
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoupling of α-bromoamides and unactivated alkenes to form γ-lactams through EDA and photocatalysis†
Sean M.
Treacy
a,
Daniel R.
Vaz
 a,
Syed
Noman
b,
Cédric
Tard
b and
Tomislav
Rovis
*a
a,
Syed
Noman
b,
Cédric
Tard
b and
Tomislav
Rovis
*a
aDepartment of Chemistry, Columbia University, New York, NY 10027, USA. E-mail: tr2504@columbia.edu
bLaboratoire de Chimie Moléculaire (LCM), CNRS, École Polytechnique, Institut Polytechnique de Paris, 91120 Palaiseau, France
First published on 20th January 2023
Abstract
γ-Lactams are prevalent in small-molecule pharmaceuticals and provide useful precursors to highly substituted pyrrolidines. Despite numerous methods for the synthesis of this valuable motif, previous redox approaches to γ-lactam synthesis from α-haloamides and olefins require additional electron withdrawing functionality as well as N-aryl substitution to promote electrophilicity of the intermediate radical and prevent competitive O-nucleophilicity about the amide. Using α-bromo imides and α-olefins, our strategy enables the synthesis of monosubstituted protected γ-lactams in a formal [3 + 2] fashion. These species are poised for further derivatization into more complex heterocyclic scaffolds, complementing existing methods. C–Br bond scission occurs through two complementary approaches, the formation of an electron donor–acceptor complex between the bromoimide and a nitrogenous base which undergoes photoinduced electron transfer, or triplet sensitization with photocatalyst, to furnish an electrophilic carbon-centered radical. The addition of Lewis acids allows for further increased electrophilicity of the intermediate carbon-centered radical, enabling tertiary substituted α-Br-imides to be used as coupling partners as well as internal olefins.
N-heterocycles provide essential cores to a number of biologically active molecules. As a privileged heterocyclic scaffold, γ-lactams are found not only in natural products and pharmaceuticals, but are also valuable intermediates in the synthesis of highly functionalized amines as well as other saturated N-heterocycles.1–3 Despite numerous conventional methods for the construction of γ-lactams, current technologies often limit the synthetic versatility of their products due to requisite substitution in starting materials to enable reactivity.4–8 Synthesizing γ-lactams in an intermolecular fashion from widely available precursors therefore provides a significant opportunity in organic methods development.
Atom-transfer radical addition (ATRA) reactions through both photoredox and Cu catalysis have been well studied for a variety of haloalkanes, especially α-Br-carbonyl compounds.9–11 In the case of α-Br-amide derivatives, efficient addition to unactivated olefins require electron withdrawing groups such as gem-difluoro substitution to increase the electrophilicity of the intermediate radical.12–15 This limitation thus mandates that γ-lactam syntheses from α-Br-amides require α,α substitution, eliminating valuable synthetic derivatization of the protected lactam products16–18 (Fig. 1A).
