1, NQ-COFA1-PSM, and NQ-COFA1-OPR was calculated from the Tauc plots to be 2.64, 2.36, and 2.15 eV, respectively (Fig. 3a). The superior light absorption of NQ-COFA1-OPR may arise from more efficient in-plane π-conjugation.46 Due to the positive nature of the slopes of the plots obtained at different frequencies by Mott–Schottky (MS) measurements, the flat-band potential (Efb) of the n-type COFA1, NQ-COFA1-PSM, and NQ-COFA1-OPR was determined from the x-axis intercepts of the MS plots to be −1.03, −1.01, and −0.99 V (vs. Ag/AgCl), respectively (Fig. 3b, supplementary Fig. 18†). Considering that the CB potential of n-type semiconductors is almost equal to the flat-band potential, the conduction band (CB) potentials of COFA1, NQ-COFA1-PSM, and NQ-COFA1-OPR are equal to −0.83, −0.81, and −0.79 V (vs. NHE), respectively. Combining the Eg and CB values, the valence band (VB) positions of COFA1, NQ-COFA1-PSM, and NQ-COFA1-OPR were calculated to be 1.81, 1.55, and 1.36 V (vs. NHE), respectively. Accordingly, all these three COFs have the ability of photogeneration of superoxide radicals (O2˙−) from O2 (Fig. 3c).
 | ||
| Fig. 3 (a) UV-vis DRS. The inset shows the corresponding Tauc plots. (b) Mott–Schottky plots of NQ-COFA1-OPR. (c) Energy band structure diagram. (d) Transient photocurrents. (e) EIS spectra. (f) EPR spectra of the DMPO-O2˙− adduct in the dark and under visible light irradiation. | ||
Furthermore, several other techniques were utilized to verify the superior photogenerated carrier transfer efficiency. In the transient photocurrent response measurements, NQ-COFA1-OPR also has the stronger photocurrent intensity (Fig. 3d). On the other hand, a smaller arc radius was obtained for NQ-COFA1-OPR than NQ-COFA1-PSM by electrochemical impedance spectroscopy (EIS) measurements (Fig. 3e). Based on these initial results, we attempted to evaluate the reduction ability of O2 to O2˙− driven by photogenerated electrons using paramagnetic spectra (EPR), and it turned out that the characteristic peaks of O2˙− could be observed when both suspended samples in CH3CN in the presence of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) were irradiated for 6 min by a 300 W Xe lamp. NQ-COFA1-OPR has the stronger ability for O2˙− generation, which should be conducive to its higher efficiency in O2˙−-mediated photocatalytic organic reactions (Fig. 3f).
Inspired by the superior potential of NQ-COFA1-OPR to reduce O2 to the reactive oxygen species (ROS) O2˙−, we attempted to apply NQ-COFs as photocatalysts in the O2˙−-mediated coupling of aryl diamine and aryl aldehyde for generation of the versatile building block benzimidazole.47 Phenylenediamine (1a) and benzaldehyde (2a) were selected as the model reactants for the initial reaction condition optimization (Fig. 4a). The desired product 2-phenyl-1H-benzo[d]imidazole (3a) could be obtained in 97% yield in the mixed solvent system EtOH/H2O with NQ-COFA1-OPR as the photocatalyst and an 18 W blue LED (460–465 nm) lamp as the light source under an O2 atmosphere. When the reaction time was prolonged to 36 h, the yield remained the same, and no product was detected in the absence of oxygen or light, proving the pivotal role of photogenerated ROS. In sharp contrast, 50% and 69% yields of the target product were afforded when COFA1 and NQ-COFA1-PSM were used as the photocatalysts under the same reaction conditions. Firstly, this revealed that the quinoline linkage helps to address the limited in-plane π-electron delocalization. More importantly, the more ordered framework of NQ-COFA1-OPR is indeed more beneficial to the transfer of photogenerated electrons and thus the generation of ROS is accelerated in comparison to NQ-COFA1-PSM, which correlates with the EPR results. With the optimized reaction conditions in hand, a variety of substituted phenylenediamines were tested to explore the coupling scope (Fig. 4b), and it was found that phenylenediamines 3,4-substituted with electron-deficient (3,4-F2, 3,4-Cl2 and 3-F) or electron-deficient (3-Me) groups were tolerated in the reaction, and the desired products 3b–3e could be obtained in good yields. Furthermore, the coupling reaction also ended in a good conversion efficiency when benzaldehyde was substituted at the ortho-, meta or para-position (p-Br, p-OH, p-CN, p-NO2, p-OCH3, o-Me, and m-Me) (3f–3q) under the standard conditions.
