
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal complexes for catalytic and photocatalytic reactions in living cells and organisms
Hugo
Madec†
a,
Francisca
Figueiredo†
b,
Kevin
Cariou
*b,
Sylvain
Roland
*a,
Matthieu
Sollogoub
 *a and
Gilles
Gasser
*b
*a and
Gilles
Gasser
*b
aSorbonne Université, CNRS, Institut Parisien de Chimie Moléculaire, Paris, France. E-mail: sylvain.roland@sorbonne-universite.fr; Web: http://www.ipcm.fr/-Glycochimie-Organique
bChimie ParisTech, PSL Université, CNRS, Institute of Chemistry for Life and Health Sciences, Paris 75005, France. E-mail: kevin.cariou@chimieparistech.psl.eu; gilles.gasser@chimieparistech.psl.eu; Web: http://www.gassergroup.com
First published on 2nd December 2022
Abstract
The development of organometallic catalysis has greatly expanded the synthetic chemist toolbox compared to only exploiting “classical” organic chemistry. Although more widely used in organic solvents, metal-based catalysts have also emerged as efficient tools for developing organic transformations in water, thus paving the way for further development of bio-compatible reactions. However, performing metal-catalysed reactions within living cells or organisms induces additional constraints to the design of reactions and catalysts. In particular, metal complexes must exhibit good efficiency in complex aqueous media at low concentrations, good cell specificity, good cellular uptake and low toxicity. In this review, we focus on the presentation of discrete metal complexes that catalyse or photocatalyse reactions within living cells or living organisms. We describe the different reaction designs that have proved to be successful under these conditions, which involve very few metals (Ir, Pd, Ru, Pt, Cu, Au, and Fe) and range from in cellulo deprotection/decaging/activation of fluorophores, drugs, proteins and DNA to in cellulo synthesis of active molecules, and protein and organelle labelling. We also present developments in bio-compatible photo-activatable catalysts, which represent a very recent emerging area of research and some prospects in the field.
 Hugo Madec | Hugo Madec completed his MSc in Molecular chemistry at Sorbonne University in 2021. He is currently a PhD candidate under the supervision of Sylvain Roland and Matthieu Sollogoub at Sorbonne University. His research is focused on the development of biocompatible encapsulated copper photocatalysts. |
 Francisca Figueiredo | Francisca Figueiredo graduated from the Université de Paris and completed her MSc in Chemistry for Life Sciences at PSL University in 2021. She is currently doing her PhD under the supervision of Gilles Gasser and Kevin Cariou at Chimie ParisTech, on copper complexes to perform intracellular photocatalysis for anticancer purposes. |
 Kevin Cariou | Kevin Cariou received his PhD in 2006 from Sorbonne Université under the supervision of Max Malacria and Louis Fensterbank. From 2007 to 2009, he worked as a postdoctoral researcher in the group of Alison Frontier at the University of Rochester (NY, USA). He was appointed as a CNRS Researcher in 2009 at the Institut de Chimie des Substances Naturelles in the team led by Robert Dodd. He obtained his HDR in 2015 and moved to Chimie ParisTech in 2020 and was appointed the Director of Research in 2021. His interests lie in the development of synthetic methods to access biologically active molecules. |
 Sylvain Roland | Sylvain Roland is an Associate Professor at Sorbonne Université in Paris. He holds a MSc in Organic and Bioorganic Chemistry (1991) and received his PhD in 1995 (Organic Chemistry) from Pierre and Marie Curie University under the supervision of Prof. J.-P. Genêt at the Ecole Nationale Supérieure de Chimie de Paris. In 1995–1996, he joined Prof. W. B. Motherwell's group at University College London as a postdoctoral fellow. His recent research interests are in the area of metal–NHC complexes and encapsulated metals with applications in homogeneous catalysis and application of cyclodextrin-NHC–metal complexes in bioorganometallic chemistry and in vivo photocatalysis. |
 Matthieu Sollogoub | Matthieu Sollogoub is Professor of Molecular Chemistry at Sorbonne University, where he conducts his research on various aspects of organic, biological and supramolecular chemistry. He focuses mainly on cyclodextrin functionalization for catalysis, supramolecular assembly and molecular motors. He obtained his PhD at the Ecole Normale Supérieure (ENS) in Paris in 1999 with Prof. P. Sinaÿ. He carried out postdoctoral research at the University of Southampton with Prof. T. Brown and joined the faculty of ENS and Sorbonne in 2001. In 2011, he received the Carbohydrate Research Award for Creativity in Glycoscience, and in 2020 he became a Chemistry Europe Fellow. |
 Gilles Gasser | Gilles Gasser started his independent scientific career at the University of Zurich (Switzerland) in 2010 before moving to Chimie ParisTech, PSL University (Paris, France) in 2016 to take a PSL Chair of Excellence. Gilles was the recipient of several fellowships and awards including the Alfred Werner Award from the Swiss Chemical Society, an ERC Consolidator Grant, the European BioInorganic Chemistry (EuroBIC) medal and the Pierre Fabre Award for therapeutic innovation from the French Société de Chimie Thérapeutique. Gilles' research interests lay in the use of metal complexes in different areas of medicinal and biological chemistry. |
1. Introduction
While Nature has long used metal catalysts, such as metallo-enzymes, to perform transformations with exquisite selectivity and efficiency, the heyday of metal catalysis is still relatively recent. The last few decades have seen transition metal complexes rise from lab curiosities to indispensable tools for synthesis. Quite logically, the implementation of metal-based catalysis in biorelevant environments became a bonafide field of research.1–3 One of the key aims is to develop metal complexes capable of selective and safe catalysis in cellulo and in vivo to modulate cellular functions and bring about new-to-nature transformations into living organisms.4–6 Apart from its fundamental aspect, this approach opens new perspectives for the development of more efficient and selective therapeutic treatments and in vivo imaging processes.Metal-based catalysis in complex biological systems faces several challenges. Once metal complexes enter a living organism, they are exposed to natural chelators and high concentrations of nucleophiles such as thiols and amines, in particular glutathione.7 These species can deactivate or capture the metal compounds/metal ions and accelerate their elimination from the body. Another potential problem is that some metals used in catalysis may have unwanted adverse toxicity towards cells, although it is important to remember that very high doses of some metal complexes are used on an every-day basis in medicine (e.g., Ga-based MRI agents).8 Therefore, it is generally important to develop catalysts that are active at low concentrations to minimise the risk of unwanted toxicity and side effects. Finally, in cellulo monitoring of metal-based catalysis may encounter some barriers due to the complex biological environment of cells and the lack of direct quantitative analytical methods.9 For instance, the exact proportion of the catalyst which enters the cells or the amount that is trapped inside membranes remains difficult to precisely quantify. As a consequence, the evaluation of turnover numbers and catalytic activity, which are classical tools to compare the efficiency of metal complexes in catalysis, is also difficult.
The field has evolved in the last few years towards a broader range of transformations with an increasing number of metals (including non-precious metals) and towards photocatalysis, which is a new challenge for cellular applications. Various strategies have been developed including in cellulo prodrug activation, profluorophore and protein activation or small molecule (NO) release through metal-(photo)catalysed decaging, in cellulo synthesis of active molecules, labelling through metal-catalysed conjugation reactions or exploiting intracellular reagents (NAD(P)H and GSH) to disturb cell processes. Some of these strategies allow for specific targeting of cells and cell components and thus may favour the development of future treatments with restricted side effects. Photocatalysis is a particularly relevant tool in this area,10 enabling precise light-activation of a catalyst in a spatio-temporal controlled manner. Overall, the in cellulo use of metal-based catalysts has already demonstrated its pertinence and potential for developing new tools for therapeutic and biomedical applications,11 but many catalytic reactions and metals remain to be explored.
Since it is an emergent field, several excellent reviews, which are dedicated, for example, to catalysis in biologically relevant conditions using nanoparticles3,12 or discrete organometallic complexes13,14 have been (recently) published. Also, some reviews focus on the applications of metal-based catalysts as therapeutics in medicine.15–18 While these reviews are organised by metal ions,11,19–21 applications or the type of reaction22–25 in biological habitats but not within living organisms, herein we present the recent breakthroughs in the biological applications of metal-based catalysts and photocatalysts within living cells and organisms. Of note, we only focus on catalytic transformations performed inside cells, bacteria and vertebrates (mainly mice or zebrafish). Finally, this review describes homogeneous catalytic systems using discrete metal complexes.
2. Ruthenium
Ruthenium catalysts have been widely used in homogeneous catalysis for a wide range of transformations ranging from CH activation26 to olefin metathesis.27 Furthermore, Ru complexes are classically used as photosensitizers in photoredox processes.28–30 Concerning in vivo applications, several Ru-based compounds have been tested for their anticancer properties,31 some of them having reached the clinical study phase (KP1339,32 NAMI-A,33 and TLD1443 (ref. 34 and 35)). It is therefore logical that Ru was one of the first metals to be used as a catalyst and photocatalyst in cellulo.2.1 Catalysis
As early as 2006, Meggers and co-workers showed that Ru complexes could induce uncaging of Alloc-protected substrates in living cells. This was initially reported in mammalian cells with a ruthenium(II) complex [Cp*Ru(cod)Cl] (1) (Cp* = pentamethylcyclopentadienyl, cod = 1,5-cyclooctadiene) (Fig. 1).36 In a preliminary study, it was shown that the RuII complex 1 was tolerant to air and water but that thiols (thiophenol) were essential for catalytic activity. The uncaging reaction was evaluated inside mammalian HeLa cells after incubation with 100 μM of a caged rhodamine 110 dye 2, which is a profluorophore with low fluorescence. After incubation with the Ru catalyst (1) (20 μM) and thiophenol, a 10-fold increase of fluorescence was observed inside cells indicating intracellular fluorophore 3 production through Alloc deprotection. By comparison, a 3.5-fold increase was measured in the control experiments without the catalyst. Control experiments without the supplementation of thiophenol also showed a limited fluorescence increase (3.5-fold) demonstrating the importance of thiols as nucleophiles to favour the reaction. Cell-staining experiments suggested that the fluorescence occurs within cells rather than on the membrane, thus implying that compound 1 can diffuse through membranes to induce intracellular catalysis. Furthermore, cytotoxicity experiments with the catalyst alone displayed no decrease in cell viability.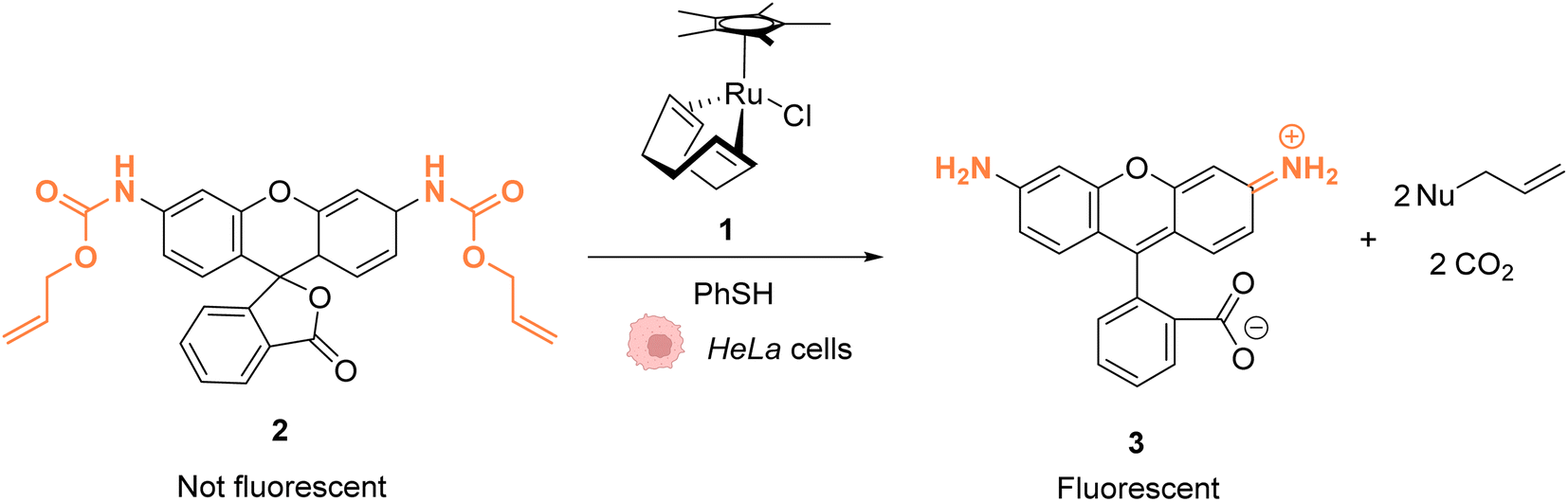 | ||
| Fig. 1 Ruthenium(II)-catalysed Alloc deprotection of the caged fluorophore to rhodamine 110 in HeLa cells in the presence of thiols.36 | ||
In 2014, the same group developed an organometallic ruthenium(IV) complex (4) catalysing the uncaging of Alloc-protected amines with high turnover numbers (TONs), reaching 270 cycles in aqueous medium in the presence of thiols (Fig. 2).37 Inside living HeLa mammalian cells preincubated with Alloc-caged rhodamine 110 5, an average of a 90-fold increase of intracellular fluorescence was observed after 10 minutes of treatment with the cationic complex 4, a clear improvement compared to the initial study with the RuII complex 1 (Fig. 1). The ability of the RuIV complex 4 to diffuse through the cell membrane was demonstrated. The authors also investigated the influence of cytoplasmic thiols on the activity of the catalyst. They discovered that thiols induced the activation of the RuIV(allyl) complex, leading to improved efficiency in the decaging reaction, in contrast to previous Ru catalysts, which were deactivated by thiols. Finally, the uncaging of the anticancer drug doxorubicin 6 inside HeLa cells was developed (Fig. 2B) by using an N-Alloc protected doxorubicin prodrug 5, exhibiting significantly reduced overall affinity for DNA compared to the drug itself. Cells incubated with the prodrug (50 μM or 100 μM) and the RuIV catalyst 4 (20 μM) showed a dramatic decrease in cell viability (reduced to 7% and 2%, respectively), whereas cells treated with the prodrug or complex 4 only were not affected. This demonstrated the efficiency of the Ru precatalyst 4 to induce uncaging of doxorubicin 6 inside cells, leading to apoptosis.
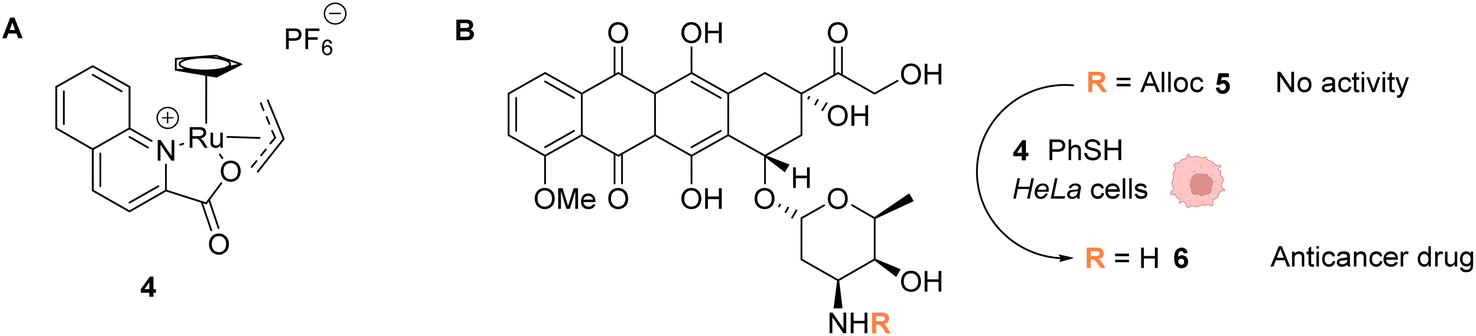 | ||
| Fig. 2 (A) Structure of the cationic ruthenium(IV) precatalyst 4; (B) Ru-induced uncaging of the anticancer drug doxorubicin inside HeLa cells.37 | ||
In 2014, Mascareñas and coworkers reported a Ru-based decaging strategy for the in cellulo control of the selective targeting of double stranded DNA by small molecules, such as ethidium bromide or DAPI (4′,6-diamidino-2-phenylindole).38 External control of DNA binding to these molecules is interesting for regulating DNA transcription and thus gene expression. Adding Alloc protecting groups to DAPI and ethidium bromide was first shown to reduce the affinity to DNA and thus to deactivate their function. These two DNA binding agents have intrinsic fluorescence, allowing for the monitoring of their cellular distribution and activity. The uncaging reaction was tested inside chicken embryo fibroblast (CEF) cells with 2.5 μM of caged DAPI and 2.5 μM of the ruthenium(II) catalyst [RuCp*(COD)Cl] (1). Before incubation with 1, fluorescence microscopy showed whole cell staining as DAPI did not interact with DNA and was therefore spread throughout the cell. After 20 minutes of treatment, the typical blue DAPI staining shifted from the cytoplasm to the cell nucleus, strongly supporting that the RuII catalyst 1 did induce intracellular uncaging, leading to DAPI release and DNA binding. As previously observed, the presence of thiol (PhSH, 100 μM) is required for efficient Alloc deprotection. Furthermore, the di-Alloc DAPI derivative was found to induce better intracellular staining than DAPI, suggesting a better internalisation inside cells. Finally, the effect of uncaging of DNA binding molecule 7 into 8 on cancer cell growth was studied. Cisplatin-resistant A2780 cells (human ovarian carcinoma) were incubated with both protected (7) and unprotected (8) phenyl azapentamidine derivatives (Fig. 3). The inhibitory effect of the caged derivative 7 (IC50 = 5.0 μM) decreased by 10-fold compared to the uncaged molecule 8 (IC50 = 0.4 μM). Overall, this study demonstrated the potential of in cellulo Ru-based uncaging reactions for controlling the activation of small DNA-binding molecules in biological media.