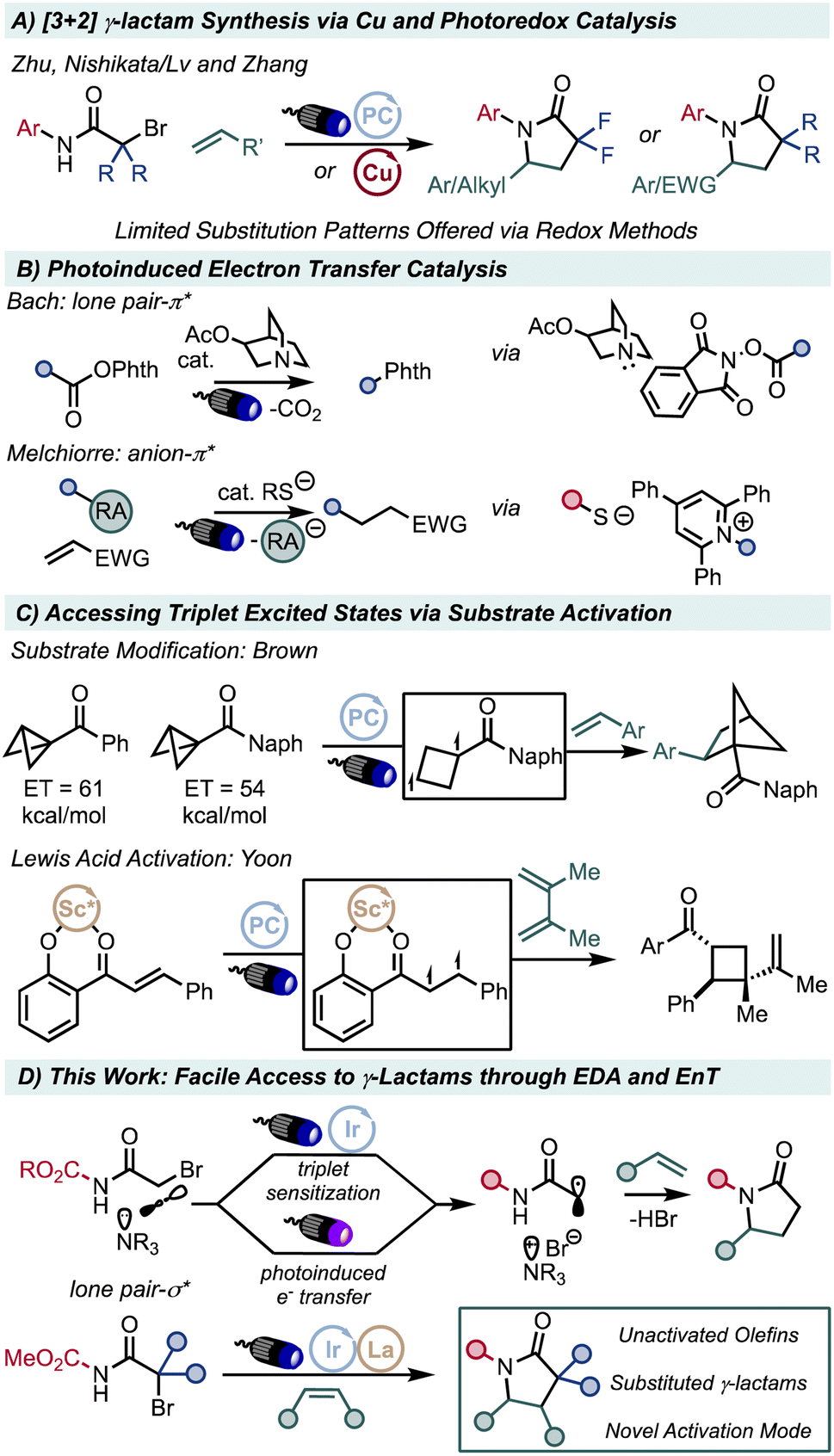 | ||
| Fig. 1 EDA complexation and Lewis acid activation permits access to highly electrophilic α-amido radicals. | ||
Electron donor–acceptor (EDA) complexes have gained prominence due to the useful reactive species that can be generated simply via photoirradiation.19,20 Excitation of these intermediates with an exogenous photosensitizer may likewise proceed via triplet energy transfer (EnT). EDA complexation has been utilized towards the activation of various C–halogen bonds to form corresponding C-centered radicals.21–23 Whereas many strategies have leveraged the bathochromic shift of complex formation to enable electron transfer under visible light,24–29 only recently have groups demonstrated using the donor or acceptor component in a catalytic fashion.30–37 (Fig. 1B).
