DOI:
10.1039/D2SC06036A
(Edge Article)
Chem. Sci., 2023,
14, 4375-4389
Manganese(II) complexes stimulate antitumor immunity via aggravating DNA damage and activating the cGAS-STING pathway†
Received
2nd November 2022
, Accepted 22nd March 2023
First published on 24th March 2023
Abstract
Activating the cyclic GMP-AMP synthase-stimulator of the interferon gene (cGAS-STING) pathway is a promising immunotherapeutic strategy for cancer treatment. Manganese(II) complexes MnPC and MnPVA (P = 1,10-phenanthroline, C = chlorine, and VA = valproic acid) were found to activate the cGAS-STING pathway. The complexes not only damaged DNA, but also inhibited histone deacetylases (HDACs) and poly adenosine diphosphate-ribose polymerase (PARP) to impede the repair of DNA damage, thereby promoting the leakage of DNA fragments into cytoplasm. The DNA fragments activated the cGAS-STING pathway, which initiated an innate immune response and a two-way communication between tumor cells and neighboring immune cells. The activated cGAS-STING further increased the production of type I interferons and secretion of pro-inflammatory cytokines (TNF-α and IL-6), boosting the tumor infiltration of dendritic cells and macrophages, as well as stimulating cytotoxic T cells to kill cancer cells in vitro and in vivo. Owing to the enhanced DNA-damaging ability, MnPC and MnPVA showed more potent immunocompetence and antitumor activity than Mn2+ ions, thus demonstrating great potential as chemoimmunotherapeutic agents for cancer treatment.
Introduction
The recurrence, metastasis, and drug resistance of tumors pose big challenges for conventional antitumor drugs.1 Immunotherapy is an effective strategy to eradicate tumor cells by harnessing or boosting the immune system of patients.2,3 In the past decade, multiple cancer immunotherapies including immune checkpoint inhibitors, oncolytic virus, and chimeric antigen receptor T (CAR-T) cells were applied in clinical practice to enhance adaptive antitumor immunity.4–6 However, primary and adaptive drug resistance, inadequate immune activation and loss of tumor-specific antigen targets frequently undermine the efficacy of immunotherapy.7,8 Adaptive antitumor immunity is highly dependent on robust innate immunity.8 Besides molding and sustaining adaptive antitumor immunity, the innate immune system as the first barrier for defending host cells takes key responsibility for the recognition of tumor cells.9 Unfortunately, during tumor progression, malignant cells often evade from immune surveillance and eventually develop into “cold” tumors.10,11 Thus, activating innate immunity and facilitating its interplay with adaptive antitumor immunity may reinforce the immune responses to tumors and improve the therapeutic effect of immunotherapy.
The cyclic GMP-AMP synthase-stimulators of interferon genes (cGAS-STING) constitute the vital components of innate immunity, which have emerged as promising targets for cancer immunotherapy.12,13 The activation of the cGAS-STING pathway triggers a series of downstream signaling events, including stimulation and recruitment of TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3), which induce the release and secretion of type I interferons (IFN-Is) and proinflammatory factors IL-6 and TNF-α.13,14 IFNs subsequently promote the maturation and migration of dendritic cells (DCs), enhance the natural killer (NK) cell-mediated cytotoxic effect and cross-prime tumor-specific T cells, thus orchestrating the innate and adaptive immunity to regulate the behavior of aggressive tumors.15,16 At present, the therapeutic profits of small molecule oral agonist MSA-2,17 natural agonist cyclic dinucleotides (CDNs),18 and some nano-systems19–23 have been tested in murine tumor models to intervene the cGAS-STING pathway.
Manganese (Mn) is a nutritional trace element that plays important roles in many physiological processes including antitumor immune responses.24,25 Recently, Mn2+ ions were discovered to enhance the sensitivity of a double strand DNA (dsDNA) sensor cGAS and trigger the production of a secondary messenger cGAMP, thereby increasing STING activity through augmenting cGAMP-STING binding affinity.12,26 Moreover, Mn2+ ions indirectly inhibit tumor progression by promoting the function of CD8+ T cells via inducing IFN production in vivo.27 Nevertheless, direct administration of Mn2+ ions in vivo cannot guarantee effective concentration in the tumor microenvironment23 and excessive Mn2+ ions may induce system toxicities, such as irreversible neurotoxicity and cardiovascular toxicity.28 Mn complexes are more stable and inert to biomolecules due to the sheltering effect of ligands and hence could alleviate the toxicity of Mn2+ ions. However, no Mn complex has been used to activate the cGAS-STING pathway so far.
Epigenetic modifications reversibly alter gene expressions, which may contribute to the initiation and progression of cancers and suppression of antitumor immunity. The reversible nature of epigenetic modification permits malignant cells to return to a normal state.29,30 Histone deacetylases (HDACs) mediate protein acetylation, chromatin dynamics, protein turnover and DNA damage responses. HDAC inhibitors are a class of potent epigenetic modulators, with chromatin as their target, acting upon most or all tumor types.31 The inhibition of HDACs at least partly contributes to histone acetylation, resulting in altered formation and repair of DNA double strand break (DSB),32,33 which may contribute to abolishing the drug resistance in cancer cells.34 The relaxed chromatin structure induced by HDAC inhibitors may promote chemotherapeutic agents to access DNA more easily, thereby aggravating DNA damage,35 which is beneficial for the activation of the cGAS-STING pathway. Some HDAC inhibitors—such as valproic acid (VA)—affect class I and II HDACs (HDAC1/2), which sensitize cancer cells to DNA damaging therapies.36
Poly adenosine diphosphate-ribose polymerases (PARPs) are essential proteins involved in cancer resistance to chemotherapies. These enzymes are crucial for the repair of DNA single-strand breaks (SSBs),37 and participate in multiple cellular processes, such as apoptosis, immune response, gene transcription, and inflammation.38–40 PARP inhibitors prevent the repair of DNA lesions, leading to the enrichment of cytosolic DNA, which is recognized by cGAS to activate the cGAS-STING pathway.41,42 A series of anticancer metal complexes including those of platinum, ruthenium, and gold have been found to inhibit the PARP activity.43,44
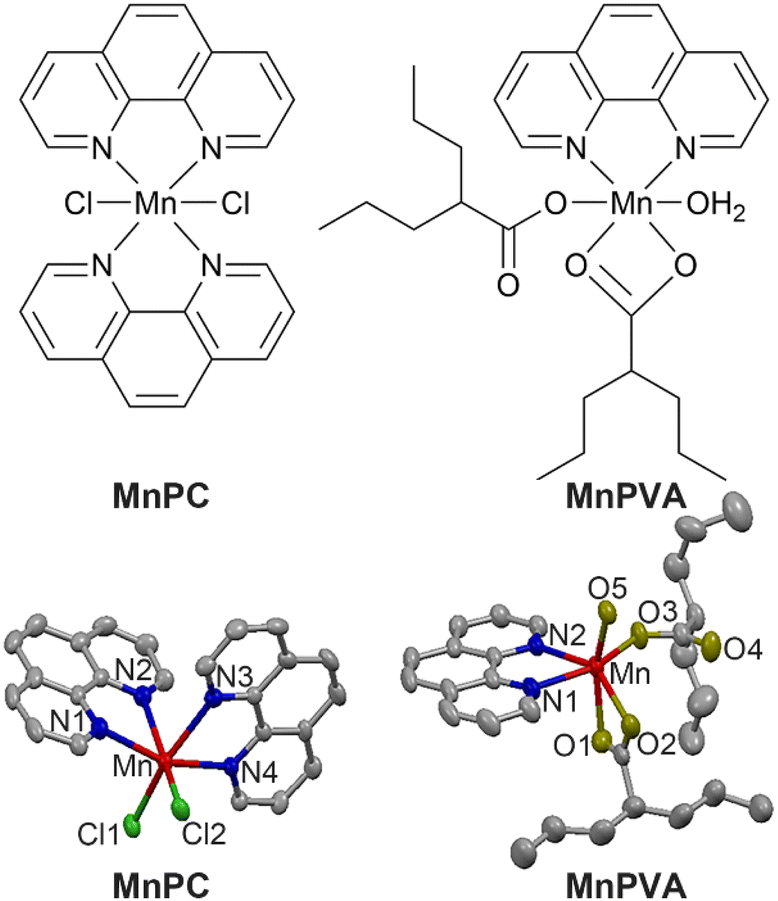
Herein we report the immunostimulating and antitumor properties of two MnII complexes MnPC and MnPVA. The chemical and crystal structures of the complexes are shown in Fig. 1. In these complexes, 1,10-phenanthroline (P) is a nontoxic DNA intercalator, and VA is an inhibitor of HADCs.31 VA could promote the acetylation of DNA-conjugated histone proteins, enhance the accessibility of DNA within relaxed chromatin, and reactivate dormant tumor suppressor genes.33,35 We suppose that the incorporation of 1,10-phenanthroline and/or VA into MnPC or MnPVA may endow the complexes with antiproliferative activity through inducing DNA damage and stimulating antitumor immunity. A series of experiments showed that MnPC and MnPVA effectively damaged DNA, activated the cGAS-STING pathway in tumor and immune cells, and increased the secretion of IFNs and pro-inflammatory cytokines. In particular, MnPVA inhibited the activity of HDAC1/2 and PARP1, thus activating the cGAS-STING pathway more effectively than MnPC. All the results were confirmed both in vitro and in vivo. To the best of our knowledge, MnPC and MnPVA are the first multifunctional MnII complexes that suppress tumor cells mainly through activating antitumor immunity via the DNA damage-initiated cGAS-STING pathway.
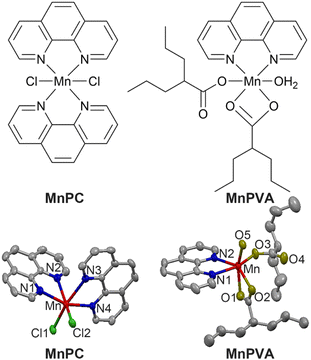 |
| | Fig. 1 Chemical and crystal structures of MnPC and MnPVA. Hydrogen atoms are omitted for clarity. | |
Results and discussion
Chemical and physical properties
MnPC and MnPVA were prepared according to literature methods45,46 and fully characterized by elemental analysis, IR spectroscopy (Fig. S1†), electron paramagnetic resonance (Fig. S2,† EPR), and X-ray crystallography. The crystal structure parameters and selected bond lengths and angles are presented in Tables S1 and S2 (see the ESI†). The Mn atom in these complexes exhibited a distorted octahedral geometry with a coordinate number of 6. MnPC crystallized in the monoclinic crystal system with the space group P21/c; MnII formed four Mn–N bonds with two bidentate 1,10-phenanthroline ligands and two Mn–Cl bonds. The Mn–N bond lengths vary between 2.281 and 2.369 Å, that is, the bond length of Mn–N1, Mn–N2, Mn–N3, and Mn–N4 is 2.369(4), 2.281(3), 2.341(3), and 2.287(3) Å, respectively, and the angle of N1–Mn–N2, N1–Mn–N4, and N2–Mn–N3 is 71.15(11)°, 97.86(11)°, and 89.00(11)°, respectively. This structure is somewhat different from that reported by Abbas et al.,45 where MnPC crystallized in the triclinic crystal system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Accordingly, the corresponding bond lengths and angles differ in these cases. Similar to a reported structure,46 MnPVA crystallized in the monoclinic crystal system with the space group C2/c, having a N2O4 donor set. MnII coordinated with two N atoms from 1,10-phenanthroline, and four O atoms from one water molecule and two VA ligands, respectively. One of the VA reacted as a monodentate ligand, while the other as a bidentate ligand. The bond length of Mn–N1 and Mn–N2 is 2.265(19) and 2.298(2) Å, respectively, and that of Mn–O1, Mn–O2, Mn–O3, and Mn–O5 is 2.2476(19), 2.260(2), 2.0639 (18), and 2.1471 (17) Å, respectively. As a strong N,N-chelating ligand, 1,10-phenanthroline stabilizes Mn complexes against demetallation.
. Accordingly, the corresponding bond lengths and angles differ in these cases. Similar to a reported structure,46 MnPVA crystallized in the monoclinic crystal system with the space group C2/c, having a N2O4 donor set. MnII coordinated with two N atoms from 1,10-phenanthroline, and four O atoms from one water molecule and two VA ligands, respectively. One of the VA reacted as a monodentate ligand, while the other as a bidentate ligand. The bond length of Mn–N1 and Mn–N2 is 2.265(19) and 2.298(2) Å, respectively, and that of Mn–O1, Mn–O2, Mn–O3, and Mn–O5 is 2.2476(19), 2.260(2), 2.0639 (18), and 2.1471 (17) Å, respectively. As a strong N,N-chelating ligand, 1,10-phenanthroline stabilizes Mn complexes against demetallation.
The stability of MnPC and MnPVA in phosphate buffered saline (PBS, with 0.5% v/v DMSO, pH 7.4, and 37 °C) and cell culture media (containing 10% FBS) was investigated by UV-visible spectroscopy (Fig. S3†). The time-dependent UV spectra demonstrate that these complexes are stable within 72 h.
Antiproliferative activity
The antiproliferative activities of the compounds against human triple-negative breast cancer (MDA-MB-231), human pancreatic cancer (PANC-1), human hepatocellular cancer (HepG2), mouse breast cancer (4T1) cell lines, and the human normal renal tubular epithelial (HK-2) cell line were evaluated by the MTT assay. MnCl2, VA, cisplatin (CDDP), and 1,10-phenanthroline (P) were used as reference compounds. The half maximal inhibitory concentrations (IC50) at 72 h are listed in Table 1. MnPC and MnPVA showed potent antiproliferative activity toward all the tested cancer cell lines, particularly to MDA-MB-231 and PANC-1 cells that are insensitive to CDDP, displaying about 8- and 11-times higher activity, respectively. The antiproliferative activity of MnPC and MnPVA against HepG2 and 4T1 is similar to that of CDDP. Interestingly, both MnPC and MnPVA showed a decreased toxicity towards HK-2 cells, with IC50 values being slightly higher than that of CDDP. By contrast, MnCl2, VA, and P were almost nontoxic towards these cell lines, which are in agreement with the literature.13,36 According to these results, the following investigations were focused on the action of MnPC and MnPVA on the MDA-MB-231 cells.
