
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGermanium hydrides as an efficient hydrogen-storage material operated by an iron catalyst†
Yoshinao
Kobayashi
a and
Yusuke
Sunada
 *ab
*ab
aDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 4-6-1, Komaba, Meguro-ku, Tokyo 153-8505, Japan
bInstitute of Industrial Science, The University of Tokyo, 4-6-1, Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: sunada@iis.u-tokyo.ac.jp
First published on 20th January 2023
Abstract
The use of metal hydrides such as NaBH4 as hydrogen-storage materials has recently received substantial research attention on account of the worldwide demand for the development of efficient hydrogen-production, -storage, and -transportation systems. Here, we report the quantitative production of H2 gas from a germanium hydride, Ph2GeH2, mediated by an iron catalyst at room temperature via dehydrogenative coupling, concomitant with the formation of (GePh2)5. Of particular importance is that Ph2GeH2 can be facilely recovered from (GePh2)5 by contact with 1 atm of H2 or PhICl2/LiAlH4 at 0 °C or 40 °C, respectively. A detailed reaction mechanism for the iron-catalyzed dehydrogenative coupling of Ph2GeH2 is proposed based on the isolation of four intermediate iron species.
Introduction
Economic hydrogen production/storage is the key issue preventing the application of hydrogen as an energy carrier in the context of a global low-carbon strategy to address the ever-increasing energy challenges that face humanity. Recently, a number of hydrogen storage materials, such as metal hydrides, liquid organic hydrogen carriers, ammonia and ammonia borane, were applied for the development of effective hydrogen production/storage systems.1 Among them, metal hydrides have attracted great attention recently as potential efficient hydrogen-storage materials. Sodium borohydride (NaBH4) is one of the most studied hydrides for this purpose, as it is able to deliver H2 gas under mild conditions, e.g., at room temperature, via hydrolysis.2 However, one major issue with this approach is that the regeneration of NaBH4 from the hydrolytic products formed after H2 production generally carries a high energy penalty and thus requires, e.g., high temperatures (400–500 °C). Thus, the development of hydrogen-production/-storage systems in which the generation of H2 can proceed under mild conditions and hydrogen-storage materials that can be regenerated easily is highly desirable.In this paper, we focus on the use of group-14 hydrides as potentially reusable hydrogen-storage materials. It is well known that group-14 hydrides such as hydrosilanes, hydrogermanes, and hydrostannanes undergo dehydrogenative coupling reactions in the presence of appropriate transition-metal catalysts to form E–E (E = Si, Ge, Sn) bond(s), concomitant with the generation of H2.3 A pioneering study in this area has been published by Harrod and co-workers, who reported that hydrosilanes such as PhSiH3 effectively undergo multiple Si–Si-bond-forming reactions in a dehydrogenative manner to afford polysilanes and/or a mixture of oligosilanes.4 This study revealed that hydrosilanes can act as reagents for hydrogen-production and E–E-bond-formation reactions; however, the regeneration of the starting hydrosilanes is considered to be challenging because high-molecular-weight polymeric compounds are obtained as the major products after the production of H2. Although polymeric products have mainly been obtained in catalyst systems based on early transition metals,5 it has been reported that late-transition-metal catalysts often tend to afford relatively short-chain oligomers in the dehydrogenative coupling of group-14 hydrides.6 For instance, Rosenberg et al. have indicated that the dehydrogenative coupling of Ph2SiH2 catalyzed by Rh(PPh3)3Cl affords the disilane Ph2(H)Si–Si(H)Ph2 selectively under the optimal reaction conditions;5b however, no attempts to regenerate Ph2SiH2 from Ph2(H)Si–Si(H)Ph2 have been reported in the same study. Although some examples of the reverse reaction of the dehydrogenative coupling of hydrosilanes, i.e., hydrogenolysis of disilanes to afford hydrosilanes, have been reported for Ni or Pt catalysts,7 reversible and reusable hydrogen-production/-storage systems with group-14 hydrides have not been explored so far.
In this work, we developed a catalytic system based on iron for the reversible production/storage of H2 using germanium hydrides as the potential hydrogen-storage material because iron is the least expensive and least toxic late transition metal (Scheme 1). We found that the combination of the iron precursor [Fe(mesityl)2]2 (mesityl = 2,4,6-Me3-C6H2) with N-heterocyclic carbene (NHC) ligands promotes the effective production of H2 in the dehydrogenative coupling of R2GeH2 (R = Ph or Et) (hydrogen content: ca. 0.87 wt% for Ph2GeH2 and ca. 1.51 wt% for Et2GeH2). The quantitative generation of H2 at room temperature using Ph2GeH2 as a hydrogen-storage material was confirmed, along with the formation of (GePh2)5 as the dehydrogenative coupling product. It is noteworthy that the starting Ph2GeH2 can be readily regenerated via the hydrogenation of (GePh2)5 under 1 atm of H2 at 0 °C catalyzed by the same iron catalyst system. Alternatively, Ph2GeH2 can also be recovered from (GePh2)5 by treatment with PhICl2 followed by LiAlH4 at 40 °C. These results show the promising potential of germanium hydrides as prospective reusable hydrogen-production/-storage materials that can evolve H2 at room temperature using an iron catalyst. Moreover, four possible intermediary iron species formed in this catalysis were structurally characterized, based on which, a detailed reaction mechanism is proposed. The present catalyst system is also applicable to the dehydrogenative coupling of other group-14 hydrides, such as hydrosilanes and hydrostannanes R2EH2 (E = Si, Sn).