We envisioned that EDA complexation could enable activation of α-halo-amides through donor catalysis with sufficient energy irradiation or via EnT with an appropriate photocatalyst. Whereas α-halo-amides require additional electron withdrawing functionality at carbon to enable electrophilic radical formation via single electron transfer, energy transfer strategies offer an orthogonal activation mechanism. EnT from excited state photosensitizers has been leveraged through both substrate modification as well as Lewis acid activation in previous studies.38–48 (Fig. 1C) The formation of a Brønsted acid-base adduct should promote complexation between a donor base and an acceptor acid in the case of sufficiently acidic amides, poising it for photoinduced electron transfer or photocatalytic EnT. We designed our reagent with a traceless electron-withdrawing substituent at N, tert-butoxy carbonyl, to both facilitate ATRA with unactivated olefins and promote intramolecular N-cyclization of the intermediate alkyl halide via facile deprotonation of the acidic N–H bond (Fig. 1D).49,50
We began our studies by reacting tert-butyl (2-bromoacetyl)carbamate 1a with 1-hexene (2a) via photocatalysis with catalytic loading of N,N,-diisopropylethylamine (DIPEA). Subjecting 2-bromoacetamide and N-phenyl-2-bromoacetamide under identical conditions resulted in complete recovery of starting material (see Table ESI 1†). Having achieved high conversion of the ATRA product we found that basic workup with excess K3PO4 in wet MeCN furnishes our desired Boc-protected γ-lactam 3aa in good yield (88%, Fig. 2). Evaluation of the reaction controls revealed the necessity of visible light, Ir photocatalyst, and the tertiary amine base for high yields (Fig. 2, entries 2–4). Likewise, an excess of the α-Br-acyl carbamate 1a was found to be essential for efficient reactivity (Fig. 2, entries 5–6).
 | ||
| Fig. 2 Selected optimization trials (see ESI† for complete details). All reactions were conducted on 0.1 mmol scale. Yields were determined by 1H NMR integration relative to 0.5 equiv. dibromomethane as an internal standard. Standard conditions A: 1% Ir = (Ir[dF(CF3)ppy]2(dtbpy))PF6, 20% DIPEA = N,N-diisopropylethylamine, 440 nm irradiation in 0.4 M DCE. Standard Conditions B: 20% 2-Me-pyridine, 390 nm irradiation in 0.4 M DCE. Both conditions are followed by the addition of K3PO4 in MeCN (0.1 M). | ||
Interestingly, the reaction proceeds modestly in the absence of photocatalyst (Fig. 2, entry 3) suggesting an alternative activation mode enabled solely by irradiation. The use of higher energy light (390 nm) leads to a more efficient transformation to deliver synthetically useful yields of the ATRA product. Screening of amine bases (Table ESI 6†) showed 2-methyl-pyridine to be the optimal donor catalyst in the absence of exogenous photosensitizer (Fig. 2, entry 8). A larger excess of 1a (3 equiv. vs. 2 equiv.) is required to achieve comparable yields of the ATRA product without the addition of photocatalyst.
With optimized conditions in hand, we set to explore the scope of olefin coupling partners under both sets of conditions (Fig. 3). We found α-olefins to be effective coupling partners with various alkyl and aryl substituents (3aa–3ag, 3an) undergoing coupling in high yield. Of note, olefin substrates 2c and 2d undergo the ATRA reaction with comparable yields to other substrates, but the α-tertiary and α-quaternary positions of the intermediate alkyl bromide impart steric hindrance on the subsequent intramolecular alkylation, lowering the yield of the desired γ-lactam. In exploring functional group tolerance, we found that alkenes bearing esters (3ah, 3ai), ketones (3am), and protected alcohols (3ai, 3al) tolerate the reaction conditions. Pleasingly, the incorporation of polar electrophiles in the substrate such as alkyl halides (3aj) and alkyl tosylates (3ak) also deliver the desired lactam in good yields. Finally, imides (3ao) and various amides (3ap–3as) all undergo efficient coupling using our protocol. In many cases, the direct sensitization of the EDA complex was shown to be competitive with the photocatalytic reaction. We posited that the direct irradiation protocol would be amenable to larger scale reactions due to the low cost and availability of 2-Me-pyridine as a catalyst. Indeed, the procedure scales up to 1 mmol with similar efficiency (76% vs. 74% for 3ah), demonstrating the additional synthetic utility of this method.
