
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-specific covalent metalation of DNA oligonucleotides with phosphorescent platinum(II) complexes†
Felix
Boisten
a,
Iván
Maisuls
 ab,
Tim
Schäfer
a,
Cristian A.
Strassert
*abc and
Jens
Müller
*ac
ab,
Tim
Schäfer
a,
Cristian A.
Strassert
*abc and
Jens
Müller
*ac
aWestfälische Wilhelms-Universität Münster, Institut für Anorganische und Analytische Chemie, Corrensstr. 28/30, 48149 Münster, Germany. E-mail: ca.s@uni-muenster.de; mueller.j@uni-muenster.de
bWestfälische Wilhelms-Universität Münster, Center for Nanotechnology (CeNTech), Heisenbergstr. 11, 48149 Münster, Germany
cWestfälische Wilhelms-Universität Münster, Center for Soft Nanoscience (SoN) and Cells in Motion Interfaculty Centre (CiMIC), Corrensstr. 28/30, 48149 Münster, Germany
First published on 3rd February 2023
Abstract
Phosphorescent Pt(II) complexes, composed of a tridentate N^N^C donor ligand and a monodentate ancillary ligand, were covalently attached to DNA oligonucleotides. Three modes of attachment were investigated: positioning the tridentate ligand as an artificial nucleobase via a 2′-deoxyribose or a propane-1,2-diol moiety and orienting it towards the major groove by appending it to a uridine C5 position. The photophysical properties of the complexes depend on the mode of attachment and on the identity of the monodentate ligand (iodido vs. cyanido ligand). Significant duplex stabilization was observed for all cyanido complexes when they are attached to the DNA backbone. The luminescence strongly depends on whether a single or two adjacent complexes are introduced, with the latter showing an additional emission band indicative of excimer formation. The doubly platinated oligonucleotides could be useful as ratiometric or lifetime-based oxygen sensors, as the green photoluminescence intensities and average lifetimes of the monomeric species are drastically boosted upon deoxygenation, whereas the red-shifted excimer phosphorescence is nearly insensitive to the presence of triplet dioxygen in solution.
Introduction
Nucleic acids represent excellent scaffolds for the predictable arrangement of functional entities in three-dimensional space, making use of their modular composition, their facile modification and their superb self-assembly.1 This has enabled, amongst others, the field of DNA-templated organic synthesis.2 Organic chromophores represent another prominent type of moieties assembled using nucleic acids.3–6 The site-specific incorporation of transition metal ions is of interest, too, because it can equip the DNA with metal-based properties such as luminescence. It is typically achieved by introducing metal-mediated base pairs.7–9 In this type of artificial base pairs, two ligand-based nucleosides are located on opposing positions within a duplex composed of otherwise complementary oligonucleotides. The respective metal ion then site-specifically binds to this high-affinity binding site.10,11 While the resulting metal-modified DNA may be fluorescent (depending on the identity of ligand and metal ion),12 phosphorescent metal complexes have not yet been incorporated into the base pair stack of nucleic acid duplexes. A few examples are known where a luminescent complex is covalently attached to the uridine C5-position, positioning it in the major groove of the duplex.13,14 In the past, a variety of luminescent intercalating metal complexes have been established, with fascinating applications in optical microscopy.15–18 Moreover, terminally appended photo-reactive Ru(II) and Rh(III) complexes have been prominently applied,19e.g. in the study of charge transfer along DNA duplexes.20 Several organometallic metal-mediated base pairs have also been reported in the context of Hg(II) and Pd(II) complexes.21–28 Their advantage over regular metal-mediated base pairs is the increased metal–ligand bond strength, so that they persist even at low concentrations.We report herein on the site-specific covalent incorporation of phosphorescent Pt(II) complexes into DNA duplexes (Chart 1). The coordination compounds are