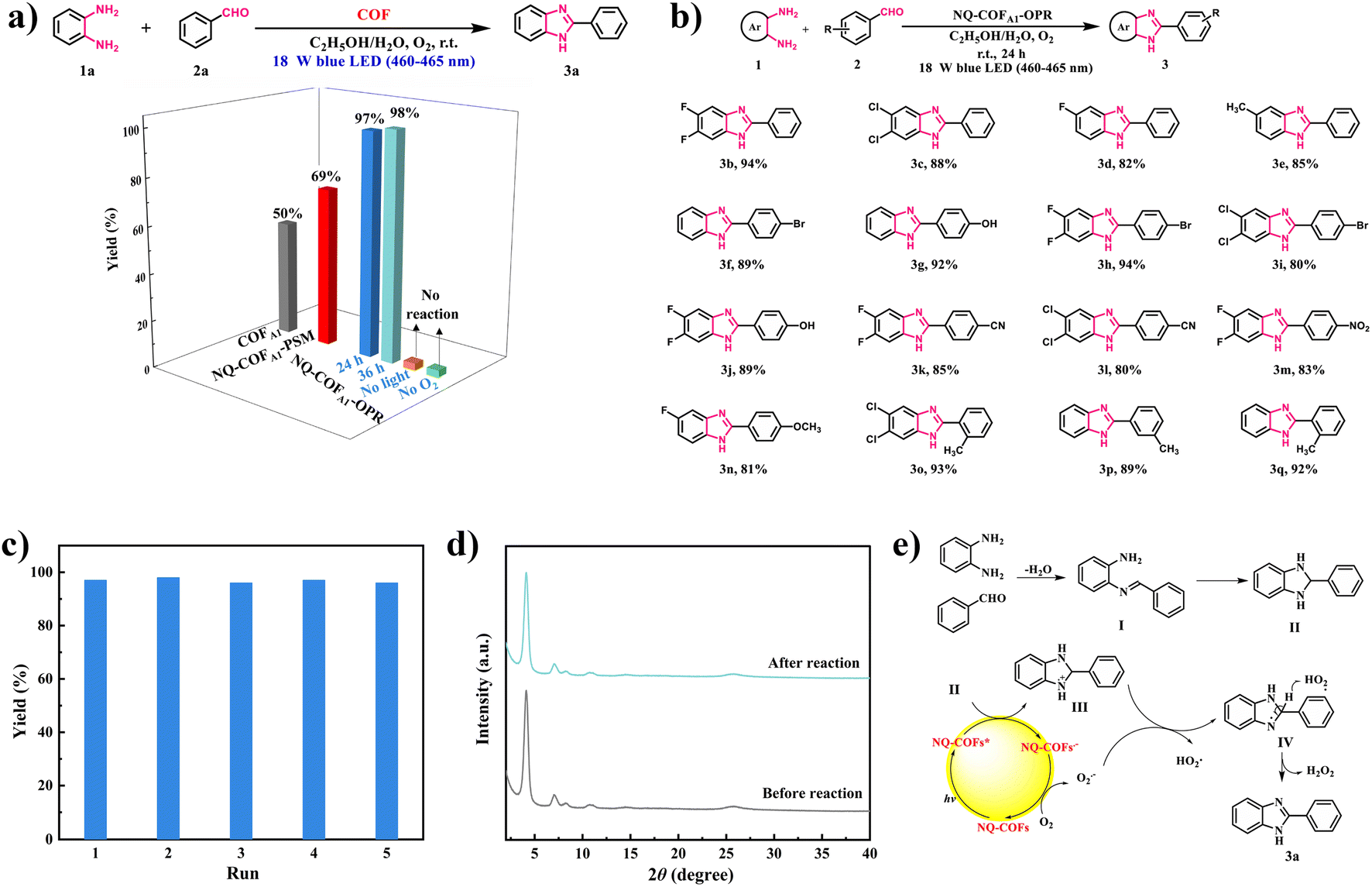 | ||
| Fig. 4 (a) Screening and comparison of reaction conditions for the synthesis of the model compound 2-phenyl-1H-benzo[d]imidazole. Standard conditions: 1a (0.5 mmol), 2a (0.5 mmol), COF (5 mg), EtOH/H2O (1 mL/1 mL), 18 W blue LED (460–465 nm), and 24 h. (b) Substrate scope for the synthesis of 2-benzimidazole using NQ-COFA1-OPR as the photocatalyst under standard reaction conditions. (c) Long-term stability obtained by recycling experiments. (d) PXRD comparison before and after five runs of the photocatalytic reaction. (e) Proposed mechanism for the photocatalytic reaction. | ||
Another advantage of heterogeneous photocatalytic reaction is the facile recycling of the photocatalyst for repeated runs without activity decay. Taking the model reaction for the generation of 3a as an example, NQ-COFA1-OPR could be easily separated by filtration after the reaction ended every time. After being washed and dried, the recycled NQ-COFA1-OPR was directly used for the next run, and it turned out that a 96% yield of 3a was still obtained after the fifth recycling reaction, proving its high photochemical stability (Fig. 4c). Meanwhile, the high stability of NQ-COFA1-OPR was further supported by PXRD and FT-IR data, which confirmed that the chemical structure and robust framework could be retained after the reaction (Fig. 4d and supplementary Fig. 19†).