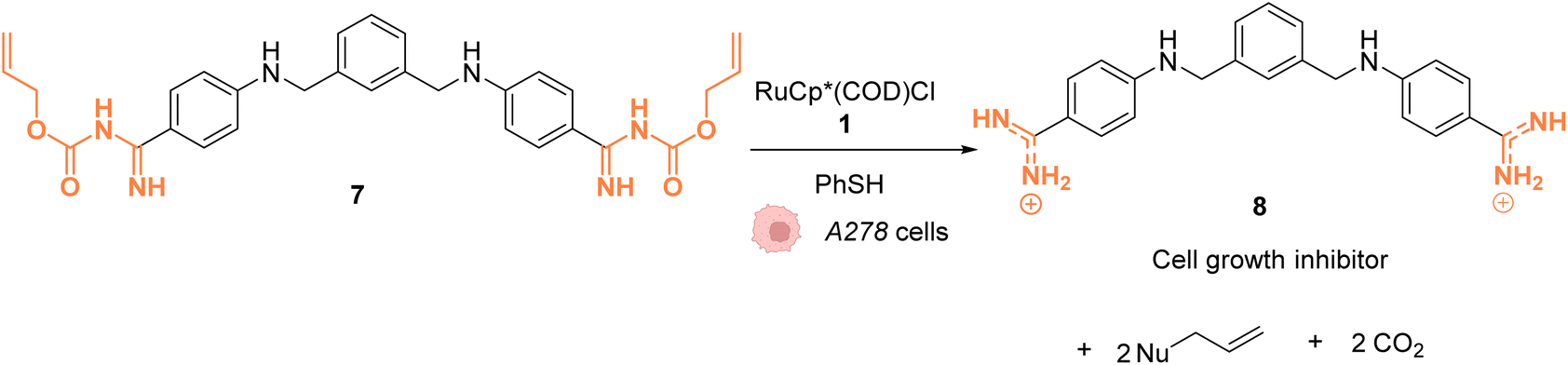 | ||
| Fig. 3 Ru-catalysed decaging of double stranded DNA binding agents by [RuCp*(COD)Cl] (1) inside A278 cancer cells.38 | ||
The same authors reported in 2016 a RuIV(allyl) complex 9a that accumulates inside the mitochondria of living mammalian cells to catalyse the targeted uncaging of allyl/Alloc protected substrates in this organelle (Fig. 4).39 An active RuII species is likely to be generated in cellulo through the reduction of the RuIV(allyl) species by intracellular thiols. The specific localisation of the catalyst and its retention in mitochondria were guaranteed by using a triphenylphosphonium (TPP) delivery vector linked to the ruthenium complex via a hydrophobic alkyl chain. TPP cations have the property to diffuse through the mitochondrial inner membrane. Cellular tests were performed in HeLa cells, for which catalyst 9a did not show any toxicity even at concentrations >100 μM. Incubation with Alloc-protected rhodamine 2 together with 9a showed an intracellular increase of fluorescence, mainly concentrated inside mitochondria. To monitor the Ru-catalysed deprotection reaction by fluorescence, an analogue of 9a possessing a pyrene unit (9b) was developed (Fig. 4A). To test the catalytical capacity of this new complex, cells were treated with 100 μM of a caged rhodamine 110 dye 2 and 50 μM of catalyst 9b. The pattern of the green fluorescence of the free rhodamine 3 was found to match the blue fluorescence of 9b and the red staining identifying the mitochondria. These results, demonstrating that catalysis did take place in mitochondria, were reproduced in A549 cancer cells. This work shows that it is possible to have an abiotic catalyst, not initially present in living organisms, acting in a precise subcellular compartment.
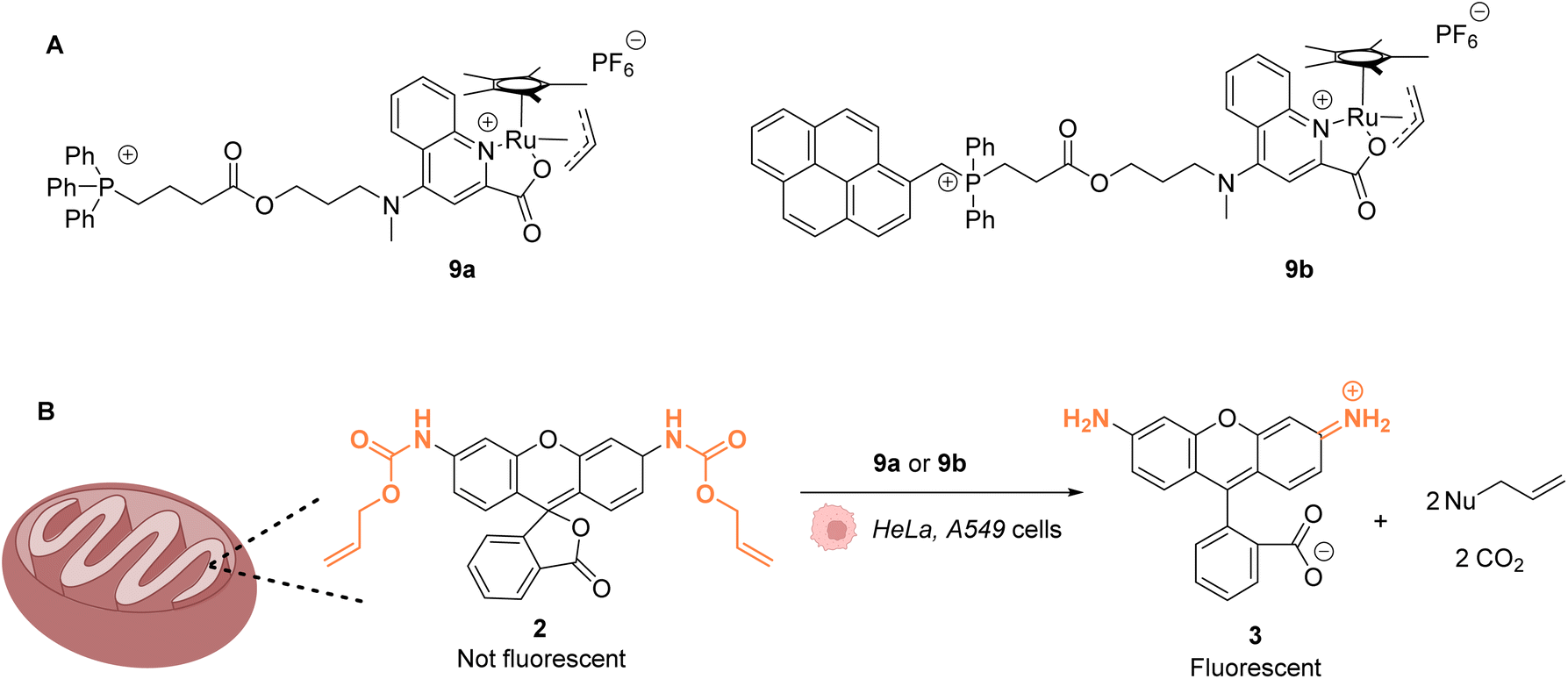 | ||
| Fig. 4 (A) Structure of mitochondria targeting RuIV complexes 9a and 9b; (B) catalysis of the uncaging of fluorophore rhodamine 110 inside HeLa and A549 cells, preferentially occurring inside mitochondria.39 | ||
Ward and co-workers reported in 2018 a cell-penetrating ruthenium-based Artificial Metalloenzyme (ArM) 10 catalysing Alloc deprotection in epithelial human cells.40 This ArM 10 was built from the biotin N-binding protein streptavidin, combining a biotin N-linked ruthenium(II) complex 11 that catalyses the uncaging reaction of the Alloc-protected substrate 14 and a biotinylated cell-penetrating moiety having a fluorescent probe 12 (Fig. 5A). Cellular tests were performed in HEK-293T cells, which were successively incubated with ArM 10 and the Alloc-protected prosubstrate 13. When incorporated into cells, 13 is hydrolysed by endogenous esterases to give the alloc-protected substrate 14. The uncaging of 14 by ArM 10 releases the hormone 15 that activates a metabolic cascade (Fig. 5B). The cells were previously transfected with two genes that change the outcome of the metabolic cascade when the hormone is deprotected, leading to cell fluorescence and allowing for reaction monitoring. The increase in fluorescence in cells confirmed that ArM 10 catalysed Alloc deprotection to release the hormone. This was not observed in cells treated with the ruthenium catalyst only. Furthermore, no cytotoxicity induced by the catalyst was detected at concentrations below 500 nM. The optimisation of the ArM was performed by site-directed mutagenesis at two positions close to the catalytic centre, and the best TONs were obtained for the mutation of a serine in position 112 into an alanine.
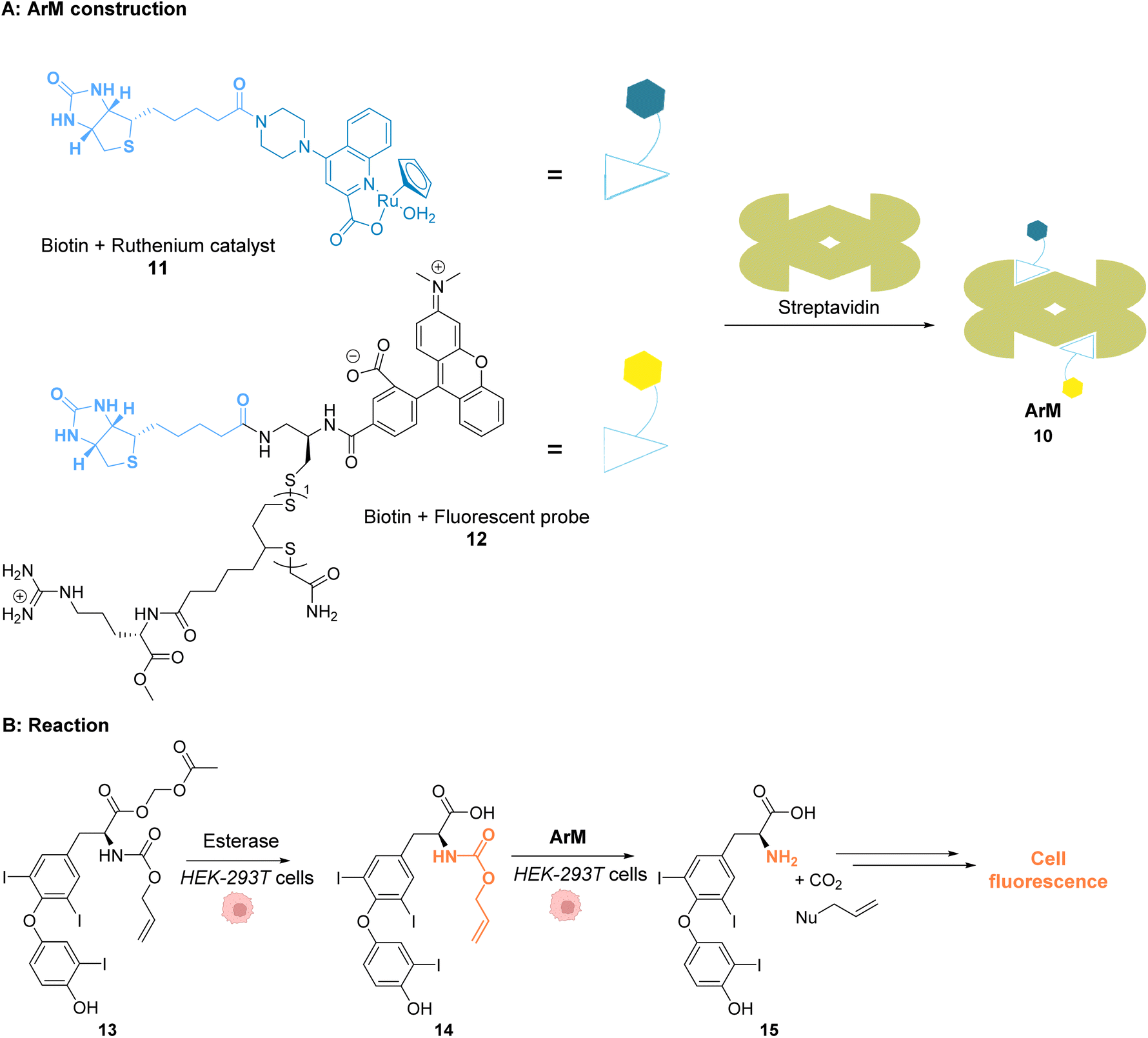 | ||
| Fig. 5 (A) Assembly of a cell-penetrating Ru-based artificial metalloenzyme (ArM). The ruthenium catalyst and the fluorescent probe/cell penetrating peptide are both attached to biotin units interacting with streptavidin to create the artificial metalloprotein; (B) the ArM catalyses the Alloc deprotection of compound 14, generating the hormone 15. The HEK-293T cells have been modified to become fluorescent once 15 is liberated.40 | ||
Mao and co-workers reported in 2022 a ruthenium-based catalyst 16, which is attached to a HER2 antibody that targets the receptor on the membrane of HER2-positive cancer cells.41 Complex 16 catalyses the activation of a gemcitabine prodrug 17via Alloc deprotection to form the free primary amine 18, which enters the cell to cleave DNA. Cell death is also induced by blocking of the HER2 signalling pathway by the ruthenium(IV)-antibody complex (Fig. 6). Cell tests performed with SKBR-3 HER2-positive cancer cells confirmed, after incubation with the prodrug 17 and RuIV catalyst 16, that the reaction was catalysed by the ruthenium complex at the cell surface. The specificity of this treatment was confirmed by comparing cell death in a co-culture of SKBR-3 cells and MCF-10A non-tumorigenic cells incubated with both the RuIV complex 16 and the alloc-protected prodrug 17. 75% cell death was observed for the HER2-positive cells, whereas HER2-negative cells showed only 15% cell death. Next, 3D spheroids of SKBR-3 cells were developed and incubated with 40 μM of prodrug 17 and 2 μM of the RuIV complex 16. This treatment led to a decrease of cell viability in spheroids, reaching 32%. Finally, in vivo tests were performed in zebrafish larvae grafted with SKBR-3 cells, in which 2 μM of the RuIV complex 16 and 8 μM of the alloc-protected prodrug 17 were injected. Fluorescence images revealed a decrease in SKBR-3 cell fluorescence in the zebrafish larvae by ca. 78%, indicating significant cell death. To the best of our knowledge, this study described the first ruthenium-antibody catalyst capable of performing HER2-targeted chemotherapy.
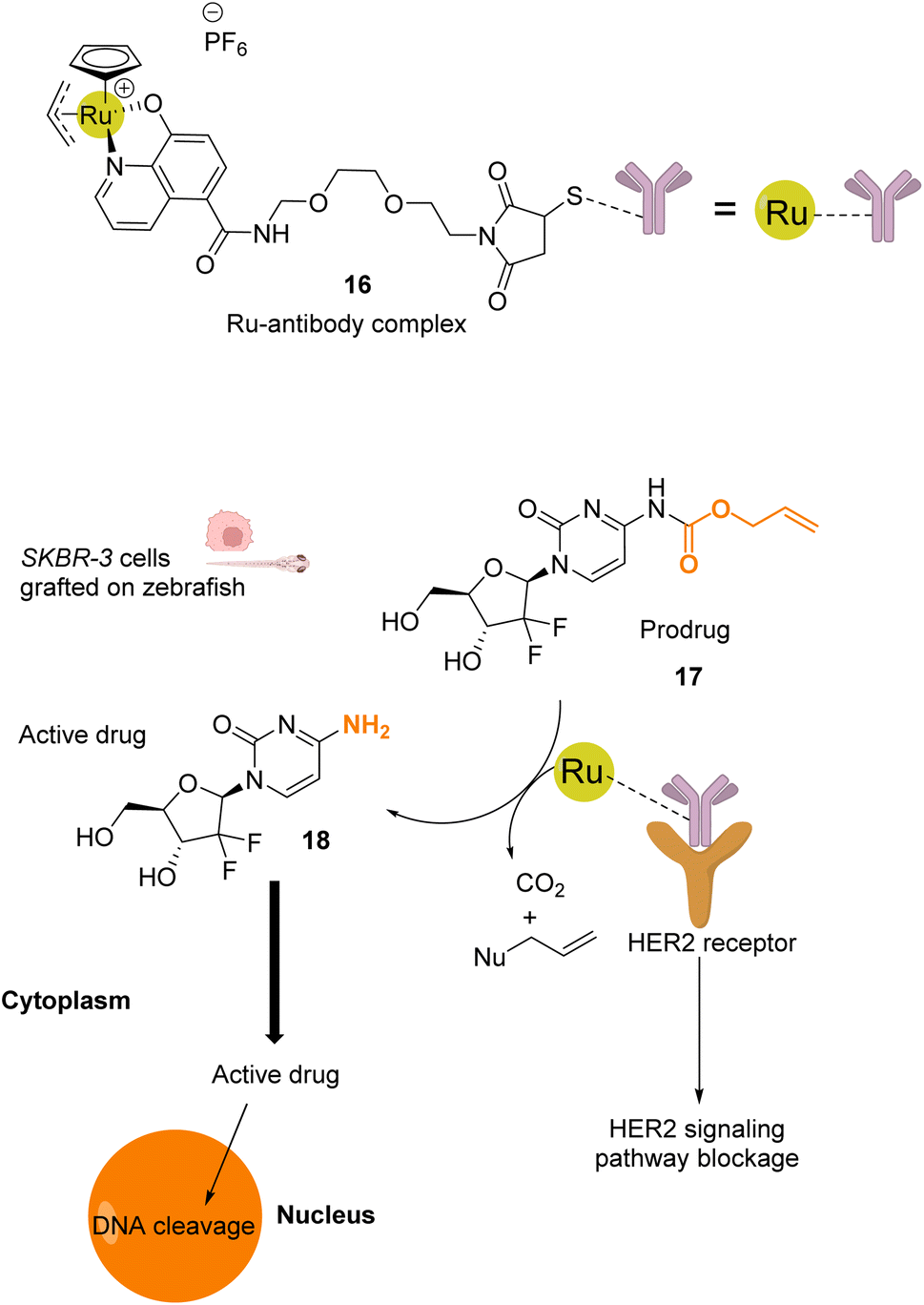 | ||
| Fig. 6 Schematic illustration of HER2-targeted chemotherapy using a gemcitabine-based prodrug and the RuIV complex 16 as the catalyst.41 The prodrug is activated by the catalyst near the membrane and is taken up by the cell as an active anticancer drug. The reaction was performed in vivo inside zebrafish larvae grafted with SKBR-3 cells. | ||
Beside Alloc deprotection reactions, other types of Ru-catalysed reactions have been developed in eukaryotic and prokaryotic cells. Sadler and co-workers described in 2015 a system based on Noyori-type RuII arene complexes and formate ions that disrupt the NAD+/NADH balance in cells by reducing NAD+ to NADH, inducing selective cancer cell death (Fig. 7).23 The authors showed that complex 19 bearing a sulfonylethyldiamine ligand catalysed transfer hydrogenation inside living cells in the presence of non-toxic concentrations of formate acting as a hydride donor. The formation of Ru–H hydride species by mixing the RuII complex 19 and formate ions was evidenced by 1H NMR and mass spectroscopy. In A2780 human ovarian cancer cells, the catalyst 19 alone exhibited an antiproliferative activity comparable to that of cisplatin (IC50 2.2 and 1.2 μM, respectively). However, in the presence of formate ions, the antiproliferative activity was found to be greatly increased (up to 50-fold), supporting a direct contribution of formate in the intracellular catalytic cycle of transfer hydrogenation. The comparison of antiproliferative activity towards normal cells (MRC5) and A2780 cancer cells also showed that formate increased the selectivity factor from 3.6 to 5. Complex 19 is mainly distributed in the cytosol (51%), as shown by ICP-MS experiments. Experiments carried out on A2780 human ovarian cancer cells revealed that the mechanism of cell death via reductive stress caused by the disruption of the NAD+/NADH balance was different from other anticancer modes of action. According to the authors, this means that this new approach could be efficient against cisplatin-resistant cancer cell lines.