derived from a recently established family of Pt(II) complexes containing a tridentate N^N^C donor ligand and an ancillary monodentate ligand. These species show robust phosphorescence, with an emission wavelength essentially independent of the identity of the monodentate ligand.29–31 While their green phosphorescence (lifetimes and intensities) is quenched by triplet dioxygen (3O2), their dimers and higher aggregates appear with a red-shifted yet oxygen-insensitive luminescence portraying excimeric character supported by metal–metal interactions (i.e., coupling between dz2-orbitals protruding out of the coordination plane). The site-specific incorporation of a Pt(II) complex into DNA is complicated by the high affinity of Pt(II) for purine N7 positions, as is well-known from the mode of action of the antitumor drug cisplatin.32 In the past, two pre-platinated building blocks for automated DNA solid-phase synthesis were reported to tackle this challenge.33,34 Similarly, the solid-phase synthesis of a terminally Pt(II)-modified oligonucleotide was reported.35 However, none of these approaches can be applied for the introduction of the Pt(II) complex under consideration here, because they make use of exclusively monodentate ligands. Instead, we decided to use the Cu(I)-catalysed azide–alkyne Huisgen cycloaddition to achieve the desired site-specific attachment of the Pt(II) complexes. A similar approach was recently reported in the context of (non-luminescent) cis-Pt(II)-modified triplex-forming oligonucleotides.36
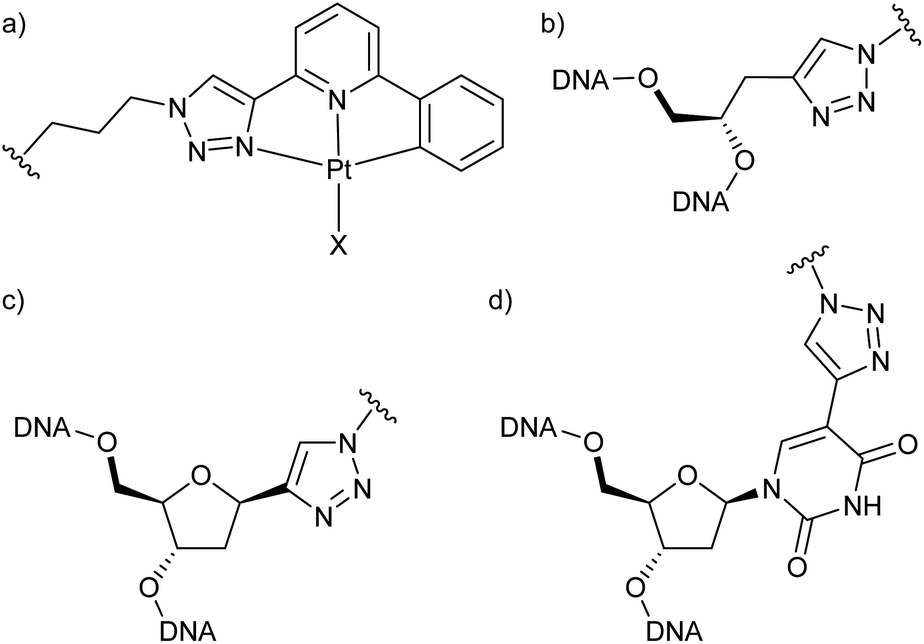 | ||
| Chart 1 (a) Structure of the Pt(II) complexes used in this study (X = I, CN) and their different modes of attachment to the oligonucleotide via (b) a GNA moiety, (c) a 2′-deoxyribose, (d) C5-modified 2′-deoxyuridine. | ||
Results and discussion
Synthesis and nomenclature
To be able to compare different locations of the Pt(II) complex within the DNA duplex, three points of attachment were evaluated: (a) via a glycol nucleic acid (GNA) moiety, (b) via a 2′-deoxyribose and (c) via a C5-alkynylated 2′-deoxyuridine moiety (Chart 1). The Pt(II) precursor complexes PtI and PtCN were prepared via oxidative addition (and subsequent ligand exchange in the case of PtCN) using a suitably iodinated ligand precursor (Scheme 1). These precursor complexes were attached to the respective oligonucleotides in a post-synthetic Cu(I)-catalysed azide–alkyne Huisgen cycloaddition (see ESI† for details). Table 1 summarizes the DNA duplexes under investigation in this study. It is essentially one duplex sequence with a variable central base pair. All four canonical nucleobases are placed opposite the artificial one. Taking into consideration the two Pt(II) complexes (PtI, PtCN) and the three modes of attachment (GNA, Chart 1b; DNA, Chart 1c; Uri, Chart 1d), 20 duplexes with a single site-specific Pt(II) modification were prepared, plus eight reference duplexes in which the Pt(II)-containing GNA or DNA building blocks were replaced by an unsubstituted 1H-1,2,3-triazole-4-yl base (tri). Throughout the herein reported work, the identity of the Pt(II) complex and its mode of attachment are designated using a superscript denotation. For example, IIPtCN,GNA represents duplex II (with a cytosine opposite the artificial nucleobase) bearing a PtCN complex attached via a GNA moiety. Similarly, ODN1Tri,DNA represents a single-stranded oligonucleotide with a central non-platinated 1H-1,2,3-triazole attached via a 2′-deoxyribose. In addition, duplex V was prepared, bearing two centrally located consecutive PtCN complexes (connected via a 2′-deoxyribose) and two complementary guanine residues.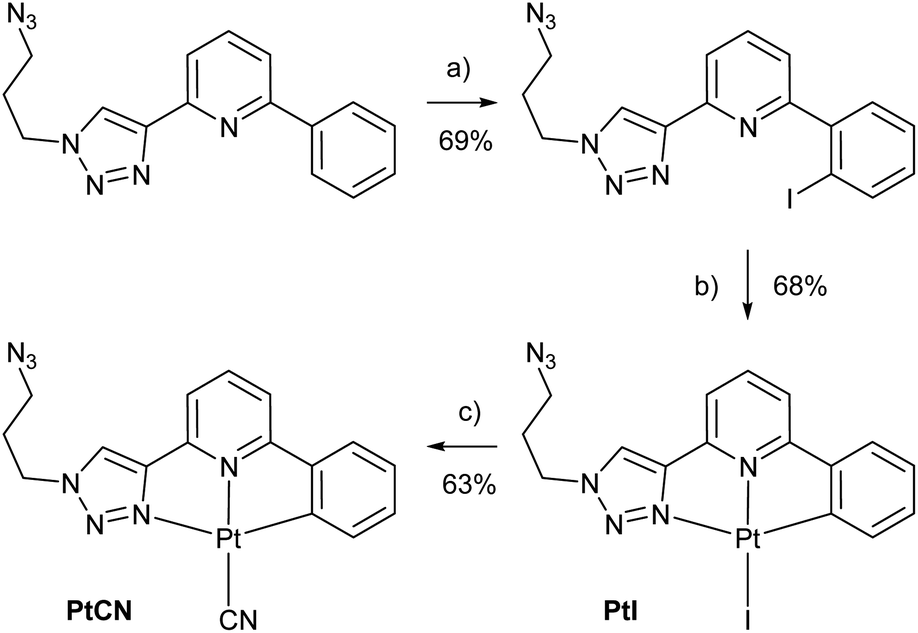 | ||
Scheme 1 Synthesis of the Pt(II) precursor complexes PtI and PtCN for the subsequent cycloaddition to a suitably alkyne-modified oligonucleotide. (a) NIS, Pd(OAc)2, CH3CN, 90 °C, 2 d; (b) Pt2(dba)3, THF, 50 °C, 90 min; (c) KCN, CH3OH/CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 90 °C, 3 h. For further synthetic details, see ESI.† :1), 90 °C, 3 h. For further synthetic details, see ESI.† | ||
