
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning P-type bi-stable overcrowded alkene-based chiroptical photoswitches†
Jinyu
Sheng
a,
Wojciech
Danowski
 *ab,
Stefano
Crespi
ac,
Ainoa
Guinart
a,
Xiaobing
Chen
a,
Cosima
Stähler
a and
Ben L.
Feringa
*a
*ab,
Stefano
Crespi
ac,
Ainoa
Guinart
a,
Xiaobing
Chen
a,
Cosima
Stähler
a and
Ben L.
Feringa
*a
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, Groningen 9747 AG, Netherlands. E-mail: B.L.Feringa@rug.nl
bUniversity of Strasbourg CNRS, ISIS UMR 7006, 8 Allée Gaspard Monge, Strasbourg F-67000, France. E-mail: danowski@unistra.fr
cDepartment of Chemistry-Ångström Laboratory, Uppsala University, Box 523, Uppsala, 751 20, Sweden
First published on 13th March 2023
Abstract
Overcrowded alkene based molecular motors and switches constitute a unique class of photo-responsive systems due to their intrinsic chirality near the core C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, making them highly suitable candidates for the construction of light-switchable dynamic systems, i.e., for controlling molecular motion, modulation of material chiroptical properties and supramolecular assembly. However, the lack of general design principles, along with the challenging synthesis of these molecules, precludes full exploitation of their dynamic structures. Therefore, systematic investigations of the key parameters are crucial for the further development of these systems. Here we provide a facile alternative synthetic route, elucidate the influence of substituents on the photochemistry of overcrowded alkene-derived bistable chiroptical photoswitches, and show nearly quantitative bidirectional photoswitching. The established structure–property relationship constitutes a practical guideline for the design of these photochromes tailored to a specific application.
C bond, making them highly suitable candidates for the construction of light-switchable dynamic systems, i.e., for controlling molecular motion, modulation of material chiroptical properties and supramolecular assembly. However, the lack of general design principles, along with the challenging synthesis of these molecules, precludes full exploitation of their dynamic structures. Therefore, systematic investigations of the key parameters are crucial for the further development of these systems. Here we provide a facile alternative synthetic route, elucidate the influence of substituents on the photochemistry of overcrowded alkene-derived bistable chiroptical photoswitches, and show nearly quantitative bidirectional photoswitching. The established structure–property relationship constitutes a practical guideline for the design of these photochromes tailored to a specific application.
Introduction
The development of molecular photoswitches has opened the way for waste-free and reversible control of molecular geometry with high spatiotemporal precision.1–11 In recent years, the family of accessible synthetic photoswitches1,2 has undergone a major expansion from classic motifs such as azobenzenes,12–15 spiropyrans,16,17 stiff-stilbenes18,19 and diarylethenes20,21 to new structures including hydrazone-22,23 and acylhydrazone-24 based switches,25 donor–acceptor Stenhouse adducts (DASAs),4,26 hemithioindigos,27 iminothioindoxyls,28 phenylimino indolinones,29 and indirubins.30 Among these, overcrowded alkene-derived photoswitches and molecular motors combine reliable and simple design with unidirectional motion derived from a sequence of alternating photochemical E/Z and thermal isomerizations powered by light and heat.31–35 Precise structural modifications which involve the engineering of the steric hindrance in the fjord region allow their rotation rate, properties and functions to be fine-tuned.35 In particular, structures derived from fluorene-tetrahydrophenanthrene scaffolds (Fig. 1) show increased thermal stability of the metastable isomer to the point of practical suppression of the thermal isomerization step which in essence makes these compounds multi-wavelength-responsive P-type photoswitches with large geometric changes between two states and distinct helical chirality.36,37 Hence, in contrast to other classical photoswitchable scaffolds such as azobenzenes or dithienylethenes these compounds allow precise control over distinct molecular helicities, and are in practice thermally stable, offering unique prospects for the control of more complex supramolecular systems and responsive materials.38 In combination with the typically large spectral band separation between stable and metastable isomers (usually >60 nm between the two diastereomers) and high photostationary state distributions, these features make the overcrowded alkene-derived bi-stable switches particularly suitable molecular building blocks for construction of light-responsive adaptive systems39 including reconfigurable catalysts40–43 and porous materials.44 | ||
| Fig. 1 (a) Main features of the overcrowded alkene photoswitches and their derivatives studied in this work. The experimental UV/Vis spectra of the folded isomer (stable) and twisted isomer (metastable). The latter was extrapolated from the PSD mixture after 365 nm light irradiation in dichloromethane. (b) Resonance structures of a 2,7-disubstituted switch derivative. Only dimethoxy-switch 7 is depicted here, demonstrating that the substituents are directly conjugated to the alkene core. | ||
Systematic studies on light-driven molecular motors functionalized with a range of electron-donating (EDG) and electron-withdrawing (EWG) substituents on the “upper part” of the motor indicated that the quantum yield (QY) of the photoisomerization is commensurate with the increasing electron withdrawing character of the substituent without any noticeable influence on the rate of thermal isomerization.45 Transient absorption and time-resolved fluorescence spectroscopies correlated these findings with the energy level and lifetime of the excited dark-state.45 More recently, femtosecond stimulated Raman spectroscopy studies linked the higher QY of photoisomerization to an increasingly more pronounced sp3 character of the central double bond in the excited dark state.46 On the other hand, studies concerning functionalization of the stator of molecular motors are scarce. The few available reports suggest negligible impact of the substituents' electronic character on the rate of thermal isomerization and inconclusive influence on the QYs of photoisomerization.47–49
In contrast, almost no data is available for the bistable counterparts of the overcrowded alkene-based molecular switches. While it can be expected that functionalization of the “top part” of these switches should have a similar influence on the efficiency of the photochemical step, as for the related motors, the lack of systematic studies focused on the derivatization of the “bottom part” precludes any extrapolation. Consequently, the structure–property relationship of these compounds remains virtually unknown, which poses a significant challenge for the design of overcrowded olefin-based bistable chiroptical switches for selected functions and as such constitutes a major limitation to their applications and future development.50
Here we present a systematic study on the functionalization of the fluorene-derived “bottom part” of overcrowded alkene-based bistable chiroptical photoswitches (Fig. 1a). A library of ten compounds bearing a variety of EDG and EWG substituents was synthesized and the impact of the substituents on the photochemistry of the switches was evaluated with electronic absorption UV/Vis and 1H NMR spectroscopies. We show that the increasingly electron-donating or -withdrawing nature of the substituents appended to the “bottom part” decreases the QY of the photochemical formation of the metastable and stable isomers of the photoswitch. However, the favourable interplay of the QYs and molar absorptivities of the respective diastereomers enables high photochemical selectivity and essentially quantitative bidirectional photoswitching in derivatives appended with EWG groups. These overall trends in the photochemical properties allow us to identify several guidelines for effective connectivity patterns that enable integration of these switches into more complex structures, which serve as a manual for the design of photoresponsive molecular systems and materials based on overcrowded alkene photoswitches.