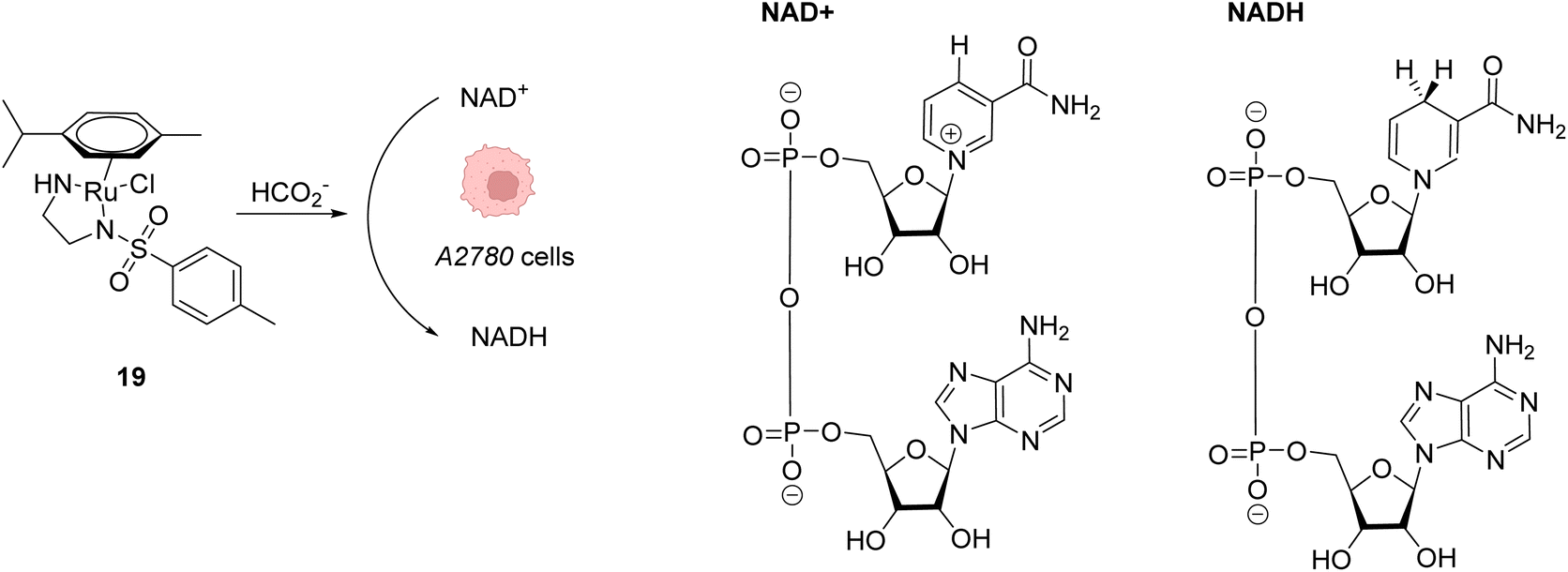 | ||
| Fig. 7 Induction of reductive stress in cancer cells by the RuII arene complex 19 in the presence of formate ions, leading to selective cancer cell death.23 | ||
Mascareñas and co-workers described in 2019 RuIV complexes catalysing the intracellular isomerisation of allylic alcohols into saturated carbonyl derivatives by intramolecular hydride transfer.42 To understand whether the reaction could be carried out inside cells, the authors designed a ruthenium complex 20 and a substrate 21, whose isomeric form 22 is fluorescent (Fig. 8A and B). The reactions were studied in human A549 and HeLa cells as well as animal Vero cell lines (Fig. 8C). The treatment of cells with only 10 μM of the RuIV complex 20 and 100 μM of the non-fluorescent substrate 21 generated significant fluorescence, building up in the cytosol. A TON of over 22 was obtained after 6 h. In this time span, none of the compounds reduced cell viability. Looking for biological applications for this approach, the authors considered to generate in situ glutathione depleting agents such as ketone 24 by isomerisation. Inside cells, the incorporation of 100 μM of allylic alcohol 23 and 50 μM of the Ru catalyst 20 led to the intracellular generation of the GSH depleting agent 24. After 6 h, the authors showed that the formation of 24 was associated with a consumption of over 40% of the intracellular glutathione. They concluded that metal catalysis could be used to generate bioactive molecules inside cells by other strategies not involving uncaging reactions.
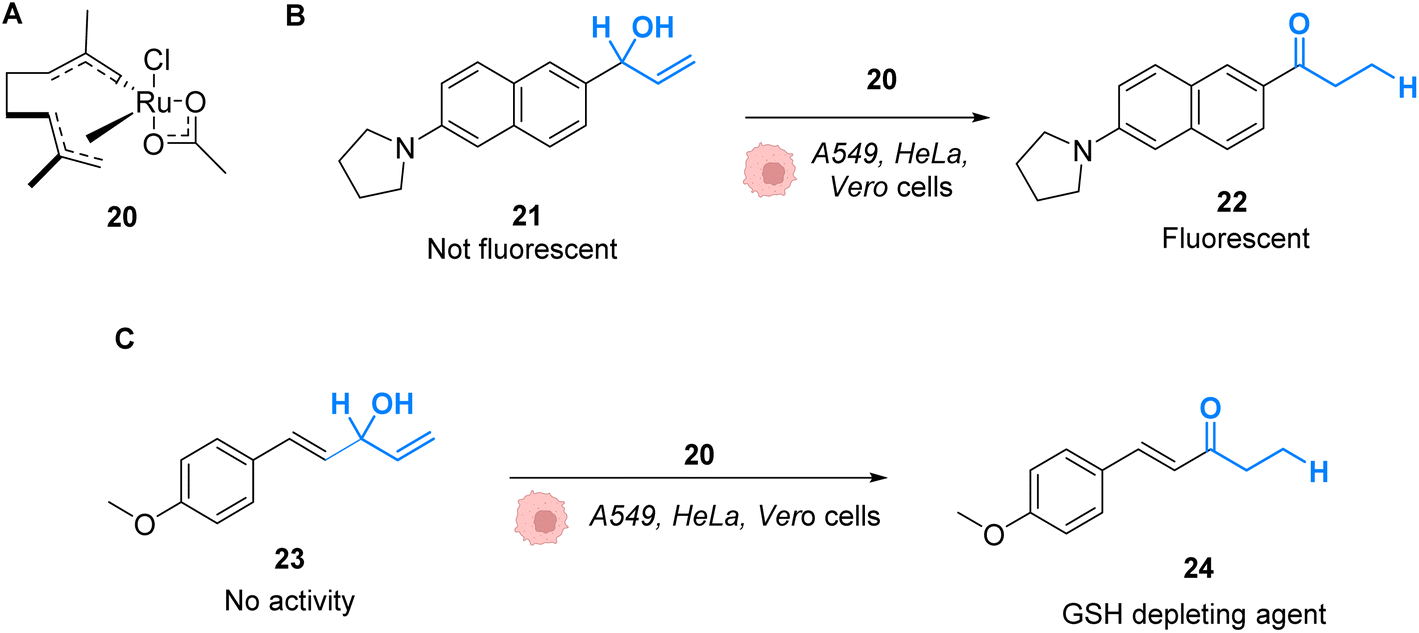 | ||
| Fig. 8 (A) Ruthenium(IV) complex 20 inducing intracellular isomerisation of allylic alcohols. (B) and (C) Generation of functional unsaturated ketones inside A549, HeLa and Vero cells.42 | ||
In 2020, Mascareñas and co-workers reported the transposition of intramolecular and intermolecular [2 + 2 + 2] cycloaddition reactions inside living mammalian cells using the ruthenium complexes 1, 25 and 26 (Fig. 9).43 Both the neutral RuII complex 1 and cationic RuIV complexes 25 and 26 were found to be active inside HeLa cells and biocompatible, as no change in the cell morphology was observed after the experiments. The presence of the Ru catalyst inside cells was demonstrated by ICP-MS after the incubation of HeLa cells with 50 μM of the neutral RuII complex 1. The production of a fluorescent probe 28 from substrate 27 by an intramolecular [2 + 2 + 2] cycloaddition reaction was used for the tracking of the reaction inside cells. The incubation of the cells with the Ru complexes 1 or 26 (50 μM) followed by triyne 28 (100 μM) showed that cells incubated with the cationic RuIV complex 26 exhibited three times more fluorescence than those incubated with the neutral RuII complex 1. These results were explained by the good cellular uptake and low toxicity at 50 μM of the RuIV complex 25 that was previously observed by the authors.39 The transfer of the [2 + 2 + 2] cycloaddition reaction inside cells made possible the in cellulo synthesis of molecules, which could not have been internalised without the permeabilisation of the cell membrane. This strategy was applied to the intracellular production of anthraquinones, which are secondary metabolites that cannot be generated by mammalian cells. For instance, the anthraquinone 31, which presents aggregation-induced emission properties, was synthesised inside HeLa cells by using 50 μM of the RuIV complex 25, 50 μM of the precursor 29 and 150 μM of 30 through an intermolecular cycloaddition reaction. The formation and the localisation of the anthraquinone 31 were determined by fluorescence microscopy. Finally, the authors showed that the spatial localisation of the as-formed fluorescent molecule 31 could be controlled by using different catalysts. With the ruthenium complex 25, the product displayed a cytosolic distribution, whereas it was mainly localised inside mitochondria by using the ruthenium complex 26, a complex known to preferentially accumulate inside mitochondria.
 | ||
| Fig. 9 (A) Ru complexes 1 and 25–26 used for intracellular [2 + 2 + 2] cycloaddition reactions; (B) intramolecular and (C) intermolecular cycloaddition reactions promoted by ruthenium complexes inside HeLa cells for the generation of a fluorescent probe (28) or an anthraquinone (31), respectively.43 | ||
In 2021, Weng and co-workers developed aqueous-stable organo-ruthenium(II) complexes able to perform intracellular reduction of O2 to H2O2 to induce targeted bacteria death through the subsequent generation of ROS such as hydroxyl radicals (HO˙) (Fig. 10).44 The initial strategy proposed by the authors depends on the exploitation of endogenous formate as a hydride source to generate Ru–H species for transfer hydrogenation to reduce O2. A library of 480 ruthenium complexes was screened to test their ability to generate H2O2 in the presence of air/HCOONa on a model reaction in aqueous medium. Ten lead complexes were then selected for evaluating their antimicrobial activity on six bacterial strains, including formate abundant strains S. aureus (Gram+) and E. coli (Gram−) and formate deficient strains Mycobacterium smegmatis (Gram+), B. subtilis (Gram+) and P. aeruginosa (Gram−). A comparison with normal mammalian cells (HEK293) was also performed. Cell viability assays showed that the ruthenium complex 32 presented the best antimicrobial activity, especially against Gram positive bacterial cells. This suggests that 32 boosts ROS production in Gram+ compared to Gram− bacteria and normal mammalian cells. The fact that 32 is active against both formate abundant and formate deficient Gram+ bacteria suggested two different modes of action. The authors showed that the catalyst 32 used two different pathways to reduce O2 to H2O2 inside bacteria involving either hydrogen transfer through a Ru–H intermediate 33 (formate abundant strain) or single electron transfer (SET) via the formation of O2˙ radicals (formate deficient strains) (Fig. 27). Finally, the lead organoruthenium complex 32 was shown to be efficient against methicillin-resistant S. aureus (MRSA), paving the way for the development of alternative antimicrobial agents to treat antimicrobial resistance.
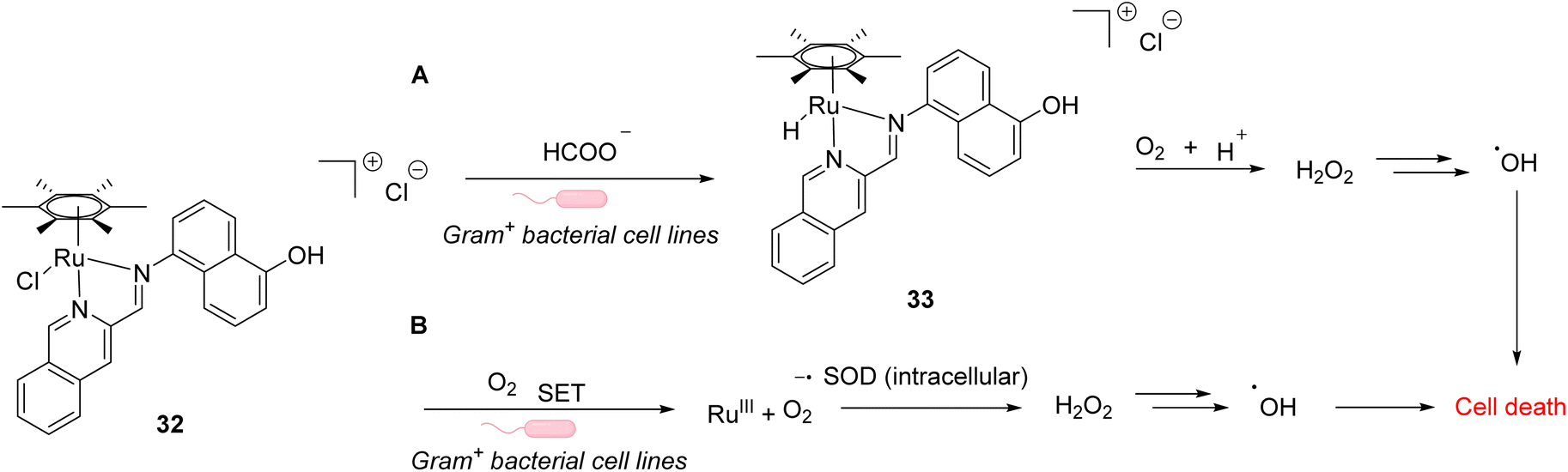 | ||
| Fig. 10 ROS generation by a ruthenium(II) complex inside bacterial cells: (A) hydride transfer mechanism in formate abundant Gram+ strains and (B) SET mechanism in formate deficient strains.44 | ||
The main reaction performed in cellulo by Ru complexes was initially limited to the decaging of an alloc protecting group. In the initial reports, this was only achieved by adding exogenous thiols in addition to the catalyst and substrate. This was later improved by the development of systems that only required endogenous thiols for the decaging to occur. More recent reports have significantly expanded the scope of transformations that could be enabled by ruthenium complexes in cellulo, adding isomerization or cycloadditions, for example. On a more critical note, intermolecular reactions between two distinct exogenous substrates, such as the [2 + 2 + 2] cycloaddition, despite their conceptual interest, might be very challenging to be transposed in the clinic, due to the use of several independent molecules together.
2.2 Photocatalysis
As stated above, Ru complexes are well-known for their photochemical properties, including in a biological environment,45 paving the way to their application as a photocatalyst in cellulo. Following their initial work with [Cp*Ru(cod)Cl] (1) for intracellular Alloc deprotection under “classical” thermal conditions, Meggers and colleagues reported in 2012 a ruthenium(II) aryl complex 34 able to induce Alloc cleavage under light activation inside HeLa cells.46 Upon irradiation at λ ≥ 330 nm, the RuII complex 34 (Fig. 11) forms a [Cp*Ru(solvent)3]+ complex, which is catalytically active, by releasing the pyrene. The presence of thiols as nucleophiles is mandatory to perform the cleavage reaction. The catalyst 34 is more active in the presence of thiophenol than aliphatic thiols. Inside HeLa cells, 34 can perform the Alloc cleavage reaction to generate fluorescent rhodamine 3 from Alloc-protected rhodamine 2 (Fig. 11). HeLa cells were incubated with the caged rhodamine 2, washed to remove the non-internalised, protected rhodamine, and submitted to the action of RuII complex 34 with or without external thiols. After irradiation of the cells for 10 min at λ ≥ 330 nm, the appearance of fluorescence due to rhodamine 3 was observed. Cells incubated with the catalyst 34, 2 and thiophenol were found to exhibit a fluorescence intensity 70-fold higher than those without thiophenol. Without thiophenol, only a small increase of fluorescence was observed, thus demonstrating the need for external additional nucleophilic thiol.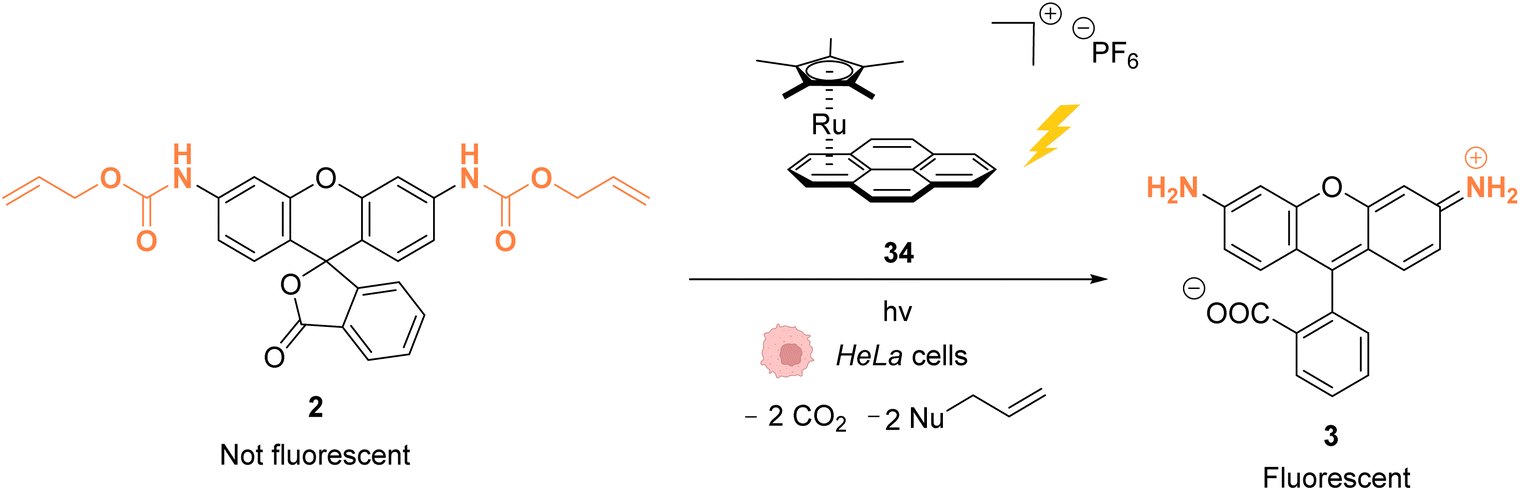 | ||
| Fig. 11 Allylcarbamate (Alloc) cleavage by photo-activatable ruthenium(II) precatalyst 34 inside HeLa cells allowing spatial and temporal control of the reaction.46 | ||
Winssinger and co-workers described in 2012 an abiotic photoreduction of azide-based immolative ligands by a [RuII(bipy)3]-type complex bearing a modified phenanthroline (Fig. 12A). The reduction leads to the intracellular uncaging of a rhodamine fluorophore 35 after visible light irradiation.47 The photoreduction of the azide to an aniline, according to the mechanism described in Fig. 12A,48 entails a decomposition of the immolative linker. The strategy uses one set of protein ligands that bind to an oligomeric receptor. Each protein ligand has been modified by conjugation with a different reactive partner: a rhodamine-based substrate or the RuII catalyst. The ligands have high affinity for the oligomeric receptor, bringing the two reactive partners into proximity upon binding and inducing an increase of the reaction rate (Fig. 12B). This was achieved with three sets of ligands having affinity for three oligomeric receptors: biotin, desthiobiotin and raloxifene. The system was shown to release rhodamine upon visible light irradiation and in the presence of sodium ascorbate, which makes it suitable for in cellulo imaging. The authors studied the reaction inside PA01 bacteria, having an acetyl CoA carboxylase (ACC) that uses biotin as a cofactor as the oligomeric receptor. When bacteria were irradiated with a 455 nm LED lamp for 30 min, an 8-fold increase in fluorescence was observed, thus demonstrating the fluorophore release. Finally, studies were performed on human HER2-driven oncogenic cell lines where ACC is known to be upregulated. It is also known that ACC is more abundant in BT-474 cells than MCF-7 cells. This was an opportunity to uncover whether it was possible to determine the cell type depending on the fluorescence intensity. The authors showed that, after irradiation at 455 nm, there was a higher fluorescence intensity for BT-474 cells than for MCF-7 cells. As a conclusion, it was suggested that this strategy could be more widely applied to uncage active biomolecules and pharmaceutical compounds in a specific target. For instance, it would be possible to target cells that overexpress certain oligomeric receptors.