| Duplex | Sequence | |
|---|---|---|
| a The letter X represents the identity of the Pt(II) complex and its point of attachment (PtI, PtCN, GNA, DNA, Uri). In Pt(II)-free reference duplexes, an unsubstituted 1,2,3-triazole moiety (Tri) was used. See text for more details. | ||
| IX | ODN1X | 5′-d(CTT TCT XTC CCT C)-3′ |
| ODN2G | 3′-d(GAA AGA GAG GGA G)-5′ | |
| IIX | ODN1X | 5′-d(CTT TCT XTC CCT C)-3′ |
| ODN2C | 3′-d(GAA AGA CAG GGA G)-5′ | |
| IIIX | ODN1X | 5′-d(CTT TCT XTC CCT C)-3′ |
| ODN2A | 3′-d(GAA AGA AAG GGA G)-5′ | |
| IVX | ODN1X | 5′-d(CTT TCT XTC CCT C)-3′ |
| ODN2T | 3′-d(GAA AGA TAG GGA G)-5′ | |
| V | ODN3X | 5′-d(CTT TCT XXC CCT C)-3′ |
| ODN4 | 3′-d(GAA AGA GGG GGA G)-5′ | |
Characterization of duplex stability and conformation
DNA melting studies were initially performed with all duplexes in which the Pt(II) complexes are attached directly to the DNA backbone, i.e., via the GNA or the 2′-deoxyribose linker. All duplexes bearing a PtI complex show a broad melting transition and a melting temperature Tm significantly below that of the corresponding Pt(II)-free reference duplexes (Table 2 and Fig. S1†). Nevertheless, the duplexes mainly adopt a regular B-DNA-type geometry, as indicated by their CD spectra (Fig. S2†). The destabilization is even more pronounced for the duplexes in which a 2′-deoxyribose linker is used to attach the Pt(II) complex. The broad melting transition could be explained by the loss of the iodido ligand, followed by a non-specific complexation of one of the canonical nucleobases, either in an intrastrand or an interstrand fashion. As numerous nucleo-bases are available for this platination, different products are expected, all of which melt at different temperatures. The resulting duplexes are expected to be distorted, thereby explaining the decrease in Tm. The fact that the iodido ligand is easily substituted is also apparent from the mass spectra of ODN1PtI,GNA and ODN1PtI,DNA (Fig. S3 and S4†), where the iodido ligand is found to be cleaved off.| Duplex | T m | ΔTm | Duplex | T m | ΔTm |
|---|---|---|---|---|---|
| a Experimental conditions: 1 μM DNA duplex, 5 mM MOPS buffer (pH 7.0), 150 mM NaClO4, 2.5 mM Mg(ClO4)2. | |||||
| I Tri,GNA | 26.1 | n.a. | I Tri,DNA | 30.5 | n.a. |
| IPtI ,GNA | 19.9 | −6.2 | IPtI ,DNA | 13.8 | −16.7 |
| II Tri,GNA | 24.3 | n.a. | II Tri,DNA | 28.1 | n.a. |
| IIPtI ,GNA | 19.0 | −5.3 | IIPtI ,DNA | 11.6 | −16.5 |
| III Tri,GNA | 28.2 | n.a. | III Tri,DNA | 31.3 | n.a. |
| IIIPtI ,GNA | 20.4 | −7.8 | IIIPtI ,DNA | 12.2 | −19.1 |
| IV Tri,GNA | 25.0 | n.a. | IV Tri,DNA | 27.7 | n.a. |
| IVPtI ,GNA | 19.1 | −5.9 | IVPtI ,DNA | 11.8 | −15.9 |
Hence, in a second set of experiments, the iodido ligand was exchanged by a cyanido unit, which is expected to bind more tightly to the Pt(II) ion due to its strong σ-donor and π-acceptor character. This was again confirmed by mass spectrometry, where a non-dissociated Pt(II) complex was observed for ODN1PtCN,GNA and ODN1PtCN,DNA (Fig. S5 and S6†). Indeed, the PtCN-modified duplexes show a more regular melting behaviour, as illustrated in Fig. 1 based on duplexes IIIX. In the presence of the Pt(II) complex, a significant increase in Tm is observed with respect to the triazole-containing reference duplexes. The increase is largely independent of the complementary nucleobase (Fig. S7†), with duplex IIPtCN,GNA being a prominent exception. In general, the melting transition is steeper for duplexes in which the Pt(II) complex is attached via a 2′-deoxyribose, compared to the GNA linker. This is in agreement with the distortion expected upon the incorporation of the non-canonical backbone fragment. Again, the CD spectra indicate no major structural changes upon the introduction of the Pt(II) complex (Fig. 1b and S7†), except for the fact that the Pt(II)-free reference duplexes show an unusually strong positive Cotton effect at ∼280 nm. However, as the wavelengths of the Cotton effects do not significantly shift in the presence of the Pt(II) complex, major structural changes can be ruled out.