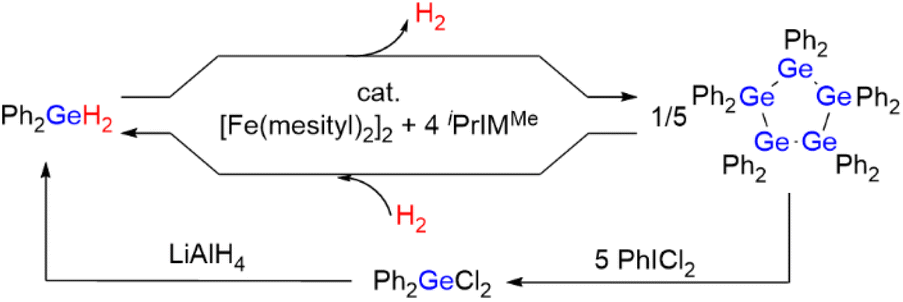 | ||
| Scheme 1 Overall scheme of this work; Ph2GeH2 as a reusable hydrogen-storage material in the presence of an iron catalyst. | ||
Results and discussion
Hydrogen production from secondary germanes
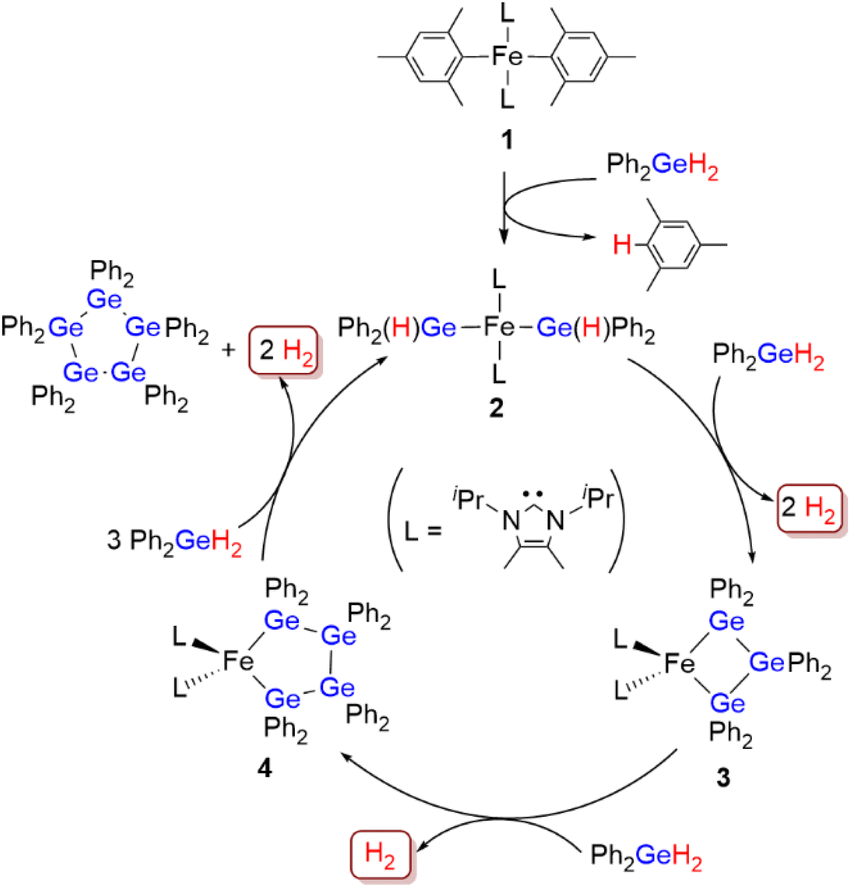
First, the diphenylgermane Ph2GeH2 was used as the substrate, and the dinuclear iron(II) complex [Fe(mesityl)2]2 (mesityl = 2,4,6-trimethylphenyl) was used as the catalyst precursor, because it has been reported to effectively activate group-14-element–hydrogen bonds.8 The dehydrogenative coupling of Ph2GeH2 proceeded effectively at room temperature in THF under an N2 atmosphere when a mixture of 2.5 mol% of [Fe(mesityl)2]2 (5 mol% based on Fe) and 10 mol% of the N-heterocyclic carbene iPrIMMe (iPrIMMe = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) was used as the catalyst (Scheme 2). This reaction furnished cyclic pentagermane (GePh2)5 as the sole product in 62% isolated yield after 40 h. The formation of (GePh2)5 was confirmed based on a comparison of the 1H and 13C NMR spectra of the product with those of an authentic sample.9 It is noteworthy that the formation of (GePh2)5 was accompanied by the generation of H2 gas, which was confirmed by 1H NMR spectroscopy. The quantity of H2 gas produced was determined using a gas burette; when the dehydrogenative coupling of 1 mmol of Ph2GeH2 was performed at room temperature under the catalytic conditions described above, ca. 21 mL of gaseous product(s) were obtained in the gas burette. These results indicate that the dehydrogenation of Ph2GeH2 to afford (GePh2)5 proceeded effectively at room temperature to give a quantitative amount of H2 gas.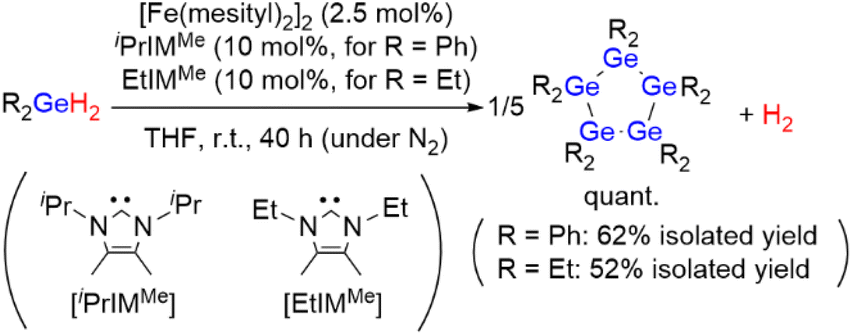 | ||
| Scheme 2 Catalytic dehydrogenative coupling of Ph2GeH2 or Et2GeH2 catalyzed by the [Fe(mesityl)2]2/NHC catalyst system. | ||
It should be emphasized here that this catalysis is highly sensitive to the electronic and/or steric environment of the ligands used. Thus, iPrIMMe was found to be the only ligand that selectively furnished (GePh2)5 for a quantitative conversion of Ph2GeH2, and that other NHC ligands used in this study with different substituents on the nitrogen atoms or the ligand backbone barely generated any (GePh2)5. Although the use of certain NHC ligands led to a high conversion of Ph2GeH2, the formation of a mixture of oligogermanes other than (GePh2)5 was suggested by the 1H NMR spectra of the corresponding crude products (for details, see Table S2 in the ESI†). Subsequently, we examined the effect of the solvent on the reaction of Ph2GeH2 catalyzed by [Fe(mesityl)2]2/iPrIMMe, which revealed that the use of ethereal solvents such as Et2O and THF afforded (GePh2)5 in medium to good yield.