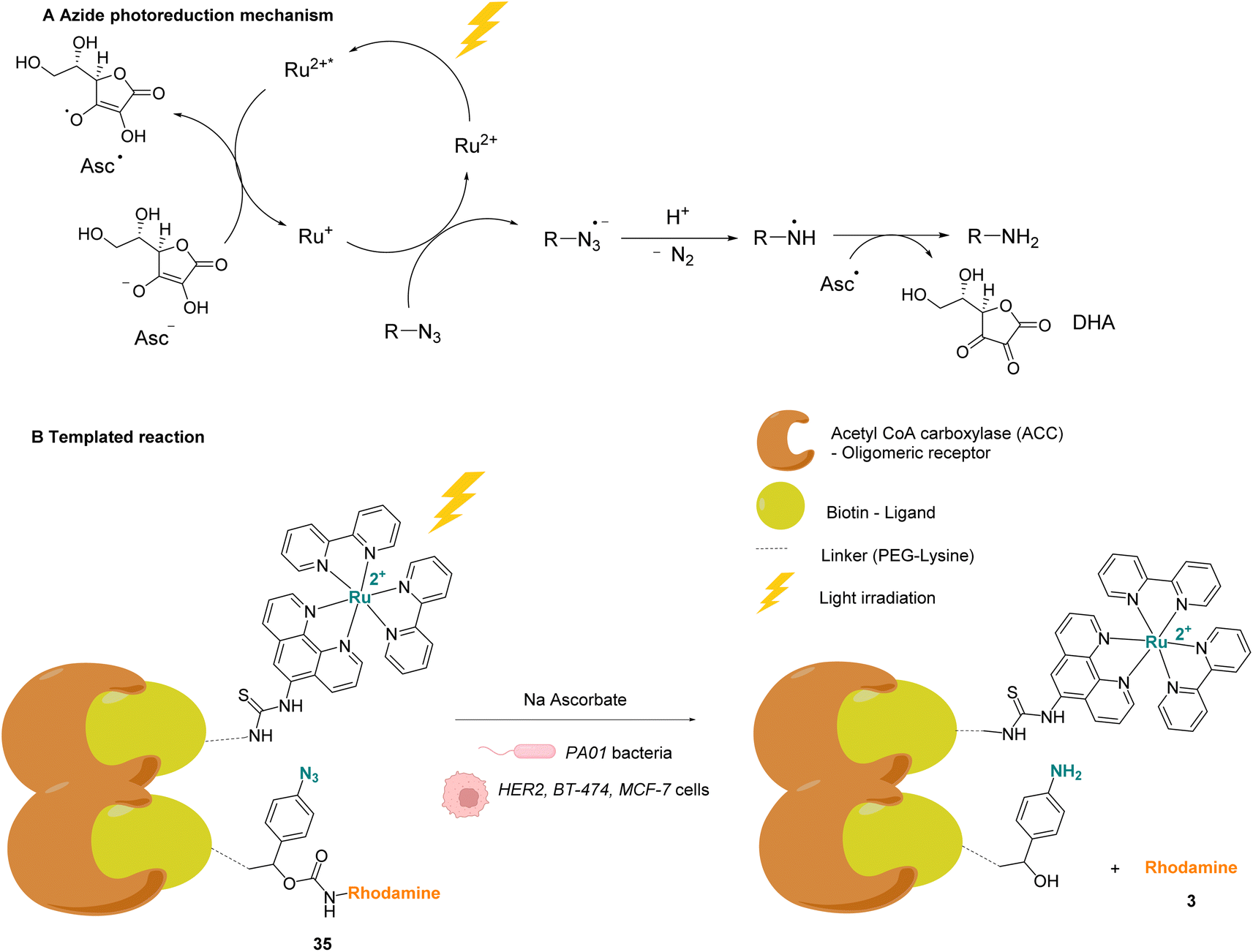 | ||
| Fig. 12 (A) Mechanism of the photoreduction of the azide. (B) [RuII(bipy)3]-type complexes and azide/rhodamine substrates linked to protein ligands for an intracellular protein templated reaction induced by visible light irradiation. Photoreduction of the azide to the aniline leads to immolative linker decomposition and uncaging of rhodamine.47 | ||
In 2013, Wissinger and coworkers further showed that the photocatalysed reduction of azide-triggered immolative linkers with [Ru(bpy)3]2+ complexes could be applied in cells to detect and image miRNAs.49 miRNAs are responsible for the regulation of almost 30% of human genes, and the alteration of their activity is the origin of the development of some cancers, and hence the interest in their imaging and location. This study focused on miRNAs miR-21 and miR-31 as they are overexpressed in certain types of cancers. The imaging probe consisted of two strands of peptide nucleic acid (PNA) that are complementary to two different portions of the targeted miRNA. One PNA bears a lysine residue and the luminescent RuII complex on its C-terminus. The N-terminus side of the other PNA was conjugated to a molecule containing an azide and a caged rhodamine. The hybridisation of both PNAs and the targeted miRNA would place the Ru complex and the caged fluorophore in proximity and therefore accelerate the reaction of uncaging by the Ru (Fig. 13). For both miRNAs, the authors also made equivalent mismatched probes, where the PNA strand does not correspond to the miRNA targeted strand, as a control. This approach was tested inside BT474 cancer cells that overexpress miR-21 and HeLa cells that overexpress miR-31. In both cases, an increase in fluorescence inside the cells was observed after visible light irradiation, thereby demonstrating that the nucleic acid templated reaction takes place in cells leading to the release of the active fluorophore. Tests performed with the mismatched probe or inside cells with no expression of the target miRNAs revealed no fluorescence, indicating that the PNA strands did not bind to the miRNAs in the control experiments and therefore did not bring the reagents into proximity. The authors believe that this technique could be expanded to the release of masked bioactive molecules.
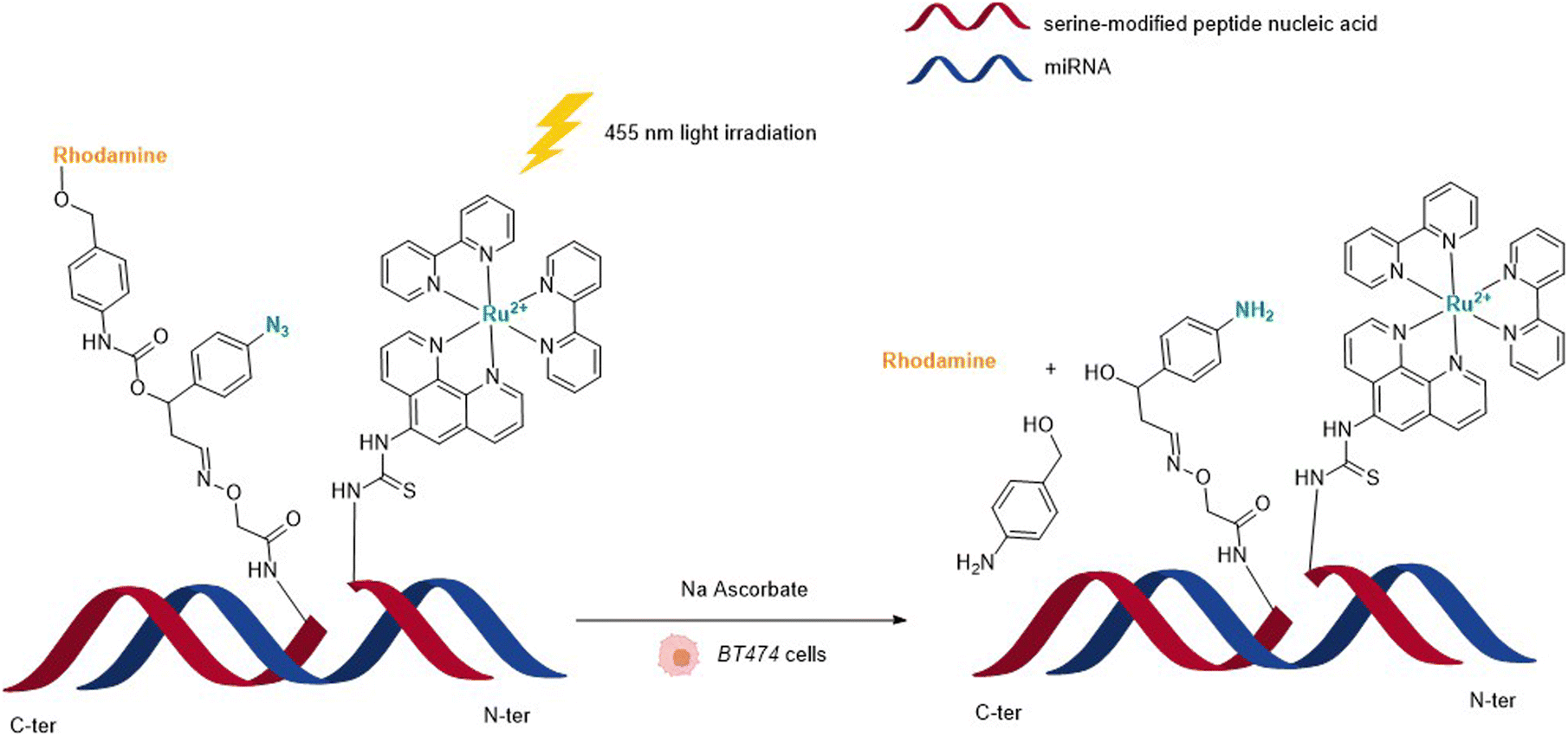 | ||
| Fig. 13 Nucleic acid templated reaction catalysed by [RuII(bipy)3]-type complexes under visible light irradiation for imaging of miRNAs in BT474 and HeLa cancer cells.49 | ||
In 2015, Wissinger and co-workers extended this strategy by using the Ru photocatalyst 36, to generate a quinazoline precipitating dye 38 that can be imaged in cells (HEK293T, MCF-7 and human bone cancer cells) upon two-photon excitation (730 nm) (Fig. 14).50 In this study, complex 36 was linked through phenanthroline to a ligand for specific targeting of proteins either in the intracellular membrane, in the cytosol or in the nucleus. After incubation with cells, the ruthenium-tagged proteins could be localised in their specific cell compartment through luminescence imaging. Thanks to specific protein targeting by the catalyst, the photo-induced (at 450 nm) azide reduction reaction can occur in several chosen subcellular locations. When R is a ligand targeting oestrogen receptor inside MCF-7 cells, the receptor induces nuclear translocation of the Ru photocatalyst 36, which once located inside the nucleus of the cells can still perform the photo-induced azide reduction of 37 to the quinazoline precipitating dye 38 inducing luminescence in the nucleus. Other subcellular locations such as mitochondria can be targeted by using O6-benzylguanine as the ligand.
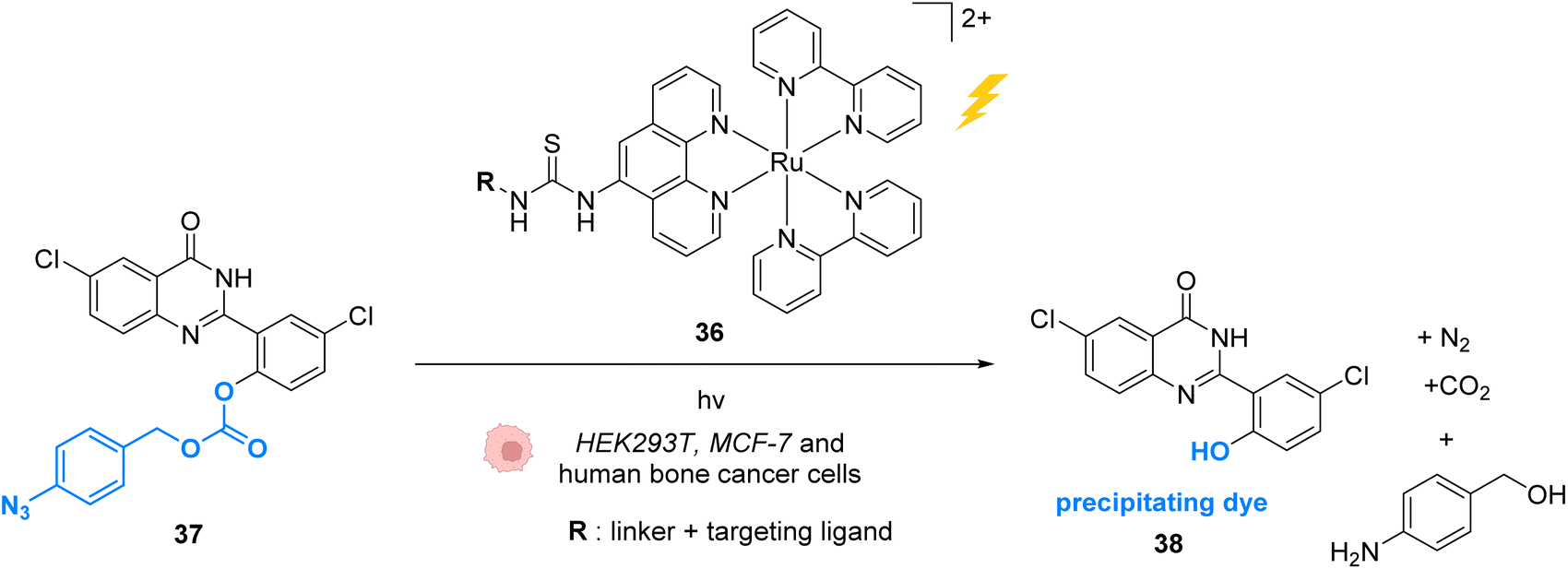 | ||
| Fig. 14 Azide reduction of a pro-fluorescent probe inside different cell lines by the ruthenium(II) complex 36, which is conjugated with a protein ligand R, allowing specific localisation of the reaction in targeted parts of the cell by two-photon excitation at 730 nm.50 | ||
In 2013, Nakamura and co-workers developed a new strategy to selectively modify a protein based on local Single Electron Transfer (SET) catalysis induced by the [RuII(bpy)3]2+ complex 39 (bpy = bipyridine) after visible light irradiation inside mouse erythrocytes.51 The strategy lies on the linkage of the Ru photocatalyst to a ligand of the protein of interest. After visible-light irradiation, the activated RuII* performs a SET reaction onto a tyrosine residue of the protein to form the tyrosyl radical and is oxidised to RuIII (Fig. 15). The resulting tyrosyl radical can then react with a fluorescent tyrosyl radical trapping (TRT) agent 40 to label the protein. By using the RuII complex 39 conjugated with benzene sulfonamide (a ligand of carbonic anhydrase CA) in mouse erythrocytes, the labelling reaction of CA was possible, without modification of other proteins. These results demonstrated that this visible light-induced SET reaction was suitable for bioorthogonal intracellular protein modifications. In order to overcome the limitations of the fluorescence labelling, biotin-conjugated TRT could also be used to enable streptavidin-based detection methods.