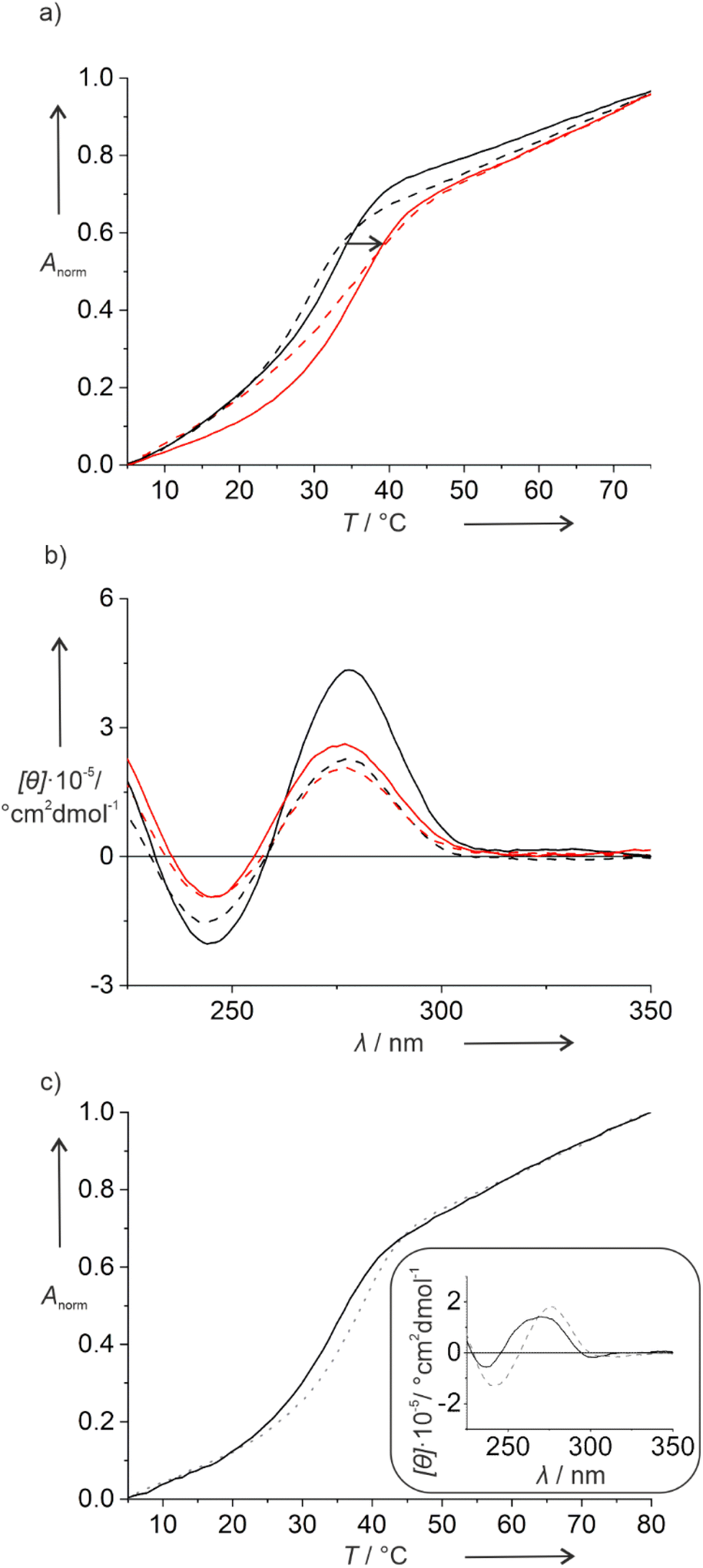 | ||
| Fig. 1 (a) Melting curves and (b) CD spectra of duplexes IIITri,GNA, IIIPtCN,GNA, IIITri,DNA and IIIPtCN,DNA. Broken lines represent the use of the GNA linker, solid lines of the DNA linker. Data for PtCN-containing DNA are shown in red, for the Pt(II)-free reference strands in black. (c) Melting curves of duplexes IPtCN,DNA (dotted line) and V (solid line). The inset shows the corresponding CD spectra. Experimental conditions: 1 μM DNA duplex, 5 mM MOPS buffer (pH 7.0), 150 mM NaClO4, 2.5 mM Mg(ClO4)2. | ||
Table 3 lists the melting temperatures of the duplexes in the presence of PtCN. The reason for the increased stabilization of duplex IIPtCN,GNA in comparison to the other duplexes with a PtCN, GNA modification remains unclear. While it could be speculated that the complex optimally fits into a duplex with a complementary 2′-deoxycytidine, such an explanation would not be in agreement with the shallow melting transition (vide supra) or the data for PtCN, DNA.