 | ||
| Fig. 3 Scope of γ-lactam synthesis. All reactions performed on 0.3 mmol scale. All yields are from isolation. aSee ESI† for reation details. | ||
In order to augment the derivatization available to our lactam products, we explored the possibility that other carbamate protecting groups could be utilized in the formal [3 + 2] coupling with high efficiency. This protocol proves to be general in this regard, with methoxycarbonyl (3ba), 2,2,2-trichloroethoxycarbonyl (Troc) (3ca), and 2-(trimethylsilyl)ethoxycarbonyl (Teoc) (3da) all furnishing the desired protected γ-lactam products in high yields. Likewise, a tosyl imide results in product 3ea albeit with a modified protocol (see ESI†). These protecting groups offer opportunities for orthogonal deprotection strategies and may be carried through as intermediates towards further valuable N-containing complex products.
Focusing our attention on expanding the scope of α-Br-imide coupling partners, we found that additional alkyl substitution at the site of electrophilic radical generation was detrimental to the ATRA step. We attribute this attenuated reactivity to the reduced electrophilicity of the intermediate radical, which should react more slowly with α-olefins leading to slightly diminished yields of the desired γ-lactam product and in undesirable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoselectivity (3fa). Furthermore, tertiary alkyl bromides lead to quantitative recovery of unreacted starting material under identical conditions (1g), indicating that activation of the substate is also inhibited by insufficient electrophilicity. We hypothesized that the formation of a Lewis acid adduct with the substrate could enable single electron transfer (SET) to the substrate, enhance the electrophilicity of the intermediate radical, and further enable the substrate to act as a halogen atom donor under a radical chain paradigm. The combination of photoredox catalysis and Lewis acid activation has become a powerful tool in organic synthesis in recent years.43,45–47,51,52 Due to the instability of imide 1g in the presence of various Lewis acids, we shifted to imide 1h. Screening and optimization (Table ESI 7–9†) showed La(OTf)3 to be the most effective Lewis acid, improving the yield of the desired product 3ha from 0% to 59% in a single step with slight excess of DIPEA (Fig. 4). Evaluation of this protocol on the corresponding primary α-Br-imide 1b showed limited cyclization (<15%) after the first step under identical conditions, suggesting that a Thorpe–Ingold effect likely facilitates the formation of the desired final product without the need for additional inorganic base. Evaluation of the scope of tertiary alkyl halides proved fruitful, as fused ring systems are readily furnished via this protocol, including cyclohexyl (3ia), cyclopentyl (3ja), and cyclobutyl (3ka) substituents. Additionally, heterocycles such as tetrahydropyran (3la) prove to be well tolerated. Furthermore, the addition of La(OTf)3 enables ATRA with internal olefins such that symmetric internal olefins could be added to the protocol albeit with modest diastereoselectivity (3bt and 3bu). The successful reaction of these substrates supports the role of the Lewis acid in enhancing the electrophilicity of the intermediate radical to promote addition to hindered olefins (Fig S8†).
:1 diastereoselectivity (3fa). Furthermore, tertiary alkyl bromides lead to quantitative recovery of unreacted starting material under identical conditions (1g), indicating that activation of the substate is also inhibited by insufficient electrophilicity. We hypothesized that the formation of a Lewis acid adduct with the substrate could enable single electron transfer (SET) to the substrate, enhance the electrophilicity of the intermediate radical, and further enable the substrate to act as a halogen atom donor under a radical chain paradigm. The combination of photoredox catalysis and Lewis acid activation has become a powerful tool in organic synthesis in recent years.43,45–47,51,52 Due to the instability of imide 1g in the presence of various Lewis acids, we shifted to imide 1h. Screening and optimization (Table ESI 7–9†) showed La(OTf)3 to be the most effective Lewis acid, improving the yield of the desired product 3ha from 0% to 59% in a single step with slight excess of DIPEA (Fig. 4). Evaluation of this protocol on the corresponding primary α-Br-imide 1b showed limited cyclization (<15%) after the first step under identical conditions, suggesting that a Thorpe–Ingold effect likely facilitates the formation of the desired final product without the need for additional inorganic base. Evaluation of the scope of tertiary alkyl halides proved fruitful, as fused ring systems are readily furnished via this protocol, including cyclohexyl (3ia), cyclopentyl (3ja), and cyclobutyl (3ka) substituents. Additionally, heterocycles such as tetrahydropyran (3la) prove to be well tolerated. Furthermore, the addition of La(OTf)3 enables ATRA with internal olefins such that symmetric internal olefins could be added to the protocol albeit with modest diastereoselectivity (3bt and 3bu). The successful reaction of these substrates supports the role of the Lewis acid in enhancing the electrophilicity of the intermediate radical to promote addition to hindered olefins (Fig S8†).