To gain deep insight into the mechanism of the coupling reaction, experiments featuring ROS trapping were designed. When the singlet oxygen (1O2) scavenger β-carotene (3 mg) was added into the reaction, a 96% yield of 3a in the model reaction was obtained without other conditions changed. On the other hand, no product was detected in the presence of the O2˙− scavenger p-benzoquinone (3 mg), proving that O2˙− was the dominant active species. Based on the above observations and a previous report, a plausible reaction mechanism was proposed, as shown in Fig. 4e. NQ-COFA1-OPR is converted to the photoexcited NQ-COFA1-OPR*, and the cyclized intermediate II of imine I acts as the electron donor to NQ-COFA1-OPR* for the generation of NQ-COFA1-OPR•− and intermediate III. Next, O2 is activated by NQ-COFA1-OPR•− to afford the active species O2˙− along with the ground state NQ-COFA1-OPR, and hydrogen abstraction of III aided by O2˙− occurs to form HO2· and intermediate IV, which finally transformed to the final product 3a and H2O2. To further verify the possible mechanism, the by-product H2O2 was detected by iodometric experiments (supplementary Fig. 20†).
Conclusions
In summary, we developed a highly efficient Rh-catalyzed one-pot cascade strategy to synthesize robust non-substituted quinoline linked COFs directly from simple or readily available precursors. It was found that the dehydrating agent MgSO4 played a pivotal role in adjusting the reaction balance between formation of reversible imine and irreversible annulation. In comparison with the “stop-and-go” PSM strategy developed in our previous work, the OPR strategy presented herein not only avoided the isolation of intermediates and featured operational friendliness, but also enabled COFs to be obtained with higher crystallinity and specific surface areas. The later merits led the NQ-COFs prepared by OPR to exhibit stronger visible-light absorption and carrier transfer ability, which contributed to the facile generation of the active species O2˙−. As a result, the NQ-COFs prepared by OPR were ascertained to be more efficient photocatalysts in the O2˙−-mediated coupling reaction for synthesizing a variety of 2-benzimidazole derivatives. Furthermore, the general applicability of this OPR strategy was demonstrated by constructing 12 other NQ-COFs with different topologies and pore sizes. The approach reported in this work will reduce the difficulty in synthesizing stable COFs and thus promote COFs to find wide application in the future.Experimental section
Typical procedure for the one-pot synthesis of NQ-COFA1-OPR
A Pyrex glass tube (10 mL) was charged with aldehyde A (19.5 mg, 0.05 mmol), amine 1 (17.6 mg, 0.05 mmol), vinylene carbonate (38.7 mg, 0.45 mmol), acetic acid (HOAc, 0.05 mL), MgSO4 (72 mg), mesitylene (0.5 mL), and 1,4-dioxane (1.0 mL). Subsequently, the tube was sonicated for 10 minutes, degassed by three freeze–pump–thaw cycles (liquid nitrogen), and sealed under vacuum. After being heated in an oven at 120 °C for 3 days, the cooled suspension was centrifuged to separate the solid, which was repeatedly washed with THF and water until the solvent was colourless. NQ-COFA1-OPR was finally obtained as a brown powder (36 mg, 95%) after being dried under vacuum at 80 °C.Typical procedure for the synthesis of 2-phenyl-1H-benzo[d]imidazole
A 10 mL Schlenk tube equipped with a stir bar was charged with o-phenylenediamine 1 (0.5 mmol), aldehyde 2 (0.5 mmol), NQ-COFA1-OPR (5 mg), EtOH (1.0 mL), and H2O (1.0 mL). After being subjected to evacuation/purging with O2 for 10 min, the reaction mixture was irradiated in the photoreactor (18 W blue LEDs, 460–465 nm) with a cooling fan for 24 h under an O2 atmosphere. After the reaction ended, the solid was collected by filtration, and the concentrated filtrate was purified by flash chromatography on silica gel with PE/EA as the eluent to afford the products.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Y.-G. Xiang conceived and designed the whole project, and H.-J. Pang conducted the synthesis of COFs and photocatalytic activity evaluation. Y.-Q. Zhu and X.-D. Zhao helped in parts of the structural characterization, and H.-J. Pang, D.-K. Huang, and Y.-G. Xiang participated in the writing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support from the National Natural Science Foundation of China (21975090).References
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- S. Karak, K. Dey and R. Banerjee, Adv. Mater., 2022, e2202751 CrossRef PubMed.