| Duplex | T m | ΔTm | Duplex | T m | ΔTm |
|---|---|---|---|---|---|
| a Experimental conditions: 1 μM DNA duplex, 5 mM MOPS buffer (pH 7.0), 150 mM NaClO4, 2.5 mM Mg(ClO4)2. | |||||
| IPtCN ,GNA | 34.1 | 8.0 | IPtCN ,DNA | 37.5 | 7.0 |
| IIPtCN ,GNA | 39.0 | 14.7 | IIPtCN ,DNA | 35.7 | 7.6 |
| IIIPtCN ,GNA | 33.8 | 5.6 | IIIPtCN ,DNA | 35.1 | 3.8 |
| IVPtCN ,GNA | 34.1 | 9.1 | IVPtCN ,DNA | 34.2 | 6.5 |
To evaluate the effect of two consecutive Pt(II) complexes within a DNA double helix, duplex V was synthesized. It is derived from IPtCN,DNA (the PtCN-containing duplex with the highest Tm) by formally replacing the T:A pair adjacent to the X:G pair by second X:G pair. Duplex V melts at 34.0 °C and hence at a slightly lower Tm as IPtCN,DNA (Fig. 1c). Its CD spectrum resembles that of B-DNA. Nonetheless, slightly blue-shifted maxima with respect to those in the CD spectrum of duplex IPtCN,DNA indicate a slight structural change upon the incorporation of the second Pt(II) complex (Fig. 1c).
The general applicability of our approach of a post-synthetic modification of nucleic acids with organometallic complexes was confirmed by applying the Pd(II) complex PdCN analogous to PtCN. As expected, the behaviour of the resulting duplexes is essentially identical, as exemplified by a comparison of the respective melting temperatures (Fig. S8, Table S1†).
Time-resolved photoluminescence spectroscopy
The photoluminescence spectra of the Pt(II)-containing oligonucleotides show an emission band peaking around 510 nm with a clear vibrational progression indicative of an emission from metal-perturbed ligand-centred states, in agreement with the results obtained for comparable complexes outside the DNA context (for representative examples, see Fig. S20–S24†).29–31 The photoluminescence lifetimes are summarized in Table 4. They are in the order of μs and confirm that phosphorescent oligonucleotides were obtained. As the lifetimes also depend on the microenvironmental shielding of the Pt(II) complex from physical quenching by water and 3O2,37–39 they provide valuable structural information.| DNA | Air-equilibrated | Argon-purged | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PtI,GNA | PtI,DNA | PtCN,GNA | PtCN,DNA | PtCN,Uri | PtI,GNA | PtI,DNA | PtCN,GNA | PtCN,DNA | PtCN,Uri | |
| a Experimental conditions: 1 μM DNA (single-stranded in the case of ODN1X, double-stranded for IX, IIX, IIIX, IVX), 5 mM MOPS buffer (pH 7.0), 150 mM NaClO4, 2.5 mM Mg(ClO4)2, room temperature, experimental uncertainty ± 0.1 μs. The corresponding original data, together with the fitting parameters, are given in the ESI (Fig. S25–S74). | ||||||||||
| ODN1X | 5.3 | 5.9 | 4.3 | 4.9 | 6.2 | 11.6 | 13.2 | 19.5 | 20.7 | 19.8 |
| IX | 9.9 | 9.6 | 13.3 | 19.0 | 14.0 | 13.6 | 14.6 | 24.5 | 24.8 | 22.0 |
| IIX | 7.7 | 9.3 | 13.6 | 16.6 | 11.6 | 13.8 | 14.4 | 23.9 | 23.6 | 21.4 |
| IIIX | 9.8 | 10.9 | 12.8 | 18.0 | 14.5 | 13.8 | 15.1 | 23.1 | 22.7 | 22.2 |
| IVX | 9.2 | 10.7 | 13.6 | 17.1 | 15.1 | 13.9 | 15.1 | 23.9 | 23.5 | 21.9 |
In the following, we will first discuss the behaviour in air-equilibrated solutions. Here, duplexes bearing PtCN entities in a complete DNA context have the longest amplitude-weighted average lifetimes, compared to PtCN attached via a GNA linkage and all PtI-containing duplexes. This means that in duplexes containing the oligonucleotide ODN1PtCN,DNA, the Pt(II) complex is best shielded from water and dioxygen. The average lifetimes of the PtCN complexes attached via the uracil C5 position are shorter by 20–30% (see Fig. S9† for their melting curves and CD spectra and Table S2† for their Tm). Here, the Pt(II) complex is protruding into the major groove. It is therefore expected to be less efficiently shielded from water, which is in agreement with the shorter lifetimes. The poorer shielding in the single-stranded oligonucleotides ODNX is reflected by their even further shortened lifetimes.
Upon Ar-purging, i.e., in the absence of 3O2, the lifetimes clearly reflect the different structural shielding from physical quenching. In general, they are always longer upon deoxygenation. The systems with PtI moieties show the shortest lifetimes, in agreement with a faster deactivation rate related to a lower ligand field splitting. The average lifetimes of PtCN-bearing duplexes are very similar, irrespective of the identity of the nucleobase in the complementary position and whether they are attached via a 2′-deoxyribose or a GNA linker. The duplexes with PtCN facing the major groove (i.e., attached to uracil) display a somewhat shorter average lifetime (by 3–11%). Still, the duplex lifetimes remain longer than those of the corresponding single-stranded oligonucleotides, indicating that the complex is better shielded from quenching in the duplex. Taken together, these data are in agreement with the localization of the PtCN complexes either in the base pair stack or as groove binders. Even the shorter average lifetimes of all the DNA duplexes bearing PtI moieties reflect the different solvent and 3O2 accessibility of their Pt(II) centres, in agreement with the non-specific cross-linking to other DNA strands as proposed above on the basis of the melting profiles.