 | ||
| Fig. 4 Scope of γ-lactam synthesis through Lewis acid activation. All reactions performed on 0.3 mmol scale. All yields are from isolation. aSee ESI† for experimental details. | ||
In order to understand the mechanism of initial radical formation, as well as radical propagation, we conducted a number of electrochemical and photochemical studies. Cyclic voltammetry was conducted on 1h, to reveal an irreversible 1-electron wave corresponding to the reductive cleavage of the C–Br bond occurring at E1/2 = −1.47 V vs. SCE (Fig S2†), meaning that reduction by the reduced state of the photocatalyst (Ered = −1.37 V vs. SCE53) would be endothermic and therefore unfavorable at room temperature. Similarly, the employment of photocatalysts with greater reduction potentials in their excited state do not furnish high yields of the ATRA product (Table ESI 2†). Further cyclic voltammetry studies demonstrated that SET to 1h could be made even more facile through addition of La(OTf)3, to shift the reduction potential as low as at E1/2 = −1.32 V vs. SCE with an irreversible 2-electron wave (Fig S2†) indicating that the addition of Lewis acid may indeed change the mechanism of activation of the C–Br bond in the starting material from EnT to SET. Higher yields are achieved with excess DIPEA relative to starting material 1h, indicating that the concentration of reduced photocatalyst may additionally enable this change in mechanism as there may be a higher concentration of the more reducing reduced state photocatalyst. Lewis acids have been shown to promote EnT as well as produce complexes more amenable to SET.47,48
UV-vis spectroscopy of the starting material 1a reveals a significant absorption in the UV region with λmax = 288 nm (Fig S1†). UV-vis characterization of mixtures of DIPEA and starting material 1a reveal a growing shoulder absorption tailing into the visible range with λmax = 344 nm (Fig. 5C). We ascribe this absorption to an EDA complex of the tertiary amine base with the α-Br-imide, which enables direct photoinduced electron transfer without the need for photocatalyst under sufficiently high energy irradiation. When present, the photocatalyst sensitizes the EDA complex under visible light irradiation to enable initial radical formation via an EnT mechanism (ET = 61.7 kcal mol−1).53 Similar studies were undertaken with 2-Me-pyridine as the donor to find a similar red shift in the UV-vis absorption (Fig S3†).