Results and discussion
All the studied photoswitches were synthesized via a Barton–Kellogg olefination using a diazo compound derived from the tetralone “top-part” and the thioketone derived from the “bottom-part”, i.e., 2,7-dibromo- or 2,7-dimethoxy-fluorenone (Scheme 1a). The classical stepwise Haworth's tetralone synthesis of the “top-part” (Scheme 1b) could be readily simplified following a recently51 reported oxidative ring opening/cyclization method involving conversion of substituted cyclobutanol S1 to S2. We also found that addition of hexamethylphosphoramide (HMPA) during the methylation of the enolate suppresses the formation of the di-methylated side product, whose presence necessitates tedious purification procedures to access the desired product S3 (Scheme 1c). This new approach only takes three simple steps with very mild and fast reactions, which opens up new opportunities for the development of practical and scalable routes to previously unexplored photoswitches (Scheme 1d) which are suitable for further functionalisation or conjugation with extended π-systems,52,53 which leads to bathochromically shifted absorption spectra. In this study, the various functional groups were appended to the “bottom-part” using metal-catalysed reactions, generating a library of functionalised overcrowded alkenes with electron withdrawing and donating substituents (see the ESI† for synthetic details).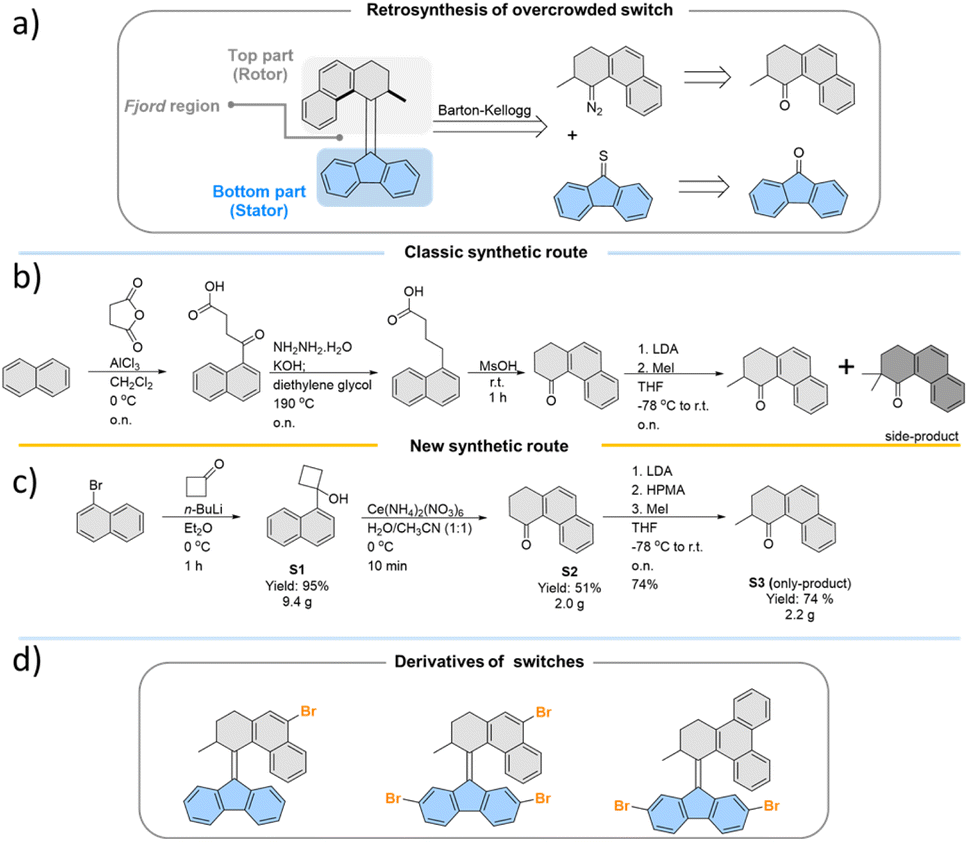 | ||
| Scheme 1 (a) Retrosynthesis route towards overcrowded alkene photoswitches; optimization of top-part synthesis from (b) the classic route to (c) the new route; (d) target Br-substituted overcrowded alkene molecules synthesized by applying the new synthetic route suitable for ready functionalization. | ||
In total, ten derivatives (1–10, see Fig. 2 and the ESI† for structures) of bistable chiroptical overcrowded alkene-based switches functionalized at the 2- and 7-positions of the fluorene-derived “bottom part” with substituents spanning a wide range of Hammett σ parameter values were synthesized. Their photoswitching behaviour was studied with electronic absorption UV/Vis and 1H NMR spectroscopies. The overcrowded alkenes 1–10 (the stable isomers are referred to as e.g.1st, while the metastable isomers are referred to as e.g.1mst) can be roughly divided into two groups based on the similarities in the electronic absorption UV/Vis spectra of the stable isomers in dichloromethane (1st–9st) or in THF (10st). It should be noted that pyridine54 and its derivatives are known to react with dichloromethane in an SN2 fashion and furthermore from our observations this reaction seems to be significantly accelerated by irradiation, therefore for all the studies with 10, a THF solution was used.55 In 1st–3st and 7st–9st, the lowest-energy absorption band (Fig. 2b, d, e and i–k) tentatively ascribed to the π → π* electronic transition is well separated from the higher energy bands. In contrast, in derivatives 4st–6st and 10st which bear additional aromatic rings (Fig. 2f–h and l), this band overlaps to various degrees with the other transitions.