Subsequently, the dehydrogenative coupling of Et2GeH2 was performed using [Fe(mesityl)2]2/iPrIMMe. Although the conversion of Et2GeH2 reached >99% in THF after 40 h, the 1H NMR spectrum of the crude product suggested the formation of a complex mixture of oligogermanes, and almost no formation of cyclopentagermane (GeEt2)5 was observed. This result stands in stark contrast to the fact that the selective production of (GeEt2)5 with >99% conversion (52% isolated yield) of Et2GeH2 was confirmed when EtIMMe was used instead of iPrIMMe (Scheme 2). These results indicate that sophisticated control of the steric environment around the iron center on the catalytically active species can be expected to be crucial to achieving the dehydrogenative coupling of secondary germanes in a selective manner to produce the corresponding cyclopentagermanes (GeR2)5 (R = Ph, Et) as a single product. It should also be noted here that although cyclooligogermanes are usually generated via the Wurtz-type reductive coupling of R2GeX2 (R = alkyl, aryl; X = halide) using a stoichiometric amount of alkali or alkaline earth metals,10 the development of alternative synthetic methods would be highly desirable due to the harsh reaction conditions required. The dehydrogenative coupling of secondary germanes could be a potential alternative to such Wurtz-type reductive coupling reactions in terms of the formation of Ge–Ge bonds, although previously reported examples of the catalytic dehydrogenative coupling of secondary germanes only furnished dimers. To the best of our knowledge, the results shown here thus represent the first example of the formation of cyclooligogermanes via the sequential catalytic dehydrogenative coupling of secondary germanes under the formation of multiple Ge–Ge bonds.
Regeneration of Ph2GeH2 from (GePh2)5
We discovered that the starting material Ph2GeH2 can be readily regenerated from (GePh2)5 by treatment with 1 atom of H2 in the presence of [Fe(mesityl)2]2/iPrIMMe in THF at 0 °C for 24 h, i.e., Ph2GeH2 was formed in 51% yield. It was also confirmed that no reaction occurred when (GePh2)5 was exposed to 1 atm of H2 in the absence of an iron catalyst. The regeneration of Ph2GeH2 was accompanied by the formation of germoxane (Ph2GeO)3 in 49% yield; this is presumably due to the rapid hydrolysis of the formed Ph2GeH2 induced by iPrIMMe and unremovable traces of H2O. It should also be noted here that hydrosilanes such as Ph2SiH2 undergo fast hydrolysis in the presence of a catalytic amount of N-heterocyclic carbenes.11 Although the quantitative formation of Ph2GeH2 from (GePh2)5 and H2 gas was hampered despite many attempts to remove the aforementioned trace amounts of H2O, we found that Ph2GeH2 could be readily recovered from (GePh2)5via an alternative method; (GePh2)5 can be converted quantitatively into Ph2GeCl2 (64% isolated yield) by treatment with PhICl2 in THF at 40 °C, before Ph2GeCl2 can be quantitatively transformed into Ph2GeH2 by reaction with LiAlH4 in THF at room temperature (82% isolated yield) (Scheme 3). Thus, we concluded that the germanium hydride Ph2GeH2 was easily recovered and reused via two independent reaction protocols. These results corroborate the notion that Ph2GeH2 is an effective and reusable hydrogen-storage material that can quantitatively produce H2 gas at mild operating temperatures and that can be readily regenerated from (GePh2)5 through hydrogenation under 1 atm of H2 gas or sequential treatment with PhICl2 and LiAlH4.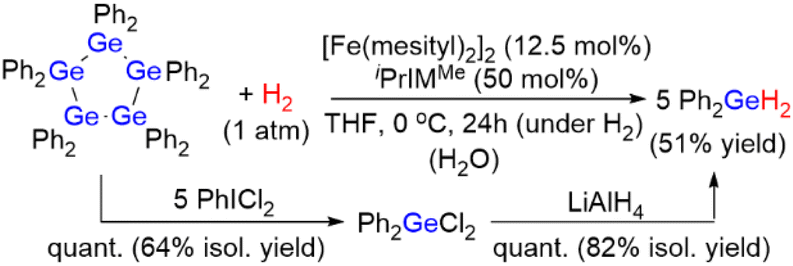 | ||