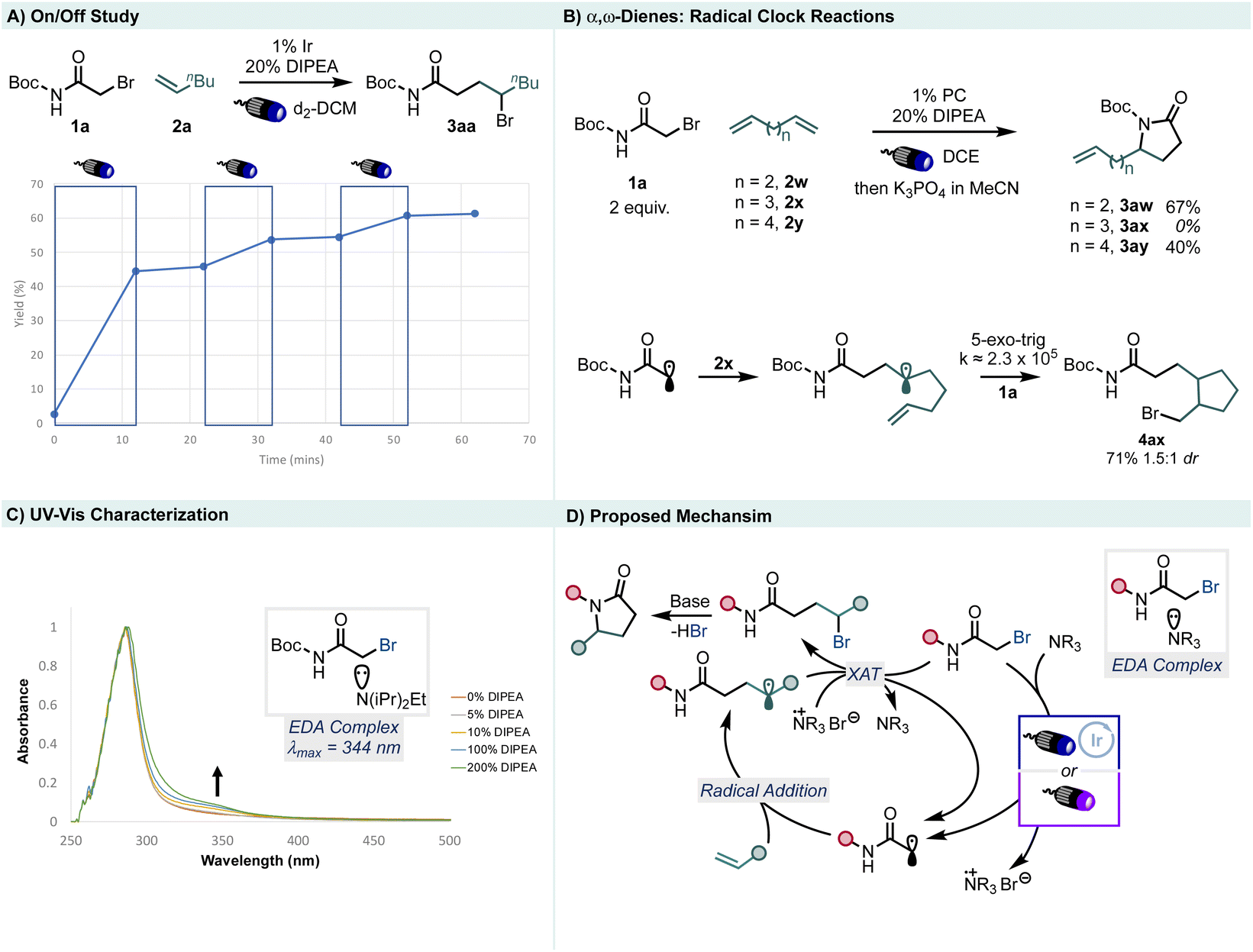 | ||
| Fig. 5 Mechanistic investigations. | ||
When α-ω-dienes 2w, 2x, and 2y are used as coupling partners, we note radical cyclization only in the case of 1,6-heptadiene (2x), which permits a rough estimate of the rate of alkyl radical halogenation when compared to the analogous known radical cyclization rate constants54 (Fig. 5B). This shows that halogen atom transfer must occur at a rate roughly between 5.5 × 103 and 2.3 × 105 s−1 under the reaction conditions, as the 5-exo-trig cyclization is the only cyclization fast enough to cyclize prior to intermolecular halogen atom transfer. Since the 6-exo-trig cyclization product is not detected, the rate of intermolecular halogen transfer under our conditions must exceed the rate constant of the cyclization which is 5.5 × 103 s−1.54 To test whether the reaction is merely initiated, proceeding primarily through a radical chain, we conducted an on-off study which revealed that the reaction is photocontrolled (Fig, 5A). However, experimental determination of the quantum yield (Φ = 14, see ESI†) showed that radical chains are indeed operative, although they are relatively short lived and likely attenuate as the reaction progresses.