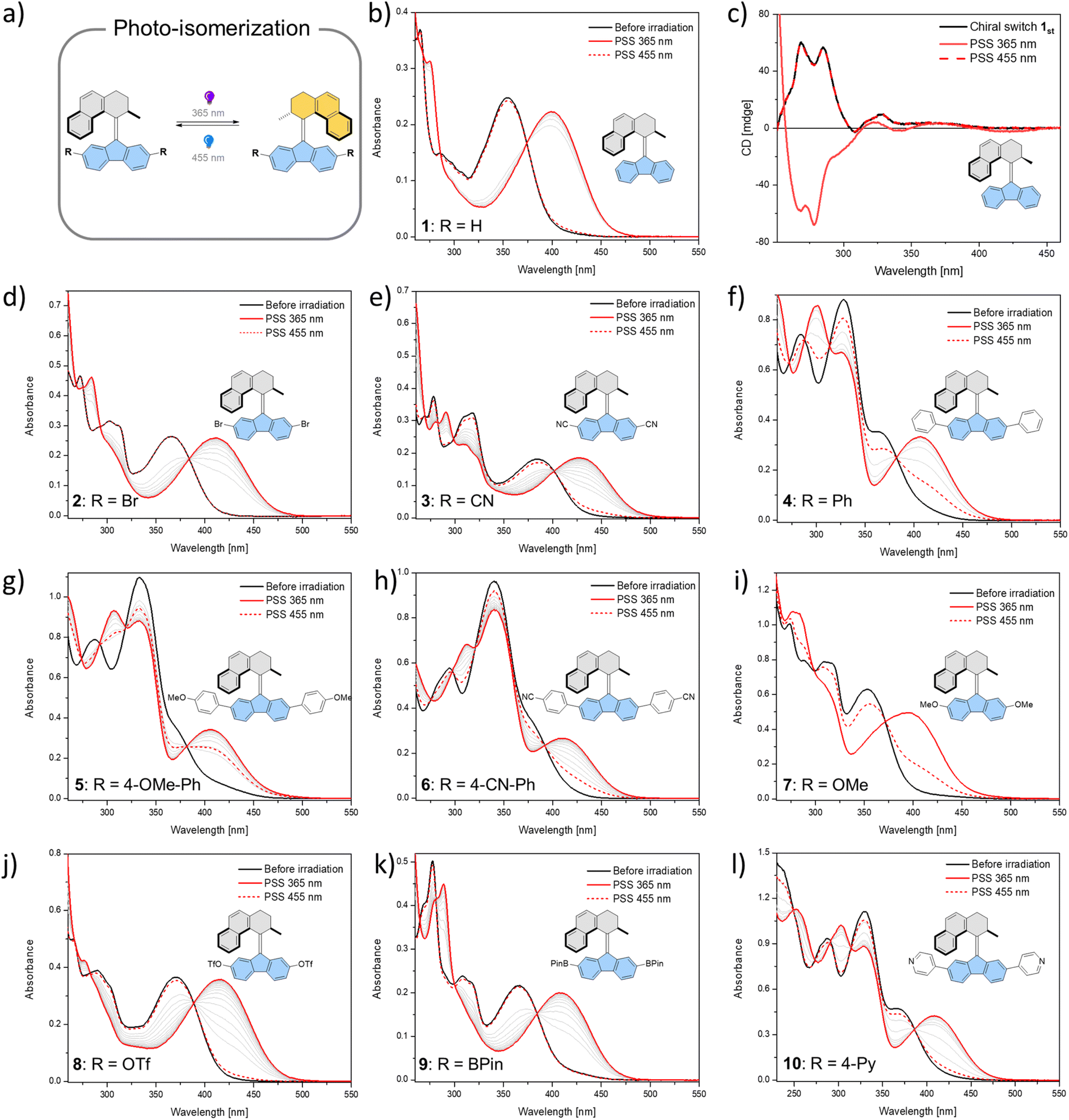 | ||
| Fig. 2 Photochemical isomerization of overcrowded alkenes 1–10. (a) Isomerization from the stable isomer to a metastable isomer by 365 nm light irradiation, followed by isomerization back to the stable isomer by 455 nm light irradiation. (b–l) UV/Vis or CD spectra of compounds 1–10 (298 K, CH2Cl2, 17 μM for 1 (UV spectra), 42 μM for 1 (CD spectra), 17 μM for 2, 13 μM for 3, 23 μM for 4, 24 μM for 5, 16 μM for 6, 47 μM for 7, 24 μM for 8, 63 μM for 9 and 37 μM for 10) from the stable isomer (black solid line) upon 365 nm irradiation to the photostationary state (PSS365, red solid line) and then upon 455 nm irradiation to the photostationary state (PSS455, red dashed line). | ||
Irradiation of 1st–10st in the appropriate solvent at 365 nm led to bathochromic shifts of the electronic absorption spectra, consistent with the formation of the metastable isomers of the respective overcrowded alkenes. The isomerization of enantiopure 1st (see the ESI† General Information for details on the preparative method) was followed by electronic circular dichroism (ECD) spectroscopy. ECD spectra revealed inversion of the Cotton effect in the 250–300 nm region, indicative of the inversion of the overall helicity of the molecule.37,56 Accordingly, back-isomerization to the stable isomer induced upon irradiation at 455 nm led to near complete recovery of the initial spectrum, demonstrating chiroptical switching (see Fig. S8†). Furthermore, for all switches, clear isosbestic points (Fig. 2b and d–l) were maintained, indicating clean and unimolecular photochemical processes. Upon subsequent irradiation of these photostationary state mixtures at 420 nm, we observed hypsochromic shifts of the electronic absorption UV/Vis spectra, accompanied, in many cases, by partial recovery of the original spectra. This phenomenon is consistent with reverse photoisomerization. Comparison of the UV/Vis spectra indicated that, overall, the introduction of EWGs led to a significant bathochromic shift of the spectrum while almost no shift was observed for the methoxy substituted derivative 7 (Fig. 2i). Irrespective of the EDG/EWG nature, substituents with additional phenyl rings induced the largest bathochromic shifts, which can be attributed to the extension of the π-system.