| Scheme 3 Regeneration of Ph2GeH2 from (GePh2)5. | ||
Isolation of four possible intermediary iron species
To obtain insight into the reaction mechanism of the [Fe(mesityl)2]2/iPrIMMe-catalyzed dehydrogenative coupling of Ph2GeH2 to produce (GePh2)5, several investigations were carried out to isolate any potential intermediary iron species. Initially, the reaction of [Fe(mesityl)2]2 and 4 equivalents of iPrIMMe was carried out in Et2O at room temperature for 16 h, which provided mononuclear trans-Fe(mesityl)2(iPrIMMe)2 (1) in 52% isolated yield (Scheme 4A). A single-crystal X-ray diffraction (XRD) analysis revealed that the iron center in 1 adopts a square-planar geometry with two iPrIMMe ligands located at the trans-positions, which is similar to that observed in a previously reported analogous Fe(mesityl)2(iPmIMH)2 complex.8b Subsequently, isolated complex 1 was treated with 2 equivalents of Ph2GeH2, which produced a trace amount of orange crystals of trans-Fe(GePh2H)2(iPrIMMe)2 (2), together with a white powder of (GePh2)5. Although the isolation of pure 2 from this reaction was hampered by the concomitant formation of (GePh2)5, 2 was isolated using an alternative reaction protocol. Namely, the reaction of FeCl2 with in situ-generated LiGe(H)Ph2 in the presence of 2 equivalents of iPrIMMe in THF led to the isolation of 2 in 43% yield (Scheme 4B). The molecular structure of 2 was determined by single-crystal XRD analysis (Fig. 1A). Similar to that in complex 1, the iron center in 2 adopts a square-planar coordination geometry wherein two iPrIMMe ligands occupy trans-positions. To the best of our knowledge, complex 2 is the first example of an Fe(II)-germyl complex with a four-coordinate square-planar coordination geometry. The Fe–Ge bond distances of 2.4488(8) Å are longer than those in previously reported iron(II) germyl complexes with octahedral structures (Cp*Fe(CO)2(GeMe2SPh): 2.3633(4) Å;12 CpFe(CO)(pyridine)(GeEt3): 2.4055(11) Å;13 Cp(CO)2[Ge(C2F5)3]: for 2.3232(3) Å;14). This is presumably due to the strong trans-influence of the germyl moieties. In the IR spectrum of 2, an absorption band observed at 1848 cm−1 implied the presence of Ge–H bonds in 2.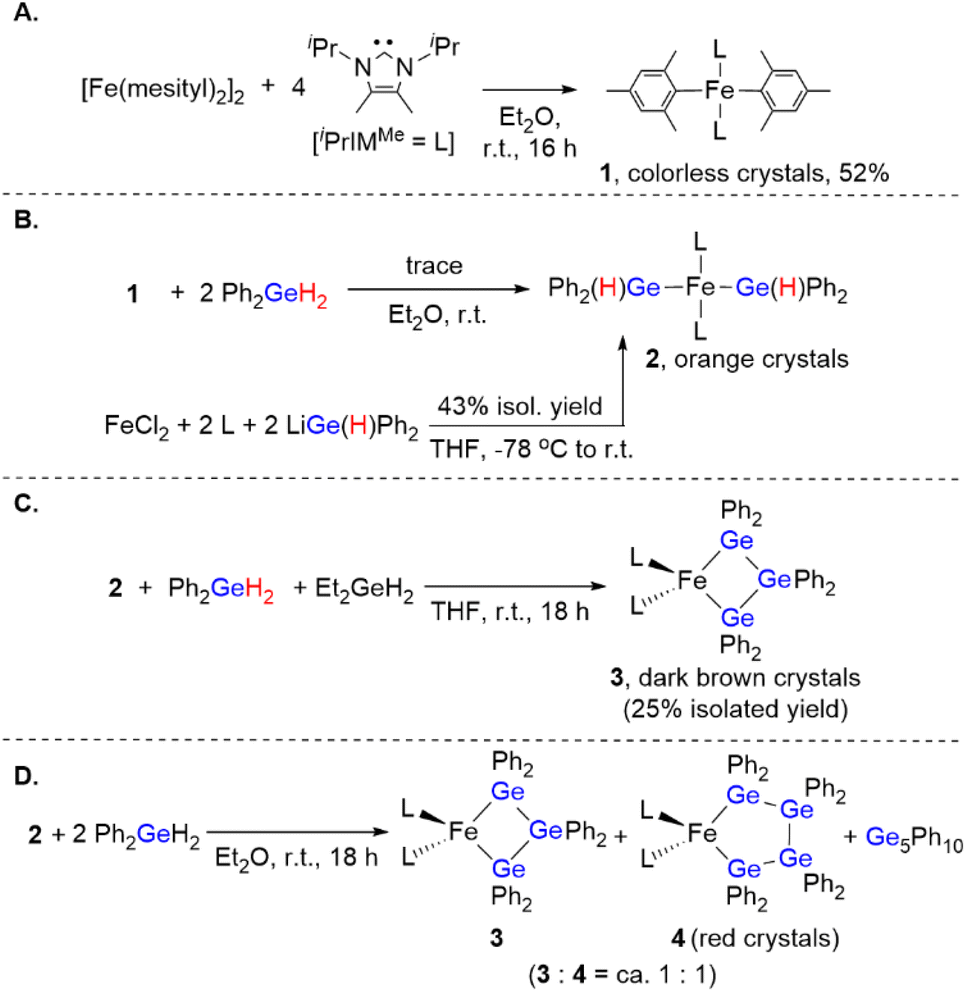 | ||
Scheme 4 Synthesis of iron complexes 1–4. (A) Synthesis and isolation of 1. (B) Two reaction schemes for the synthesis of 2. (C) Synthesis and isolation of 3. (D) Synthesis of ca. a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of iron complexes 3 and 4. :1 mixture of iron complexes 3 and 4. | ||
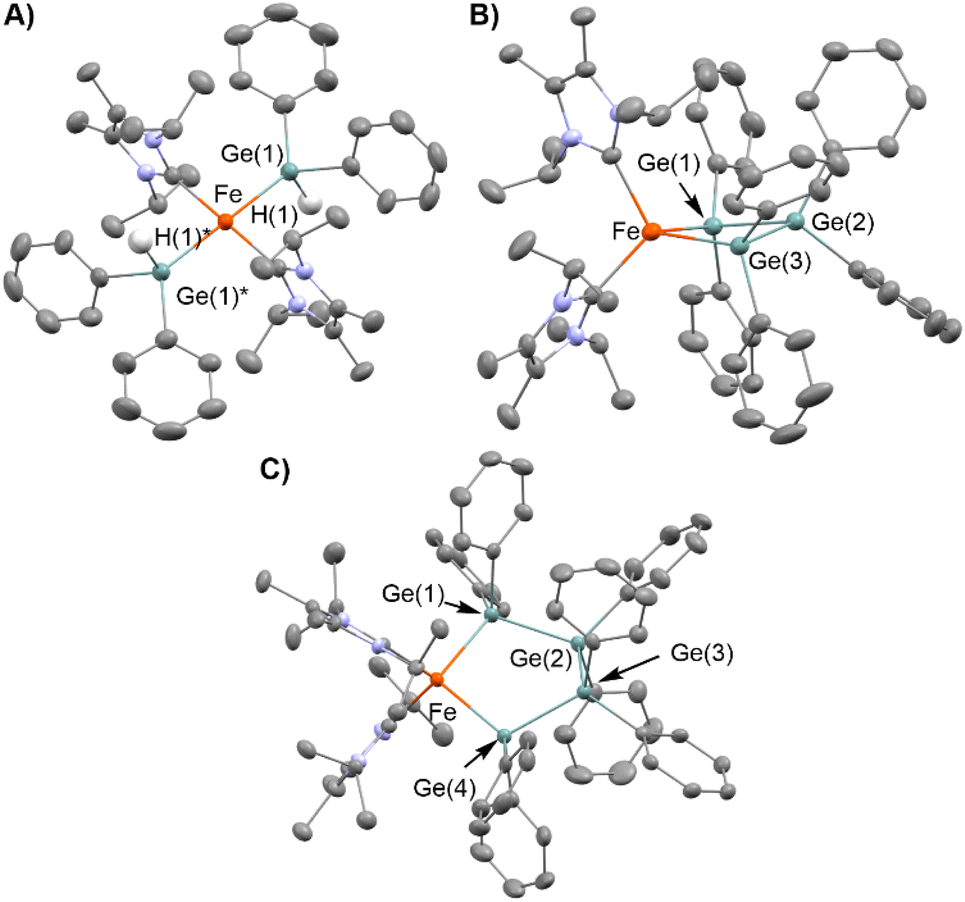 | ||
| Fig. 1 Molecular structures of 2, 3 and 4 with thermal ellipsoids at 50% probability; hydrogen atoms, except for the hydrides in 2, have been omitted for clarity. (A) Molecular structure of 2. (B) Molecular structure of 3. (C) Molecular structure of 4. | ||