Table 1 IC50 values (μM) of MnPC and MnPVA against different cell lines at 72 h, with MnCl2, VA, CDDP, and P as references. Data are shown as mean ± standard deviation (SD, n = 3)
| Complex |
MDA-MB-231 |
PANC-1 |
HepG2 |
4T1 |
HK-2 |
| MnPC |
5.62 ± 0.41 |
4.21 ± 0.49 |
6.11 ± 0.37 |
5.57 ± 0.76 |
8.02 ± 0.95 |
| MnPVA |
4.76 ± 0.41 |
5.37 ± 0.58 |
4.98 ± 0.45 |
4.35 ± 0.36 |
7.51 ± 0.58 |
| MnCl2 |
>64 |
>64 |
>64 |
>64 |
>64 |
| VA |
>64 |
>64 |
>64 |
>64 |
>64 |
| CDDP |
38.34 ± 3.16 |
55.46 ± 1.48 |
4.34 ± 0.32 |
5.65 ± 0.37 |
7.45 ± 0.19 |
| P |
37.41 ± 2.84 |
35.55 ± 3.93 |
>64 |
>64 |
>64 |
Cellular uptake and DNA damage
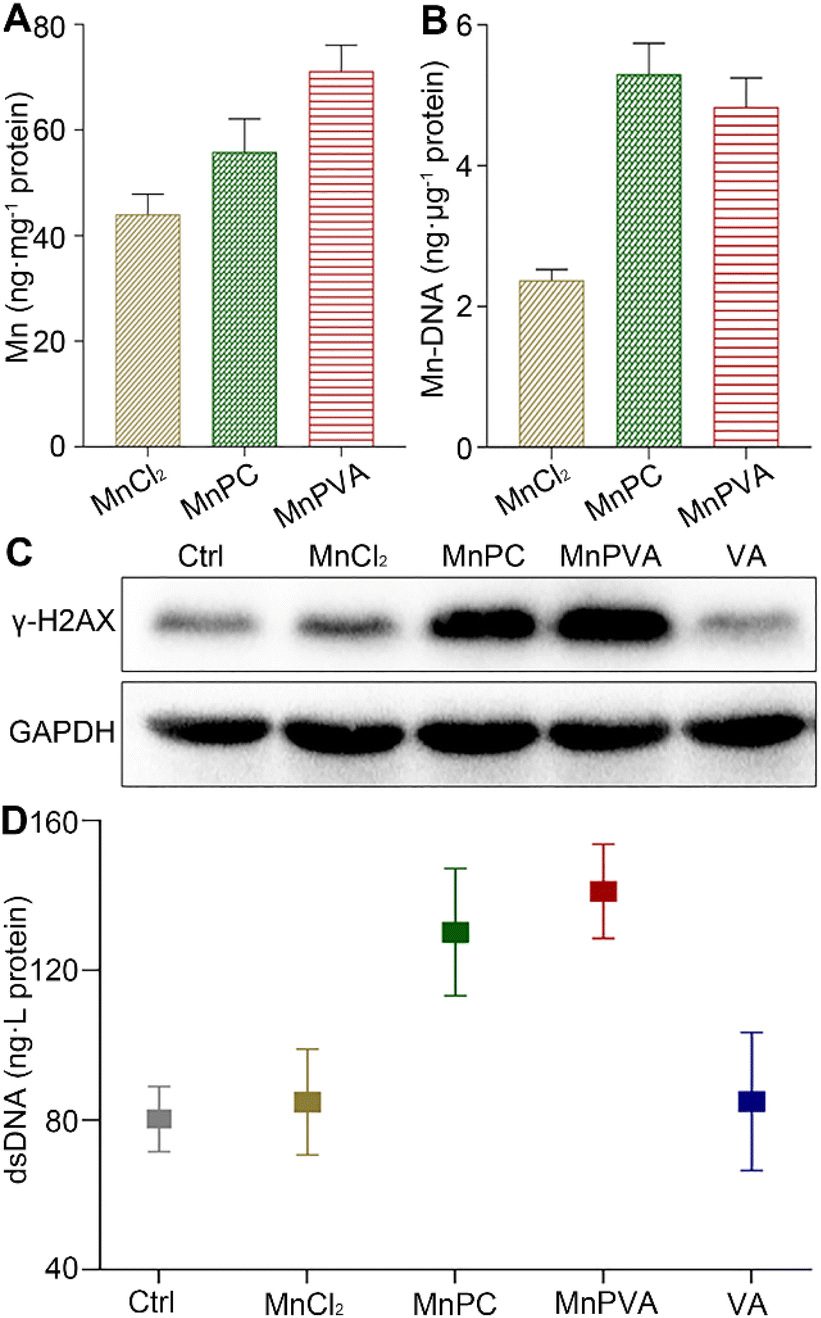
The accumulation of the complexes in MDA-MB-231 cells in terms of Mn was measured by ICP-MS after co-incubation for 24 h. As shown in Fig. 2A, the accumulation follows an order of MnPVA > MnPC > MnCl2, keeping in line with their antiproliferative activity. We further determined the DNA-bound Mn in MDA-MB-231 cells by ICP-MS. As shown in Fig. 2B, MnPC and MnPVA exhibited greater DNA binding ability than MnCl2 possibly due to their higher cellular uptake.
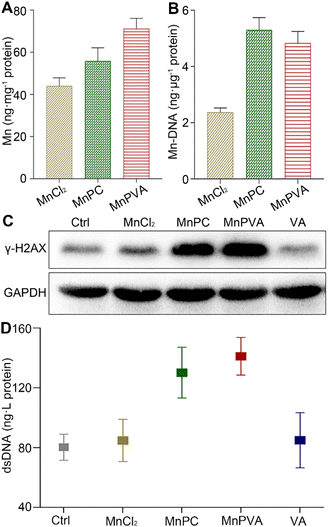 |
| | Fig. 2 Cellular uptake (A) and DNA-bound Mn (B) determined by ICP-MS, expression of the DNA damage marker γ-H2AX determined by western blotting (C), and extracellular dsDNA determined by using the ELISA kit (D) after MDA-MB-231 cells were treated with MnCl2, MnPC, MnPVA, and VA (6 μM) respectively for 24 h. | |
Phosphorylated histone γ-H2AX is a sensitive marker of DNA double strand breaks (DSBs).47 To evaluate the DNA damage induced by the Mn complexes, we detected the expression of γ-H2AX by immunoblotting. As shown in Fig. 2C, MnPC and MnPVA increased the expression of γ-H2AX in MDA-MB-231 cells at 24 h. It is known that metal complexes of P are effective DNA intercalators;48,49 however, P alone hardly caused DNA damage (Fig. S4†). The expression of γ-H2AX in tumor tissues of 4T1 tumor-bearing mice was also investigated by immunofluorescence after treatment with each compound (1.3 mg Mn per kg) once every 2 days for 16 days. MnPC and MnPVA effectively damaged DNA in the tumor tissue, while MnCl2 and PBS barely affected γ-H2AX (Fig. S5†). The dsDNA released from MDA-MB-231 cells to the culture medium supernatant was determined by using an ELISA kit after treatment with the Mn complexes. As presented in Fig. 2D, the content of dsDNA in the MnPC- or MnPVA-treated cell culture medium was evidently higher than that in other groups. The results suggest that MnPC and MnPVA can elevate the instability of genomic DNA and the level of cytosolic DNA.
The nature of the DNA damage was first studied by measuring the fluorescence changes of a calf thymus DNA-ethidium bromide (EB) system. EB is an intercalator that gives a significant increase in fluorescence when bound to DNA, while the fluorescence decreases when it is displaced.50 MnPC and MnPVA moderately reduced the fluorescence intensity (Fig. S6†), suggesting that these complexes could intercalate into the DNA grooves to replace the bound EB due to the planar structure of P. However, the interaction is not a robust covalent binding.
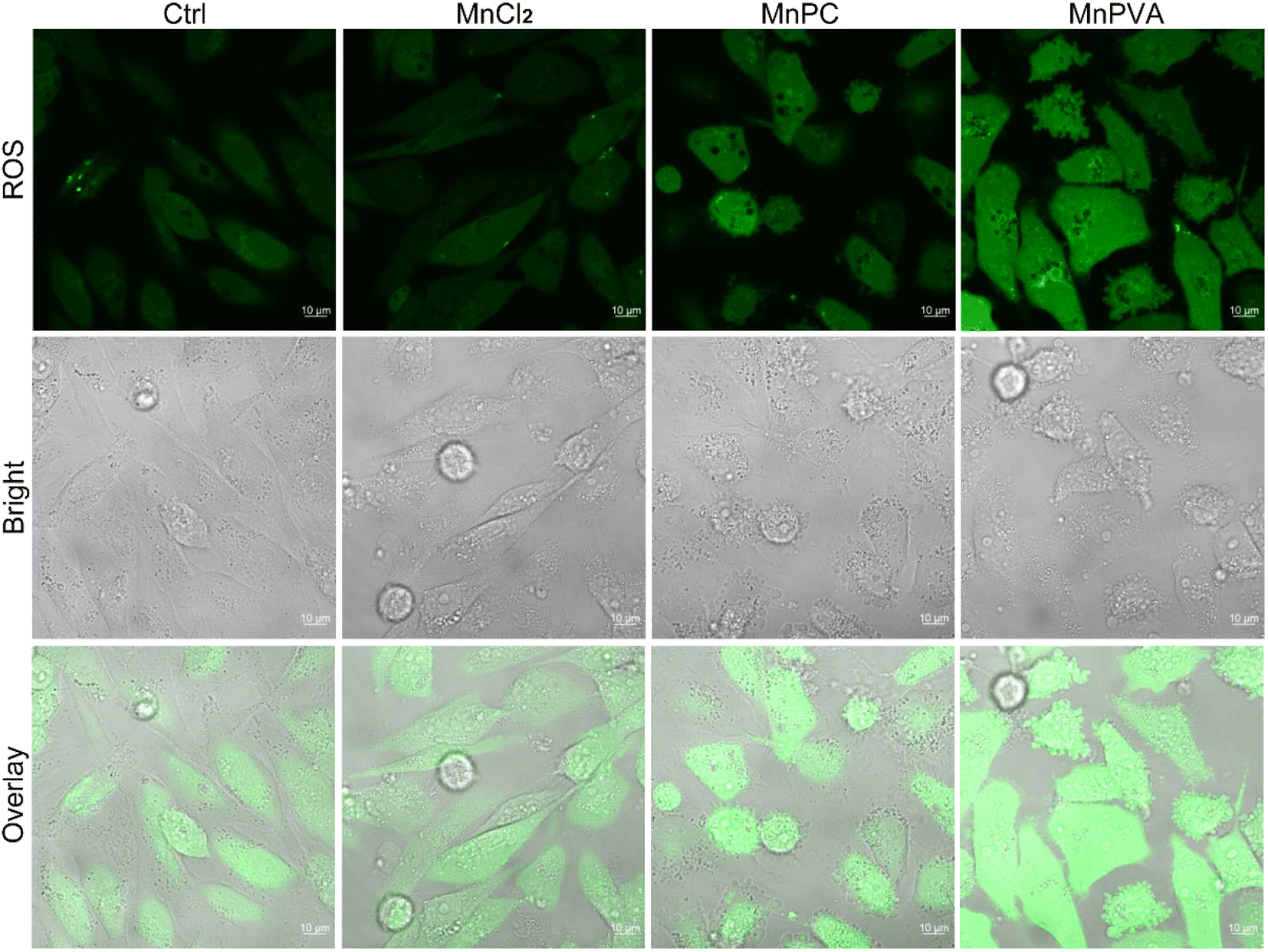
Some Mn complexes could generate intracellular reactive oxygen species (ROS) and induce oxidative stress,51 and we hence detected the ROS in MDA-MB-231 cells by confocal imaging using 2′,7′-dichloro-fluorescein diacetate (DCFH-DA) as a ROS fluorescence probe after exposure to the Mn complex for 18 h. As shown in Fig. 3, MnPC and MnPVA intensified the intracellular ROS fluorescence as compared to the control or MnCl2, especially MnPVA, suggesting that they may cause oxidative damage to cellular DNA. The damage to DNA not only contributes to the killing action on cancer cells, but also stimulates antitumor immunity through activating the cGAS-STING pathway.14,52
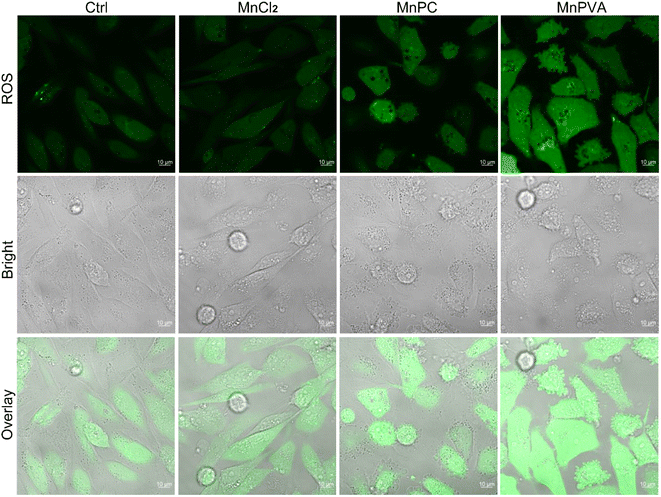 |
| | Fig. 3 Fluorescence confocal imaging of ROS generated by the Mn complexes (12 μM) in MDA-MB-231 cells after incubation for 18 h and staining with DCFH-DA (10 μM) for 30 min. | |
Cell cycle arrest and apoptosis
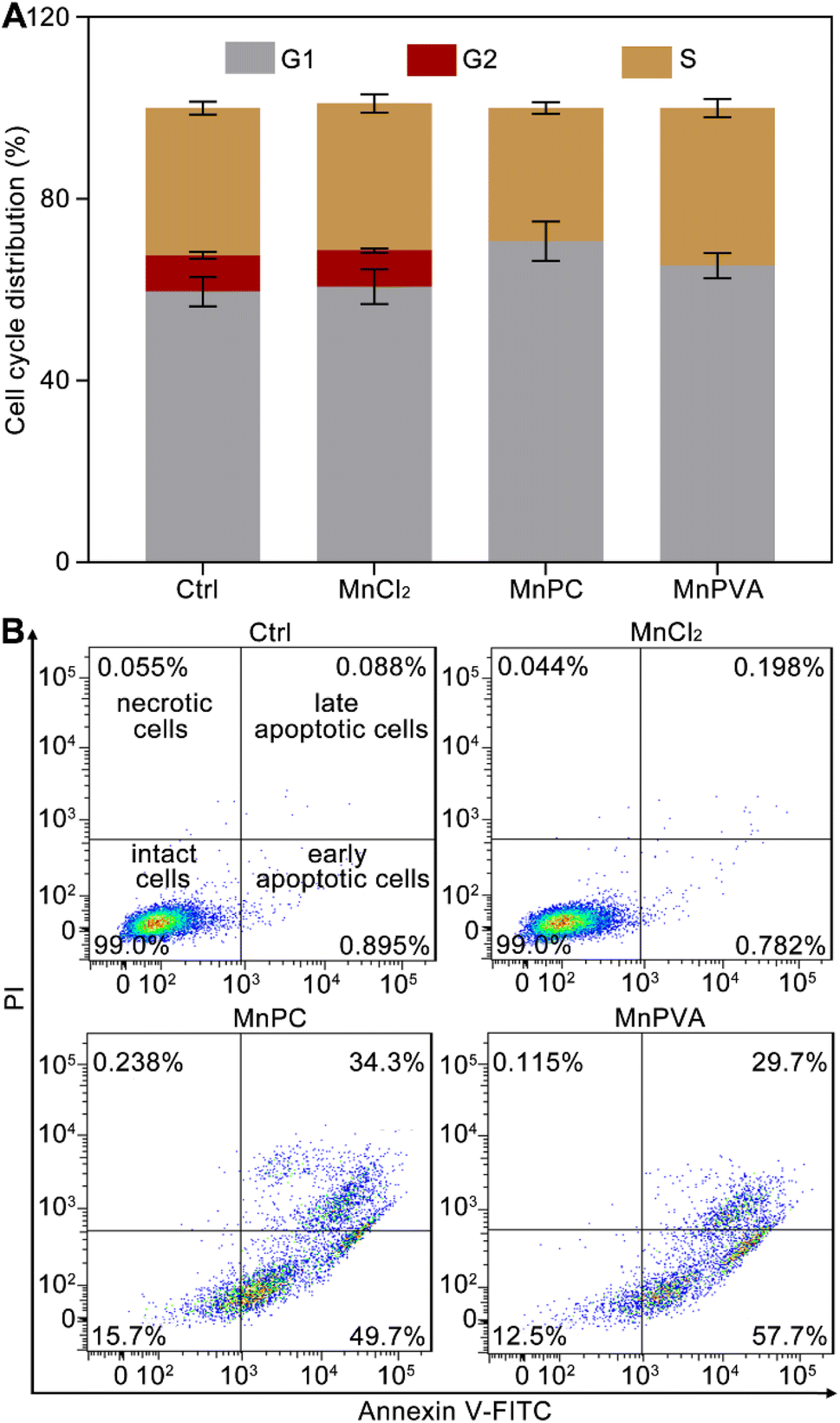
Cellular DNA damage response (DDR) is associated with the signaling that drives the checkpoint capture of the cell cycle.53 The impact of Mn complexes on the cell cycle of MDA-MB-231 cells was analyzed by flow cytometry. As shown in Fig. 4A, MnPC and MnPVA mildly increased the G1 phase arrest while caused a complete collapse of the G2 phase as compared with the control and MnCl2. The results indicate that the DNA damage induced by MnPC and MnPVA seriously blocked the later stage of DNA synthesis. Hence, the antiproliferative mechanism of MnPC and MnPVA differs from that of MnCl2. The death mode of MDA-MB-231 cells was investigated by flow cytometry after treatment with the complexes for 72 h and staining with annexin V-FITC and propidium iodide (PI). As shown in Fig. 4B and S7,† in comparison with the control and MnCl2, the apoptosis (early + late) rate induced by MnPC and MnPVA reached 84.0% and 87.4% respectively. The results indicate that MnPC and MnPVA have strong pro-apoptotic ability, which closely correlates with their antitumor activity.