- Y. Yusran, Q. Fang and V. Valtchev, Adv. Mater., 2020, 32, e2002038 CrossRef PubMed.
- Y. Zhang, R. L. Zhong, M. Lu, J. H. Wang, C. Jiang, G. K. Gao, L. Z. Dong, Y. Chen, S. L. Li and Y. Q. Lan, ACS Cent. Sci., 2021, 7, 175–182 CrossRef CAS PubMed.
- Z. Guo, H. Wu, Y. Chen, S. Zhu, H. Jiang, S. Song, Y. Ren, Y. Wang, X. Liang, G. He, Y. Li and Z. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202210466 CAS.
- J. A. Martin-Illan, J. A. Suarez, J. Gomez-Herrero, P. Ares, D. Gallego-Fuente, Y. Cheng, D. Zhao, D. Maspoch and F. Zamora, Adv. Sci., 2022, 9, e2104643 CrossRef PubMed.
- G. Das, B. Garai, T. Prakasam, F. Benyettou, S. Varghese, S. K. Sharma, F. Gándara, R. Pasricha, M. Baias, R. Jagannathan, N. i. Saleh, M. Elhabiri, M. A. Olson and A. Trabolsi, Nat. Commun., 2022, 13, 3904 CrossRef CAS PubMed.
- F. Yu, J. H. Ciou, S. Chen, W. C. Poh, J. Chen, J. Chen, K. Haruethai, J. Lv, D. Gao and P. S. Lee, Nat. Commun., 2022, 13, 390 CrossRef CAS PubMed.
- D. Bessinger, L. Ascherl, F. Auras and T. Bein, J. Am. Chem. Soc., 2017, 139, 12035–12042 CrossRef CAS PubMed.
- W. Dong, Y. Yang, Y. Xiang, S. Wang, P. Wang, J. Hu, L. Rao and H. Chen, Green Chem., 2021, 23, 5797–5805 RSC.
- R. Chen, J. L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS PubMed.
- M. Traxler, S. Gisbertz, P. Pachfule, J. Schmidt, J. Roeser, S. Reischauer, J. Rabeah, B. Pieber and A. Thomas, Angew. Chem., Int. Ed., 2022, 61, e202117738 CrossRef CAS PubMed.
- S. Trenker, L. Grunenberg, T. Banerjee, G. Savasci, L. M. Poller, K. I. M. Muggli, F. Haase, C. Ochsenfeld and B. V. Lotsch, Chem. Sci., 2021, 12, 15143–15150 RSC.
- A. Jati, K. Dey, M. Nurhuda, M. A. Addicoat, R. Banerjee and B. Maji, J. Am. Chem. Soc., 2022, 144, 7822–7833 CrossRef CAS PubMed.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- E. Q. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. H. Chen and D. L. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- S. Xu, H. Sun, M. Addicoat, B. P. Biswal, F. He, S. Park, S. Paasch, T. Zhang, W. Sheng, E. Brunner, Y. Hou, M. Richter and X. Feng, Adv. Mater., 2021, 33, e2006274 CrossRef PubMed.
- S. Bi, F. Meng, D. Wu and F. Zhang, J. Am. Chem. Soc., 2022, 144, 3653–3659 CrossRef CAS PubMed.
- X. Guan, H. Li, Y. Ma, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, Nat. Chem., 2019, 11, 587–594 CrossRef CAS PubMed.
- C. Zhao, H. Lyu, Z. Ji, C. Zhu and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142, 14450–14454 CrossRef CAS PubMed.
- J. Maschita, T. Banerjee, G. Savasci, F. Haase, C. Ochsenfeld and B. V. Lotsch, Angew. Chem., Int. Ed., 2020, 59, 15750–15758 CrossRef CAS PubMed.