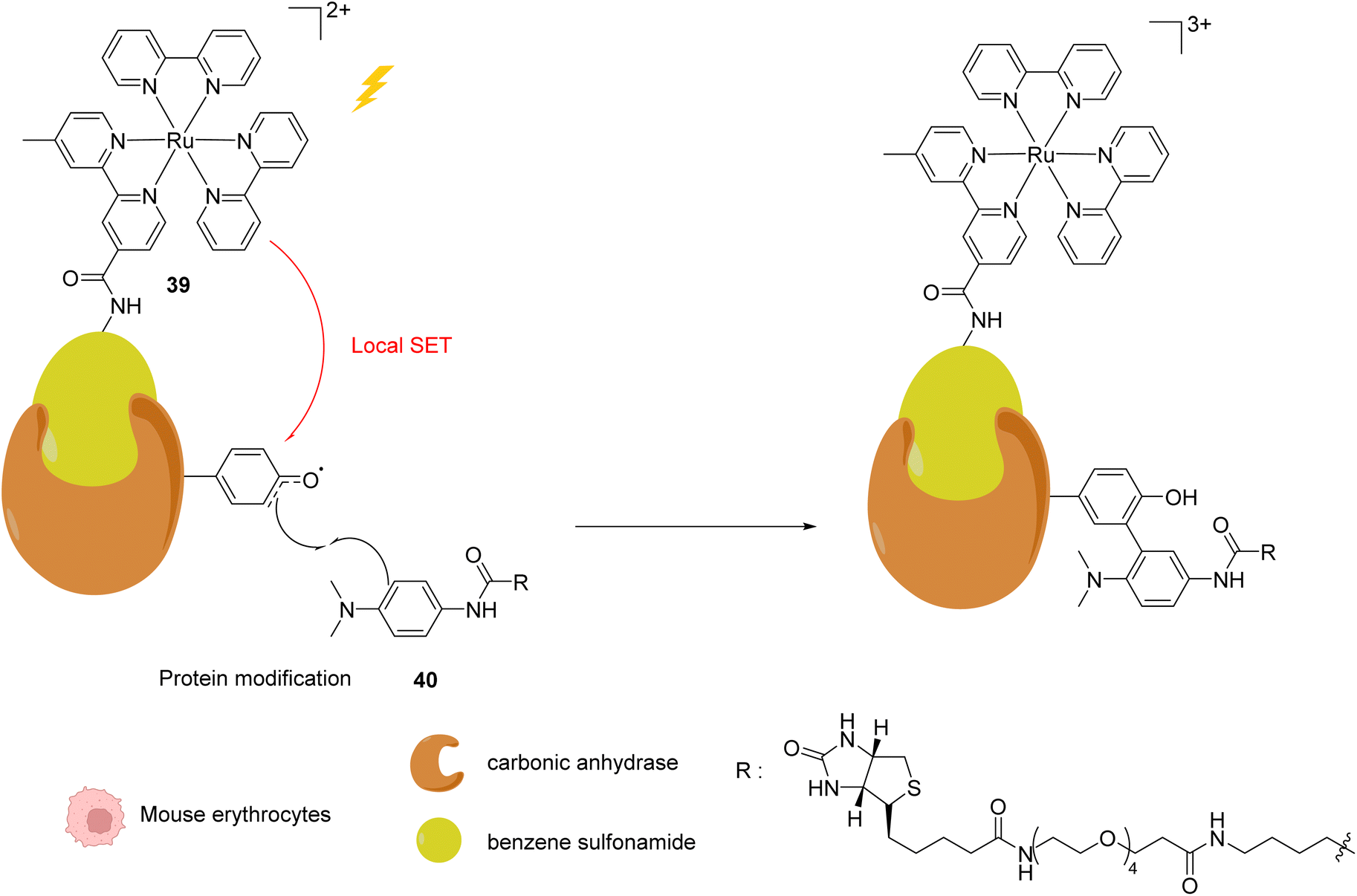 | ||
| Fig. 15 Intracellular visible light-induced targeted protein modification by Single Electron Transfer (SET) catalysed by [RuII(bpy)3]2+ complex 39 in mouse erythrocytes. Labelling of carbonic anhydrase (CA) by using a benzene sulfonamide-conjugated Ru photocatalyst and a biotin N-modified tyrosyl radical trapping agent.51 | ||
In 2015, the same authors reported a similar strategy for inactivating intracellular proteins through a visible light stimulus, using RuII complexes conjugated with ligands for selective protein targeting.52 Upon irradiation but in the absence of tyrosyl radical trapping (TRT) agents, [RuII(bpy)3]2+ type complexes such as 41 were shown to generate singlet oxygen 1O2 leading to the oxidation of histidine, tryptophan and methionine residues in the vicinity of the protein ligand site, leading to the subsequent and selective inactivation (i.e. knock-down) of the protein (Fig. 16). The formation of 1O2 was detected by using a singlet oxygen green sensor (SOSG). This knockdown reaction was applied inside A431 cells with the RuII complex 41, which was conjugated to gefitinib (in red, Fig. 16), a ligand for the epidermal growth factor receptor (EGFR) (overexpressed in A431 cell membranes). The incubation of A431 cells with the Ru-gefitinib compound 41 followed by visible light irradiation was shown, by immunoblotting analysis, to induce a decrease of the intracellular EGFR level without affecting other proteins (such as Akt, PkCα, tubulin and actin). The 1O2-mediated oxidation of EGFR residues likely leads to cross linking of the protein with other proteins or to the oligomerisation of the EGFR, as suggested by the observation of a ladder of bands of higher molecular weights by immunoblotting analysis with an anti EGFR antibody. The addition of TRT in the cell medium resulted in the recovery of the labelling properties of the [RuII(bpy)3]2+ type complex 41 along with the inhibition of its knockdown properties.
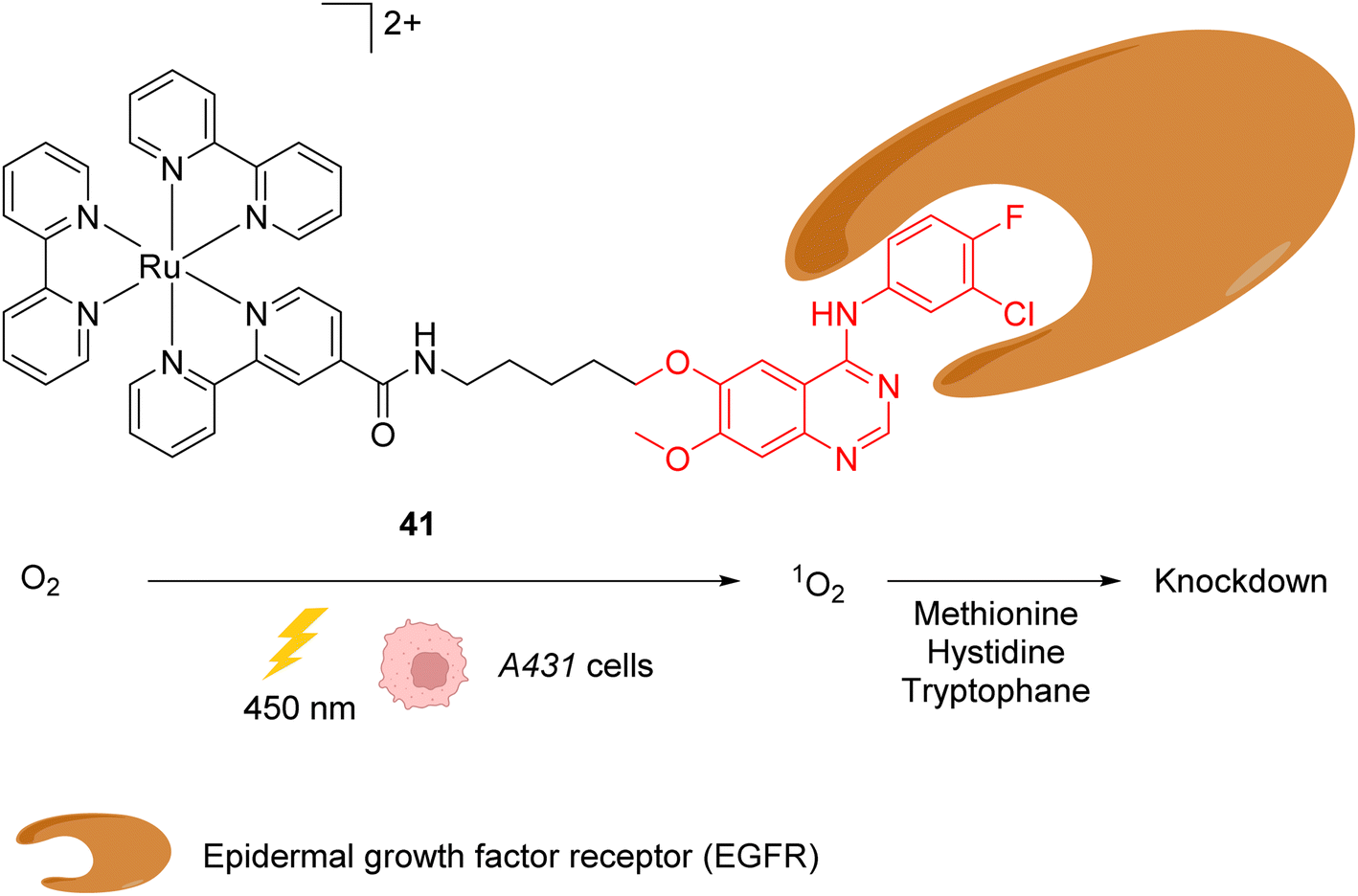 | ||
| Fig. 16 Ru-gefitinib photocatalyst 41 reported by Nakamura et al. for targeted knockdown of the epidermal growth factor receptor (EGFR) protein within A431 cells.52 | ||
In 2021, Mascareñas and co-workers reported a series of ruthenium-arene sandwich complexes 34, 42 and 43 able to catalyse azide-thioalkyne cycloadditions (RuAtAC) under UV light irradiation at 365 nm in biorelevant media such as PBS buffer, cell culture milieu (DMEM) and HeLa cell lysates (Fig. 17).53 Complex 34 can be used as a precatalyst to perform RuAtAC even at diluted concentrations (250 μM). This method was used to modify peptides and oligonucleotides having an azide function by reaction with thioalkyne-substituted molecules. Using this strategy, a fluorophore such as rhodamine was introduced on a DNA fragment. Furthermore, experiments were performed in DMEM-HEPES containing HeLa cells, by mixing thioalkyne, azide, and ruthenium catalysts (with irradiation for 15 min). After cell treatment, the cyclisation product was observed both in the extracellular media and in the cellular content. However, the authors considered that it is likely that at least part of the product internalises after being formed. We note that, in 2012, Meggers et al. used also the ruthenium complex 34 to perform the cleavage of allylcarbamates inside HeLa cells, suggesting that the reaction could take place inside cells.
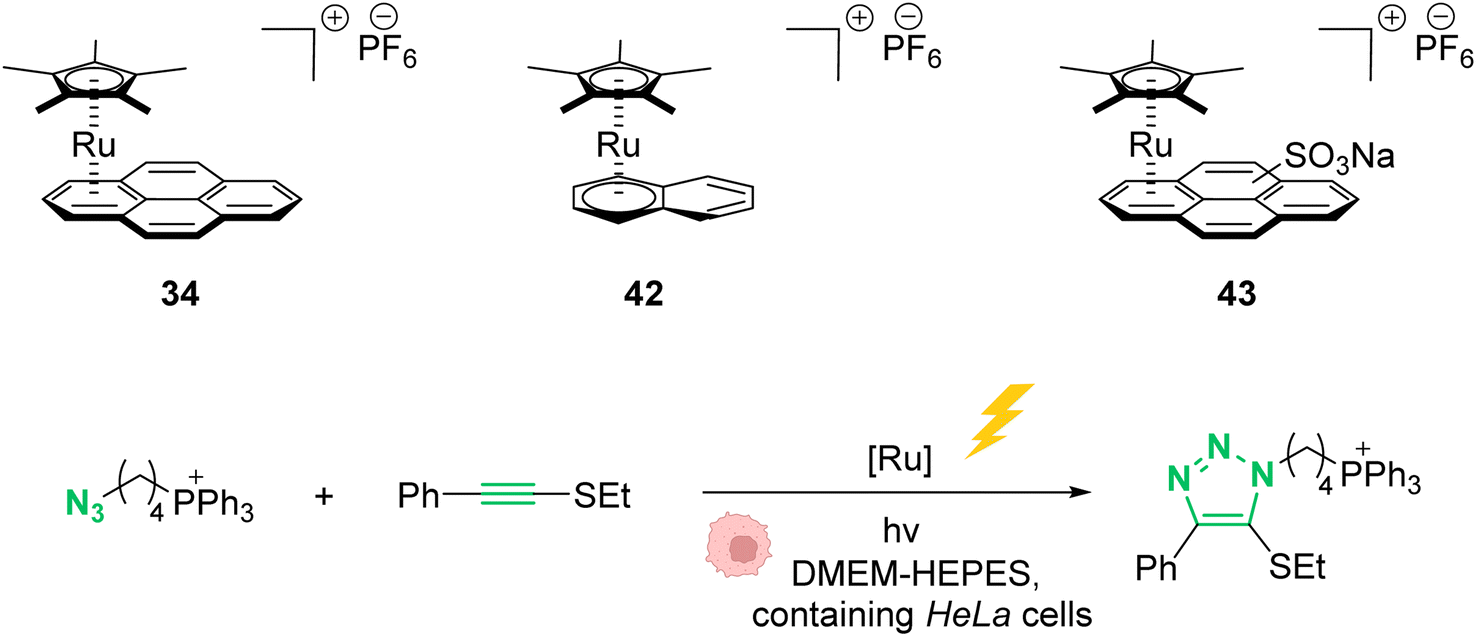 | ||
| Fig. 17 Photo-induced azide-thioalkyne cycloaddition promoted by ruthenium(II) complexes 34 and 42–43 in HeLa cells.53 | ||
The development of photocatalysis allowed the reaction spectrum available by Ru catalysts to be broadened even further. The use of azides that can be photoreduced or coupled with an alkyne in a dipolar cycloaddition has been of particular interest. The spatial and temporal addressing that is brought about by the use of light as the trigger is of course a key feature of these transformations, which can be as diverse as protein modifications with exogenous substrates or photodynamic therapy using endogenous oxygen.
3. Iridium
Iridium complexes have been found to have very interesting properties as anticancer agents and photosensitizers for photodynamic therapies.54–57 Furthermore, some iridium(III) complexes can interact with intracellular NAD(P)H to form hydride complexes. This feature was exploited by several research groups to develop IrIII-based catalytic and photocatalytic systems in normal and cancer cells to unbalance cell redox equilibrium through the generation of reactive oxygen species (ROS) or to reduce carbonyl functions on organic substrates. Furthermore, the properties of some IrI complexes to generate η3-allyl intermediates have been exploited in cells for decaging allyloxycarbonyl-protected profluorophores and prodrugs in the presence of nucleophiles such as GSH.3.1 Catalysis
In 2014, Sadler and coworkers reported two organoiridium(III) complexes 44–45 able to increase the level of reactive oxygen species (ROS) inside cancer cells (Fig. 18).58 Their anticancer activity against human ovarian A2780 cancer cells was first tested. An IC50 of 120 nM was measured for complex 45, which was found to be 6 times more active than the complex 44 and almost 10 times more than cisplatin against this cell line. A screening of the anticancer activity on approximately 60 human cancer cell lines showed that complex 45 exhibited a high efficiency against a wide range of cancer cells. By using the NCI COMPARE algorithm, which quantitatively compares the selectivity for cancer cells of a compound to a database, no correlation was observed between the IrIII complexes 44 and 45 and reported platinum complexes, suggesting a different mode of action. A reaction pathway was proposed, in which the initial complexes 44–45 are hydrolysed into active [IrIII(OH2)]+ species, which can then perform redox catalysis in cancer cells by hydride transfer from NADH to iridium and then to dioxygen (Fig. 18). IrIII complexes 44–45 can be deactivated by reaction with GSH at different rates, complex 45 being less sensitive to deactivation. By using an inhibitor of γ-glutamylcysteine synthetase to decrease the GSH concentration, an increase of ROS was observed by flow cytometry, which could be correlated with an increase of anticancer activity of 44–45.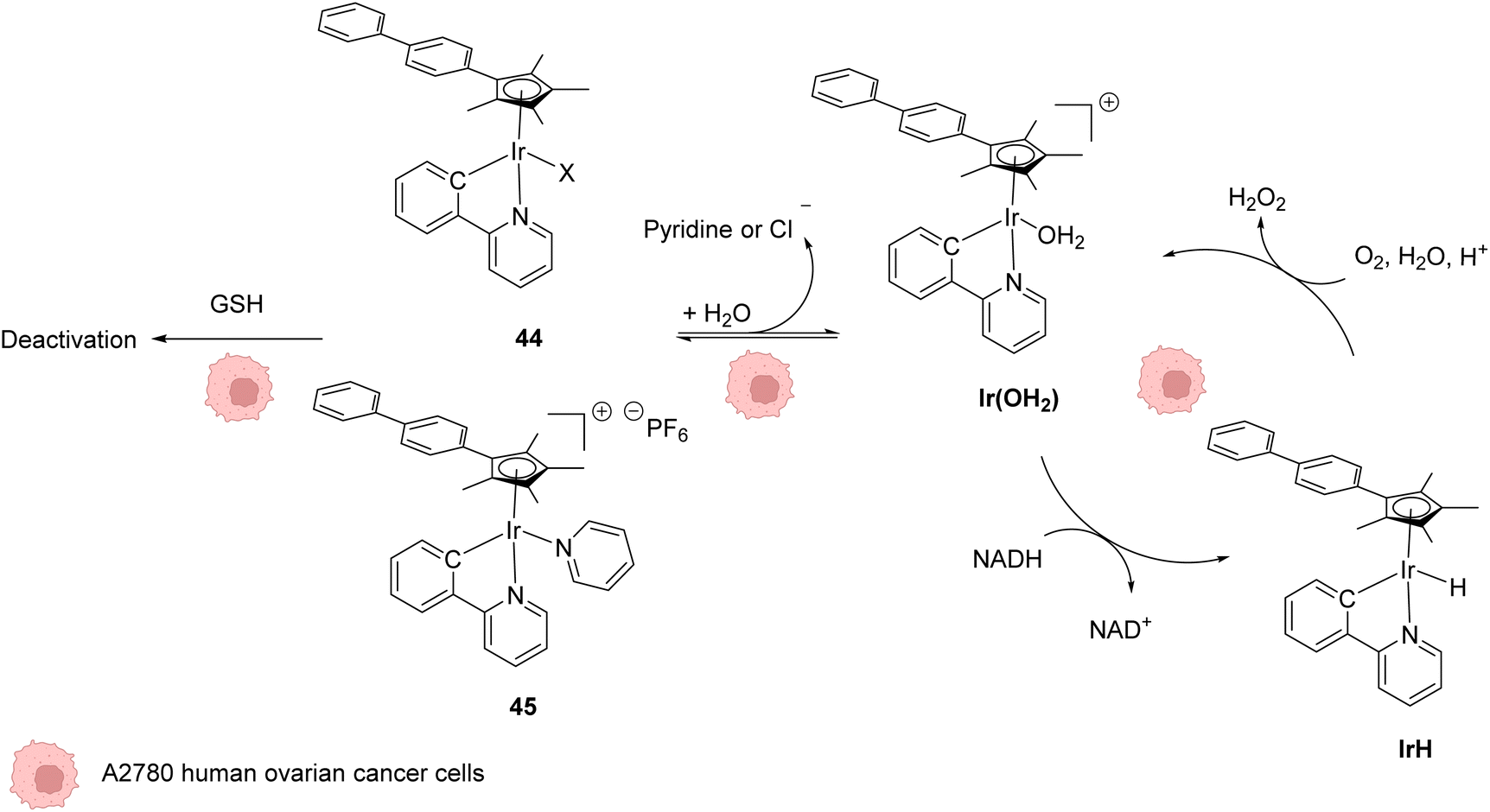 | ||
| Fig. 18 Structures of IrIII complexes 44–45 inducing ROS production inside cancer cells and proposed reaction pathways for ROS generation and activation/deactivation of Ir complexes 44–45 inside mammalian cells.58 | ||
In 2017, Do and coworkers designed an iridium(III) catalyst 46, which can perform hydride transfer from NADH to aldehydes, inside NIH-3T3 mouse embryo fibroblast cells, to generate alcohols (Fig. 19).59 The reaction was monitored inside cells by using a fluorescent probe Bodipy-CHO 47 presenting visible light absorbance (λmax = 480 nm) and having a reduced form (Bodipy-CH2OH, 48), which is about five times more emissive than the aldehyde. Fluorescence enhancement was observed when incubating the cells with both the probe and the complex, indicating the successful reduction of the probe, whereas control experiments (Bodipy-CHO without 46) showed no effect. Moreover, the morphologies of the cells after exposure to complex 46 and cytotoxicity studies showed that 46 was nontoxic at the concentration used (20 μM). The need for NADH to perform the reaction was studied by using sodium pyruvate to inhibit NADH generation. Under these conditions, no fluorescence enhancement was observed, clearly indicating that no reaction can take place without NADH.