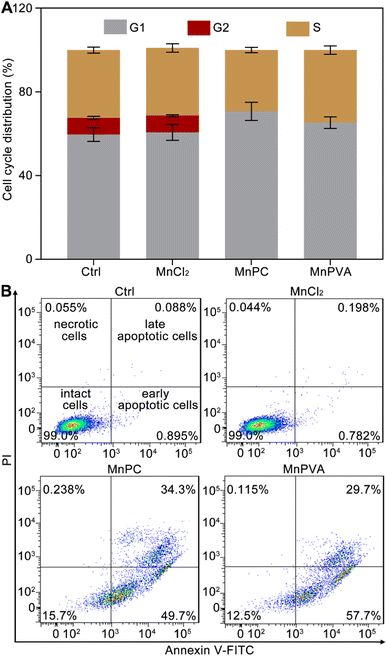 |
| | Fig. 4 Cell cycle arrest of MDA-MB-231 cells after incubation with different compounds (6 μM) for 24 h (A), and apoptotic analysis of MDA-MB-231 cells by flow cytometry after incubation with different compounds (6 μM) for 72 h (B). | |
Mitochondrial membrane potential
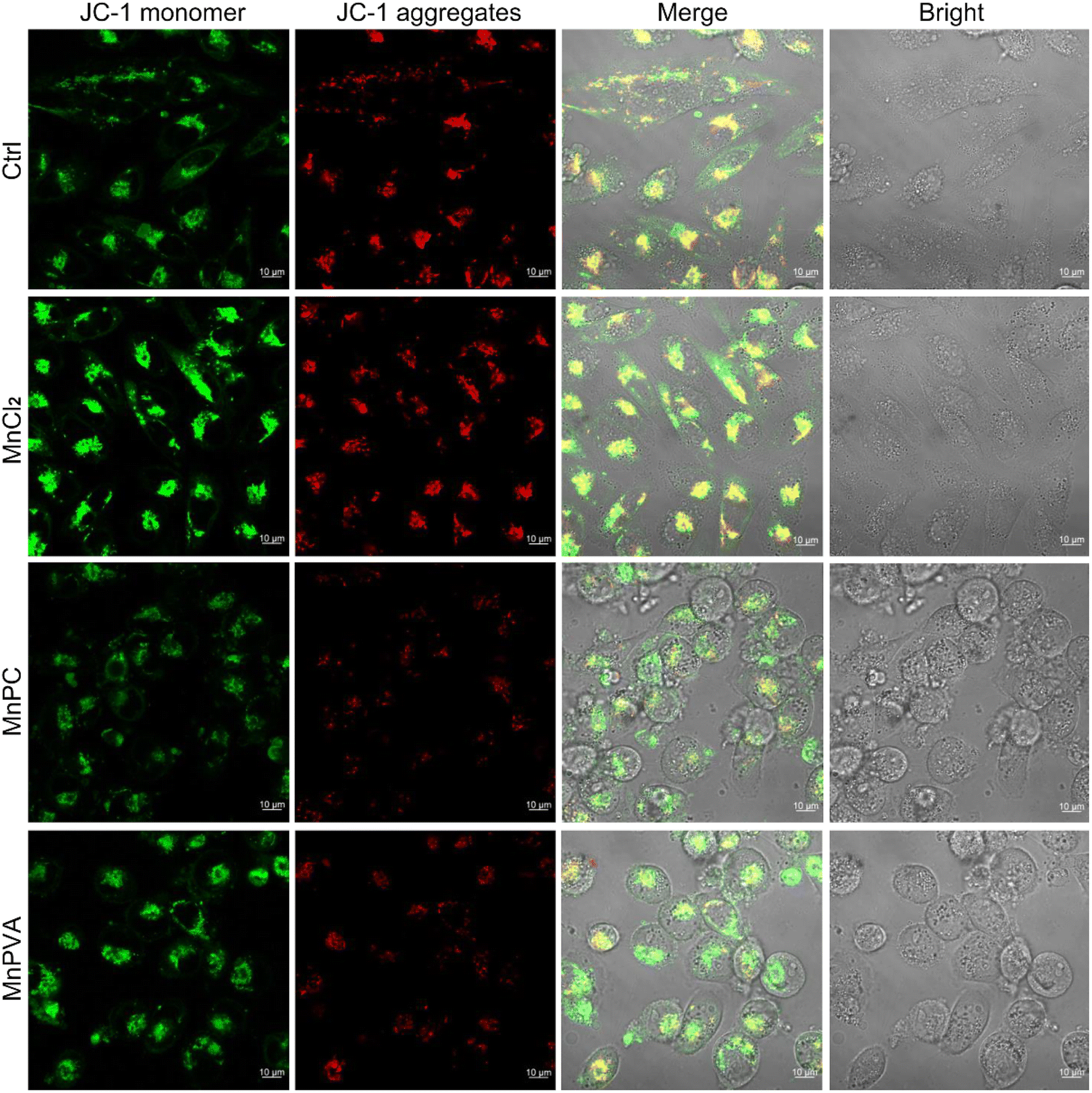
Mitochondria are central participants in innate immunity, and mitochondrial DNA (mtDNA) is recognized as an agonist of the innate immune system.54 It was reported that both cytosolic DNA and mtDNA bound to pattern-recognition receptors and triggered the cGAS-STING signaling pathway.55 To explore the possible mtDNA release from mitochondria in the presence of MnPC and MnPVA, we checked the integrity of the mitochondrial membrane by confocal imaging using JC-1 as a fluorescent probe, which forms aggregates and displays red fluorescence in normal mitochondria with a high ΔΨm, while exists as a monomer and gives off green fluorescence in damaged ones with low ΔΨm.56 As shown in Fig. 5, MnPC and MnPVA greatly reduced the intensity of red fluorescence in MDA-MB-231 cells stained with JC-1. The “green (G) to red (R)” fluorescence ratio for MnPC- and MnPVA-treated cells is 4.91 and 5.66, respectively, significantly higher than that for the control (2.35) and MnCl2-treated cells (1.75). The observations suggest that the integrity of the mitochondrial membrane was damaged by MnPC and MnPVA, which may facilitate the leakage of mtDNA into the cytosol to trigger the cCAS-STING pathway.
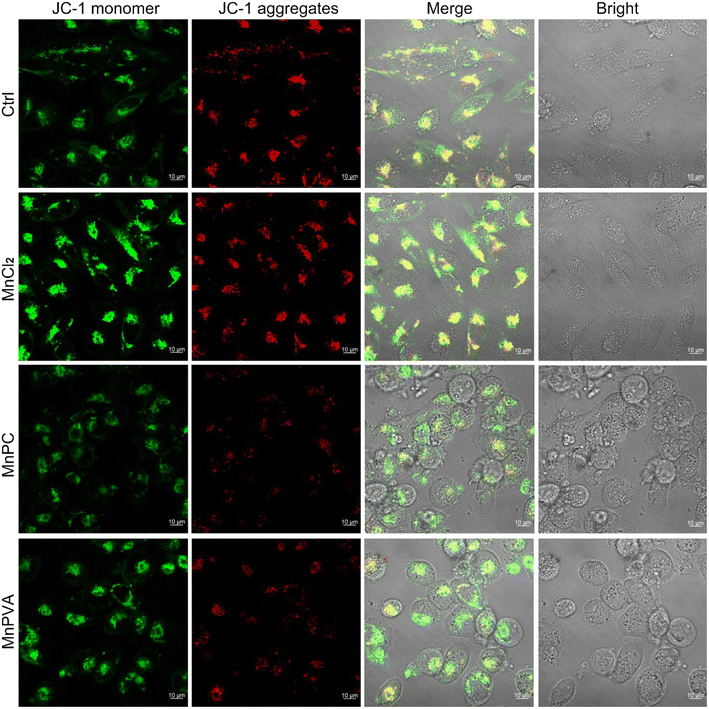 |
| | Fig. 5 Fluorescence images of MDA-MB-231 cells after incubation with MnCl2, MnPC, and MnPVA (6 μM) respectively for 36 h and staining with the JC-1 fluorescent probe. | |
Activity and expression of HDACs
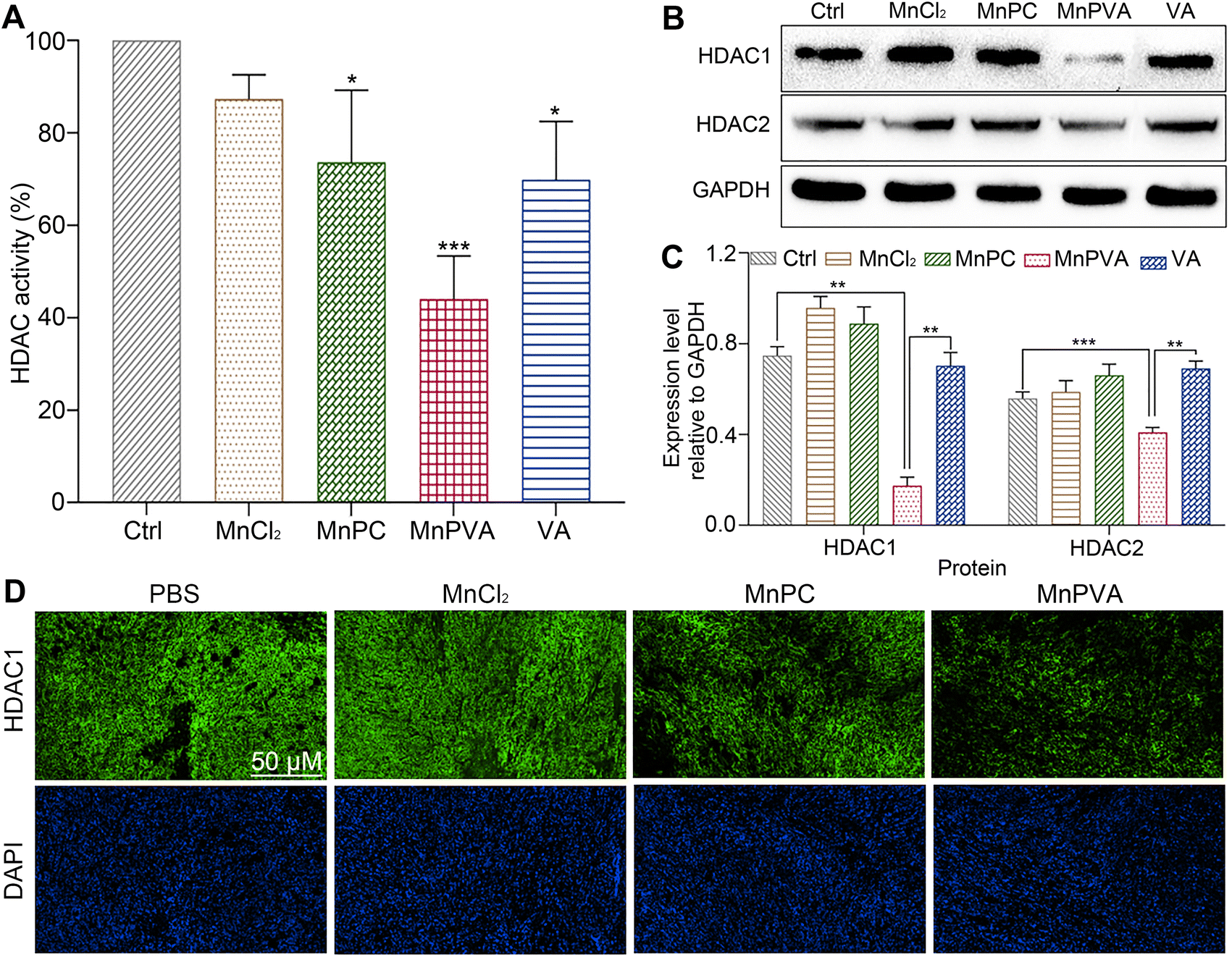
HDAC inhibitors sensitize cancer cells to DNA-damaging therapies by altering the chromatin structure and impeding DNA repair.33,35 As a class I HDAC inhibitor, VA selectively targets the HDAC1 and HDAC2 enzymes, modulating DNA damage signaling to destroy genomic stability and inhibit tumor proliferation in vivo.31 The incorporation of VA in MnPVA may reinforce the inhibition of HDAC and induction of DDR more efficiently. Therefore, we determined the effect of Mn complexes on the total HDAC activity in MDA-MB-231 cells after co-incubation for 48 h. As shown in Fig. 6A, MnPVA significantly inhibited the total HDAC activity, with inhibitory activity being about 1.5 times higher than that of VA. We further studied the expression of HDAC1/2 proteins in MDA-MB-231 cells by immunoblotting. As shown in Fig. 6B and C, MnPVA obviously downregulated the expression of HDAC1 and HDAC2 as compared with the control. Although MnPC inhibited the HDAC activity, it hardly influenced the expression of HDAC1/2 proteins. Unexpectedly, VA as a known HDAC inhibitor did not show obvious inhibition on HADC1/2 at this concentration (6 μM), thus highlighting the essential role of Mn coordination in intensifying the inhibition of VA on HADC. The results indicate that MnPVA is an effective inhibitor of HDAC1/2, which would promote DDR and trigger the cGAS-STING pathway, thereby stimulating antitumor immunity. We further detected the expression of HDAC in tumor tissues of 4T1 tumor-bearing mice by immunofluorescence after treatment with each compound. As shown in Fig. 6D, MnPVA evidently decreased the expression of HDAC1, while MnPC and MnCl2 only slightly or barely affected HDAC1.