Similarly, 1H NMR spectroscopy showed formation of the respective metastable isomers upon irradiation of 1st–10st in CD2Cl2 at 365 nm or in THF-d8 at 365 nm as evident from the shifts of the resonances characteristic of the aliphatic protons. The reverse isomerization could be achieved upon irradiation at 455 nm, as verified by the partial recovery of the original 1H NMR spectra. Among the synthesized switches, nearly all reach a remarkably high degree of photoisomerization in the photostationary state mixtures generated upon irradiation at 365 nm (PSS365), with the exception of 7st for which a decrease in selectivity was found (71% 7mst at PSS365). Interestingly, we discovered that compound 1 (R = H) and compound 3 (R = CN) both showed nearly quantitative reversible photoisomerization behaviour, i.e., PSD365 = 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11 (mst:st) and PSD455 = 5:95 (mst:st) for 1, and PSD365 = 89:11 (mst:st) and PSD455 = 12:88 (mst:st) for 3, respectively (Fig. 3a and b), while compound 7 (Fig. 3c) showed the lowest photoselectivity in the studied series with PSD365 = 71:29 (mst:st) and PSD455 = 30:70 (mst:st). The poor photochemical isomerization behaviour of 7st is reflected in its lowest determined QY of isomerization (0.4%) in the series. Overall, the QY365st→mst of the forward isomerization upon irradiation at 365 nm (from the stable to the metastable isomer) tends to decrease both with an increasing electron withdrawing and electron donating nature of the substituent. The highest value of QY365st of 10% was determined for the unsubstituted derivative 1st. Similar trends in the QY445mst→st were observed for the reverse (metastable to stable) isomerization, induced upon irradiation at 445 nm, with the highest QY445mst in the series 1mst–10mst of ca. 2% determined for 1mst → 1st photoisomerization. Intriguingly, for all metastable isomers the QYs of stable to metastable isomerizations (unwanted reaction, QY445st→mst) upon irradiation at 445 nm were found to be markedly higher than the corresponding QY445mst→st values. This observation indicates that the presence of excess stable isomer in the photostationary state mixture (PSS445) stems from the positive combination of the irradiation wavelength and favourable ratio of the molar absorption coefficients rather than the ratio of the QYs of the forward and reverse photoisomerization reactions (QY445mst→st/QY445st→mst). Similar phenomena were reported for the forward isomerization of some overcrowded alkene-45 and, more recently, for hemithioindigo-derived57 molecular motors, which may suggest common features in their photochemical isomerization mechanism. In contrast, for all of the studied switches, ratios of the QYs at 365 nm were found to favour the forward isomerization process (QY365mst→st/QY365st→mst). Interestingly, none of the studied compounds in the series (1–10) was found to follow Kasha's rule, and the QYs determined at both wavelengths varied significantly. While the origin of this discrepancy is currently unclear similar deviations were observed for dithienylethene and azobenzene.20,58
:11 (mst:st) and PSD455 = 5:95 (mst:st) for 1, and PSD365 = 89:11 (mst:st) and PSD455 = 12:88 (mst:st) for 3, respectively (Fig. 3a and b), while compound 7 (Fig. 3c) showed the lowest photoselectivity in the studied series with PSD365 = 71:29 (mst:st) and PSD455 = 30:70 (mst:st). The poor photochemical isomerization behaviour of 7st is reflected in its lowest determined QY of isomerization (0.4%) in the series. Overall, the QY365st→mst of the forward isomerization upon irradiation at 365 nm (from the stable to the metastable isomer) tends to decrease both with an increasing electron withdrawing and electron donating nature of the substituent. The highest value of QY365st of 10% was determined for the unsubstituted derivative 1st. Similar trends in the QY445mst→st were observed for the reverse (metastable to stable) isomerization, induced upon irradiation at 445 nm, with the highest QY445mst in the series 1mst–10mst of ca. 2% determined for 1mst → 1st photoisomerization. Intriguingly, for all metastable isomers the QYs of stable to metastable isomerizations (unwanted reaction, QY445st→mst) upon irradiation at 445 nm were found to be markedly higher than the corresponding QY445mst→st values. This observation indicates that the presence of excess stable isomer in the photostationary state mixture (PSS445) stems from the positive combination of the irradiation wavelength and favourable ratio of the molar absorption coefficients rather than the ratio of the QYs of the forward and reverse photoisomerization reactions (QY445mst→st/QY445st→mst). Similar phenomena were reported for the forward isomerization of some overcrowded alkene-45 and, more recently, for hemithioindigo-derived57 molecular motors, which may suggest common features in their photochemical isomerization mechanism. In contrast, for all of the studied switches, ratios of the QYs at 365 nm were found to favour the forward isomerization process (QY365mst→st/QY365st→mst). Interestingly, none of the studied compounds in the series (1–10) was found to follow Kasha's rule, and the QYs determined at both wavelengths varied significantly. While the origin of this discrepancy is currently unclear similar deviations were observed for dithienylethene and azobenzene.20,58