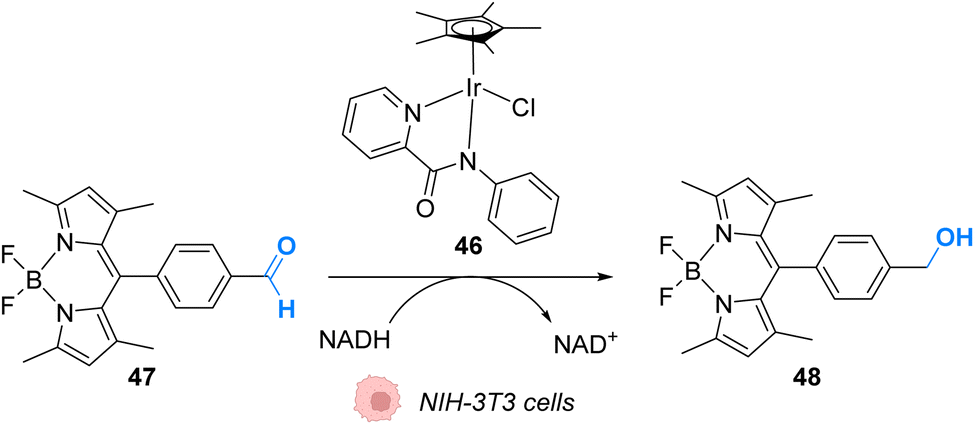 | ||
| Fig. 19 Hydride transfer reaction catalysed by IrIII complex 46 inside NIH-3T3 mouse embryo fibroblast cells inducing intracellular fluorescence enhancement.59 | ||
In 2021, Sasmal and coworkers reported organoiridium(I) complexes 49–51 (Fig. 20) capable of cleaving allyloxycarbonyl(Alloc)-protected amines 52 in phosphate-buffered saline (PBS) in the presence of GSH. The reaction also occurs in the HeLa cell lysate, and inside HeLa cells using the IrI complex 51, without any other exogenous agents (Fig. 20A).60 Within HeLa cells, the catalyst was shown to be active for the cleavage of Alloc groups on protected rhodamine 2 (Fig. 20B), which once decaged into 3 is fluorescent, and for the decaging of the Alloc-protected doxorubicin anticancer prodrug 5. The iridium complex 51 was found to be non-cytotoxic at 20 μM and to exhibit a long period of activity, suggesting negligible deactivation in the biological medium. Inductively coupled plasma mass spectrometry (ICP-MS) showed high Ir accumulation in the cytosol, suggesting that the reaction mostly occurs in the cytoplasm. The nature of the ligand L was found to be important. For instance, complex 49 was found to be inactive, which can be explained by the hydrophobicity of PPh3, leading to low solubility in water. Furthermore, the authors suggested that the bulky structure of PPh3 could cause steric hindrance, impeding substrate binding.
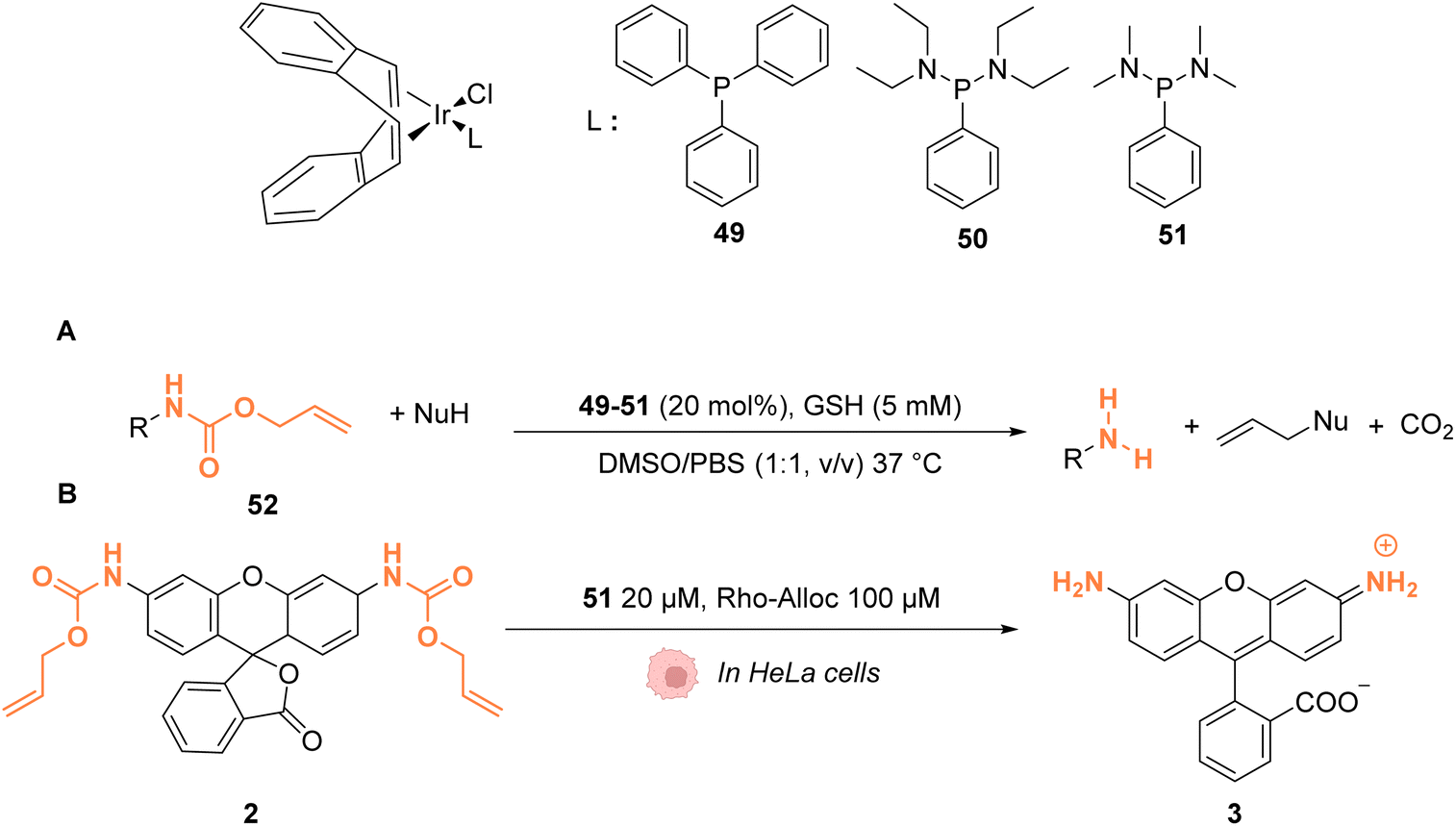 | ||
| Fig. 20 (A) Allylcarbamate (Alloc) cleavage reactions by IrI complexes 49–51 used in PBS buffer and (B) within HeLa cells for decaging of protected rhodamine (R = Alloc).60 | ||
Although there are fewer examples than with Ru, Ir complexes can also catalyse uncaging reactions without the need to add external nucleophiles for deprotection. It is quite noteworthy that these complexes can react with endogenous biomolecules such as NADH to exert their catalytical activity (e.g., ROS production or alcohol oxidation). Alleviating the need to add exogenous reagents is indeed very promising for future medical applications.
3.2 Photocatalysis
In 2019, Gasser, Chao, Sadler and coworkers reported a photocatalytically active iridium(III) complex 53 catalysing photoredox oxidation of NADH within several cancer cell lines such as NCI-H460, HeLa, HepG2 and SGC-7901 under visible light irradiation (λ = 450 nm or 465 nm). Complex 53 was found to be active both under normoxia (20% O2) and hypoxia (1% O2), where photochemotherapeutic agents used in PDT are usually less effective. The authors showed that the light-activated IrIII* catalyst acted as a strong oxidant, able to generate NAD˙ radicals through direct oxidation of NADH. Under hypoxia, cytochrome c (Fe3+, Cyt cox) acted as a co-oxidant to regenerate IrIII from IrII in the catalytic cycle but also to form NAD+ from NAD˙ (Fig. 21).61 This induces an imbalance of cell redox equilibrium, leading to cell death. In cells, 53 was found to mainly localise in mitochondria. In the dark, 53 was found to be nontoxic for cells at the active concentrations used, an important property to reduce the side effects of therapeutic treatments. 53 was also found to be photostable under visible light irradiation. Finally, 53 was shown to be active toward A549 lung cancer multicellular spheroids of ∼800 μm diameter after two-photon irradiation (760 nm), thus demonstrating its potential for the treatment of large or deep-seated tumours. By comparison, 5-ALA (a photosensitizer precursor) and cisplatin, used as controls, did not present any photocytotoxicity on the same tumor models.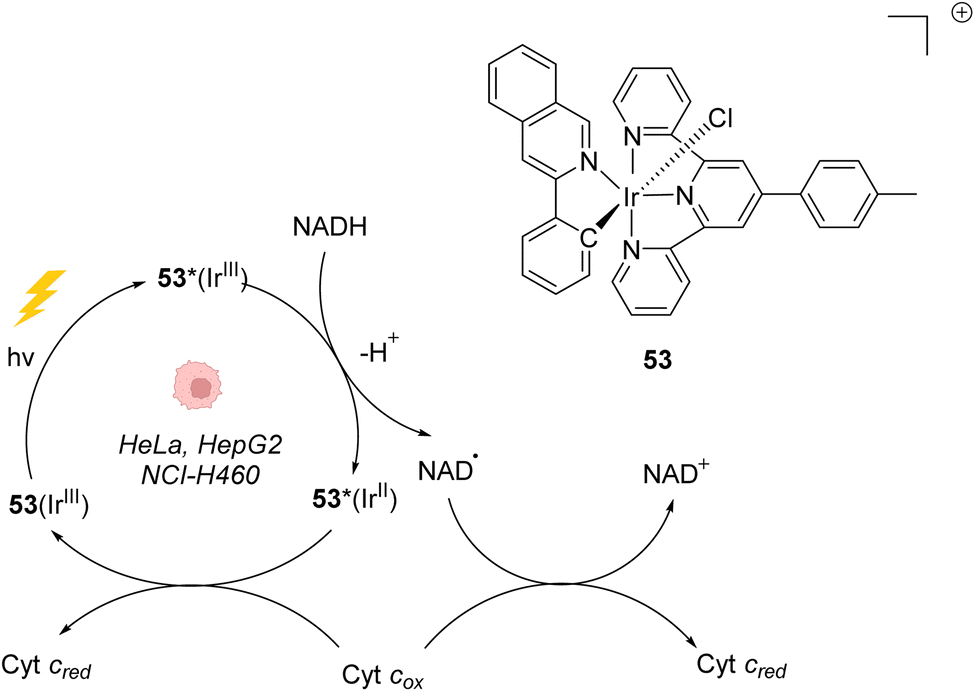 | ||
| Fig. 21 Photoredox IrIII-based catalytic cycle proposed by Sadler et al. for in cellulo light-induced NADH oxidation under hypoxic conditions.61 | ||
In 2021, Huang et al. presented an organoiridium(III) photocatalyst 54 with a coumarin-based ligand, which exhibits excellent biocompatibility (Fig. 22).62In cellulo, 54 was found to be active after visible light irradiation (λ = 465 or 525 nm) and to allow the oxidation of NADPH and amino acids via single electron transfer (SET) with a high turnover frequency. The photo-induced oxidation reaction imbalances cell redox equilibrium by oxidising34 NAD(P)H and generating ROS. It also induces changes in mitochondrial membrane potential, leading to necrosis and apoptosis of cancer cells. 54 was found to be nontoxic towards both normal and cancer cells in the dark but is highly cytotoxic after light exposure towards cancer cells including sorafenib-resistant (HepG2-SR) and cisplatin-resistant cells (A549R). In HepG2 cells, 54 was found to be three times less active under hypoxia (5% O2) than under normoxic conditions (20% O2), showing the important role of O2 in the mode of action. In contrast to previously described IrIII photocatalysts, it was proposed that the IrIII catalyst acted as a strong excited-state reductant, leading to direct intracellular O2˙− production from O2. The IrIV intermediate is then reduced to IrIII by NAD(P)H to close the catalytic cycle, generating NADP˙ (Fig. 22). Due to its hydrophilicity, the cellular uptake of 54 was found to be low (in CNE-2Z cells) but to be enhanced after photoexcitation, leading to a distribution both in the lysosome and mitochondria, determined by confocal microscopy without washing before irradiation. The photocatalytic system was shown to be highly biocompatible in vivo inducing no damage to tissues in zebrafish embryos in the dark. Finally, 54 showed light-induced activity against a mouse CT26 colon carcinoma model with a significant decrease of the tumor volume and weight after light irradiation (465 nm).
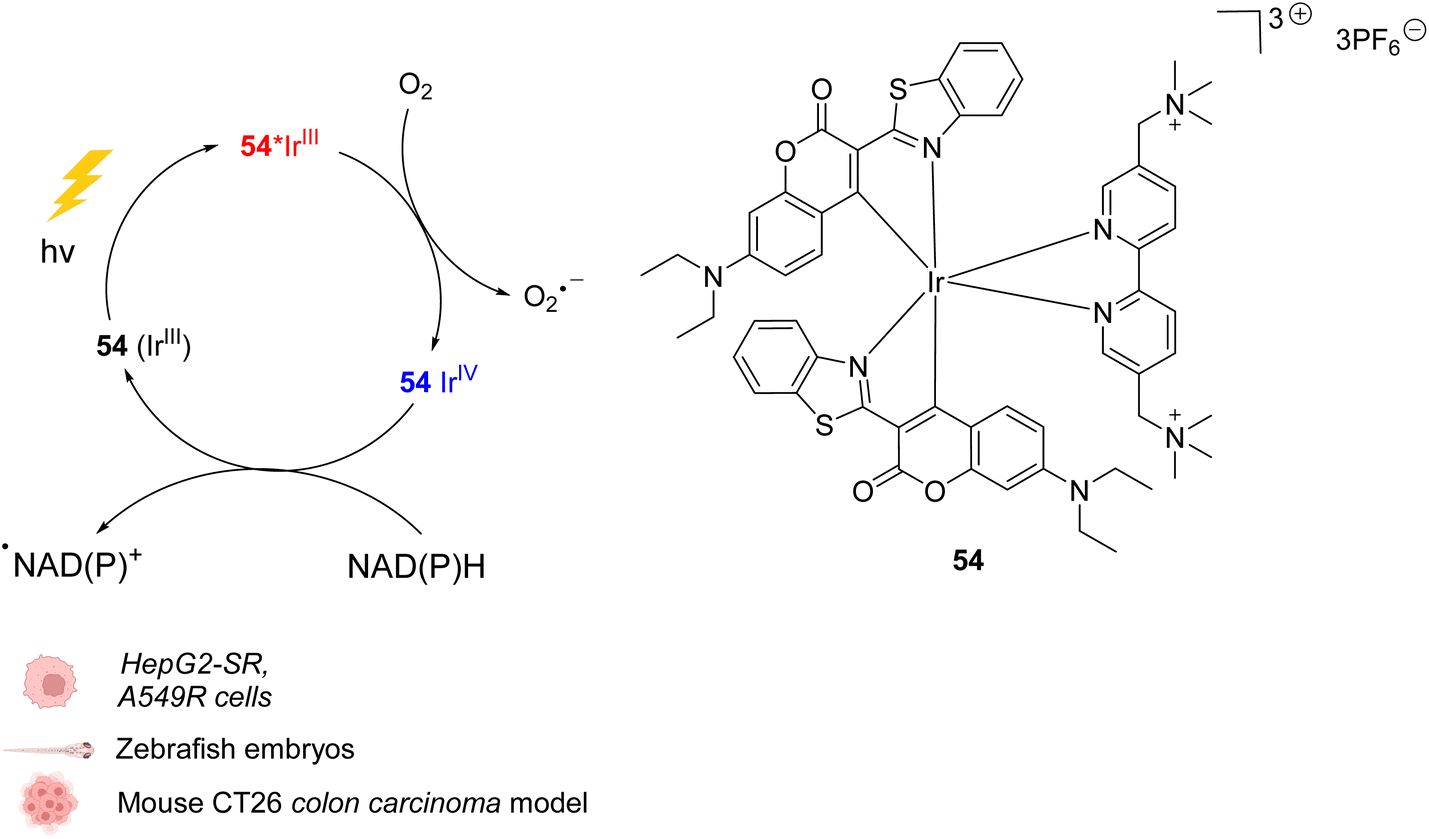 | ||
| Fig. 22 Biocompatible IrIII complex 54 developed by Huang et al. and proposed photoredox IrIII/IrIV-based catalytic cycle for in cellulo light-induced ROS generation and NAD(P)H reduction in the presence of O2.62 | ||
Iridium photocatalysis has been developed very recently and so far only for photodynamic therapy purposes. In this setting, the possible reaction with NADH greatly improves the therapeutic scope of these complexes as they can thus be applied in hypoxic environments.