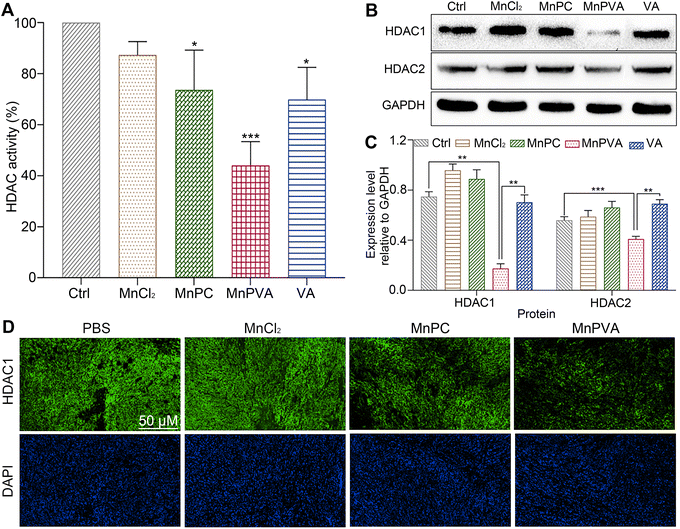 |
| | Fig. 6 (A) HDAC activity, (B) expression of HDAC1/2 proteins, and (C) the corresponding protein content relative to GAPDH in MDA-MB-231 cells after incubation with each compound (6 μM) for 48 h determined by using the ELISA kit and western blotting respectively; (D) immunofluorescence images of HDAC1 expression in the 4T1 tumor tissue of a mouse after treatment with each compound (1.3 mg Mn per kg) once every 2 days for 16 days. *p < 0.05, **p < 0.01, and ***p < 0.001. | |
Expression of PARP1 and apoptotic proteins
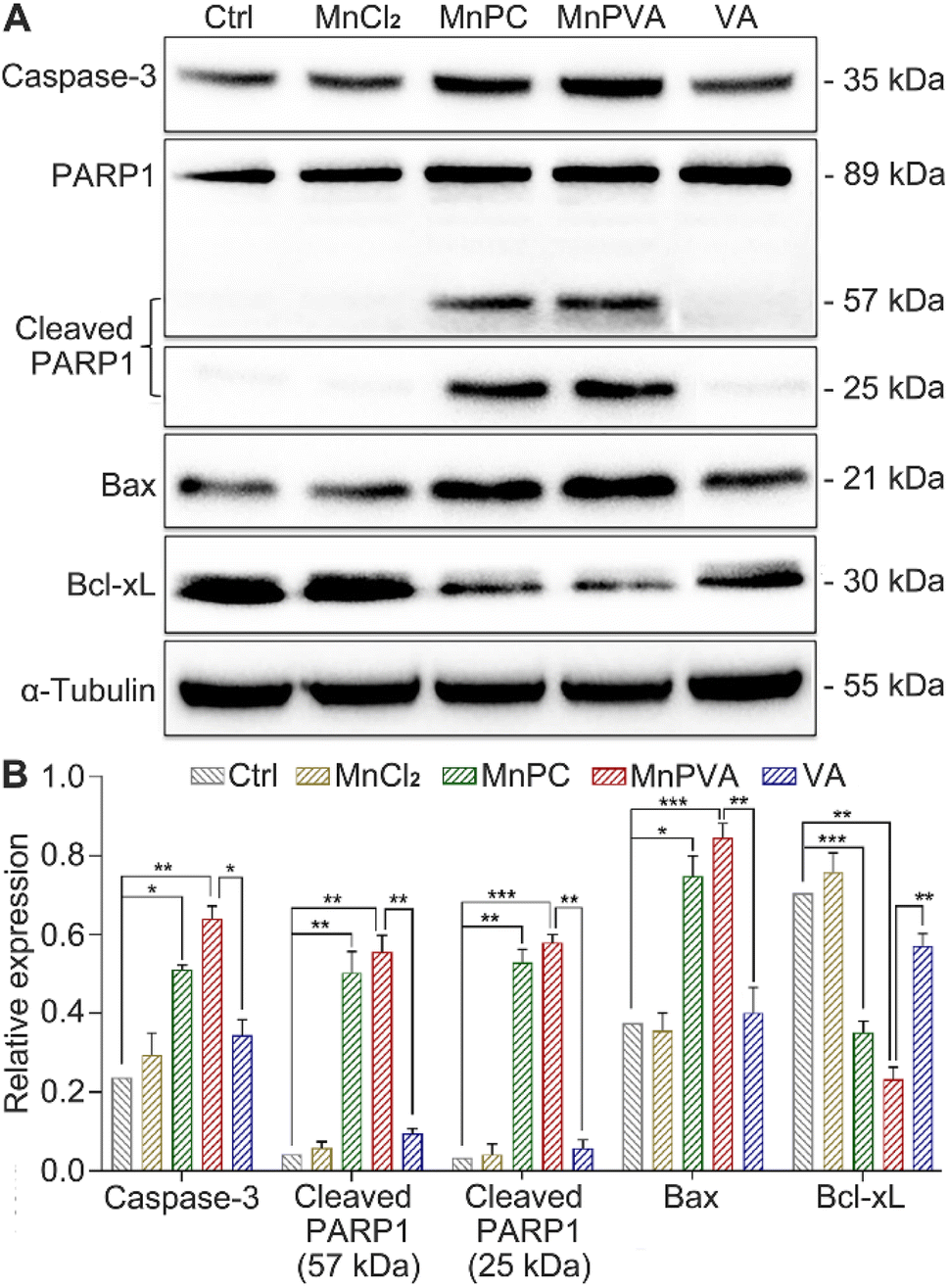
PARPs play a key role in the repair of DNA lesions. PARP inhibitors prevent DNA repair, which leads to the export of DNA fragments to the cytosol, triggering a cGAS-mediated innate immune response.42 As a sensor protein for DNA strand breaks, PARP1 localizes to the sites of DNA damage and dominates the repair of DNA, thus becoming an important target for cancer therapy. The activation of caspases, especially caspase-3, could initiate apoptosis through cleaving PARP1, which is regarded as a hallmark of apoptosis.38,57 Therefore, the expressions of caspase-3, PARP1, and cleaved PARP1 in MDA-MB-231 cells were examined by western blotting. As shown in Fig. 7, MnPC and MnPVA remarkably upregulated the expression of caspase-3 and cleaved PARP1 as compared with the control and other compounds. The cleavage of PARP1 by activated caspase-3 would separate the DNA-binding domain from the catalytic domain, thereby preventing PARP activation and repair of DNA lesions. Furthermore, the inhibition of PARP1 may increase the level of tumor-infiltrating T lymphocytes and regulate the innate immune responses.
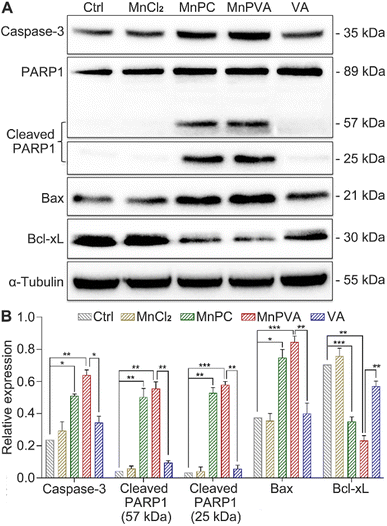 |
| | Fig. 7 Expression of DNA repair- and apoptosis-related proteins in MDA-MB-231 cells after exposure to different compounds (6 μM) for 48 h, and the corresponding protein content relative to α-tubulin. *p < 0.05, **p < 0.01, and ***p < 0.001. | |
DNA-repair dysfunction has the potential to initiate global DNA aberrations, which could trigger apoptosis. Proapoptotic Bax and antiapoptotic Bcl-xL proteins play pivotal roles in apoptosis.38 Therefore, we detected their expression in MDA-MB-231 cells after treatment with different compounds for 48 h using western blotting. MnPC and MnPVA markedly upregulated the expression of Bax and down-regulated the expression of Bcl-xL. The results indicate that MnPC and MnPVA could promote apoptosis via regulating apoptotic proteins as a PARP1-related cascade effect. Although VA can induce tumor cell apoptosis by inhibiting the expression of HDAC1/2 at high doses,58 it did not affect these apoptotic proteins significantly under this condition, signifying the superiority of Mn complexes.
Activation of the cGAS-STING pathway
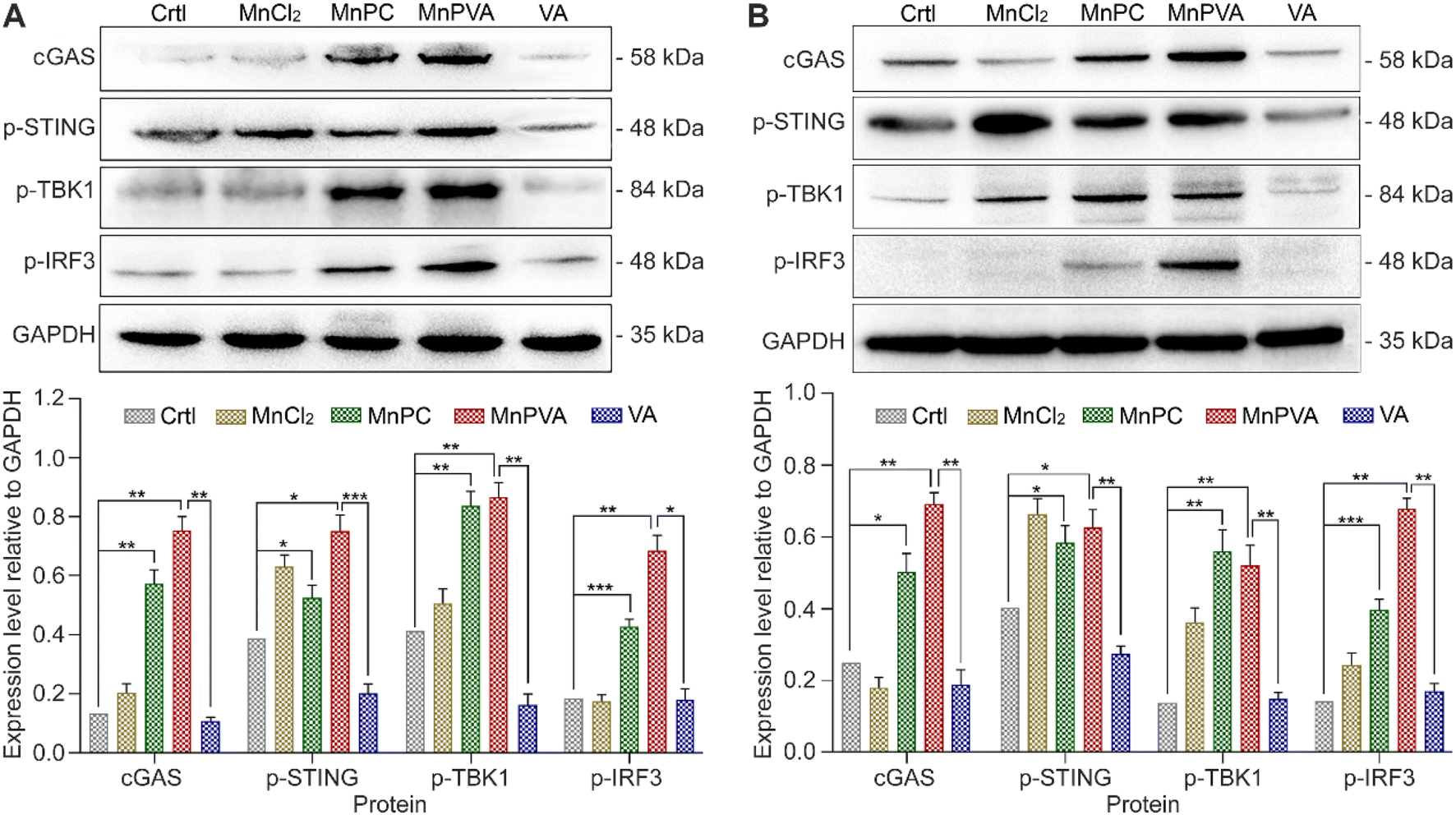
It is known that the PARP1 inhibitor can elicit a cGAS-STING signaling cascade response in cancer cells.40,59 cGAS is an innate immune sensor that recognizes a diverse array of cytosolic dsDNA, including DNA with viral, apoptotic, exosomal, mitochondrial, micronuclei, and retroelement origins.60,61 The enrichment of cytosolic DNA induced by MnPC and MnPVA created a favorable circumstance for activating the cGAS-STING pathway, which would subsequently initiate downstream signaling events via the recruitment and activation of TBK1 and IRF3.12,59 We therefore detected the expression of these proteins in MDA-MB-231 cells by immunoblotting after exposure to each compound for 24 h. As shown in Fig. 8A, MnPC and MnPVA significantly elevated the expression of cGAS, p-STING, p-TBK1, and p-IRF3. Furthermore, we investigated the effect of the complexes on the cGAS-STING pathway in human monocytic leukemia THP-1 cells by immunoblotting. As shown in Fig. 8B, MnPC and MnPVA induced a significant upregulation of cGAS, p-STING, p-TBK1 and p-IRF3, especially MnPVA, manifesting that they activated the cGAS-STING pathway, which would initiate innate immunity to block tumor escape and stimulate adaptive immunity, without harming the THP-1 cells (Table S3†). By contrast, MnCl2 only increased the level of p-STING or p-TBK1 and barely affected the expression of cGAS and p-IRF3 as compared with the control, which may be due to its inability to induce DNA damage and PARP1 cleavage at low concentrations, and thus is unable to trigger downstream signaling events such as the activation of p-IRF3. Based on these results, we conclude that MnPC and MnPVA induced nuclear and mitochondrial DNA damage within tumor cells and released the DNA fragments to cytoplasm, which then activated the cGAS-STING pathway. The activated cGAS-STING could directly activate senescence and apoptosis signaling pathways in cancer cells,62 leading to a bidirectional antitumor immune response. Nevertheless, the expression of STING, TBK1, and IRF-3 in MDA-MB-231 and THP-1 cells was barely affected (Fig. S8†).