The doubly platinated duplex V shows significantly different luminescent properties. Fig. 2 shows the photoluminescence spectra of this duplex and of the corresponding single-stranded ODN3 under different atmospheric conditions. In addition to the emission band with vibrational progression centring around 500 nm, a broad emission band is observed above 600 nm, indicating the presence of excimers. The average lifetimes of these excimeric species are significantly shorter than those of the monomers (Tables S3 and S4†). They are likewise shorter than those of the respective mono-platinated species IPtCN,DNA and ODN1PtCN (Table 4). In general, the lifetimes are reminiscent of what had been reported previously for a related Pt(II) complex interacting non-covalently with ctDNA via groove-binding.40
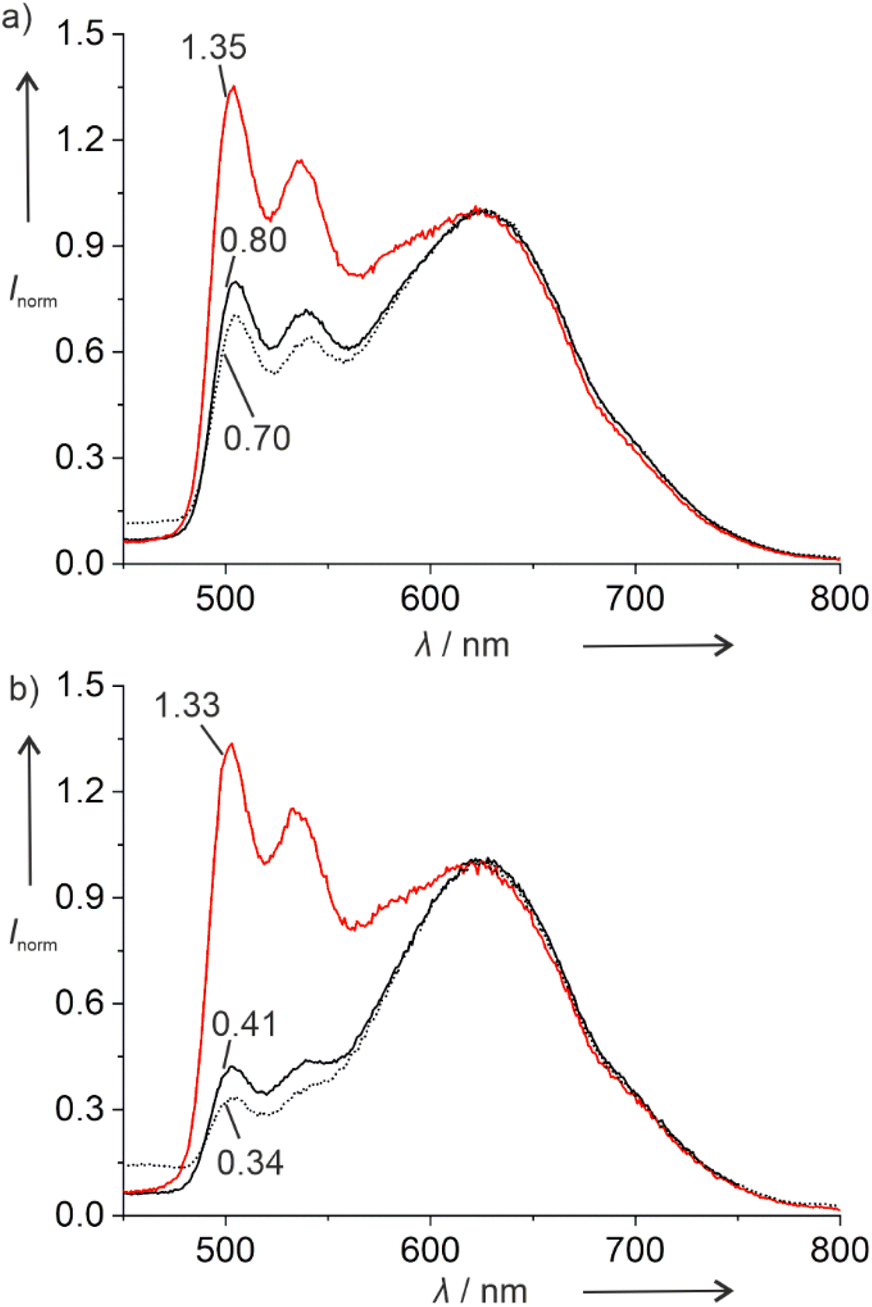 | ||
| Fig. 2 Photoluminescence spectra and relative intensities of the monomers (with respect to the red-shifted excimers) of doubly platinated (a) duplex V and (b) single-stranded ODN3. Black solid line: air-equilibrated; red solid line: Ar-purged; black dotted line: O2-saturated. Intensities normalized for clarity at (a) 625 nm and (b) 620 nm. | ||
Interestingly, duplex V and single-stranded oligonucleotide ODN3 respond differently to the presence of dissolved molecular dioxygen. The luminescence intensity of the excimers above 600 nm is much less sensitive to dissolved dioxygen than that of the monomers at 500 nm. Thus, it could be used as an internal reference to sense the concentration of dissolved 3O2, if compared with the monomeric emission (for clarity, the spectra shown in Fig. 2 have been normalized to the excimeric maximum peaking at ca. 620 nm). Interestingly, while the relative luminescence intensity of the monomer emission at 500 nm increases about 1.7-fold upon Ar-purging, this increase is much larger (3.2-fold) for the single strand, in agreement with an enhanced exposure to physical quenching. To confirm that this effect is due to the presence of dissolved 3O2, the measurements were repeated by comparing Ar-purged to O2-saturated solutions. Here, the relative monomer emission intensities increase 1.9-fold and 3.9-fold, respectively. Hence, the doubly platinated oligonucleotide ODN3PtCN (and to a lesser extent the corresponding duplex) constitutes an excellent candidate for a 3O2 sensor based on the relative phosphorescence intensities of monomer and excimer (ratiometric quantification). On the other hand, while the average lifetimes of the green monomers are drastically prolonged upon de-oxygenation, the photoluminescence decays are less sensitive if monitored at the emission maximum of the red excimers. In fact, their relative ratios mirror the qualitative trend observed for the intensities, but the multi-exponential nature (Tables S3 and S4, Fig. S75–S86†) precludes a straightforward comparison (mainly due to the manifold of co-existing conformers), thus requiring the evaluation of amplitude-weighted average lifetimes.41
Conclusions