4. Palladium
Palladium is a prime metal for organometallic catalysis, and it is therefore logical that it has been investigated for in cellulo systems. Homogeneous and heterogeneous systems have been used. For instance, palladium-based bond cleavage catalysis in living systems was performed by using Pd nanoparticles,20 which operate in the extracellular medium. Such approaches require implanting Pd resins on the reaction site. Cleavage reactions catalysed by heterogeneous catalysts can also be induced inside cells but encountered some issues such as poor cellular uptake, poor solubility, or instability of the Pd nanoparticles under biological conditions. Therefore, various well-defined PdII and Pd0 complexes have been tested for intracellular “homogeneous” catalysis. Examples of cross-coupling reactions have been reported in bacteria and on a mammalian cell surface.63–65 However, the main applications of discrete Pd complexes lie in allyl, propargyl and allenyl cleavage reactions for prodrug and profluorophore activation as well as nitric oxide (NO) release in various types of cells. No photocatalytic application of Pd complexes have been reported to date to the best of our knowledge.In 2011, Lin and coworkers reported a copper-free (Heck–Cassar) Sonogashira cross-coupling reaction in E. coli cells. The coupling reaction involved fluorescein iodide 55 and homopropargylglycine-encoded-Ubiquitin (HPG-Ub) with one HPG at its C-ter (Fig. 23A).65 After screening of several palladium ligands to perform Sonogashira cross-coupling reactions in water, the robust aminopyrimidine/PdII complex 56 was selected for evaluation inside E. coli. HPG-Ub-overexpressing M15A cells were incubated with 1 mM of palladium complex, 100 μM of fluorescein iodide and 5 mM of sodium ascorbate in PBS buffer. After 4 h at 37 °C and after washing, cells incubated with the palladium complex showed green fluorescence under 365 nm excitation while the negative control (cells not incubated with the palladium complex) did not. This result was confirmed by SDS-PAGE after cell lysis, suggesting that the cross-coupling reaction takes place inside the bacteria. We note that this cross-coupling reaction requires incubation with 50 equiv. of the Pd complex per protein substrate to be efficient. Moreover, some level of toxicity was observed when cells where incubated with PdII complex 56 without preactivation, whereas with preactivation, no toxicity was observed. This preactivation consists in stirring for 1 hour before the addition of a mixture of the fluorescein iodide 55, the PdII complex 56 and sodium ascorbate at 37 °C.
 | ||
| Fig. 23 (A) Copper-free Sonogashira cross-coupling reaction inside E. coli for fluorescent labelling of HPG-Ub protein by modified fluorescein using a discrete PdII precatalyst. (B) Sonogashira cross-coupling labeling of Myoglobin inside E. coli.65,66 | ||
Later, in 2014, Lin and coworkers reported a Sonogashira cross-coupling reaction inside E. coli and on a mammalian cell surface involving an alkynyl-modified protein and an aryl-iodide using the Pd-based complex 56.66E. coli DH10B cells, which express the modified myoglobin protein containing modified lysine bearing a butynyl group (57), were incubated with a solution of the active Pd complex 56 freshly preformed in DMSO from Pd(OAc)2, an aminopyrimidine ligand, fluorescein iodide 55 and sodium ascorbate (Fig. 23B). The C–C coupling reaction was monitored by in-gel fluorescence and SDS-PAGE of the modified protein (58).
In 2013, Chen and coworkers also developed a Sonogashira cross-coupling reaction between a green fluorescent protein (GFP) bearing an alkyne group 59 and a fluorophore (Fluor 525) linked to an iodophenyl group 60, inside E. coli and pathogenic Shigella bacterial cells (Fig. 24).67 The activity of twelve commercially available Pd complexes was studied, showing that Pd(NO3)2 was the most active catalyst. A ligand-free approach presents assets because the catalyst can be transposed from in vitro to in vivo catalytic reactions without looking at the ligand cell permeability. No loss of fluorescence was observed in the presence of Pd(NO3)2, suggesting no denaturation of fluorescent GFP. Similarly, the fluorescence of the more fragile protein luciferase remained intact in the presence of Pd(NO3)2, suggesting no denaturation. Moreover, Pd(NO3)2 can pass through the cellular membrane of E. coli and Shigella bacterial cells and was shown to be non-cytotoxic towards these cells at the concentration used (C = 200 μM), thus demonstrating its biocompatibility. However, comparison with previous results from the literature led the authors to suggest that the Pd(NO3)2-induced reaction might involve Pd nanoparticles that could be formed in the presence of sodium ascorbate. It is important to note that the use of Pd(NO3)2 allowed efficient coupling reactions to take place with only 2 equiv. of Pd per protein substrate. The catalytic system was applied to develop bioorthogonal labeling and tracking of toxin proteins in Shigella pathogens.
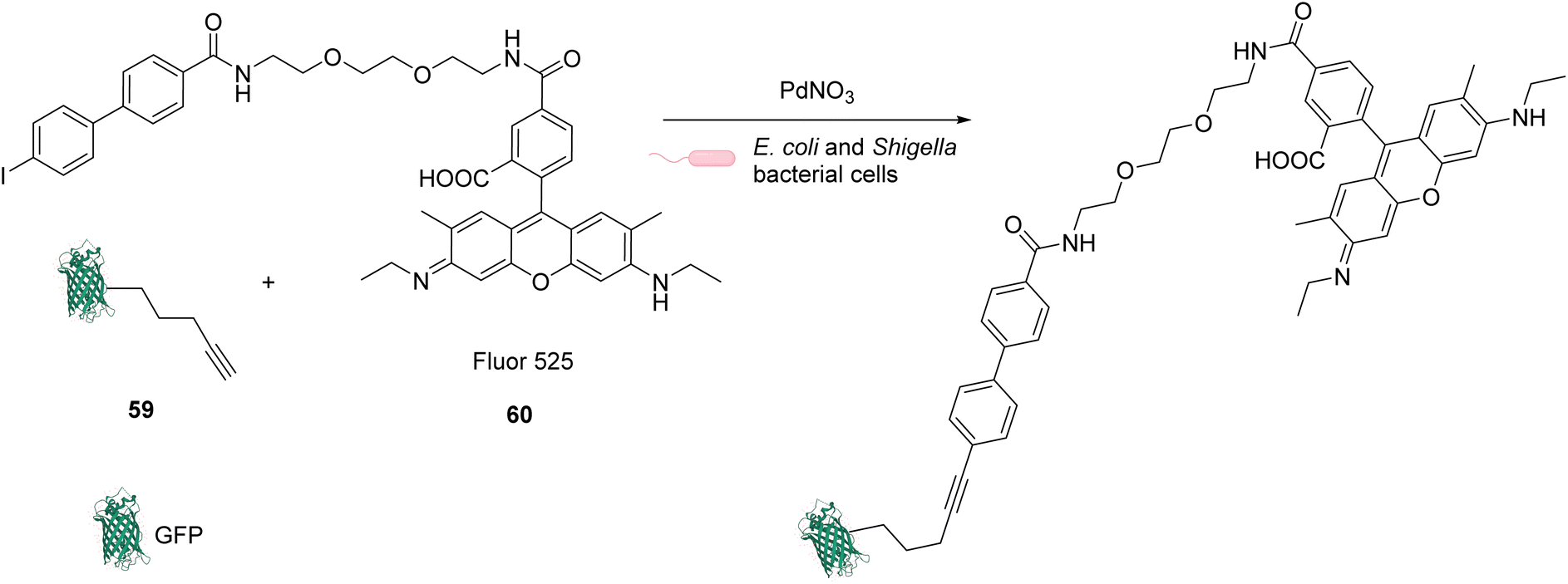 | ||
| Fig. 24 Sonogashira cross-coupling inside E. coli or Shigella cells with Pd(NO3)2 as a precatalyst for coupling of fluorescent protein GFP with Fluor 525 and bioorthogonal labeling of toxin proteins in living pathogenic Shigella cells.67 | ||
In 2014, Chen and coworkers reported that palladium complexes were able to induce cleavage reactions of allyl or propargyl carbamate groups inside cells such as HeLa, HEK293T, NIH3T3 and A549 cells.68 The cleavage activity of a series of commercially available Pd0/PdII/PdIV precatalysts 62–67 was studied using the protected profluorophore 61 and the Alloc-protected rhodamine 2 as probes (Fig. 25). Pd(dba)2 (65) (Pd0) and Pd2(allyl)2Cl2 (66) (PdII) were found to be the most active complexes of the series and were both more active for propargyl than allyl cleavage. The addition of an organic ligand (68–70) was found to decrease complex activity. In HeLa cells, complexes 65 and 66 were found to be non-cytotoxic at 10 μM, while still being active. Moreover, the reaction could be performed on different proteins without denaturation. Pd2(allyl)2Cl2 (66) was also shown to be active inside HeLa cells for the cleavage of propargyl groups on modified proteins (such as GFP), protected by a propargyl group on a lysine residue as in 71. After the incubation of the cells with Pd2(allyl)2Cl2 (66) and the modified protein, the propargyl cleavage was quantified by gel fluorescence through labeling of the unreacted protein using a click reaction, showing an efficiency of 31%. The authors suggested that the low yields could be correlated with low concentration of the protein of interest and of the nonspecific absorption of palladium complexes, leading to low effective catalyst concentration in cells. Despite this disadvantage, the Pd-mediated propargyl cleavage reaction was used for restoring the activity of inactive modified (enzymatic) proteins. For instance, in Shigella type III cells, a phosphothreonine lyase (OspF) induces the dephosphorylation of phosphorylated Erk (pErk). The mechanism involves Lys 134 as a residue. The substitution of this residue by propargyl-Lys (ProcLys) induces the loss of enzyme activity. The cleavage of the propargyl on ProcLys-134 by Pd(OAc)2 (62) occurred with a yield of 28% within host cells (followed as before by gel fluorescence after the click reaction), inducing the restoration of over 80% of enzyme activity compared to the wild type for pErk dephosphorylation. This process was shown to take place in the cell nucleus.
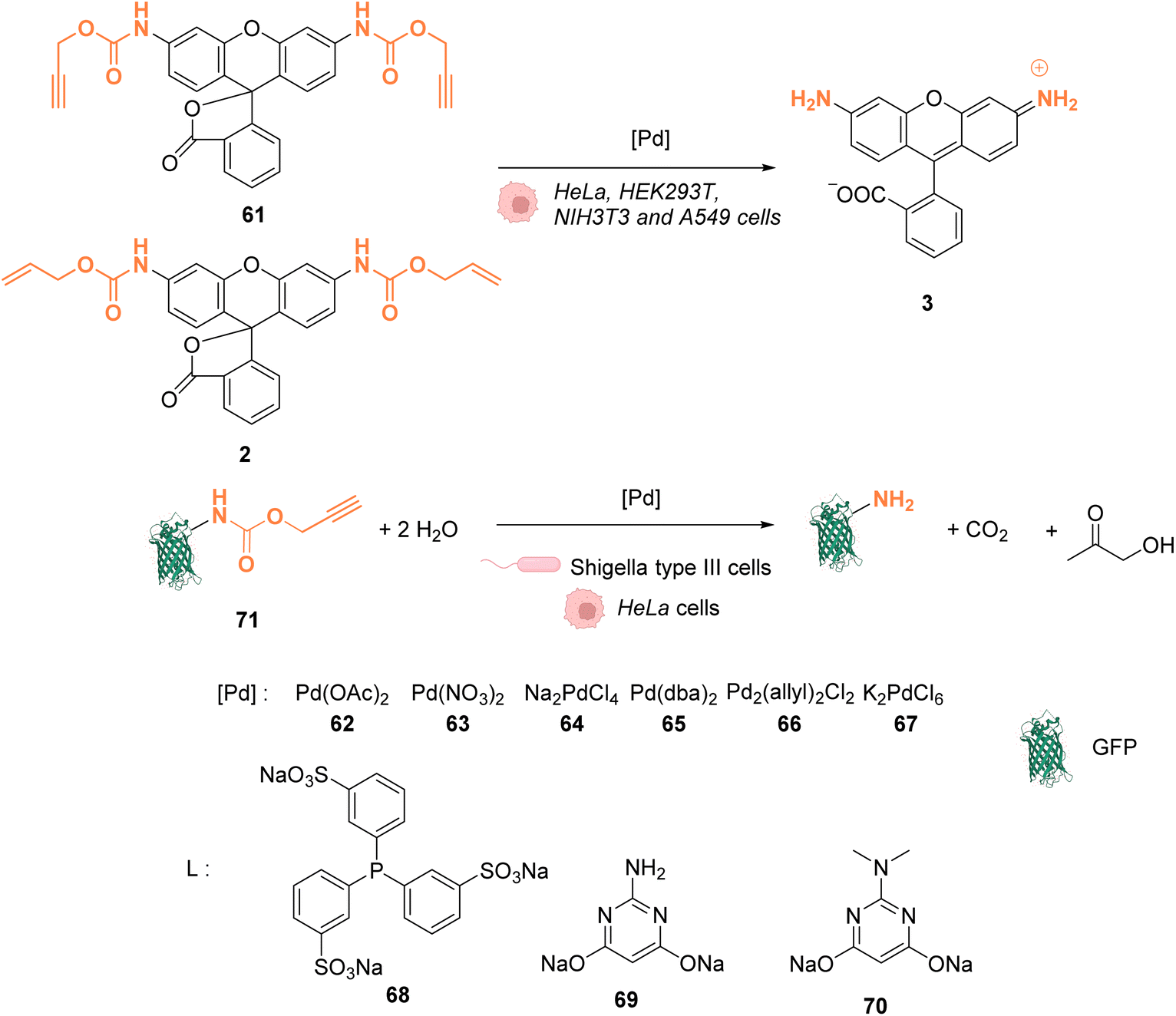 | ||
| Fig. 25 Alloc and propargyloxycarbonyl deprotection of profluorophores by palladium complexes inside cells.68 | ||
In 2016, Chen and coworkers reported the cleavage of allenyl groups on tyrosine residues on several proteins inside cells by using palladium complexes Pd(dba)2 (65), Pd2(allyl)2Cl2 (66), Pd(TPPTS)4 (72) and Pd(TAPAd)4 (73) (Fig. 26).69 The bond cleavage reaction was studied in vitro on purified proteins and inside HEK293T cells. The authors showed that complexes 65, 66, 72, and 73 could catalyse the cleavage of a GFP modified with an allene group on the Tyr149 residue (GFP149AlleY) at a concentration of 10 μM. Pd(TAPAd)4 (73) was found to be the most active compound in vitro, whereas Pd2(allyl)2Cl2 (66) was the most active in living cells. This difference of activity was linked to a difference in the cellular uptake of the complexes that was quantified by ICP-MS. Allenyl cleavage was also performed on other proteins inside HEK293T cells: Pd(dba)2 (65)and Pd2(allyl)2Cl2 (66) could rescue enzyme activity by cleaving allenyl groups on the tyrosine residue involved in enzyme activity. The activities of a phosphorylase (Src) and a protease (anthrax lethal factor) that had been inactivated were rescued by incubating cells with 20 μM of complex 65 or 66.
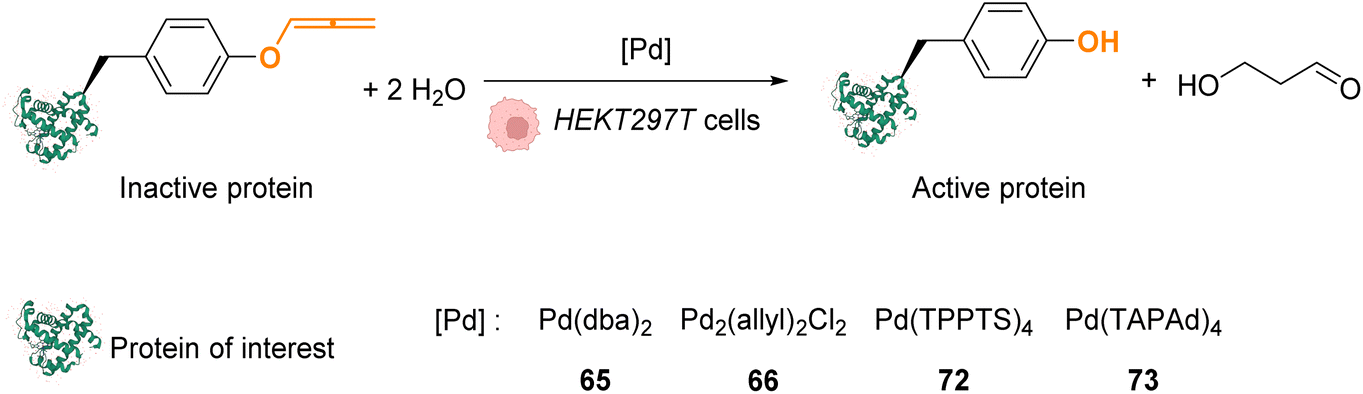 | ||
| Fig. 26 Allenyl group cleavage on tyrosine residue inside HEKT297T cells to restore the activity of Tyr-dependent enzymes.69 | ||
In 2017, Bradley and coworkers developed a discrete organopalladium catalyst able to catalyse propargyl-cleavage of Proc-rhodamine 61 inside human PC-3 cancer cells.70 Complex 74 is composed of an NHC ligand linked to a polycationic cell penetrating peptide (tri-lysine), which is itself bound to a Cy5 fluorophore (Fig. 27). Inside cells, 74 is mainly distributed in the cytosol but was also detected inside the nucleus. This distribution was monitored by fluorescence thanks to the rhodamine probe. 74 showed a low cytotoxicity toward PC-3 cells: no cytotoxicity was observed even at 200 μM. For in cellulo catalysis, PC-3 cells were incubated with 30 μM of 74, the extracellular catalyst was washed, and 50 μM of Proc-rhodamine 61 was added. The formation of rhodamine 3 was verified by flow cytometry, indicating that 74 was catalytically active for the propargyl-cleavage of Proc-rhodamine 61 inside PC-3 cells.