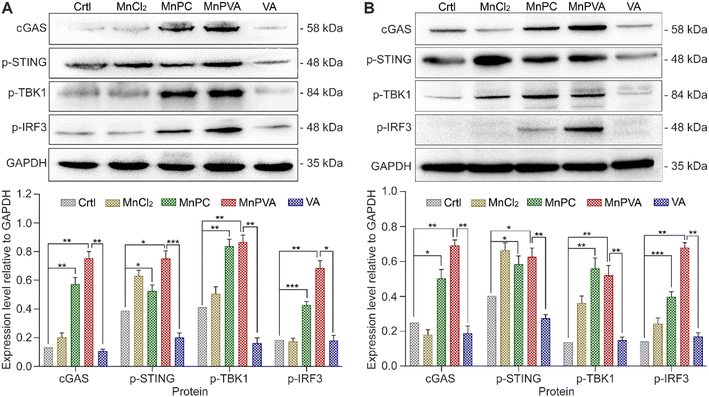 |
| | Fig. 8 Expression of proteins involved in the cGAS-STING pathway determined by western blotting after MDA-MB-231 (A) and THP-1 (B) cells were treated with 6 and 3 μM of different compounds for 24 h, respectively, and the corresponding protein content relative to GAPDH. *p < 0.05, **p < 0.01, and ***p < 0.001. | |
Interferons and proinflammatory cytokines
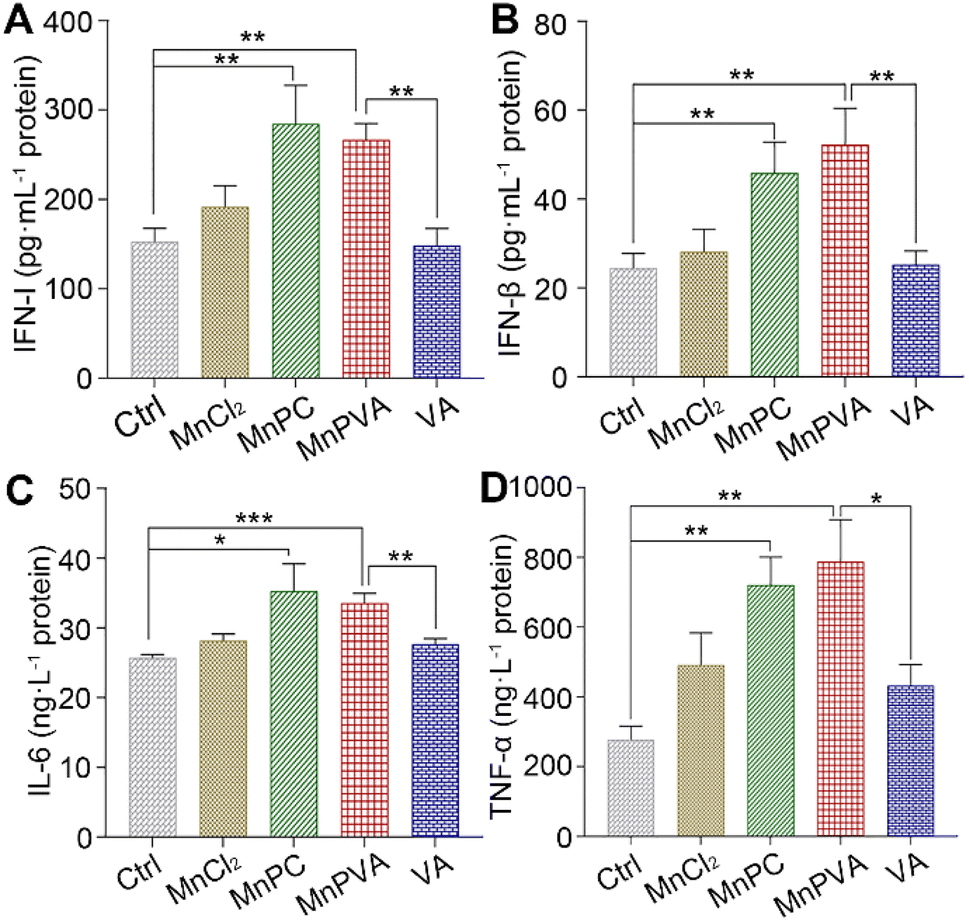
The activation of TBK1 and IRF3 could induce the expression and secretion of IFNs and proinflammatory cytokines, such as IFN-β, TFN-α, and IL-6.59,63 Thus, the extracellular IFN-I released by THP-1 cells and the IFN-β, TNF-α, and IL-6 released by MDA-MB-231 cells were measured by ELISA assay after incubation with different compounds for 24 h. As shown in Fig. 9, MnPC and MnPVA dramatically enhanced the secretion of IFN-I relative to the control, MnCl2, and VA. At the same time, they also triggered the secretion of IFN-β and TNF-α. However, the level of IL-6 was only moderately increased. IFN-I (IFN-α and IFN-β) plays multiple immunostimulatory roles in antitumor immunity, such as promoting maturation and antigen presentation of DCs, and cross-priming tumor-specific T cells to kill tumor cells by bridging the innate and adaptive immunity.13 TNF-α is involved in the cyclic dinucleotide (CDN)-mediated acute cancer necrosis and adjustment of the immune infiltrate.59 The results signify that MnPC and MnPVA could stimulate antitumor immune responses and suggest that they may overcome the drug resistance of tumors by a chemoimmunotherapeutic mechanism.
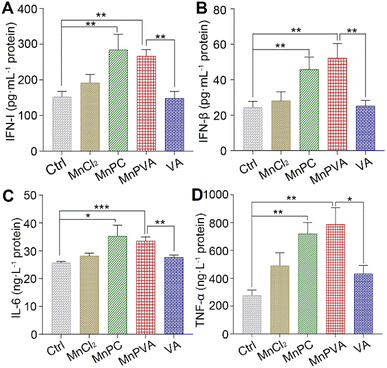 |
| | Fig. 9 Secretion of (A) IFN-I in THP-1 cells, (B) IFN-β, (C) IL-6, and (D) TNF-α in MDA-MB-231 cells after treatment with different compounds (6 μM) for 24 h. Data are shown as mean ± SD (n = 3); *p < 0.05, **p < 0.01, and ***p < 0.001. | |
Maturation of bone marrow-derived cells (BMDCs)
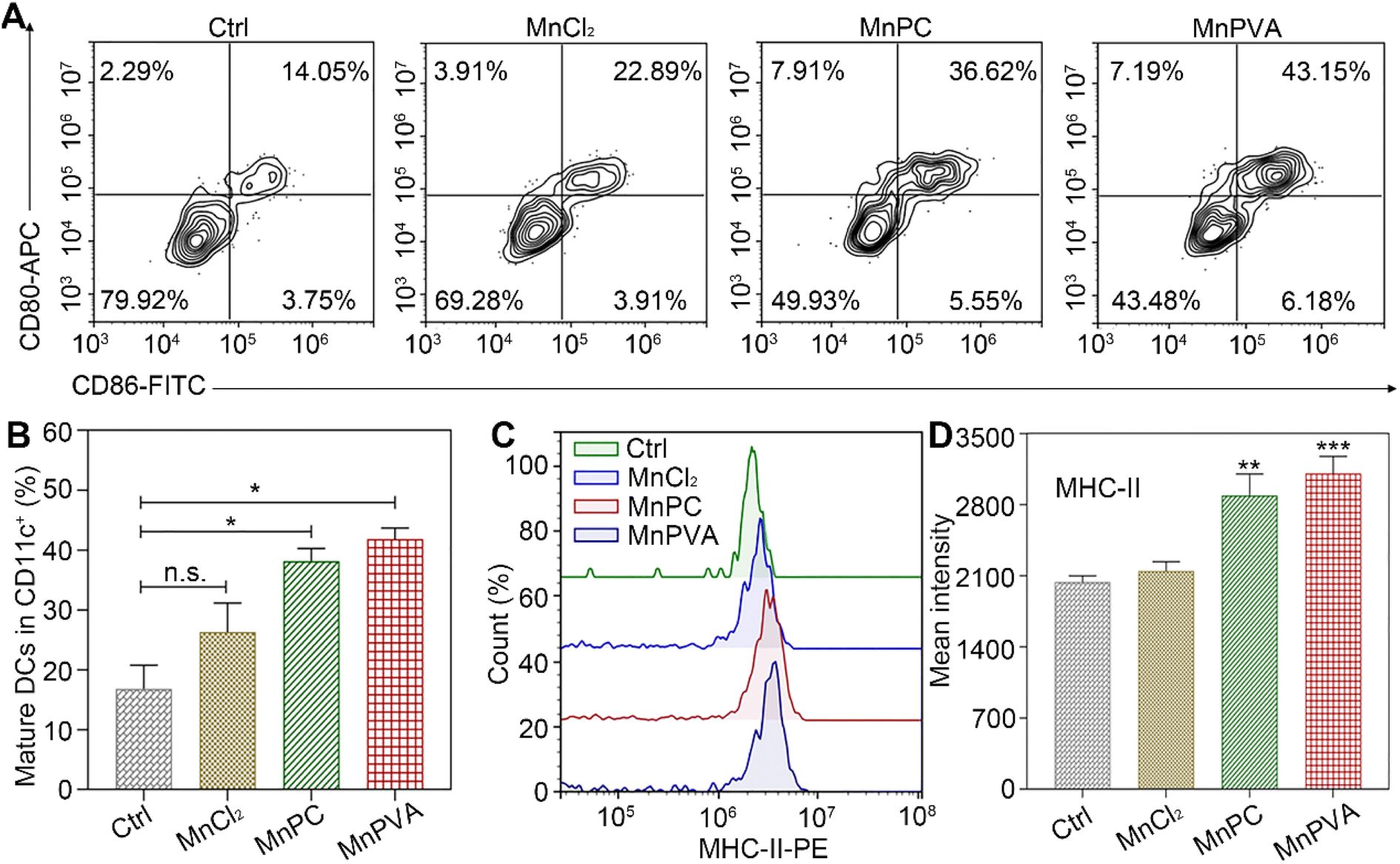
BMDCs represent heterogeneous cells of myeloid origin consisting of myeloid progenitors and immature macrophages, granulocytes and dendritic cells (DCs); they are involved in immune response by suppressing T-cell activation and tumor associated macrophage (TAM) activation.64 As the main antigen-presenting cells (APCs), DCs function as a bridge for communication between the innate and adaptive immune systems.22 Mature and activated DCs can present a specific antigen to T lymphocytes and initiate an adaptive response against tumors.65 To test whether the MnPC- and MnPVA-activated cGAS-STING pathway could induce the maturation of BMDCs, we tested the surface markers on BMDCs by flow cytometry after treatment with the supernatant of MDA-MB-231 cells incubated with different compounds for 24 h. As shown in Fig. 10A, in comparison with the control, MnPC and MnPVA notably elevated the co-expression of maturation markers CD86 and CD80, reaching 36.62% and 43.15%, respectively. However, MnCl2 only moderately surpassed that of the control. In addition, major histocompatibility complex class II (MHC-II) is constitutively expressed by APCs, which presents antigenic peptides to T cells. As a result, adaptive immune responses are initiated, maintained, and regulated.8 As shown in Fig. 10B and C, MnPC and MnPVA increased the expression of MHC-II to about 1.5-fold that of the control. The data suggest that these complexes greatly induced the maturation of BMDCs, which may trigger the immune responses of cytotoxic T lymphocytes.
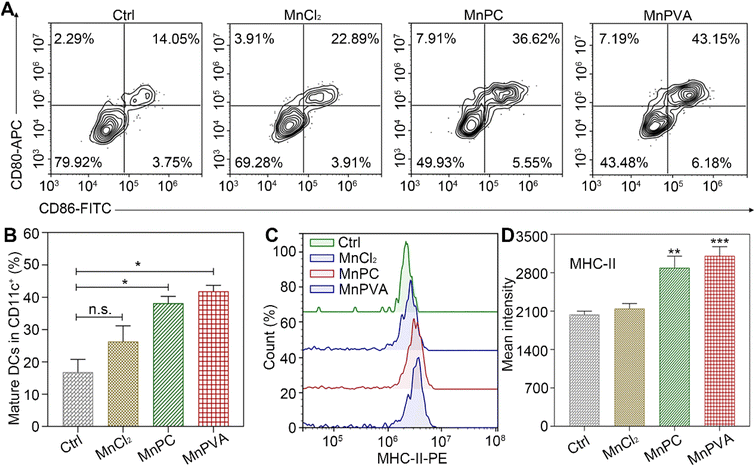 |
| | Fig. 10 (A) Co-expression of CD86 and CD80, (B) quantitative analyses of CD11c+CD86+CD80+ in BMDCs, (C) expression of MHC-II on BMDCs' surface, and (D) mean intensity of MHC-II determined by flow cytometry after treatment with the supernatant of MDA-MB-231 cells incubated with different compounds (6 μM) for 24 h. | |
Co-culture of cancer cells with PBMCs
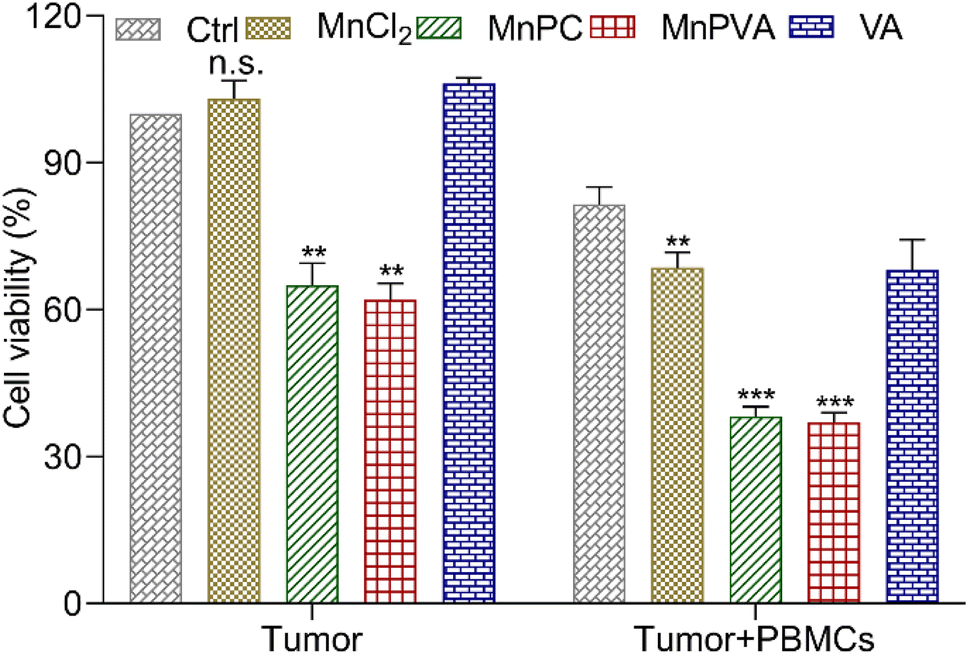
To further assess the immunomodulatory effects of the complexes, the viability of MDA-MB-231 cells in the presence of drug-activated human peripheral blood mononuclear cells (PBMCs) was evaluated by the MTT assay according to a literature method.66 Cancer cells or cancer cells with PBMCs were set as controls. As shown in Fig. 11, in the absence of PBMCs, MnPC and MnPVA suppressed the cell viability to 64.98% and 62.02%, respectively. The co-culture of the cells with PBMCs without a Mn complex only showed basal cytotoxicity with a mean cell viability of 81.49%. However, in the presence of PBMCs, MnPC and MnPVA suppressed the cell viability to 38.19% and 36.98%, respectively. At this concentration (6 μM), most PBMCs are still alive (Fig. S9†). The results demonstrate that MnPC and MnPVA can activate the immune response of PBMCs to exert cytotoxic effects on MDA-MB-231 cells, thus exhibiting a significant synergistic effect against the cancer cells.