Based on the molecular structure of 2, the first H–Ge bond activation mediated by 1 can be assumed to take place through the Fe-mesityl moiety. A similar reaction, i.e., the formation of square-planar trans-(NHC)2Fe(SnnBu3)2via the reaction of trans-(NHC)Fe(mesityl)2 with nBu3SnH, has recently been reported by Radius et al.8b Further treatment of 2 with 1 equivalent of Ph2GeH2 and 1 equivalent of Et2GeH2 led to the formation of a Ge–Ge bond on the iron center to afford complex 3, which contains a metallatrigermacycle skeleton, in 25% isolated yield (Scheme 4C). Although the actual role of Et2GeH2 in this reaction remains unclear so far, Et2GeH2 may contribute to retarding the rate of the reaction between 2 and Ph2GeH2, thus leading to the production of complex 3 as the main product. The molecular structure of 3 (Fig. 1B) revealed that the iron center adopts a tetrahedral coordination geometry, which is different from those found in 1 and 2. The Fe–Ge bond distances in 3 (2.5712(6)/2.5557(5) Å) are elongated compared to those in 2 and other previously reported iron(II) germyl complexes,12–14 although they are comparable to that of our previously reported iron(II) digermyl complex with a tetrahedral coordination geometry, (THF)2Fe[Ge(SiMe3)3]2 (2.5589(8) Å).15 The iron center and three germanium atoms form a nearly planar metallacyclic skeleton, wherein the deviation of all atoms from the least-squares plane ranges from 0.116 Å to 0.124 Å. It is worth noting here that the Ge–Ge-bond-forming reaction apparently occurs during the reaction of 2 with Ph2GeH2 to produce 3. In other words, the bond formation involving two Fe–Ge bonds and an incoming molecule of Ph2GeH2 to construct a cyclic trigermyl moiety is effectively promoted on the iron center supported by iPrIMMe ligands. The 1H NMR spectrum of isolated 3 in C6D6 at room temperature features broad resonances at δ = 27.2, 21.3, 19.8, 15.8, 14.3, 7.5, and −1.2 ppm, indicating that 3 is paramagnetic.
We found that a new iron species 4 is formed together with 3 and (GePh2)5 in the reaction between 2 and 2 equivalents of Ph2GeH2 (Scheme 4D). Although several attempts to isolate 4 failed, a subsequent recrystallization led to the generation of reddish crystals, whose 1H NMR spectrum in C6D6 indicated that they consist of a ca. 1:1 mixture of 3 and 4. A careful single-crystal XRD analysis revealed the molecular structure of 4, which consists of a five-membered metallatetragermacycle skeleton composed of four ‘GePh2’ moieties (Fig. 1C). The iron center of 4 adopts a slightly distorted tetrahedral coordination geometry with Fe–Ge bond lengths of 2.5714(10) and 2.5840(9) Å. These structural features are similar to those in 3; however, there are apparent, significant differences in the metallacycle framework. Namely, the four-membered-ring structure in 3 is almost planar, whereas that of 4 exhibits a Ge(2)TGe(3)-type twisted structure.16 The average Ge–Ge bond length (∼2.459 Å) in 4 is also comparable to that in 3 (∼2.477 Å). Based on the molecular structure of 4, one might consider that the incorporation of one additional ‘GePh2’ unit into 3 led to the generation of 4via Ge–Ge bond propagation.