Therefore, we propose the following mechanism (Fig. 5D). First, formation of an EDA complex between α-Br-imide 1a and a nitrogenous base is sensitized by the triplet excited state of the Ir photocatalyst through an EnT mechanism or through direct EDA complex absorption. The resulting electrophilic radical reacts with an olefin to form a C-centered electron rich radical. This intermediate is then poised to undergo XAT with another equivalent of 1a or an in situ formed N–Br+ species to form the ATRA product. This readily forms the desired lactam product upon treatment with excess base in acetonitrile in a single pot.
We have demonstrated a valuable method for the formal [3 + 2] synthesis of γ-lactams, enabled by complementary approaches using EDA complexation or photoredox catalysis. This strategy permits the facile synthesis of highly substituted γ-lactams from readily synthesized α-Br-imide starting materials and abundant olefins, enabling novel bond disconnections. Such opportunities will streamline synthetic routes as well as provide novel biologically active compounds for use in small-molecule pharmaceuticals.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
S. M. T., D. V., and T. R. conceived the study. S. M. T. and D. V. designed and executed the experiments. S. N., S. M. T., and C. T. conducted electrochemical investigations. All authors contributed to writing the manuscript, and given it final approval.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank NIGMS (GM125206). S. M. T. thanks the Arun Guthikonda Memorial Foundation for a fellowship. We thank Fereshteh Zandkarimi and the Mass Spectrometry Core Facility at Columbia University for high-resolution mass spectrometry analysis of the compounds.Notes and references
- J. Caruano, G. G. Muccioli and R. Robiette, Org. Biomol. Chem., 2016, 14, 10134–10156 RSC.
- L.-W. Ye, C. Shu and F. Gagosz, Org. Biomol. Chem., 2014, 12, 1833–1845 RSC.
- B. Nay, N. Riache and L. Evanno, Nat. Prod. Rep., 2009, 26, 1044–1062 RSC.
- A. Romero and K. A. Woerpel, Org. Lett., 2006, 8, 2127–2130 CrossRef CAS PubMed.
- R. B. Lettan, C. V. Galliford, C. C. Woodward and K. A. Scheidt, J. Am. Chem. Soc., 2009, 131, 8805–8814 CrossRef CAS PubMed.
- D. E. A. Raup, B. Cardinal-David, D. Holte and K. A. Scheidt, Nat. Chem., 2010, 2, 766–771 CrossRef CAS PubMed.
- K. L. Kimmel, J. D. Weaver, M. Lee and J. A. Ellman, J. Am. Chem. Soc., 2012, 134, 9058–9061 CrossRef CAS PubMed.
- R. Giri, I. Mosiagin, I. Franzoni, N. Y. Nötel, S. Patra and D. Katayev, Angew. Chem., Int. Ed., 2022, 61, e202209143 CrossRef CAS PubMed.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC.
- T. M. Williams and C. R. J. Stephenson, in Visible Light Photocatalysis in Organic Chemistry, John Wiley & Sons, Ltd, 2018, pp. 73–92 Search PubMed.
- J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160–4163 CrossRef CAS PubMed.
- T. Xu and X. Hu, Angew. Chem., Int. Ed., 2015, 54, 1307–1311 CrossRef CAS PubMed.
- W. Huang, W. Chen, G. Wang, J. Li, X. Cheng and G. Li, ACS Catal., 2016, 6, 7471–7474 CrossRef CAS.
- Z. Zhang, H. Martinez and W. R. Dolbier, J. Org. Chem., 2017, 82, 2589–2598 CrossRef CAS PubMed.
- J. He, C. Chen, G. C. Fu and J. C. Peters, ACS Catal., 2018, 8, 11741–11748 CrossRef CAS PubMed.
- Y. Yamane, K. Miyazaki and T. Nishikata, ACS Catal., 2016, 6, 7418–7425 CrossRef CAS.
- M. Zhang, W. Li, Y. Duan, P. Xu, S. Zhang and C. Zhu, Org. Lett., 2016, 18, 3266–3269 CrossRef CAS PubMed.