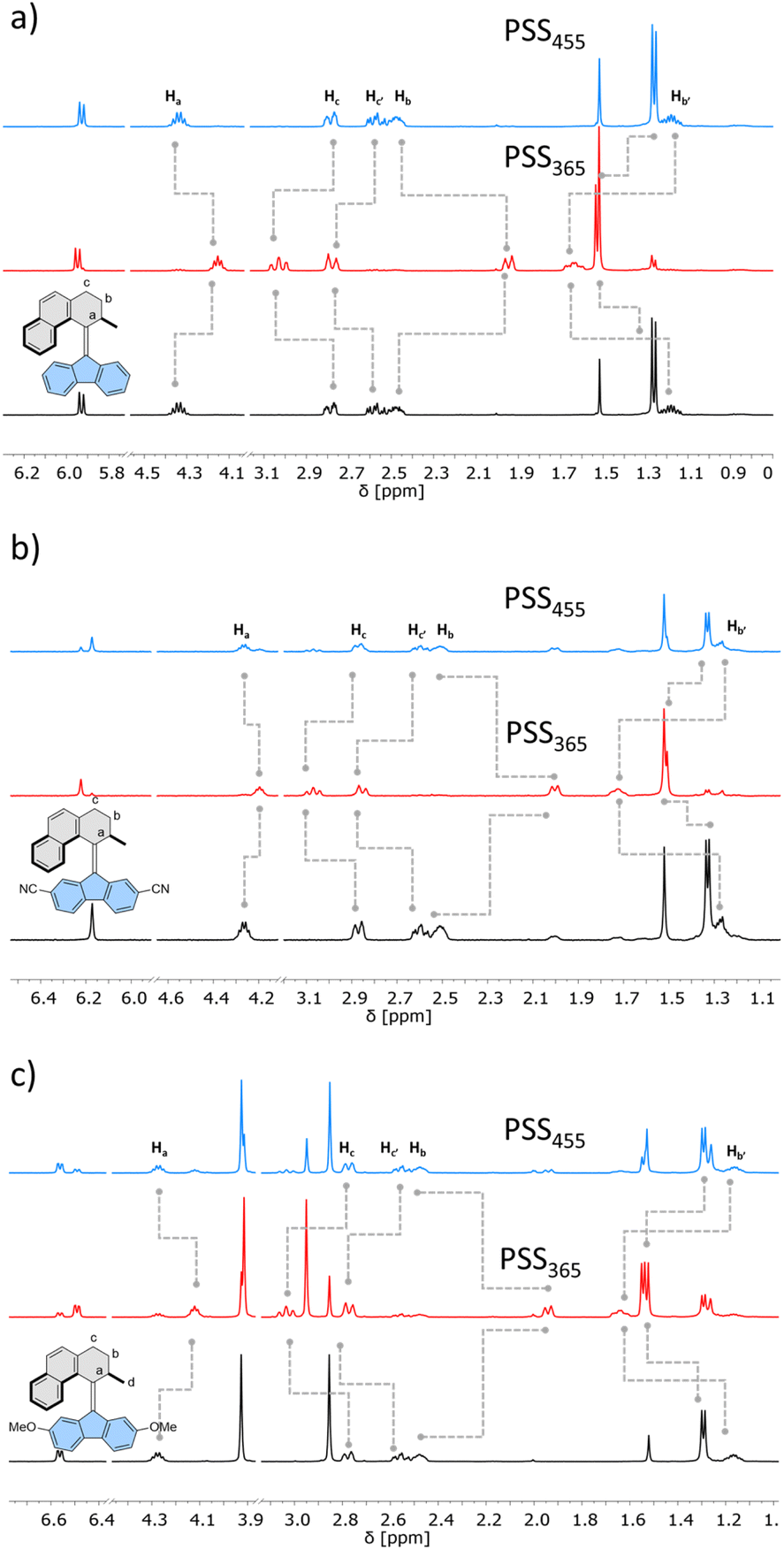 | ||
| Fig. 3 Partial 1H-NMR spectral changes of (a) 1, (b) 3 and (c) 7 (400 or 500 MHz, 298 K, CD2Cl2, 5 mM) from the stable isomer (black) upon 365 nm irradiation to the photostationary state PSS365 (red) and then upon 455 nm irradiation to the photostationary state PSS455 (blue). | ||
Computational studies, performed at the ωB97X-D3BJ/def2-QZVP//r2SCAN-3c level of theory revealed the high thermal stability of the metastable isomers, confirming the P-type nature of the photoswitches (Fig. 4). For all three studied compounds 1, 3, and 7, the barriers for the thermal E/Z isomerization (TEZI) were found to be markedly lower than the barriers for the thermal helix inversion (THI), confirming the high steric barrier in the fjord region, which is consistent with our earlier findings.37 Irrespective of the substitution, similar computed barriers were found, indicating negligible influence of the electronic nature of the substituent on the stabilisation of the transition state and rate of the thermal process. The barrier for the thermal isomerization of 1mst (1mst → 1st) was determined with UV/Vis spectroscopy by following the decay of 1mst in a range of elevated temperatures (100–110 °C, Fig. S9a†). Extrapolation of the experimental data to room temperature gave a Gibbs free energy of activation for the thermal reaction Δ‡G(20 °C) of 29.2 kcal mol−1, matching nicely the theoretical predictions (25.3 kcal mol−1) and corresponding to a half-life (t1/2) of 1.82 × 105 h at room temperature, thereby demonstrating the bistable character of these switches (Fig. S9b†).
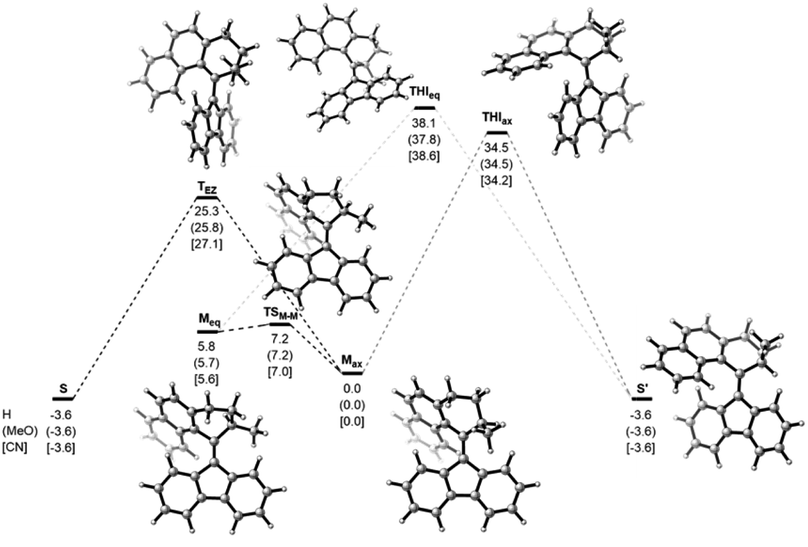 | ||
| Fig. 4 Gibbs free energies (in kcal mol−1) for the thermal pathways of H-, CN- and MeO-substituted switches 1, 3 and 7 at the ωB97X-D3BJ/def2-QZVP//r2SCAN-3c level of theory. Relative energies are given in kcal mol−1. The structures of the H-substituted compound serve as a reference for the reader. | ||
Combined with the behaviour of compounds 4–6 and 10 revealed by both UV/Vis (Fig. 2f–h and l) and 1H NMR (Fig. S2–S4 and S7†) measurements, it can be concluded that the photoisomerization behaviour of the overcrowded alkene switches is highly affected by the electronic effects of the substituents at the 2- and 7-positions. Overall, all the chiroptical switches 1st–10st display similar photochemical behaviours. All compounds can be reversibly isomerized to the corresponding metastable diastereomer upon irradiation at appropriate wavelengths. However, all compounds differ significantly in terms of the QYs of isomerization, photostationary state distributions reached upon irradiation at the specific wavelengths and absorption maxima of the respective diastereomers (Table 1). The most striking differences were found for the metastable to stable isomerization. While switches appended with EWG substituents showed high photoconversions to the corresponding stable diastereomers (2mst, 3mst and 8mst–10mst), for all the switches bearing EDG groups markedly lower photoconversions at the photostationary state were observed (4mst, 5mst and 7mst). Overall, the influence of substituents appended to the “bottom part” of these switches contrasts with the substituent effect arising from the “top-part” functionalization of the motors, for which EWGs lead to an increase and EDGs lead to a decrease of QYs, respectively.45 Nevertheless, to a certain degree these trends are consistent with the limited data on the functionalization of overcrowded alkene-based molecular motors functionalized in the 3,6-position of the fluorene stator, for which both methoxy- and cyano-substituents were shown to decrease the QY of isomerization compared to the unsubstituted structures.47
| Alkene | R | σ p | λ st | ε λ st ·104d | QY365st→msth | ε 365st·104e | PSD365f | QY445mst→sth | ε 445mst·104e | PSD445f |
|---|---|---|---|---|---|---|---|---|---|---|
| (λmst)/nm−1 | (ελmst·104)/M−1 cm−1 | (QY365mst→st)% | (ε365mst·104)/M−1 cm−1 | (PSD365clc)g | (QY445st→mst)% | (ε445sst·104)/M−1 cm−1 | (PSD445clc)g | |||