With the four possible intermediary iron species 1–4 in hand, we would like to propose a reaction mechanism for the dehydrogenative coupling of Ph2GeH2 catalyzed by the [Fe(mesityl)2]2/iPrIMMe catalyst system as shown in Scheme 5. First, [Fe(mesityl)2]2 reacts with iPrIMMe to generate catalyst precursor 1. Then, Ge–H bond activation of two moles of Ph2GeH2 occurs via the Fe-mesityl bonds in 1 to generate the catalytically active species 2. A sequence of Ge–Ge-bond-forming and Ge–Ge-bond-propagation reactions occurs on the iron center supported by the two iPrIMMe ligands to generate 3 followed by 4, both of which feature metallacyclooligogermane frameworks. Finally, the incorporation of an additional ‘GePh2’ unit proceeds when 4 comes into contact with Ph2GeH2 to produce (GePh2)5 with concomitant regeneration of 2. In summary, ‘GePh2’ units sequentially assemble on the iron center to realize the selective production of (GePh2)5via dehydrogenative coupling. It should be noted here that the catalytic activity of the isolated complexes 1, 2 and 3 was examined independently and that both showed good catalytic activity toward the dehydrogenative coupling of Ph2GeH2 to afford (GePh2)5 in quantitative yield in THF at room temperature after 40 h (5 mol% of 1, 2 or 3). This result strongly supports the notion that the iron species 2 and 3 play a crucial role in the catalytic dehydrogenative coupling of Ph2GeH2.
 | ||
| Scheme 5 Plausible reaction mechanism for the sequential production of H2 and Ge–Ge bond propagation to afford cyclopentagermane (GePh2)5via the dehydrogenative coupling of Ph2GeH2 catalyzed by [Fe(mesityl)2]2/iPrIMMe. | ||
Dehydrogenative coupling of other group-14 hydrides
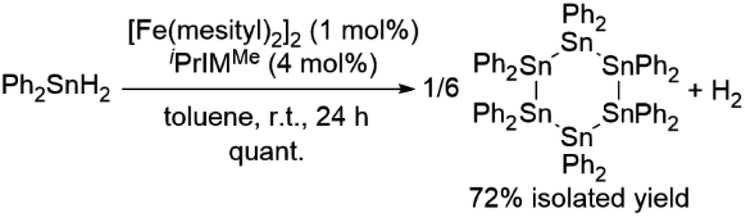
As described above, the [Fe(mesityl)2]2/NHC catalyst system is effective for H2 production via the dehydrogenative coupling of secondary germanes. We found that the dehydrogenative coupling of tertiary and primary germanes can also be achieved by this catalyst system. For instance, the dehydrogenative coupling of Ph3GeH proceeded effectively in THF at room temperature mediated by 2.5 mol% of [Fe(mesityl)2]2 in the presence of 10 mol% of MeIMEt, which contains sterically less demanding methyl groups on the nitrogen atoms of the NHC ligand, to selectively afford the dimeric product (GePh3)2. Similarly, the reaction with tBuGeH3 was briefly examined, and the formation of oligogermanes via the dehydrogenative coupling mediated by a catalytic amount of [Fe(mesityl)2]2/iPrIMMe was observed using 1H NMR and FAB-MS.Tin hydrides such as Ph2SnH2 and nBu2SnH2 were also found to be suitable for this catalytic system (hydrogen content: ca. 0.73 wt% for Ph2SnH2 and ca. 0.85 wt% for nBu2SnH2). Namely, 1 mmol of Ph2SnH2 was dehydrogenatively coupled in the presence of 1 mol% of [Fe(mesityl)2]2 and 4 mol% of iPrIMMe to exclusively produce cyclohexastannane (SnPh2)6, which was isolated in 70% yield. The quantity of H2 gas produced in the course of this reaction was determined using a gas burette, from which ca. 15.4 mL of gaseous product(s) were obtained after 2.5 h at room temperature, and then production of ca. 16.7 mL of gaseous product(s) was detected after 19 h. In this reaction, the production of H2 immediately took place once Ph2SnH2 came into contact with [Fe(mesityl)2]2/iPrIMMe, and this may cause decrease of the amount of detectable gaseous H2 compared with the theoretical value (ca. 22 mL). In contrast to the reaction with germanium congener Ph2GeH2, the reaction with Ph2SnH2 exclusively afforded cyclohexastannane. nBu2SnH2 was also found to be suitable for this catalyst system (1 mol% of [Fe(mesityl)2]2 and 4 mol% of iPrIMMe) to give a ca. 3:1 mixture of cyclopentastannane (SnnBu2)5 and cyclohexastannane (SnnBu2)6 with complete conversion of nBu2SnH2 at room temperature (Scheme 6). In this context, it should also be noted that the formation of a mixture of (SnnBu2)5 and (SnnBu2)6 has already been reported by Jousseaume et al. in the (PPh3)2PdCl2-catalyzed dehydrogenative coupling of nBu2SnH2.17
 | ||
| Scheme 6 Catalytic dehydrogenative coupling of Ph2SnH2 catalyzed by the [Fe(mesityl)2]2/NHC catalyst system. | ||
The iron-based catalyst system described here was found to be effective under mild reaction conditions for the dehydrogenative coupling of primary, secondary, and tertiary germanes as well as secondary stannanes. Although a relatively high reaction temperature was required, the dehydrogenative coupling of a secondary silane was also achieved by an [Fe(mesityl)2]2/iPrIMMe catalyst system. Neat Ph2SiH2 (hydrogen content: ca. 1.01 wt%) was treated with 2.5 mol% of [Fe(mesityl)2]2 and 10 mol% of iPrIMMe at 80 °C. The 1H NMR spectrum of the crude product indicated 82% conversion of Ph2SiH2 and the generation of the disilane Ph2(H)Si–Si(H)Ph2 and trisilane Ph2(H)Si–SiPh2–Si(H)Ph2 in 54% and 8% yield, respectively. In contrast to the reaction systems with secondary germanes and stannanes, no further propagation of the ‘SiPh2’ units took place in this reaction; instead, the generation of Ph3SiH and PhSiH3 was detected due to concomitant redistribution. It should be mentioned here that Rosenberg et al. have already reported the selective formation of Ph2(H)Si–Si(H)Ph2via the dehydrogenative coupling of Ph2SiH2 catalyzed by Rh(PPh3)3Cl. In their report, the authors mentioned that the chemoselectivity of this reaction strongly depends on the reaction conditions, including the size of the reaction vial relative to the volume of the substrate, as well as the rate and efficacy of stirring, and that trisilanes and other redistribution products are often generated under non-optimized reaction conditions. This precedent might suggest that the chemoselectivity of our reaction with Ph2SiH2 could be improved by the optimization of the reaction conditions. However, unfortunately, neither the conversion of Ph2SiH2 nor the chemoselectivity were improved under either different reaction conditions or the use of catalyst systems that contained different auxiliary ligands.