- Y. Lv, W. Pu, Q. Wang, Q. Chen, J. Niu and Q. Zhang, Adv. Syn. Cat., 2017, 359, 3114–3119 CrossRef CAS.
- C. G. S. Lima, T. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- A. Postigo, Eur. J. Org. Chem., 2018, 2018, 6391–6404 CrossRef CAS.
- Y. Shen, J. Cornella, F. Juliá-Hernández and R. Martin, ACS Catal., 2017, 7, 409–412 CrossRef CAS.
- S. Jung, S. Shin, S. Park and S. Hong, J. Am. Chem. Soc., 2020, 142, 11370–11375 CrossRef CAS PubMed.
- J. Davies, S. G. Booth, S. Essafi, R. A. W. Dryfe and D. Leonori, Angew. Chem., 2015, 127, 14223–14227 CrossRef.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed.
- J. Zhang, Y. Li, R. Xu and Y. Chen, Angew. Chem., Int. Ed., 2017, 56, 12619–12623 CrossRef CAS PubMed.
- J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS PubMed.
- J. Wu, R. M. Bär, L. Guo, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 18830–18834 CrossRef CAS PubMed.
- M. J. Cabrera-Afonso, A. Granados and G. A. Molander, Angew. Chem., Int. Ed., 2022, 61, e202202706 CrossRef CAS PubMed.
- E. Arceo, I. D. Jurberg, A. Alvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed.
- M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017, 9, 868–873 CrossRef CAS PubMed.
- I. Bosque and T. Bach, ACS Catal., 2019, 9, 9103–9109 CrossRef CAS.
- K. Matsuo, E. Yamaguchi and A. Itoh, J. Org. Chem., 2020, 85, 10574–10583 CrossRef CAS PubMed.
- E. de Pedro Beato, D. Spinnato, W. Zhou and P. Melchiorre, J. Am. Chem. Soc., 2021, 143, 12304–12314 CrossRef CAS PubMed.
- W. Zhou, S. Wu and P. Melchiorre, J. Am. Chem. Soc., 2022, 144, 8914–8919 CrossRef CAS PubMed.
- E. Le Saux, M. Zanini and P. Melchiorre, J. Am. Chem. Soc., 2022, 144, 1113–1118 CrossRef CAS PubMed.
- N. Kato, T. Nanjo and Y. Takemoto, ACS Catal., 2022, 12, 7843–7849 CrossRef CAS.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed.
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CAS.
- J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed.
- R. Guo, Y.-C. Chang, L. Herter, C. Salome, S. E. Braley, T. C. Fessard and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 7988–7994 CrossRef CAS PubMed.
- T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391–1395 CrossRef CAS PubMed.
- F. M. Hörmann, T. S. Chung, E. Rodriguez, M. Jakob and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 827–831 CrossRef PubMed.
- Z. D. Miller, B. J. Lee and T. P. Yoon, Angew. Chem., Int. Ed., 2017, 56, 11891–11895 CrossRef CAS PubMed.
- M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543–9547 CrossRef CAS PubMed.
- T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef CAS PubMed.
- M. W. Campbell, V. C. Polites, S. Patel, J. E. Lipson, J. Majhi and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 19648–19654 CrossRef CAS PubMed.
- G. Stork and R. Mah, Heterocycles, 1989, 28, 5 CrossRef.
- X. Fang, K. Liu and C. Li, J. Am. Chem. Soc., 2010, 132, 2274–2283 CrossRef CAS PubMed.
- K. N. Lee, Z. Lei and M.-Y. Ngai, J. Am. Chem. Soc., 2017, 139, 5003–5006 CrossRef CAS PubMed.
- S. Zhu and M. Rueping, Chem. Commun., 2012, 48, 11960 RSC.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- A. L. J. Beckwith and G. Moad, J. Chem. Soc., Chem. Commun., 1974, 472–473 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05973h |
| This journal is © The Royal Society of Chemistry 2023 |