| a Hammett constants were taken from ref. 59. b The Hammett constant of boronic acid was used. c Electronic absorption band maxima of a specific diastereomer corresponding to the S0–S1 transition. d Molar attenuation coefficient of a specified diastereomer at absorption maxima. e Molar attenuation coefficient of a specified diastereomer at a specified wavelength. f Photostationary state distribution determined from 1H NMR spectra upon irradiation at 365 nm and 455 nm represented as the molar fraction of the metastable or stable diastereomer, respectively. g Photostationary state distribution calculated from the determined QYs and molar attenuation coefficients represented as the molar fraction of the metastable or stable diastereomer for irradiation at 365 nm and 445 nm, respectively. h Experimental error 10%. | ||||||||||
| 1 | –H | 0.00 | 355 | 1.44 | 10 | 1.29 | 0.82 | 2 | 0.36 | 0.86 |
| (399) | (1.41) | (4) | (0.70) | (0.90) | (40) | (0.002) | (0.93) | |||
| 2 | –Br | 0.23 | 365 | 1.54 | 6 | 1.54 | 0.87 | 0.8 | 0.79 (0.02) | 0.89 |
| (410) | (1.55) | (2) | (0.55) | (0.91) | (5) | (0.90) | ||||
| 3 | –CN | 0.66 | 384 | 1.39 | 2 | 1.11 | 0.83 | 0.6 | 1.33 | 0.86 |
| (426) | (1.54) | (0.6) | (0.48) | (0.90) | (3) | (0.05) | (0.87) | |||
| 4 | –Ph | −0.01 | 364 | 1.50 | 3 | 1.49 | 0.91 | 0.4 | 0.55 | 0.71 |
| (407) | (1.42) | (0.8) | (0.60) | (0.95) | (2) | (0.05) | (0.72) | |||
| 5 | –C6H4–p–OMe | −0.08 | 373 | 1.44 | 3 | 1.65 | 0.94 | 0.3 | 0.66 | 0.47 |
| (405) | (1.51) | (0.4) | (0.80) | (0.95) | (2) | (0.10) | (0.47) | |||
| 6 | –C6H4–p–CN | — | 379 | 2.45 | 1 | 3.31 | 0.84 | 0.2 | 1.04 | 0.75 |
| (410) | (2.06) | (0.3) | (2.51) | (0.89) | (2) | (0.03) | (0.80) | |||
| 7 | –OMe | −0.27 | 353 | 1.37 | 0.4 | 1.17 | 0.69 | 0.1 | 0.40 | 0.71 |
| (1.48) | ||||||||||
| (399) | (0.3) | (0.69) | (0.72) | (0.5) | (0.03) | (0.82) | ||||
| 8 | –OTf | 0.53 | 370 | 1.48 | 2 | 1.44 | 0.85 | 0.5 | 0.99 | 0.93 |
| (414) | (1.62) | (0.8) | (0.51) | (0.86) | (2) | (0.01) | (0.94) | |||
| 9 | –Bpin | 0.12b | 366 | 0.35 | 7 | 3.54 | 0.90 | n. d. | 0.15 | n. a. |
| (407) | (0.35) | (2) | (1.45) | (0.92) | (n. d.) | (0.01) | (0.92) | |||
| 10 | -4-C5H4N | 0.44 | 371 | 1.26 | 3 | 1.27 | 0.92 | 0.4 | 0.51 | 0.87 |
| (410) | (1.17) | (0.6) | (0.53) | (0.98) | (0.9) | (0.04) | (0.85) | |||
In view of our findings, several key design aspects of these photoswitches (Fig. 5a) leading to high photostationary state distributions between diastereomers can be outlined (Fig. 5b–d). Our results indicate that the electronic factor of the substituents at the 2- and 7-positions of the fluorene-derived “bottom part” of the photoswitch has a dramatic influence on the selectivity of the photochemical isomerization of these compounds. Therefore, construction of responsive materials and functional systems by appending these photoswitches at the 2- and 7-positions should take the electronic factor of the connecting moiety into consideration. Overall, electron-donating groups should be avoided, as these substituents dramatically decrease the productive QYs and selectivity of both photoisomerizations. Hence, despite the synthetic convenience alkyl (σp-0.17) and alkoxy (σp-0.25) motifs should be discarded. While thioethers (σp-0.0)59 may be considered an attractive connecting motif, it should be noted that the presence of heavy atoms near the alkene core may lead to a significant involvement of the triplet state in the isomerization mechanism. On the other hand, weakly electron-withdrawing groups, e.g. boronic esters, which are highly suitable linkages for construction of covalent-organic frameworks, ensure high photostationary state distributions. Based on the favourable properties of EWGs, carbonyl-derived moieties such as amides, esters or imines, important from the perspective of construction of functional materials, should be connected to the overcrowded alkene core via carbonyl carbons to secure the favourable distribution of isomers at the photostationary state.
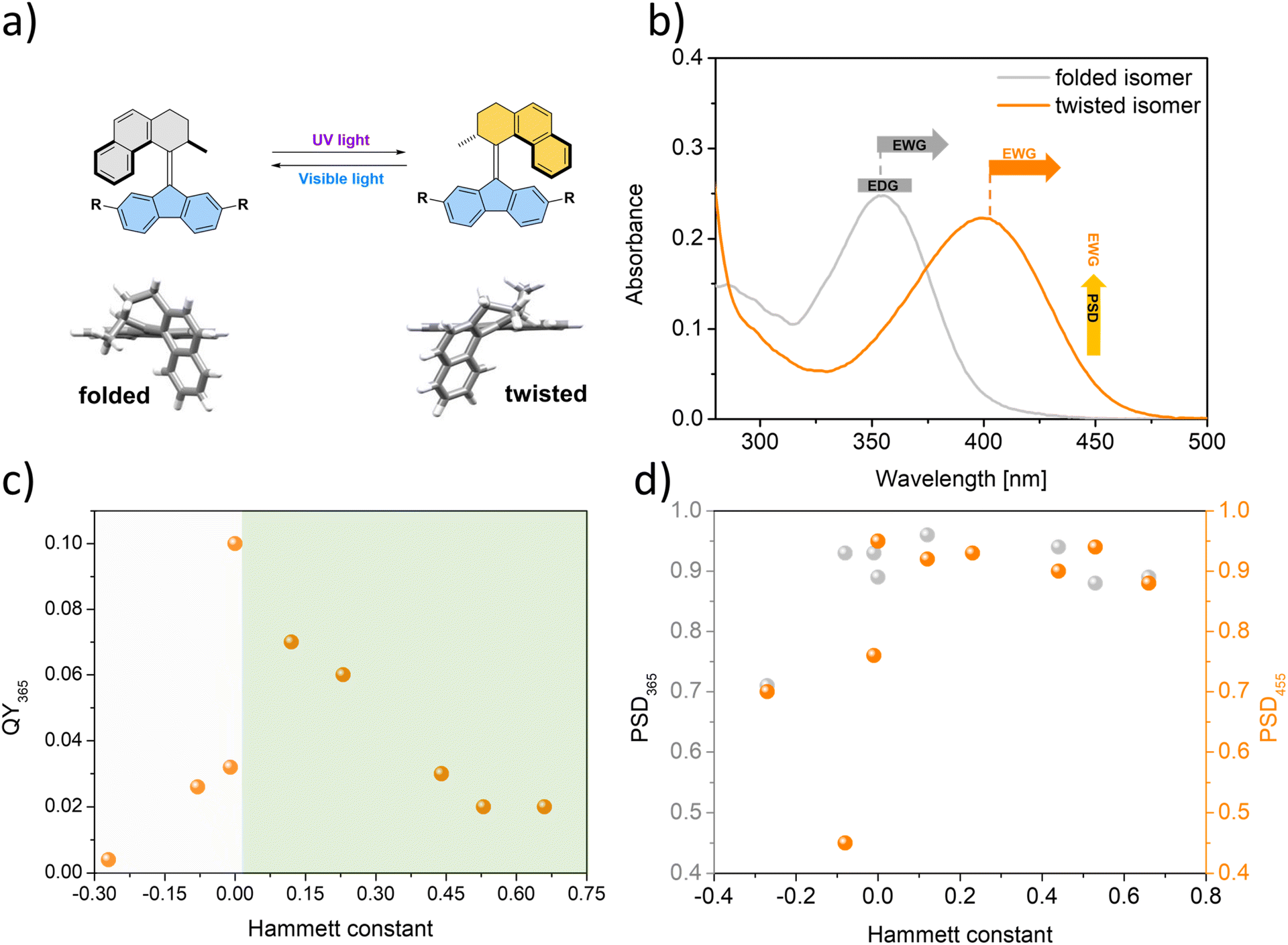 | ||
| Fig. 5 Design guidelines for overcrowded alkene based chiroptical photoswitches. (a) Isomerization of the switch between the stable folded isomer and metastable twisted isomer by UV light or visible light irradiation. (b) Influence of the absorption and PSD properties of substituents at the 2- and 7-positions of the switch. (c) Correlations between the Hammett constants and QYs of photoisomerization (st → mst) of the switches with diverse substituents. (d) Correlations between the Hammett constants and PSD365/455 of the switches with diverse substituents. | ||
Conclusions