Conclusions
Germanium hydride Ph2GeH2 can act as an efficient and reusable hydrogen-production/-storage material with the aid of iron catalysts. Hydrogen evolution from Ph2GeH2 was effectively realized at room temperature, concomitant with the quantitative formation of (GePh2)5. The regeneration of Ph2GeH2 is very facile, i.e., by simple treatment of (GePh2)5 with 1 atm of H2 at 0 °C, or by treatment with PhICl2 and LiAlH4 at 40 °C. This hydrogen-production/-storage system has some advantages compared to systems based on conventional hydrogen-storage materials such as NaBH4, in which both the hydrogen-production and -storage processes can be carried out under relatively mild operating conditions, and that they do not require precious-metal catalysts. Furthermore, germanium hydrides are generally sufficiently stable under aerobic conditions, easy-to-handle, and show less toxicity toward living organisms. We expect that the results presented in this paper will help in developing the next generation of chemical hydrogen-storage/-production systems and support efforts to use group-14 hydrides such as germanium hydrides as reusable hydrogen carriers in practical applications, which are currently in progress in our laboratory.Data availability
All experimental data are provided in the ESI.†Author contributions
Y. Kobayashi conducted all experiments. All authors analysed the data. Y. Sunada supervised this study and wrote the manuscript. All authors discussed the results and contributed to the preparation of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a project of the Kanagawa Institute of Industrial Science and Technology (KISTEC) as well as by a Grant in Aid for Scientific Research (B) (20H02751) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. The synchrotron radiation experiments were performed at the BL02B1 of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2021A1139).Notes and references
- (a) M. D. Allendorf, V. Stavila, J. L. Snider, M. Witman, M. E. Bowden, K. Brooks and T. Autrey, Nat. Chem., 2022, 14, 1214–1223 CrossRef CAS PubMed; (b) T. He, P. Pachfule, H. Wu, Q. Xu and P. Chen, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed; (c) M. R. Usman, Renew. Sustain. Energy Rev., 2022, 167, 112743 CrossRef CAS; (d) A. Kovač, M. Paranos and D. Marciuš, Int. J. Hydrog. Energy, 2021, 46, 10016–10035 CrossRef; (e) T. He, H. Cao and P. Chen, Adv. Mater., 2019, 31, 1902757 CrossRef CAS PubMed; (f) A. Schneemann, J. L. White, S. Kang, S. Jeong, L. F. Wan, E. S. Cho and V. Stavila, Chem. Rev., 2018, 118, 10775–10839 CrossRef CAS PubMed; (g) M. Paskevicius, L. H. Jepsen, P. Schouwink, R. Černý, D. B. Ravnsbæk, Y. Filinchuk and T. R. Jensen, Chem. Soc. Rev., 2017, 46, 1565–1634 RSC; (h) P. Jena, J. Phys. Chem. Lett., 2011, 2, 206–211 CrossRef CAS; (i) J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- (a) H. I. Schlesinger, H. C. Brown, A. E. Finholt, J. R. Gilbrbath, H. R. Hoekstra and E. K. Hyde, J. Am. Chem. Soc., 1953, 75, 215–219 CrossRef CAS; (b) E. Y. Marrero-Alfonso, A. M. Beaird, T. A. Davis and M. A. Matthews, Ind. Eng. Chem. Res., 2009, 48, 3703–3712 CrossRef CAS; (c) Y. Zhu, L. Ouyang, H. Zhong, J. Liu, H. Wang, H. Shao, Z. Huang and M. Zhu, Angew. Chem., Int. Ed., 2020, 59, 8623–8629 CrossRef CAS PubMed; (d) Y. Kojima, Y. Kawai, H. Nakanishi and S. Matsumoto, J. Power Sources, 2004, 135, 36–41 CrossRef CAS; (e) P. Brack, S. E. Dann and K. G. Upul Wijayantha, Energy Sci. Eng., 2015, 3, 174–188 CrossRef CAS; (f) B. H. Liu and Z. P. Li, J. Power Sources, 2009, 187, 527–534 CrossRef CAS; (g) L. Ouyang, M. Liu, K. Chen, J. Liu, H. Wang, M. Zhu and V. Yartys, J. Alloys Compd., 2022, 910, 164831 CrossRef CAS.
- (a) J. Y. Corey, Adv. Organomet. Chem., 2004, 51, 1–52 CrossRef CAS; (b) E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed; (c) F. Gauvin, J. F. Harrod and H. G. Woo, Adv. Organomet. Chem., 1988, 42, 363–405 CrossRef; (d) T. D. Tilley, Comments Inorg. Chem., 1990, 10, 37–51 CrossRef CAS; (e) T. D. Tilley, Acc. Chem. Res., 1993, 26, 22–29 CrossRef CAS; (f) R. Waterman, Chem. Soc. Rev., 2013, 42, 5629–5641 RSC; (g) T. J. Clark, K. Lee and I. Manners, Chem.–Eur. J., 2006, 12, 8634–8648 CrossRef CAS PubMed; (h) H. Yamashita and M. Tanaka, Bull. Chem. Soc. Jpn., 1995, 68, 403–419 CrossRef CAS; (i) B. Marciniec, C. Pietraszuk, P. Pawluc and H. Maciejewski, Chem. Rev., 2021, 122, 3996–4090 CrossRef PubMed; (j) B. H. Kim, M. S. Cho and H. G. Woo, Synlett, 2004, 0761–0772 CAS.