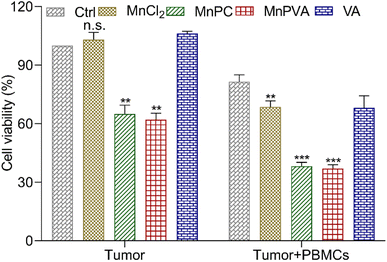 |
| | Fig. 11 Average viability (%) of MDA-MB-231 cells after co-incubation with compound-activated PBMCs (6 μM) for 48 h. Data are shown as mean ± SD (n = 3); **p < 0.01 and ***p < 0.001. | |
Acute toxicity and in vivo anticancer activity
The acute toxicity of the MnII complexes was evaluated on female Balb/c mice. The median lethal dose (LD50) and body weight changes after intravenous injection of each complex were determined. The LD50 of MnPC and MnPVA was 16.08 ± 2.16 and 25.16 ± 3.19 mg kg−1, respectively (Fig. S10†), which are much higher than that of CDDP (4.06 ± 1.02 mg kg−1).67 No significant changes in the body weight was observed in the MnPC- and MnPVA-treated mice. The results indicate that MnPC and MnPVA are low toxic to mammals. Interestingly, tethering VA to the Mn complex attenuated the general toxicity or increased the biocompatibility.
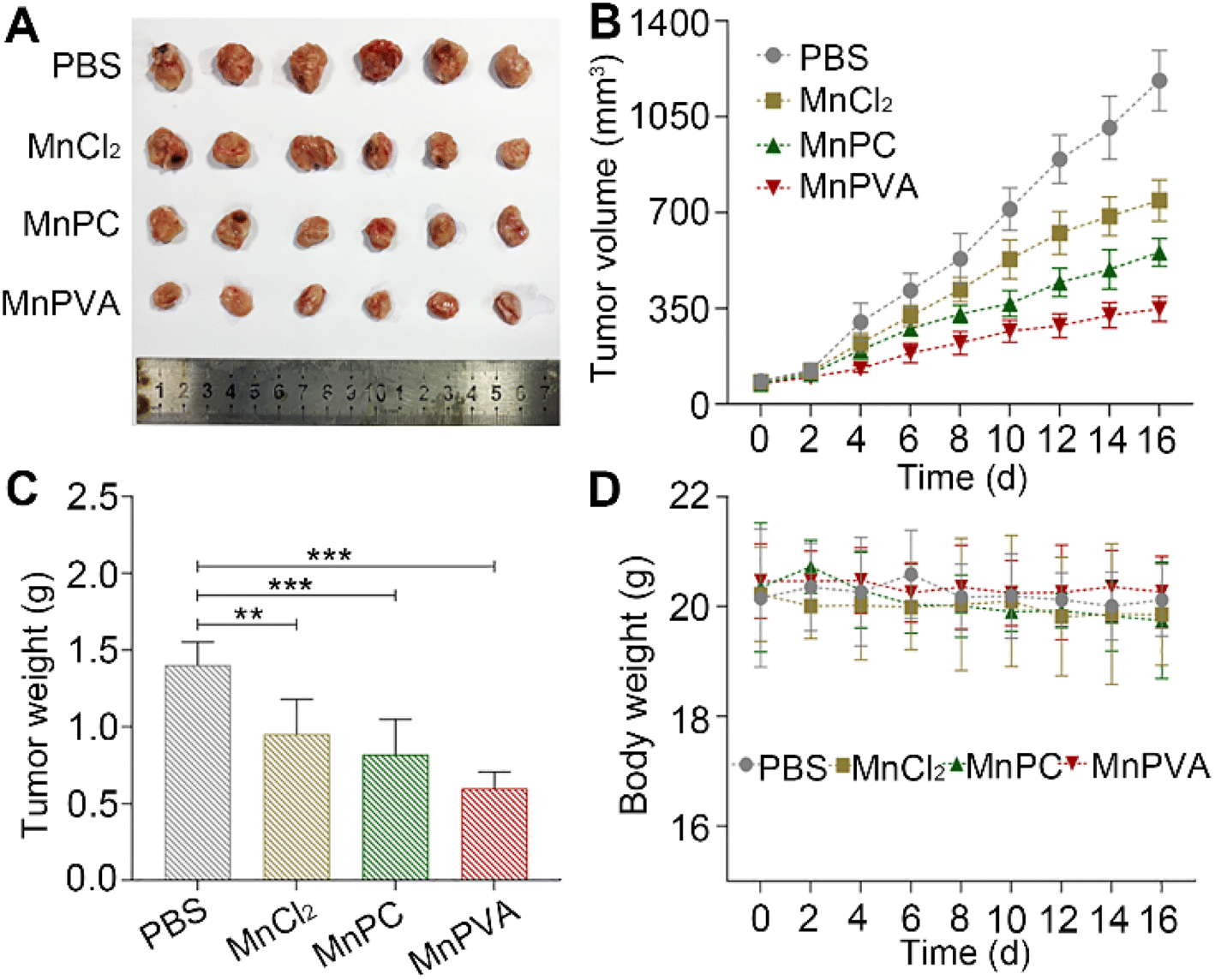
Furthermore, mouse models bearing 4T1 breast cancer were established to assess the anticancer activity of MnPC and MnPVA in vivo. As shown in Fig. 12, after treating the mice with PBS, MnCl2, MnPC, and MnPVA (1.3 mg Mn per kg) respectively for 16 days, MnPC and MnPVA showed more effective inhibition on the tumor growth than MnCl2. On day 16, the average tumor volume (A, B) was decreased to 554.78 ± 43.76 and 348.01 ± 39.61 mm3, respectively, for the MnPC- and MnPVA-treated mice, while that for the PBS- and MnCl2-treated mice was 1182.01 ± 95.87 and 784 ± 64.93 mm3, respectively. The average tumor weight (C) was 1.41 ± 0.13, 0.95 ± 0.19, 0.82 ± 0.19, and 0.59 ± 0.1 g for the PBS-, MnCl2-, MnPC-, and MnPVA-treated mice, respectively. Obviously, the tumor inhibitory effect of MnPVA was superior to that of MnPC and MnCl2, which may be due to the effective inhibition of the expression of HDACs by MnPVA. Additionally, no apparent pathological abnormality or lesion on the body weight (D), survival, and gross organ anatomy was observed (Fig. S11†). These results suggest that the MnII complexes are promising drug candidates for cancer therapy.
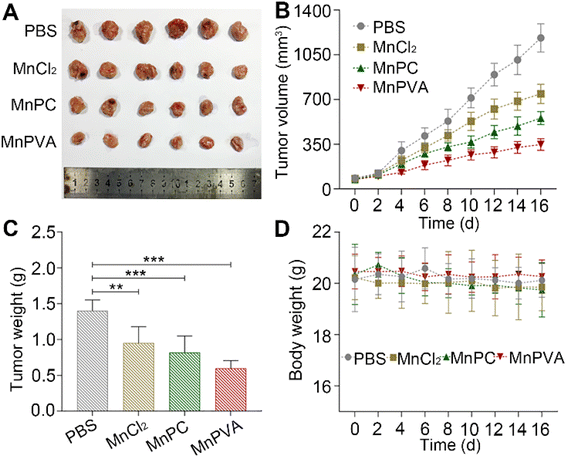 |
| | Fig. 12 Therapeutic effect of MnPC, MnPVA, and MnCl2 on 4T1 tumor-bearing mice, using PBS as a control. (A) Images of excised tumors, (B) tumor volume, (C) tumor weight, and (D) body weight of the mice after treating with PBS, MnCl2, MnPC, and MnPVA, respectively, at 1.3 mg Mn per kg once every 2 days for 16 days. **p < 0.01 and ***p < 0.001. | |
In vivo anticancer immune response
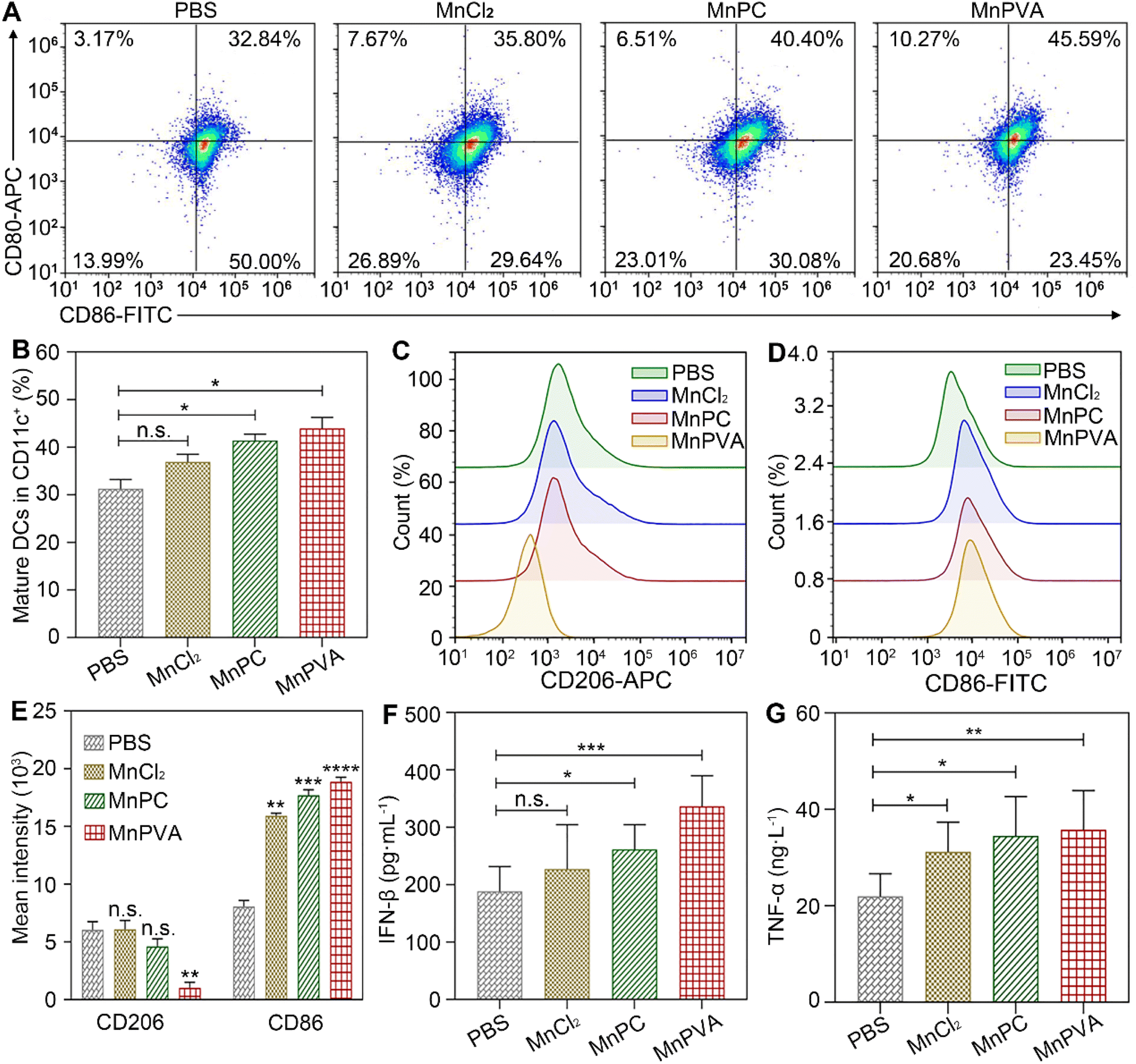
The in vivo immune-stimulating ability of the MnII complexes was tested on Balb/c mice bearing 4T1 tumors. The status of immune cells in tumor, spleen, and tumor draining lymph nodes (LNs) was analyzed by flow cytometry.21 CD8+ T cells are essential for restricting cancer initiation and malignant progression in the immune system, and the induction of cytotoxic T lymphocytes (CTLs) requires activation of immature DCs into mature ones,8,9 which were denoted as CD11c+CD80+CD86+ cells.68 As shown in Fig. 13A and B, in the MnPVA-treated mice, the percentage of mature DCs denoted by the expression of co-stimulatory receptors CD80+ and CD86+ (ref. 69) was higher (45.59%) in LNs than that treated with PBS, MnCl2, and MnPC, respectively.
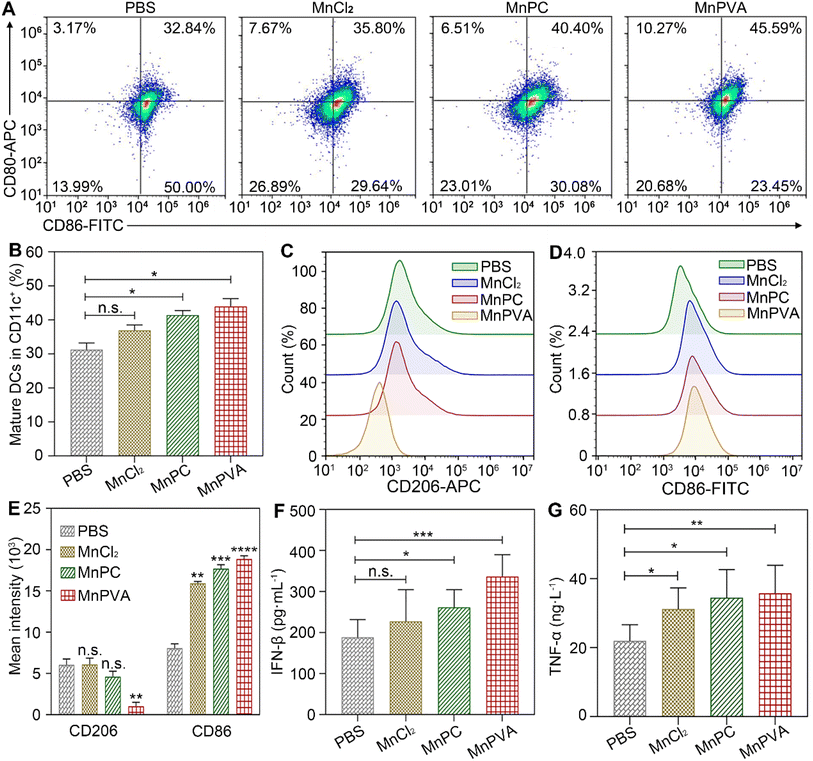 |
| | Fig. 13 Mature DCs (CD80+CD86+ gated on CD11c+) and quantitative analyses on tumor-draining lymph nodes (A and B), polarization of macrophages (CD206+CD86+ gated on CD11b+) in the 4T1 tumor tissue of a mouse (C–E) determined by flow cytometry, and ELISA analyses of IFN-β (F) and TNF-α (G) in serum from mice after treatment with each compound (1.3 mg Mn per kg) once every 2 days for 16 days. Data are presented as mean ± SD (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001. | |
Macrophages are integral parts of the immune system, modulating the tumor microenvironment.70 According to the functions, macrophages can be classified into tumoricidal M1 and protumoral M2 phenotypic activation states.71 M1 macrophages are marked by TNF-α, IL-12, CD80, CD86, and direct killing of tumor cells,70 while M2-like TAMs are characterized by high expression of macrophage mannose receptor 1 (MMR or CD206) and promotion of tumor cell growth, as well as exhibiting strong immunosuppressive activity. It is known that inhibitors of HDACs can potentiate pro-inflammation shifts in macrophage polarization and enhance the M1 phenotype in addition to preventing DNA repair.72 Therefore, we detected the polarization of macrophages in the spleen and tumor by flow cytometry after treatment with the Mn complexes. As shown in Fig. 13C–E and S12A–C,† the M2 phenotype marked by CD206+ was significantly suppressed and the M1 phenotype marked by CD86+ was remarkably increased in the MnPVA-treated mice. The results indicate that MnPVA effectively induced the polarization of macrophages from the M2 to the M1 phenotype. MnCl2 and MnPC also increased the expression of CD86, but only mildly influenced CD206 as compared to PBS. Meanwhile, IFN-β, IL-6, and TNF-α secreted by mature DCs and macrophages were measured by ELISA. As shown in Fig. 13F and G, MnPVA enhanced the levels of IFN-β and TNF-α more effectively than PBS, MnCl2 and MnPC, which may elicit T cells to kill tumor cells. However, no significant change in IL-6 was observed (Fig. S13†).