Tethering a phosphorescent tag to a nucleic acid is of high interest. Compared to commonly applied fluorophores,42 a phosphorescent tag represents a tremendous advantage in anticipated in vitro experiments because of the suppression of background fluorescence in time-gated measurements. The Pt(II)-modified nucleic acids described here represent excellent candidates for such an application. We propose that they could serve as lifetime-based or ratiometric intensity 3O2 sensors in aqueous solutions, broadening their scope by providing a dual readout for photoluminescence (lifetime imaging) microscopy. Future work will aim at establishing such an application in biological models and at introducing the Pt(II) complexes via shorter linkers, so that their location within the DNA can be predicted more precisely.Data availability
Information supporting this article has been uploaded as part of the ESI.† The datasets generated during and/or analysed during the current study are available from the authors on reasonable request.Author contributions
FB: investigation (synthesis, characterization); IM: investigation (photophysics); TS: investigation (synthesis); CAS, JM: supervision, conceptualization, funding acquisition; FB, IM, CAS, JM: Writing – original draft; Writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Marian Hebenbrock for fruitful discussions and for performing experiments at the onset of the project. We thank Dr Maria Victoria Cappellari for spectroscopic support. JM and CAS thank the Deutsche Forschungsgemeinschaft for funding (MU 1750/5-1, STR 1186/7-1, INST 211/915-1 FUGG).Notes and references
- E. Stulz and G. H. Clever, DNA in supramolecular chemistry and nanotechnology, John Wiley & Sons, Chichester, UK, 2015 Search PubMed.
- Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601–1605 CrossRef CAS PubMed.
- P. Ensslen and H.-A. Wagenknecht, Acc. Chem. Res., 2015, 48, 2724–2733 CrossRef CAS.
- H. Asanuma, T. Fujii, T. Kato and H. Kashida, J. Photochem. Photobiol., C, 2012, 13, 124–135 CrossRef CAS.
- F. Hövelmann and O. Seitz, Acc. Chem. Res., 2016, 49, 714–723 CrossRef PubMed.
- J. Gebhard, L. Hirsch, C. Schwechheimer and H.-A. Wagenknecht, Bioconjugate Chem., 2022, 33, 1634–1642 CrossRef CAS PubMed.
- Y. Takezawa, J. Müller and M. Shionoya, Chem. Lett., 2017, 46, 622–633 CrossRef CAS.
- S. Naskar, R. Guha and J. Müller, Angew. Chem., Int. Ed., 2020, 59, 1397–1406 CrossRef CAS PubMed.
- B. Lippert, J. Biol. Inorg Chem., 2022, 27, 215–219 CrossRef CAS.
- N. Santamaría-Díaz, J. M. Méndez-Arriaga, J. M. Salas and M. A. Galindo, Angew. Chem., Int. Ed., 2016, 55, 6170–6174 CrossRef PubMed.
- H. Zhao, P. Leonard, X. Guo, H. Yang and F. Seela, Chem. – Eur. J., 2017, 23, 5529–5540 CrossRef CAS PubMed.
- I. Schönrath, V. B. Tsvetkov, T. S. Zatsepin, A. V. Aralov and J. Müller, J. Biol. Inorg Chem., 2019, 24, 693–702 CrossRef PubMed.
- D. J. Hurley and Y. Tor, J. Am. Chem. Soc., 1998, 120, 2194–2195 CrossRef CAS.
- S. I. Khan, A. E. Beilstein and M. W. Grinstaff, Inorg. Chem., 1999, 38, 418–419 CrossRef CAS PubMed.
- M. D. Newton, S. D. Fairbanks, J. A. Thomas and D. S. Rueda, Angew. Chem., Int. Ed., 2021, 60, 20952–20959 CrossRef CAS.
- H. K. Saeed, S. Sreedharan and J. A. Thomas, Chem. Commun., 2020, 56, 1464–1480 RSC.