- C. Aitken and J. F. Harrod, J. Organomet. Chem., 1985, 279, C11–C13 CrossRef CAS.
- (a) J. F. Harrod, Coord. Chem. Rev., 2000, 206–207, 493–531 CrossRef CAS; (b) J. Y. Corey, X. H. Zhu, T. C. Bedard and L. D. Lange, Organometallics, 1991, 10, 924–930 CrossRef CAS; (c) B. J. Grimmond and J. Y. Corey, Organometallics, 2000, 19, 3776–3783 CrossRef CAS; (d) A. D. Sadow and T. D. Tilley, Organometallics, 2003, 22, 3577–3585 CrossRef CAS.
- (a) I. Ojima, S. I. Inaba, T. Kogure and Y. Nagai, J. Organomet. Chem., 1973, 55, C7–C8 CrossRef CAS; (b) L. Rosenberg, C. W. Davis and J. Yao, J. Am. Chem. Soc., 2001, 123, 5120–5121 CrossRef CAS PubMed; (c) S. M. Jackson, D. M. Chisholm, J. S. McIndoe and L. Rosenberg, Eur. J. Inorg. Chem., 2011, 327–330 CrossRef CAS; (d) K. A. Brown-Wensley, Organometallics, 1987, 6, 1590–1591 CrossRef CAS; (e) L. S. Chang and J. Y. Corey, Organometallics, 1989, 8, 1885–1893 CrossRef CAS; (f) M. D. Fryzuk, L. Rosenberg and S. J. Rettig, Inorg. Chim. Acta, 1994, 222, 345–364 CrossRef CAS; (g) F. G. Fontaine, T. Kadkhodazadeh and D. Zargarian, Chem. Commun., 1998, 1253–1254 RSC; (h) E. E. Smith, G. Du, P. E. Fanwick and M. M. Abu-Omar, Organometallics, 2010, 29, 6527–6533 CrossRef CAS; (i) F. G. Fontaine and D. Zargarian, Organometallics, 2002, 21, 401–408 CrossRef CAS; (j) B. P. Chauhan, T. Shimizu and M. Tanaka, Chem. Lett., 1997, 26, 785–786 CrossRef; (k) M. Tanabe, S. Iwase and K. Osakada, Organometallics, 2016, 35, 2557–2562 CrossRef CAS; (l) M. Tanabe, A. Takahashi, T. Fukuta and K. Osakada, Organometallics, 2013, 32, 1037–1043 CrossRef CAS.
- (a) B. Pribanic, M. Trincado, F. Eiler, M. Vogt, A. Comas-Vives and H. Grützmacher, Angew. Chem. Int. Ed., 2020, 59, 15603–15609 ( Angew. Chem. , 2020 , 132 , 15733–15739 ) CrossRef CAS PubMed; (b) S. I. Kalläne, R. Laubenstein, T. Braun and M. Dietrich, Eur. J. Inorg. Chem., 2016, 530–537 CrossRef; (c) D. Schmidt, T. Zell, T. Schaub and U. Radius, Dalton Trans., 2014, 43, 10816–10827 RSC; (d) J. Voigt, M. A. Chilleck and T. Braun, Dalton Trans., 2013, 42, 4052–4058 RSC.
- (a) N. P. Mankad, M. T. Whited and J. C. Peters, Angew. Chem. Int. Ed., 2007, 46, 5768–5771 ( Angew. Chem. , 2007 , 119 , 5870–5873 ) CrossRef CAS PubMed; (b) H. Schneider, D. Schmidt, A. Eichhöfer, M. Radius, F. Weigend and U. Radius, Eur. J. Inorg. Chem., 2017, 2600–2616 CrossRef CAS; (c) Y. Sunada, T. Imaoka and H. Nagashima, Organometallics, 2013, 32, 2112–2120 CrossRef CAS; (d) Y. Sunada, T. Imaoka and H. Nagashima, Organometallics, 2010, 29, 6157–6160 CrossRef CAS.
- (a) T. Tsumuraya, Y. Kabe and W. Ando, J. Organomet. Chem., 1994, 482, 131–138 CrossRef CAS; (b) L. Roß and M. Dräger, Z. Naturforsch. B, 1983, 38, 665–673 CrossRef; (c) W. P. Neumann and K. Kühlein, Justus Liebigs Ann. Chem., 1965, 683, 1–11 CrossRef CAS.
- (a) R. D. Miller and J. Michl, Chem. Rev., 1989, 89, 1359–1410 CrossRef CAS; (b) R. G. Jones and S. J. Holder, Polym. Int., 2006, 55, 711–718 CrossRef CAS; (c) R. West, J. Organomet. Chem., 1986, 300, 327–346 CrossRef CAS.
- C. Qing and C. Cui, Dalton Trans., 2017, 46, 8746–8750 RSC.
- M. Okazaki, H. Kimura, T. Komuro, H. Okada and H. Tobita, Chem. Lett., 2007, 36, 990–991 CrossRef CAS.
- M. Itazaki, M. Kamitani, K. Ueda and H. Nakazawa, Organometallics, 2009, 28, 3601–3603 CrossRef CAS.
- S. Pelzer, B. Neumann, H. G. Stammler, N. Ignat'ev and B. Hoge, Chem.–Eur. J., 2017, 23, 12233–12242 CrossRef CAS PubMed.
- Y. Kobayashi and Y. Sunada, Catalysts, 2019, 10, 29 CrossRef.
- (a) E. Westhof and M. A. Sundaralingam, J. Am. Chem. Soc., 1983, 105, 970–976 CrossRef CAS; (b) C. T. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1972, 94, 8205–8212 CrossRef CAS PubMed.
- B. Jousseaume, N. Noiret, M. Pereyre, A. Saux and J. M. Frances, Organometallics, 1994, 13, 1034–1038 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2204746, 2204747, 2204748 and 2204749. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06011f |
| This journal is © The Royal Society of Chemistry 2023 |