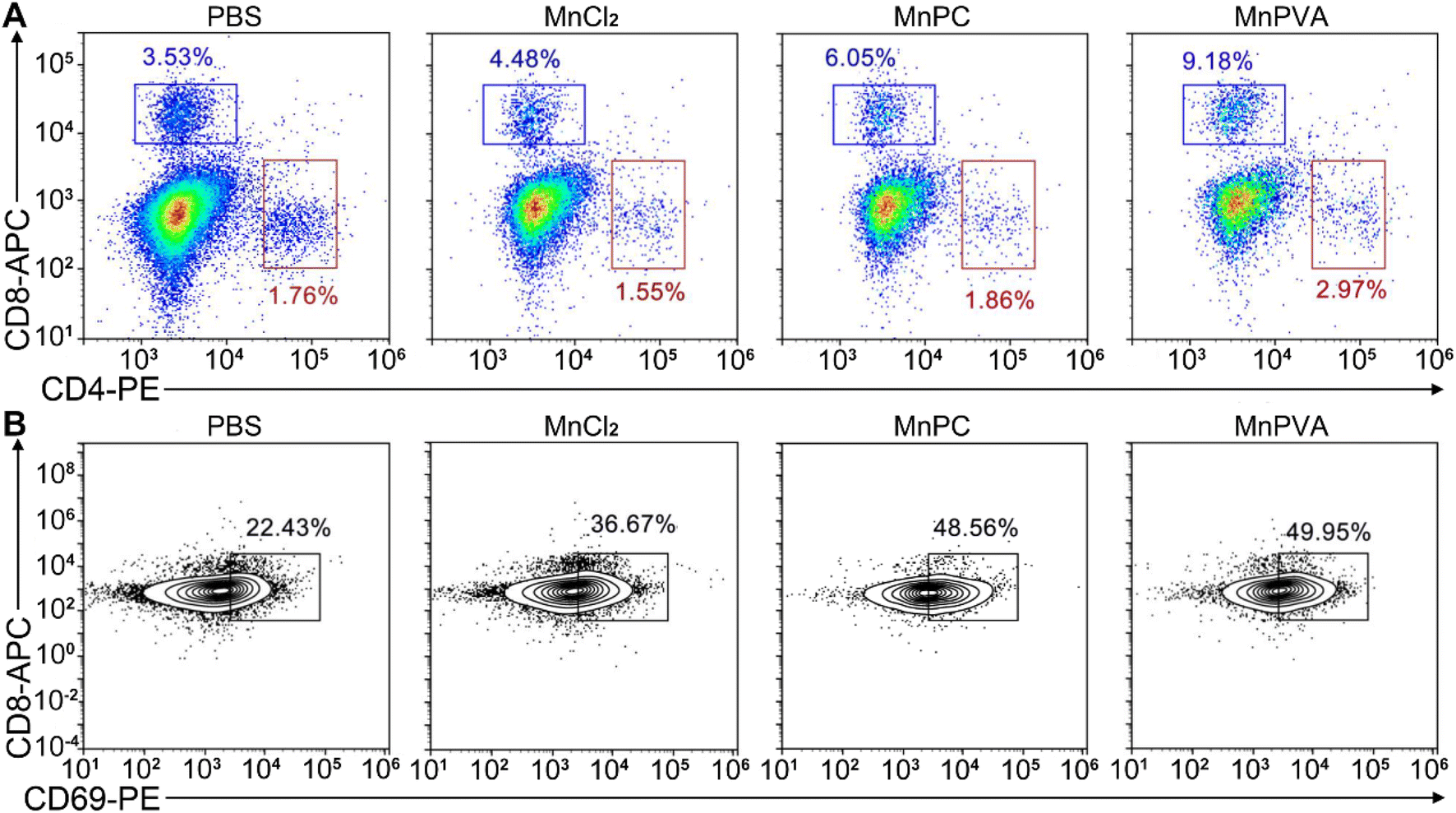
Upon activation of the cGAS-STING pathway in DCs and macrophages by MnPC and MnPVA, the secreted IFNs-I and proinflammatory cytokines could prime the cytotoxic lymphocytes.59 Therefore, we further evaluated the activation of cytotoxic T cells (CD8+) and helper T cells (CD4+) in tumor and spleen tissues, respectively. As shown in Fig. 14A and S12D–F†, the CD8+ T cells (blue square) in tumors were effectively activated (9.18%) in the MnPVA-treated mice as compared to those treated with PBS (3.53%), MnCl2 (4.48%), and MnPC (6.05%); CD4+ T cells (red square) were also activated slightly, and the results were opposite to those in the spleen after treatment with the complexes. Fig. 14B shows that the frequencies of activated (CD69+) tumor-infiltrating CD8+ T cells were dramatically increased. All these results indicate that MnPVA was able to stimulate strong immune responses in vivo due to the activation of the cGAS-STING pathway.
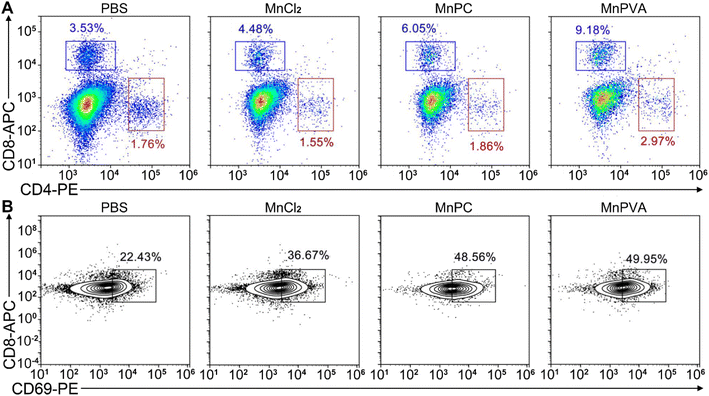 |
| | Fig. 14 CD8+ (blue square) and CD4+ T (red square) cells (A) and percentages of the CD69+ subset among CD8+ T cells (B) in the 4T1 tumor tissue of a mouse after treatment with each compound (1.3 mg Mn per kg) once every 2 days for 16 days determined by flow cytometry. | |
Mechanism of action
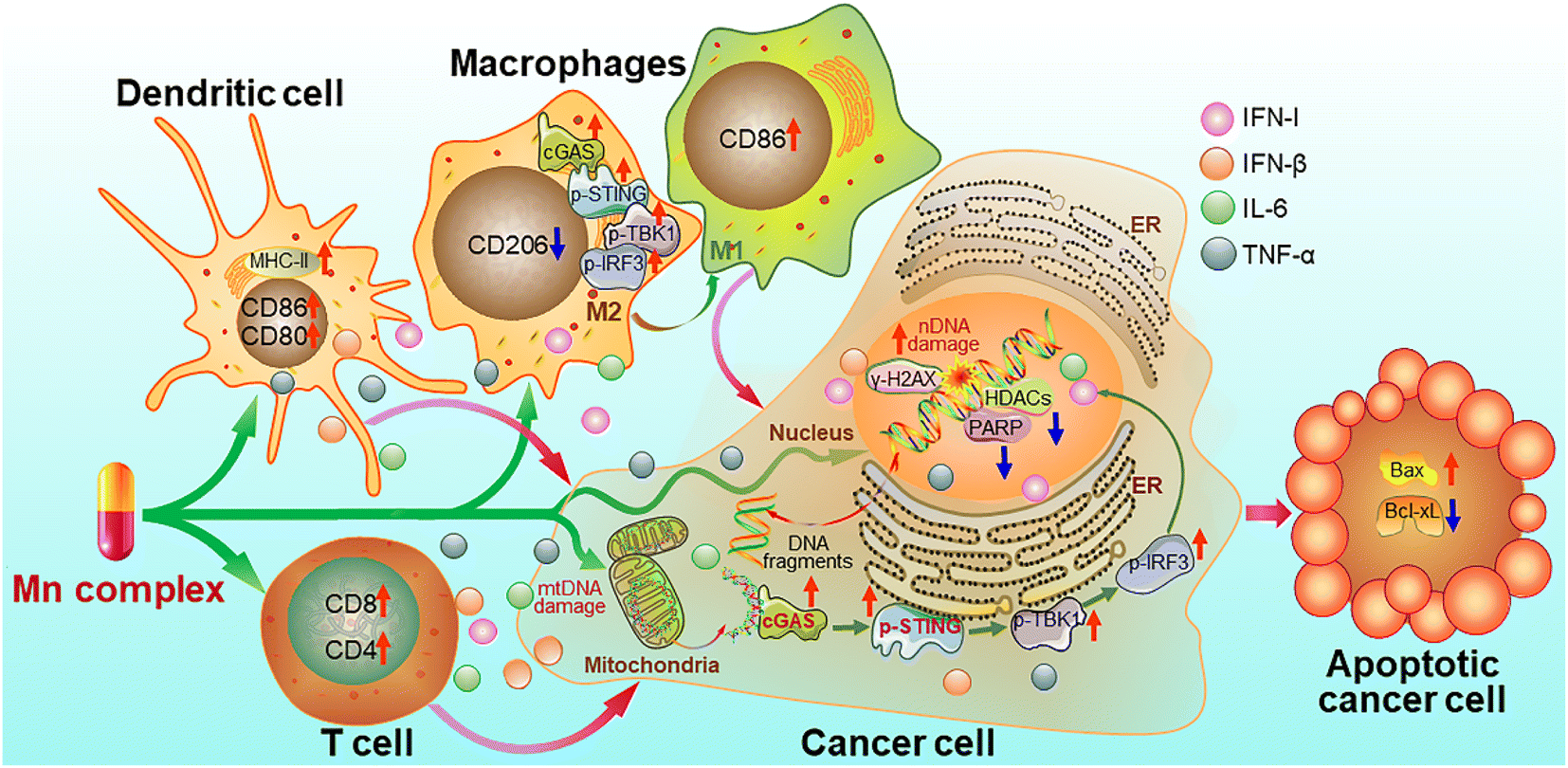
On the basis of the above experimental results, a mechanism of action for the Mn complexes as shown in Fig. 15 was proposed. In tumor cells, MnPC or MnPVA damaged nuclear and/or mitochondrial DNA, leading to the up-regulation of γ-H2AX; meanwhile, the complex inhibited the activity of HDAC1/2 and PARP1, thereby impairing the DNA repair ability and intensifying the DNA damage. The accumulated DNA fragments were released from the nucleus and/or mitochondria to cytoplasm and recognized by cGAS to activate the STING. The STING in turn stimulated the phosphorylation of TBK1 and IRF3 and translocation into the nucleus, inducing the production of IFNs, IL-6 and TNF-α. These IFNs and pro-inflammatory cytokines were then secreted from the nucleus to the cytosol and further to the tumor microenvironment, where they promoted the maturation and antigen presentation of DCs and macrophages, and further activated cytotoxic T cells to kill cancer cells. Similar events also occurred in immune cells such as macrophages and the cGAS-STING-TBK1 pathway was activated. The activation of the cGAS-STING pathway initiated innate immune responses and a two-way communication between tumor cells and neighboring immune cells to enhance the killing effect on tumors.21 Consequently, the Mn complexes showed potent antitumor activity owing to the synergy between chemotherapy and immunotherapy. Most of these processes have been proved in vitro or in vivo.
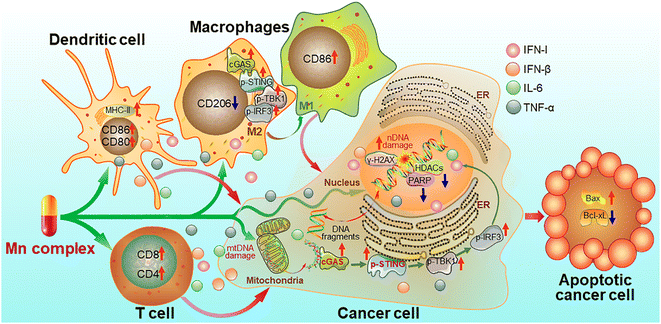 |
| | Fig. 15 Proposed mechanism of action for manganese complexes. | |
Conclusion
Increasing evidence shows that many metal complexes interact with both tumorous and immune cells to remodel immunosuppressive microenvironments besides exerting a direct cytotoxic effect on tumor cells.73 In this study, we present two MnII complexes MnPC and MnPVA as potential chemoimmunotherapeutics to restrain cancer via stimulating innate immune responses as well as breaking DNA double strands. These multifunctional complexes kill cancer cells not only by damaging DNA, but also by reactivating the dormant immune responses. Firstly, they act as DNA breakers, inducing oxidative DNA damage and producing DNA fragments; secondly, they act as inhibitors of HDACs and PARP1, preventing the repair of DNA damage and reinforcing DNA lesions; thirdly, they act as agonists of the cGAS-STING pathway, inducing the secretion of IFNs and proinflammatory cytokines to promote the maturation of DCs and macrophages, and further stimulating tumor-specific T cells to kill tumor cells. All in all, MnPC and MnPVA effectively activated the cGAS-STING pathway and restrained the growth of cancer cells in vitro and in vivo. Tethering VA to the MnII complex dramatically potentiated its inhibition on HDACs and strengthened the antiproliferative activity of the complex; and the formation of Mn complexes provides a significant lift to the antiproliferative activity of MnCl2 and 1,10-phenanthroline.
Ever since Jiang et al. first reported the stimulating activity of Mn2+ to the cGAS-STING pathway,12 no such property has been discovered in Mn complexes. This study demonstrates that the stimulating ability of MnPC and MnPVA to the cGAS-STING pathway is much higher than that of MnCl2 or Mn2+ in both tumorous and immune cells, because they induced more DNA fragments in cytoplasm to activate the cGAS-STING pathway. The findings signify that more potent cGAS-STING agonists could be obtained by constructing MnII complexes with DNA-damaging or DNA-repair-blocking ligands.