- H. Duan, K. Li, M. Xie, J. M. Chen, H. G. Zhou, X. Wu, G. H. Ning, A. I. Cooper and D. Li, J. Am. Chem. Soc., 2021, 143, 19446–19453 CrossRef CAS PubMed.
- F. Haase and B. V. Lotsch, Chem. Soc. Rev., 2020, 49, 8469–8500 RSC.
- L. Cusin, H. Peng, A. Ciesielski and P. Samori, Angew. Chem., Int. Ed., 2021, 60, 14236–14250 CrossRef CAS PubMed.
- Z. B. Zhou, X. H. Han, Q. Y. Qi, S. X. Gan, D. L. Ma and X. Zhao, J. Am. Chem. Soc., 2022, 144, 1138–1143 CrossRef CAS PubMed.
- H. L. Nguyen, C. Gropp, N. Hanikel, A. Mockel, A. Lund and O. M. Yaghi, ACS Cent. Sci., 2022, 8, 926–932 CrossRef CAS PubMed.
- H. L. Qian, F. L. Meng, C. X. Yang and X. P. Yan, Angew. Chem., Int. Ed., 2020, 59, 17607–17613 CrossRef CAS PubMed.
- P. J. Waller, S. J. Lyle, T. M. O. Popp, C. S. Diercks, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15519–15522 CrossRef CAS PubMed.
- F. Haase, E. Troschke, G. Savasci, T. Banerjee, V. Duppel, S. Dorfler, M. M. J. Grundei, A. M. Burow, C. Ochsenfeld, S. Kaskel and B. V. Lotsch, Nat. Commun., 2018, 9, 2600 CrossRef PubMed.
- V. Singh, J. Kim, B. Kang, J. Moon, S. Kim, W. Y. Kim and H. R. Byon, Adv. Energy Mater., 2021, 11, 2003735 CrossRef CAS.
- X. R. Ren, B. Bai, Q. Zhang, Q. Hao, Y. Guo, L. J. Wan and D. Wang, J. Am. Chem. Soc., 2022, 144, 2488–2494 CrossRef CAS PubMed.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef PubMed.
- X. Zhao, H. Pang, D. Huang, G. Liu, J. Hu and Y. Xiang, Angew. Chem., Int. Ed., 2022, e202208833 CAS.
- Y. Yang, L. Yu, T. Chu, H. Niu, J. Wang and Y. Cai, Nat. Commun., 2022, 13, 2615 CrossRef CAS PubMed.
- J. M. Seo, H. J. Noh, H. Y. Jeong and J. B. Baek, J. Am. Chem. Soc., 2019, 141, 11786–11790 CrossRef CAS PubMed.
- H. Y. Liu, J. Chu, Z. L. Yin, X. Cai, L. Zhuang and H. X. Deng, Chem, 2018, 4, 1696–1709 CAS.
- S. J. Lyle, T. M. Osborn Popp, P. J. Waller, X. Pei, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 11253–11258 CrossRef CAS PubMed.
- P. F. Wei, M. Z. Qi, Z. P. Wang, S. Y. Ding, W. Yu, Q. Liu, L. K. Wang, H. Z. Wang, W. K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed.
- D. A. Pyles, J. W. Crowe, L. A. Baldwin and P. L. McGrier, ACS Macro Lett., 2016, 5, 1055–1058 CrossRef CAS PubMed.
- J. Liu, T. Yang, Z. P. Wang, P. L. Wang, J. Feng, S. Y. Ding and W. Wang, J. Am. Chem. Soc., 2020, 142, 20956–20961 CrossRef CAS PubMed.
- P. L. Wang, S. Y. Ding, Z. C. Zhang, Z. P. Wang and W. Wang, J. Am. Chem. Soc., 2019, 141, 18004–18008 CrossRef CAS PubMed.
- K. Wang, Z. Jia, Y. Bai, X. Wang, S. E. Hodgkiss, L. Chen, S. Y. Chong, X. Wang, H. Yang, Y. Xu, F. Feng, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 11131–11138 CrossRef CAS PubMed.
- N. Huang, K. H. Lee, Y. Yue, X. Xu, S. Irle, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 16587–16593 CrossRef CAS PubMed.
- M. Wang, M. Ballabio, M. Wang, H. H. Lin, B. P. Biswal, X. Han, S. Paasch, E. Brunner, P. Liu, M. Chen, M. Bonn, T. Heine, S. Zhou, E. Canovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2019, 141, 16810–16816 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- S. Dadwal, M. Kumar and V. Bhalla, J. Org. Chem., 2020, 85, 13906–13919 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06044b |
| This journal is © The Royal Society of Chemistry 2023 |