In summary, we have carried out a systematic study of the photochemical properties of a range of overcrowded alkene based bi-stable chiroptical photoswitches with various substituents at the bottom part of the switch. It was established that the electronic effect of the substituents has a dramatic impact on the kinetics and selectivity of the forward and reverse photochemical isomerization. The overall trends in the photo-reactivity of these molecules revealed that the presence of electron-donating substituents in the bottom part of the switch diminishes both the QYs and selectivity of the isomerization, while electron-withdrawing substituents ensure high photoconversions in the forward and backward isomerization reactions. Combined with computational calculations, our investigation provides a deeper understanding of the substituent effects on the photochemical behaviour of this responsive motif. In addition, our new and simplified route towards the “upper-part” of the photoswitch provides an easy access to variously substituted tetralones, bypassing previous tedious multi-step procedures. The extended set of experimental data presented here constitutes a practical guideline for easy access to efficiently functionalizing switches. Unveiling the general design principles of these molecules directed towards ensuring the optimal photochemical performance in a variety of light-addressable systems thereby opens several opportunities for construction of light-responsive materials based on these chiroptical overcrowded alkenes. Owing to their thermally stable yet intrinsic and dynamic helicity these switches may offer new prospects in materials sciences, providing predictable control over helical structures with light. These compounds are particularly attractive candidates for construction of liquid crystal actuators,60 reconfigurable catalysts61 and spintronic devices.62 In addition, incorporation of these switches into extended rigid scaffolds like covalent-organic, metal organic or porous-aromatic frameworks may yield materials capable of switchable and enantioselective capture of chiral adsorbates or switchable separation of enantiomers.Data availability
All relevant data have been included in ESI.† All original data generated during this work is available from the corresponding author upon request.Author contributions
J. S., W. D. and B. L. F. conceived the study. B. L. F. guided and supervised this work. J. S., X. C. and C. S. synthesized the investigated compounds. J. S. performed NMR and UV-vis experiments. S. C. and A. G. performed DFT calculations. W. D. analysed and determined the quantum yield data. J. S., W. D. and B. L. F. wrote the paper. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support from the China Scholarship Council (CSC PhD Fellowship No. 201808330459 to J. S. and No. 202108330058 to X. C.). Financial support was received from the European Research Council (ERC; advanced Grant No. 694345 to B. L. F.) and the Dutch Ministry of Education, Culture and Science (Gravitation program No. 024.001.035). We are grateful for financial support from Marie Skłodowska-Curie Actions (Individual Fellowship No. 101027639 for W. D., 838280 to S. C. and Innovative Training Network No. 859416 to A. G.).Notes and references
- Molecular Switches, B. L. Feringa and W. R. Browne, ed. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- Molecular Photoswitches, Z. L. Pianowski, ed. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2022 Search PubMed.
- A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600–2610 CrossRef CAS PubMed.
- M. M. Lerch, W. Szymański and B. L. Feringa, Chem. Soc. Rev., 2018, 47, 1910–1937 RSC.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed.
- T. Fukaminato, J. Photochem. Photobiol., C, 2011, 12, 177–208 CrossRef CAS.
- J. Volarić, W. Szymanski, N. A. Simeth and B. L. Feringa, Chem. Soc. Rev., 2021, 50, 12377–12449 RSC.
- W. Danowski, T. Van Leeuwen, W. R. Browne and B. L. Feringa, Nanoscale Adv., 2021, 3, 24–40 RSC.
- J. Andréasson and U. Pischel, Coord. Chem. Rev., 2021, 429, 213695 CrossRef.
- W. C. Xu, S. Sun and S. Wu, Angew. Chem., Int. Ed., 2019, 58, 9712–9740 CrossRef CAS PubMed.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- D. Villarón and S. J. Wezenberg, Angew. Chem., Int. Ed., 2020, 59, 13192–13202 CrossRef PubMed.
- J. Sheng, S. Crespi, B. L. Feringa and S. J. Wezenberg, Org. Chem. Front., 2020, 7, 3874–3879 RSC.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- H. Cheng, S. Zhang, E. Bai, X. Cao, J. Wang, J. Qi, J. Liu, J. Zhao, L. Zhang and J. Yoon, Adv. Mater., 2022, 34, 2108289 CrossRef CAS PubMed.
- H. Qian, S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 9140–9143 CrossRef CAS PubMed.
- S. van Vliet, G. Alachouzos, F. de Vries, L. Pfeifer and B. L. Feringa, Chem. Sci., 2022, 13, 9713–9718 RSC.
- D. J. Van Dijken, P. Kovaříček, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS PubMed.
- B. Shao and I. Aprahamian, Chem, 2020, 6, 2162–2173 CAS.
- S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker and J. R. De Alaniz, J. Am. Chem. Soc., 2014, 136, 8169–8172 CrossRef CAS PubMed.