- H. K. Saeed, P. J. Jarman, S. Archer, S. Sreedharan, I. Q. Saeed, L. K. Mckenzie, J. A. Weinstein, N. J. Buurma, C. G. W. Smythe and J. A. Thomas, Angew. Chem., Int. Ed., 2017, 56, 12628–12633 CrossRef CAS PubMed.
- B. Önfelt, P. Lincoln and B. Nordén, J. Am. Chem. Soc., 2001, 123, 3630–3637 CrossRef.
- S. Le Gac, M. Foucart, P. Gerbaux, E. Defrancq, C. Moucheron and A. Kirsch-De Mesmaeker, Dalton Trans., 2010, 39, 9672–9683 RSC.
- C. J. Murphy, M. R. Arkin, Y. Jenkins, N. D. Ghatlia, S. H. Bossmann, N. J. Turro and J. K. Barton, Science, 1993, 262, 1025–1029 CrossRef CAS PubMed.
- D. Ukale, V. S. Shinde and T. Lönnberg, Chem. – Eur. J., 2016, 22, 7917–7923 CrossRef CAS PubMed.
- D. U. Ukale and T. Lönnberg, ChemBioChem, 2018, 19, 1096–1101 CrossRef CAS PubMed.
- D. U. Ukale and T. Lönnberg, Angew. Chem., Int. Ed., 2018, 57, 16171–16175 CrossRef CAS PubMed.
- S. K. Maity and T. Lönnberg, Chem. – Eur. J., 2018, 24, 1274–1277 CrossRef CAS PubMed.
- S. K. Maity and T. Lönnberg, ACS Omega, 2019, 4, 18803–18808 CrossRef CAS PubMed.
- D. U. Ukale, P. Tähtinen and T. Lönnberg, Chem. – Eur. J., 2020, 26, 2164–2168 CrossRef CAS PubMed.
- M. Hande, S. Maity and T. Lönnberg, J. Inorg. Biochem., 2021, 222, 111506 CrossRef CAS PubMed.
- K. Kowalski, Coord. Chem. Rev., 2021, 432, 213705 CrossRef CAS.
- M. Hebenbrock, L. Stegemann, J. Kösters, N. L. Doltsinis, J. Müller and C. A. Strassert, Dalton Trans., 2017, 46, 3160–3169 RSC.
- M. Hebenbrock, D. González-Abradelo, A. Hepp, J. Meadowcroft, N. Lefringhausen, C. A. Strassert and J. Müller, Inorg. Chim. Acta, 2021, 516, 119988 CrossRef CAS.
- I. Maisuls, F. Boisten, M. Hebenbrock, J. Alfke, L. Schürmann, B. Jasper-Peter, A. Hepp, M. Esselen, J. Müller and C. A. Strassert, Inorg. Chem., 2022, 61, 9195–9204 CrossRef CAS PubMed.
- B. Lippert, Cisplatin - Chemistry and Biochemistry of a Leading Anticancer Drug, Wiley-VCH and Verlag Helvetica Chimica Acta, Zürich, 1999 Search PubMed.
- R. Manchanda, S. U. Dunham and S. J. Lippard, J. Am. Chem. Soc., 1996, 118, 5144–5145 CrossRef CAS.
- J. Schliepe, U. Berghoff, B. Lippert and D. Cech, Angew. Chem., Int. Ed. Engl., 1996, 35, 646–648 CrossRef CAS.
- K. S. Schmidt, D. V. Filippov, N. J. Meeuwenoord, G. A. van der Marel, J. H. van Boom, B. Lippert and J. Reedijk, Angew. Chem., Int. Ed., 2000, 39, 375–377 CrossRef CAS.
- J. Hennessy, B. McGorman, Z. Molphy, N. P. Farrell, D. Singleton, T. Brown and A. Kellett, Angew. Chem., Int. Ed., 2022, 61, e202110455 CrossRef CAS PubMed.
- I. Maisuls, J. Singh, I. P. Salto, S. T. Steiner, T. M. Kirse, S. Niemann, C. A. Strassert and A. Faust, Inorg. Chem., 2021, 60, 11058–11069 CrossRef CAS PubMed.
- S. Chatnahalli Gangadharappa, I. Maisuls, I. P. Salto, S. Niemann, V. Bachtin, F. C. Herrmann and C. A. Strassert, J. Phys. Chem. C, 2021, 125, 5739–5747 CrossRef CAS.
- I. Maisuls, C. Wang, M. E. Gutierrez Suburu, S. Wilde, C.-G. Daniliuc, D. Brünink, N. L. Doltsinis, S. Ostendorp, G. Wilde, J. Kösters, U. Resch-Genger and C. A. Strassert, Chem. Sci., 2021, 12, 3270–3281 RSC.
- M. Hebenbrock, D. González-Abradelo, C. A. Strassert and J. Müller, Z. Anorg. Allg. Chem., 2018, 644, 671–682 CrossRef CAS.
- A. Sillen and Y. Engelborghs, Photochem. Photobiol., 1998, 67, 475–486 CAS.
- S. Benson, F. de Moliner, W. Tipping and M. Vendrell, Angew. Chem., Int. Ed., 2022, 61, e202204788 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional DNA melting curves and CD spectra; lists of melting temperatures; MALDI-ToF spectra; photoluminescence spectra; time-resolved photoluminescence decay curves with fitting parameters; photoluminescence lifetimes; experimental details; NMR spectra. See DOI: https://doi.org/10.1039/d2sc05916a |
| This journal is © The Royal Society of Chemistry 2023 |