Data availability
Supplementary figures and tables, as well as experimental details are provided in the ESI.†
Author contributions
Linxiang Cai designed the complexes, performed most of the experiments, and wrote the draft; Ying Wang, Yayu Chen, and Hanhua Chen finished the cell culturing and animal feeding; Tao Yang and Shuren Zhang provided the technical support; Zijian Guo contributed to the formation of concepts; Xiaoyong Wang wrote and edited the paper and supervised the research.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the National Natural Science Foundation of China (Grants 91953201, 92153303, 21877059, and 21731004).
References
- S. L. Shiao, A. P. Ganesan, H. S. Rugo and L. M. Coussens, Genes Dev., 2011, 25, 2559–2572 CrossRef CAS PubMed.
- A. Ribas and J. D. Wolchok, Science, 2018, 359, 1350–1355 CrossRef CAS PubMed.
- P. Sharma and J. P. Allison, Science, 2015, 348, 56–61 CrossRef CAS PubMed.
- M. Yi, D. Jiao, H. Xu, Q. Liu, X. Han and K. Wu, Mol. Cancer, 2018, 17, 129 CrossRef PubMed.
- O. Hemminki, J. M. Dos Santos and A. Hemminki, J. Hematol. Oncol., 2020, 13, 84 CrossRef PubMed.
- S. Yu, A. Li, Q. Liu, T. Li, X. Han and K. Wu, J. Hematol. Oncol., 2017, 10, 78 CrossRef PubMed.
- P. Sharma, S. Hu-Lieskovan, J. A. Wargo and A. Ribas, Cell, 2017, 168, 707–723 CrossRef CAS PubMed.
- A. Li, M. Yi, S. Qin, Y. Song, Q. Chu and K. Wu, J. Hematol. Oncol., 2019, 12, 35 CrossRef PubMed.
- A. Saeed, X. Ruan, J. Su and S. Ouyang, Adv. Sci., 2020, 7, 1902599 CrossRef CAS PubMed.
- L. L. van der Woude, M. A. J. Gorris, C. G. Figdor and I. J. M. de Vries, Trends Cancer, 2017, 3, 797–808 CrossRef CAS PubMed.
- D. S. Chen and I. Mellman, Nature, 2017, 541, 321–330 CrossRef CAS PubMed.
- C. Wang, Y. Guan, M. Lv, R. Zhang, Z. Guo, X. Wei, X. Du, J. Yang, T. Li, Y. Wan, X. Su, X. Huang and Z. Jiang, Immunity, 2018, 48, 675–687 CrossRef CAS PubMed.
- Y. Song, Y. Liu, H. Y. Teo, Z. B. Hanafi, Y. Mei, Y. Zhu, Y. L. Chua, M. Lv, Z. Jiang and H. Liu, Cell. Mol. Immunol., 2021, 18, 1571–1574 CrossRef CAS PubMed.
- Q. Chen, L. Sun and Z. J. Chen, Nat. Immunol., 2016, 17, 1142–1149 CrossRef CAS PubMed.
- F. McNab, K. Mayer-Barber, A. Sher, A. Wack and A. O'Garra, Nat. Rev. Immunol., 2015, 15, 87–103 CrossRef CAS PubMed.
- H. Liu, H. Zhang, X. Wu, D. Ma, J. Wu, L. Wang, F. Liu, D. Yan, C. Chen, Z. Mao and B. Ge, Nature, 2018, 563, 131–136 CrossRef CAS PubMed.
- B. S. Pan, S. A. Perera, J. A. Piesvaux, H. Woo, D. F. Wyss, S. Xu, D. J. Bennett and G. H. Addona, Science, 2020, 369, 935 CrossRef PubMed.
- R. D. Luteijn, S. A. Zaver, B. G. Gowen, S. K. Wyman, N. E. Garelis, L. Onia, S. M. McWhirter, G. E. Katibah, J. E. Corn, J. J. Woodward and D. H. Raulet, Nature, 2019, 573, 434–438 CrossRef CAS PubMed.
- L. Hou, C. Tian, Y. Yan, L. Zhang, H. Zhang and Z. Zhang, ACS Nano, 2020, 14, 3927–3940 CrossRef CAS PubMed.
- M. Gao, Y.
Q. Xie, K. Lei, Y. Zhao, A. Kurum, S. Van Herck, Y. Guo, X. Hu and L. Tang, Adv. Ther., 2021, 4, 2100065 CrossRef CAS.
- Q. Luo, Z. Duan, X. Li, L. Gu, L. Ren, H. Zhu, X. Tian, R. Chen, H. Zhang, Q. Gong, Z. Gu and K. Luo, Adv. Funct. Mater., 2021, 32, 2110408 CrossRef.
- H. Tian, G. Wang, W. Sang, L. Xie, Z. Zhang, W. Li, J. Yan, Y. Tian, J. Li, B. Li and Y. Dai, Nano Today, 2022, 43, 101405 CrossRef CAS.
- C. Wang, Z. Sun, C. Zhao, Z. Zhang, H. Wang, Y. Liu, Y. Guo, B. Zhang, L. Gu, Y. Yu, Y. Hu and J. Wu, J. Controlled Release, 2021, 331, 480–490 CrossRef CAS PubMed.
- H. Haase, Immunity, 2018, 48, 616–618 CrossRef CAS PubMed.
- K. J. Horning, S. W. Caito, K. G. Tipps, A. B. Bowman and M. Aschner, Annu. Rev. Nutr., 2015, 35, 71–108 CrossRef CAS PubMed.
- X. Sun, Y. Zhang, J. Li, K. S. Park, K. Han, X. Zhou, Y. Xu, J. Nam, J. Xu, X. Shi, L. Wei, Y. L. Lei and J. J. Moon, Nat. Nanotechnol., 2021, 16, 1260–1270 CrossRef CAS PubMed.
- M. Lv, M. Chen, R. Zhang, W. Zhang, C. Wang, Y. Zhang, X. Wei, Y. Guan, Y. C. Liu, Q. Mei, W. Han and Z. Jiang, Cell Res., 2020, 30, 966–979 CrossRef CAS PubMed.
- S. L. O'Neal and W. Zheng, Curr. Environ. Health Rep., 2015, 2, 315–328 CrossRef PubMed.
- E. Li, Nat. Rev. Genet., 2002, 3, 662–673 CrossRef CAS PubMed.
- M. Guo, Y. Peng, A. Gao, C. Du and J. G. Herman, Biomark. Res., 2019, 7, 23 CrossRef PubMed.
- A. Duenas-Gonzalez, M. Candelaria, C. Perez-Plascencia, E. Perez-Cardenas, E. de la Cruz-Hernandez and L. A. Herrera, Cancer Treat. Rev., 2008, 34, 206–222 CrossRef CAS PubMed.
- H. V. Diyabalanage, M. L. Granda and J. M. Hooker, Cancer Lett., 2013, 329, 1–8 CrossRef CAS PubMed.
- T. Robert, F. Vanoli, I. Chiolo, G. Shubassi, K. A. Bernstein, O. A. Botrugno, D. Parazzoli, A. Oldani, S. Minucci and M. Foiani, Nature, 2011, 471, 74–79 CrossRef CAS PubMed.
- J. H. Lee, M. L. Choy, L. Ngo, S. S. Foster and P. A. Marks, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14639–14644 CrossRef CAS PubMed.
- K. L. Jin, J. Y. Park, E. J. Noh, K. L. Hoe, J. H. Lee, J. H. Kim and J. H. Nam, J. Gynecol. Oncol., 2010, 21, 262–268 CrossRef CAS PubMed.
- M. Gottlicher, S. Minucci, P. Zhu, A. Schimpf and S. Giavara, EMBO J., 2001, 20, 6968–6978 CrossRef PubMed.
- J. S. Brown, B. O'Carrigan, S. P. Jackson and T. A. Yap, Cancer Discovery, 2017, 7, 20–37 CrossRef CAS PubMed.
- A. N. Weaver and E. S. Yang, Front. Oncol., 2013, 3, 290 Search PubMed.
- F. J. Bock and P. Chang, FEBS J., 2016, 283, 4017–4031 CrossRef CAS PubMed.
- S. Cerboni, N. Jeremiah, M. Gentili, U. Gehrmann, C. Conrad, S. Amigorena, F. Rieux-Laucat and N. Manel, J. Exp. Med., 2017, 214, 1769–1785 CrossRef CAS PubMed.
- C. Pantelidou, O. Sonzogni, M. De Oliveria Taveira, A. K. Mehta, A. J. L. Guerriero, G. M. Wulf and G. I. Shapiro, Cancer Discovery, 2019, 9, 722–737 CrossRef PubMed.
- J. Shen, W. Zhao, Z. Ju, L. Wang, Y. Peng, M. Labrie, T. A. Yap, G. B. Mills and G. Peng, Cancer Res., 2019, 79, 311–319 CrossRef CAS PubMed.
- N. A. Yusoh, H. Ahmad and M. R. Gill, ChemMedChem, 2020, 15, 2121–2135 CrossRef CAS PubMed.
- F. Mendes, M. Groessl, A. A. Nazarov, Y. O. Tsybin, G. Sava, I. Santos, P. J. Dyson and A. Casini, J. Med. Chem., 2011, 54, 2196–2206 CrossRef CAS PubMed.
- S. Abbas, F. Rashid, E. Ulker, S. Zaib, K. Ayub, S. Ullah, M. A. Nadeem, S. Yousuf, R. Ludwig, S. Ali and J. Iqbal, J. Biomol. Struct. Dyn., 2021, 39, 1068–1081 CrossRef CAS PubMed.
- L. Tabrizi, P. McArdle, M. Ektefan and H. Chiniforoshan, Inorg. Chim. Acta, 2016, 439, 138–144 CrossRef CAS.
- H. Zhang, T. Yang, Y. Wang, Z. Wang, Z. Zhu, Z. J. Guo and X. Y. Wang, Dalton Trans., 2021, 50, 304–310 RSC.
- R. Jastrząb, M. Nowak, M. Skrobańska, A. Tolińska, M. Zabiszak, M. Gabryel, L. Marciniak and M. T. Kaczmarek, Coord. Chem. Rev., 2019, 382, 145–159 CrossRef.
- Y. Cheng, L. Lv, L. Zhang, Y. Tang and L. Zhang, J. Mol. Struct., 2021, 1228, 129745 CrossRef CAS.
- Z. Zhu, Z. Wang, C. Zhang, Y. Wang, H. Zhang, Z. Gan, Z. J. Guo and X. Y. Wang, Chem. Sci., 2019, 10, 3089–3095 RSC.
- C. Slator, Z. Molphy, V. McKee and A. Kellett, Redox Biol., 2017, 12, 150–161 CrossRef CAS PubMed.
- T. J. Hayman, M. Baro, T. MacNeil, C. Phoomak, T. N. Aung, T. Sandoval Schaefer, B. A. Burtness, D. L. Rimm and J. N. Contessa, Nat. Commun., 2021, 12, 2327 CrossRef CAS PubMed.
- R. M. Chabanon, M. Rouanne, C. J. Lord, J. C. Soria, P. Pasero and S. Postel-Vinay, Nat. Rev. Cancer, 2021, 21, 701–717 CrossRef CAS PubMed.
- A. P. Wes and G. S. Shadel, Nat. Rev. Immunol., 2017, 17, 363–375 CrossRef PubMed.
- A. P. West, W. Khoury-Hanold, M. Staron, M. C. Tal, C. M. Pineda, S. M. Lang, M. Bestwick, B. A. Duguay, N. Raimundo, D. A. MacDuff, S. M. Kaech, J. R. Smiley, R. E. Means, A. Iwasaki and G. S. Shadel, Nature, 2015, 520, 553–557 CrossRef PubMed.
- S. Jin, Y. Hao, Z. Zhu, N. Muhammad, Z. Zhang, K. Wang, Y. Guo, Z. J. Guo and X. Y. Wang, Inorg. Chem., 2018, 57, 11135–11145 CrossRef CAS PubMed.
- N. J. Curtin and C. Szabo, Nat. Rev. Drug Discovery, 2020, 19, 711–736 CrossRef CAS PubMed.
- J. Chen, F. M. Ghazawi, W. Bakkar and Q. Li, Mol. Cancer, 2006, 5, 71 CrossRef PubMed.
- S. M. Harding, J. L. Benci, J. Irianto, D. E. Discher, A. J. Minn and R. A. Greenberg, Nature, 2017, 548, 466–470 CrossRef CAS PubMed.
- E. J. Sayour and D. A. Mitchell, J. Immunol. Res., 2017, 17, 3145742 Search PubMed.
- B. J. Francica, A. Ghasemzadeh, A. L. Desbien, D. Theodros, K. E. Sivick, A. B. Sharabi, M. L. Leong, S. M. McWhirter, T. W. Dubensky Jr, D. M. Pardoll and C. G. Drake, Cancer Immunol. Res., 2018, 6, 422–433 CrossRef CAS PubMed.
- C. Vanpouille-Box, S. Demaria, S. C. Formenti and L. Galluzzi, Cancer Cell, 2018, 34, 361–378 CrossRef CAS PubMed.
- S. R. Woo, M. B. Fuertes, L. Corrales, S. Spranger, M. J. Furdyna, M. Y. Leung, K. A. Fitzgerald, M. L. Alegre and T. F. Gajewski, Immunity, 2014, 41, 830–842 CrossRef CAS PubMed.
- C. Belli, D. Trapani, G. Viale, P. D'Amico, B. A. Duso, P. Della Vigna, F. Orsi and G. Curigliano, Cancer Treat. Rev., 2018, 65, 22–32 CrossRef CAS PubMed.
- B. U. Schraml, J. van Blijswijk, S. Zelenay, P. G. Whitney, A. Filby, S. E. Acton, N. C. Rogers, N. Moncaut, J. J. Carvajal and C. Reis e Sousa, Cell, 2013, 154, 843–858 CrossRef CAS PubMed.
- A. A. van de Loosdrecht, E. Nennie, G. J. Ossenkoppele, R. H. J. Beelen and M. M. A. C. Langenhuijsen, J. Immunol. Methods, 1991, 141, 15–22 CrossRef CAS PubMed.
- S. Zhang, X. Zhong, H. Yuan, Y. Guo, D. Song, F. Qi, Z. Zhu, X. Y. Wang and Z. J. Guo, Chem. Sci., 2020, 11, 3829–3835 RSC.
- Y. Li and E. Seto, Cold Spring Harbor Perspect. Med., 2016, 6, a026831 CrossRef PubMed.
- C. Chen, Y. Tong, Y. Zheng, Y. Shi, Z. Chen, J. Li, X. Liu, D. Zhang and H. Yang, Small, 2021, 17, e2006970 CrossRef PubMed.
- A. Mantovani, F. Marchesi, A. Malesci, L. Laghi and P. Allavena, Nat. Rev. Clin. Oncol., 2017, 14, 399–416 CrossRef CAS PubMed.
- B. Ruffell and L. M. Coussens, Cancer Cell, 2015, 27, 462–472 CrossRef CAS PubMed.
- W. He, N. Kapate, C. W. t. Shields and S. Mitragotri, Adv. Drug Delivery Rev., 2020, 17, 251–266 Search PubMed.
- B. Englinger, C. Pirker, P. Heffeter, A. Terenzi, C. R. Kowol, B. K. Keppler and W. Berger, Chem. Rev., 2019, 119, 1519–1624 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Xiaoyong
Wang
b and
Xiaoyong
Wang