- S. Wiedbrauk and H. Dube, Tetrahedron Lett., 2015, 56, 4266–4274 CrossRef CAS.
- M. W. H. Hoorens, M. Medved, A. D. Laurent, M. Di Donato, S. Fanetti, L. Slappendel, M. Hilbers, B. L. Feringa, W. J. Buma and W. Szymanski, Nat. Commun., 2019, 1–11 CAS.
- S. Crespi, N. A. Simeth, M. Di Donato, S. Doria, C. N. Stindt, M. F. Hilbers, F. L. Kiss, R. Toyoda, S. Wesseling, W. J. Buma, B. L. Feringa and W. Szymański, Angew. Chem., Int. Ed., 2021, 60, 25290–25295 CrossRef CAS PubMed.
- S. Thumser, L. Köttner, N. Hoffmann, P. Mayer and H. Dube, J. Am. Chem. Soc., 2021, 143, 18251–18260 CrossRef CAS PubMed.
- B. L. Feringa, N. P. M. Huck and H. A. van Doren, J. Am. Chem. Soc., 1995, 117, 9929–9930 CrossRef CAS.
- N. Koumura, R. W. J. Zijistra, R. A. Van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 CrossRef CAS PubMed.
- D. Roke, S. J. Wezenberg and B. L. Feringa, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9423–9431 CrossRef CAS PubMed.
- D. R. S. Pooler, A. S. Lubbe, S. Crespi and B. L. Feringa, Chem. Sci., 2021, 12, 14964–14986 RSC.
- J. Vicario, A. Meetsma and B. L. Feringa, Chem. Commun., 2005, 5910–5912 RSC.
- J. C. M. Kistemaker, S. F. Pizzolato, T. van Leeuwen, T. C. Pijper and B. L. Feringa, Chem. - Eur. J., 2016, 22, 13478–13487 CrossRef CAS PubMed.
- Y. Ren, R. Jamagne, D. J. Tetlow and D. A. Leigh, Nature, 2022, 612, 78–82 CrossRef CAS PubMed.
- D. Pijper, M. G. M. Jongejan, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2008, 130, 4541–4552 CrossRef CAS PubMed.
- S. F. Pizzolato, B. S. L. Collins, T. van Leeuwen and B. L. Feringa, Chem. - Eur. J., 2017, 23, 6174–6184 CrossRef CAS PubMed.
- S. F. Pizzolato, P. Štacko, J. C. M. Kistemaker, T. Van Leeuwen, E. Otten and B. L. Feringa, J. Am. Chem. Soc., 2018, 140, 17278–17289 CrossRef CAS PubMed.
- S. F. Pizzolato, P. Štacko, J. C. M. Kistemaker, T. van Leeuwen and B. L. Feringa, Nat. Catal., 2020, 3, 488–496 CrossRef CAS.
- X. Chen, P. Gilissen, P. Tinnemans, N. Vanthuyne, F. Rutjes, B. Feringa, J. Elemans and R. Nolte, Nat. Synth., 2022, 1, 873–882 CrossRef PubMed.
- F. Castiglioni, W. Danowski, J. Perego, F. K. C. Leung, P. Sozzani, S. Bracco, S. J. Wezenberg, A. Comotti and B. L. Feringa, Nat. Chem., 2020, 12, 595–602 CrossRef CAS PubMed.
- J. Conyard, A. Cnossen, W. R. Browne, B. L. Feringa and S. R. Meech, J. Am. Chem. Soc., 2014, 136, 9692–9700 CrossRef CAS PubMed.
- P. Roy, A. S. Sardjan, A. Cnossen, W. R. Browne, B. L. Feringa and S. R. Meech, J. Phys. Chem. Lett., 2021, 12, 3367–3372 CrossRef CAS PubMed.
- L. Pfeifer, M. Scherübl, M. Fellert, W. Danowski, J. Cheng, J. Pol and B. L. Feringa, Chem. Sci., 2019, 10, 8768–8773 RSC.
- L. Pfeifer, N. V. Hoang, M. Scherübl, M. S. Pshenichnikov and B. L. Feringa, Sci. Adv., 2020, 6, eabb6165 CrossRef CAS PubMed.
- L. Pfeifer, S. Crespi, P. van der Meulen, J. Kemmink, R. M. Scheek, M. F. Hilbers, W. J. Buma and B. L. Feringa, Nat. Commun., 2022, 13, 2124 CrossRef CAS PubMed.
- C. Stähler, L. Grunenberg, M. W. Terban, W. R. Browne, D. Doellerer, M. Kathan, M. Etter, B. V. Lotsch, B. L. Feringa and S. Krause, Chem. Sci., 2022, 13, 8253–8264 RSC.
- J. Fang, L. Li, C. Yang, J. Chen, G. J. Deng and H. Gong, Org. Lett., 2018, 20, 7308–7311 CrossRef CAS PubMed.
- T. Van Leeuwen, J. Pol, D. Roke, S. J. Wezenberg and B. L. Feringa, Org. Lett., 2017, 19, 1402–1405 CrossRef CAS PubMed.
- D. Roke, B. L. Feringa and S. J. Wezenberg, Helv. Chim. Acta, 2019, 102, e1800221 CrossRef.
- J. Sheng, Y. Wang, X. Su, R. He and C. Chen, Angew. Chem., Int. Ed., 2017, 56, 4824–4828 CrossRef CAS PubMed.
- A. B. Rudine, M. G. Walter and C. C. Wamser, J. Org. Chem., 2010, 75, 4292–4295 CrossRef CAS PubMed.
- A. Cnossen, J. C. M. Kistemaker, T. Kojima and B. L. Feringa, J. Org. Chem., 2014, 79, 927–935 CrossRef CAS PubMed.
- R. Wilcken, A. Gerwien, L. A. Huber, H. Dube and E. Riedle, ChemPhotoChem, 2022, 6, e20210023 Search PubMed.
- E. M. M. Tan, S. Amirjalayer, S. Smolarek, A. Vdovin, F. Zerbetto and W. J. Buma, Nat. Commun., 2015, 6, 5860 CrossRef PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J. Hou, G. Long, W. Zhao, G. Zhou, D. Liu, D. J. Broer, B. L. Feringa and J. Chen, J. Am. Chem. Soc., 2022, 144, 6851–6860 CrossRef CAS PubMed.
- R. Dorel and B. L. Feringa, Chem. Commun., 2019, 55, 6477–6486 RSC.
- Q. Zhu, W. Danowski, A. K. Mondal, F. Tassinari, C. L. F. Beek, G. H. Heideman, K. Santra, S. R. Cohen, B. L. Feringa and R. Naaman, Adv. Sci., 2021, 8, 2101773 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05903g |
| This journal is © The Royal Society of Chemistry 2023 |