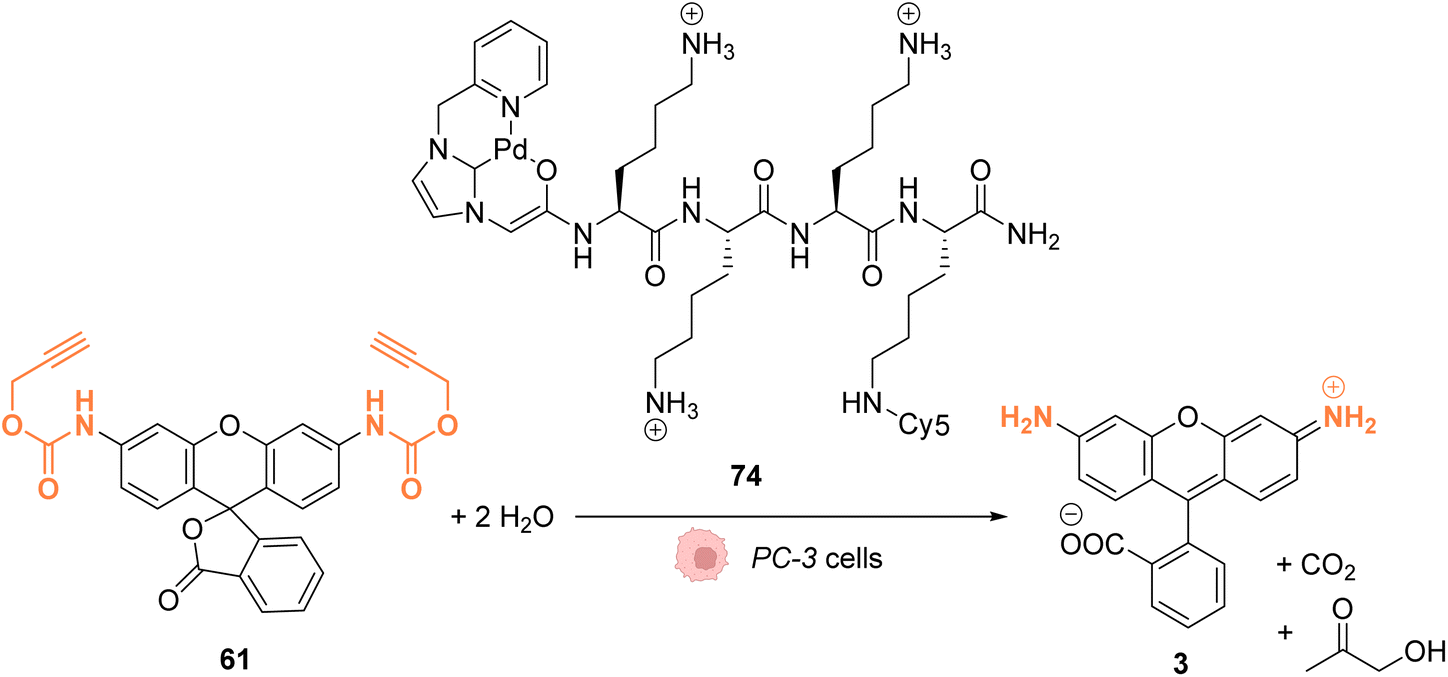 | ||
| Fig. 27 Propargyl deprotection of a profluorescent probe by a discrete palladium complex inside PC-3 cancer cells. The ligand is linked to a polycationic cell penetrating peptide to favor cellular uptake.70 | ||
In 2018, Huang and coworkers reported the use of Pd(dba)2 as a biocompatible Pd0 catalyst to induce the bond cleavage of O2-alkyl-derived diazeniumdiolates 76–84 and release nitric oxide (NO), via the intermediate 75, inside living cells to control cell growth.71O2-propargyl- (76–80), O2-allyl- (81) and O2-(4-alkyloxybenzyl)-diazeniumdiolates (82–84) have been evaluated as substrates for intracellular N–O bond cleavage inside HCT116, A549, HL-60, and OVCAR5 cancer cells (Fig. 28). Cells were preincubated with 1 μM of Pd(dba)2 and washed before incubation of 1 μM of the diazeniumdiolate derivatives (76–82). Under these conditions, 76 and 78 were found to be the most efficient substrates to release NO, inducing significant inhibition of cell growth after 48 h in the presence of Pd(dba)2 (65). Control experiments without Pd(dba)2 (65) showed that the antiproliferative activity of 76/78 was ca. 17 times lower on HCT116 and A549 cells and even lower on human normal colonic epithelial cells (IC50 = 68.1 μM for colonic cells compared to 27.2 μM and 33.9 μM, respectively, for HCT116 and A549).
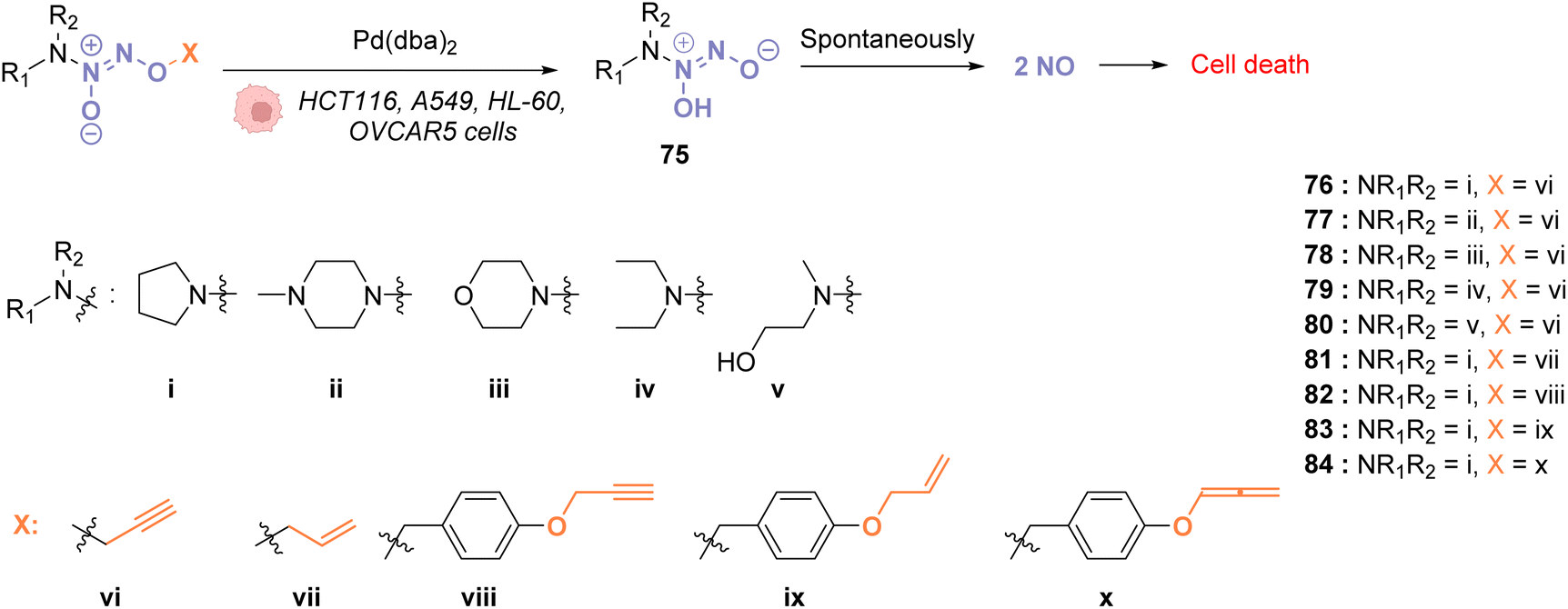 | ||
| Fig. 28 Cleavage of different protecting groups by Pd(dba)2 inducing NO release in living cancer cells.71 | ||
In 2018, Mascareñas and coworkers designed several palladium(II) complexes 87–92, bearing a variety of N, S or P ligands, which were found to be catalytically active in water for the cleavage of propargyl protecting groups on a phenol derivative 85 (Fig. 29) or for an Alloc group deprotection on rhodamine 2.72 The activity of the PdII complexes 87–92 was then tested in cell lysates and in living mammalian cells (Vero and HeLa cells). Catalysts bearing phosphine ligands (89–92) were shown to be the most active inside cells although the yields were still low for the propargyl and Alloc cleavage. This observation suggested a superior stability of the phosphine ligands in biological media (compared to N and S ligands). Moreover, the phosphine palladium complexes were found to have a superior cellular uptake, while being nontoxic at a concentration of 50 μM. Phosphine ligands are an asset in this study since they can be easily modified to modulate reactivity and cellular uptake or to target cells: a phosphonium group or a hydrophobic pyrene group can lead to a preferential accumulation of the catalyst in mitochondria.
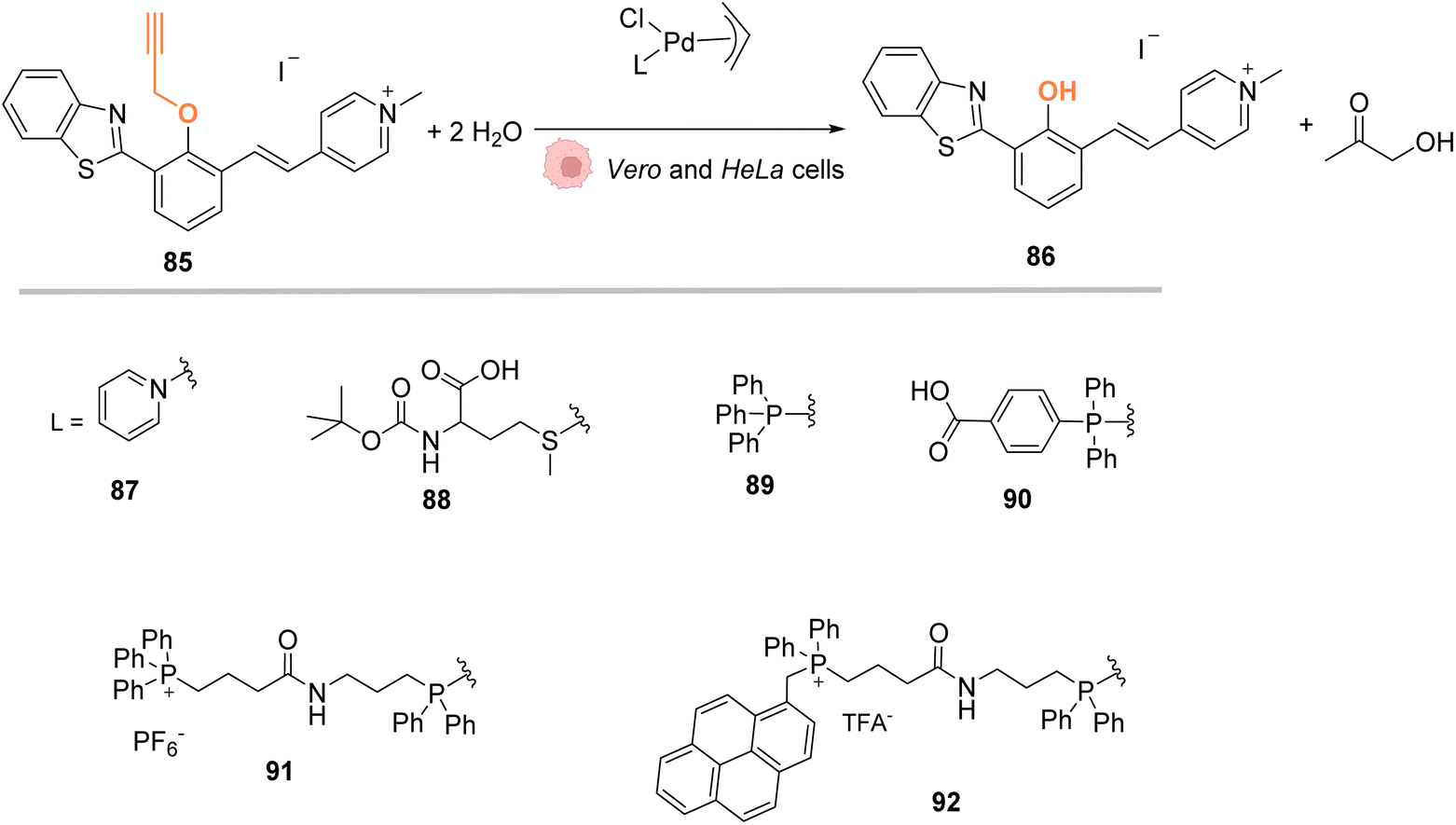 | ||
| Fig. 29 Propargyl cleavage on a profluorophore catalysed by well-defined palladium(allyl) complexes in Vero and HeLa cells.72 | ||
In 2019, Bradley, Lilienkampf and coworkers reported the propargyl cleavage of an anticancer protected prodrug 93 inside MCF-7 cells by the NHC-PdII catalyst 95 (Fig. 30).73 Complex 95 showed a good activity at 10 mol% for the cleavage of propargyl-5-FU (5-pFU, 93) in MCF-7 cells after 24 h incubation with 93. This cleavage reaction leads to the release of 5-FU (94), an anti-cancer drug. The PdII catalyst 95 was also found to be catalytically active in a 3D cancer cell culture (cancer cell spheroids) for the cleavage of the propargyl group on 5-pFU 93. In this case, the toxicity generated through the deprotection of the prodrug 93 by the PdII complex 95 was found comparable to that of the control experiment with 5-FU (94). The cleavage reaction occurred inside and outside the cells.
 | ||
| Fig. 30 Propargyl cleavage of a prodrug inside MCF-7 cells and in a tumor model using a well-defined NHC-PdII precatalyst.73 | ||
In 2022, Bernardes and coworkers demonstrated that O-protected or N-protected prodrugs and profluorophores protected by allyl ether or allyl carbamate groups could be deprotected in living cells using Na2PdCl4 as a simple source of water-soluble palladium(II) (Fig. 31).74 Na2PdCl4 is a good candidate for intracellular uptake, better than the Pd0/TPPTS system employing a water-soluble phosphane ligand. The system was shown to be operative in SKBR3 breast cancer cells. After preincubation with Na2PdCl4, the cells were submitted to sodium ascorbate and then incubated with the substrates. The PdII precatalyst is reduced in cellulo to active Pd0 species by sodium ascorbate (no decaging reaction was observed in its absence). The exact structure of intracellular catalytically active Pd0 remains to be clarified, although kinetic studies suggest the contribution of nanoparticles in the reaction. The method was applied for in cellulo bioorthogonal cleavage of the allyl group on O-protected duocarmycin 97 and of the Alloc group on N-protected doxorubicin 6 (two anticancer prodrugs) inside SKBR3 breast cancer cells. In both cases, the cytotoxic activity of the free drugs 5 and 96 was recovered after sequential incubation with Na2PdCl4 and then sodium ascorbate.
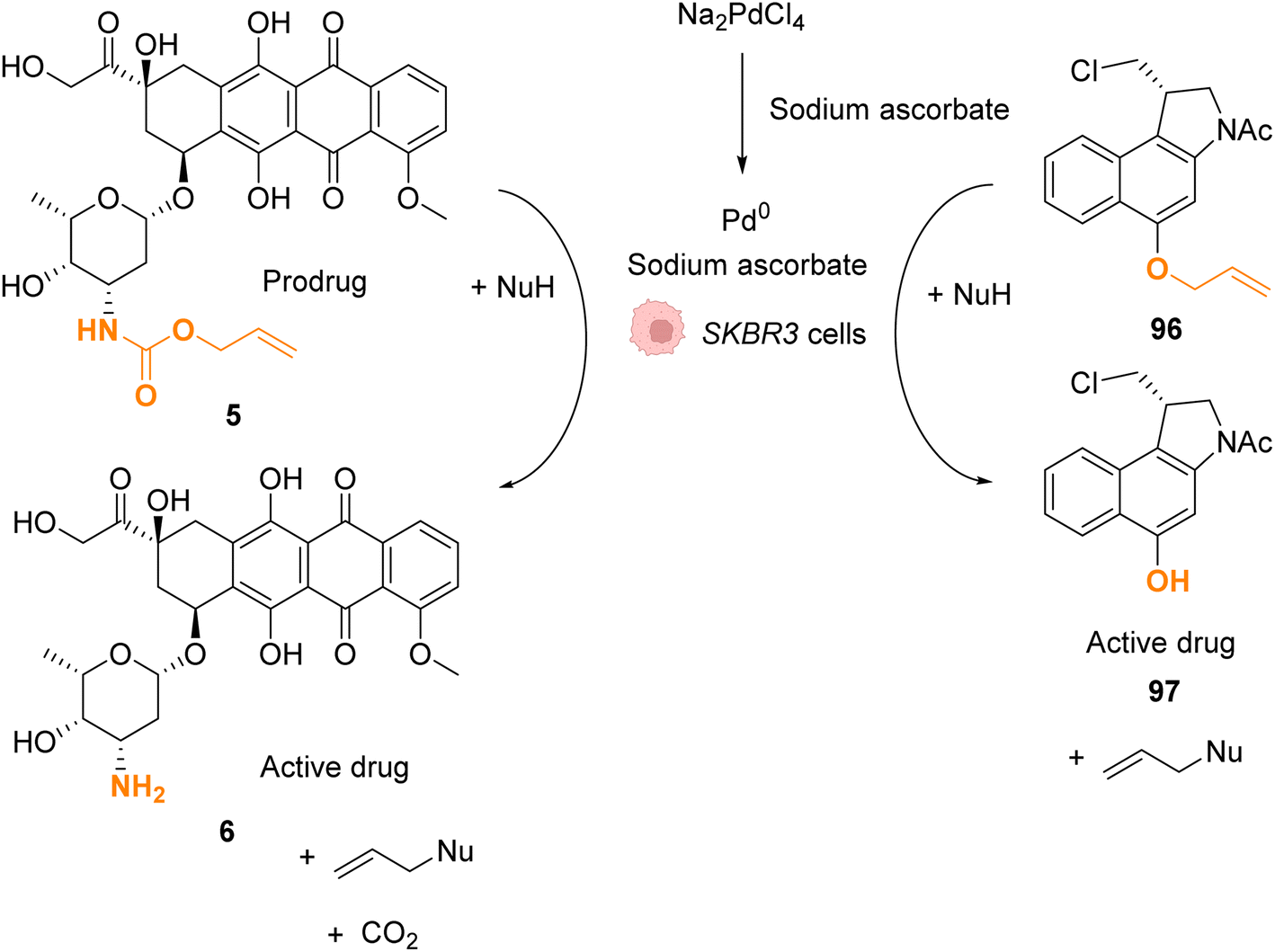 | ||
| Fig. 31 Allyl ether or allyl carbamate cleavage by a palladium complex inside SKBR3 cells.74 | ||
In 2022, Mascareñas and coworkers reported a new approach for controlling DNA binding based on a palladium(0)-mediated uncaging process under mild and physiological conditions (Tris buffer). The authors designed a dimer, based on basic leucine zippers (bZIP) linked with a phenol derivative protected with a propargyl group 98, which is cleavable on demand using Pd(allyl) precatalysts 66–90 (Fig. 32).75 The propargyl cleavage of the phenol leads to a chain reaction, releasing the two monomers (Fig. 32). The dimeric form 98 has a good affinity for DNA unlike the monomer 99. The binding of DNA could therefore be modulated by the addition of a PdII precatalyst. This on/off process has been performed under biological conditions but never in cellulo nor in vivo. Considering that this kind of propargyl-cleavage reaction has already been undertaken in cellulo with this family of PdII catalysts,9 the authors suggested that it was likely possible to be transposed in living cells.
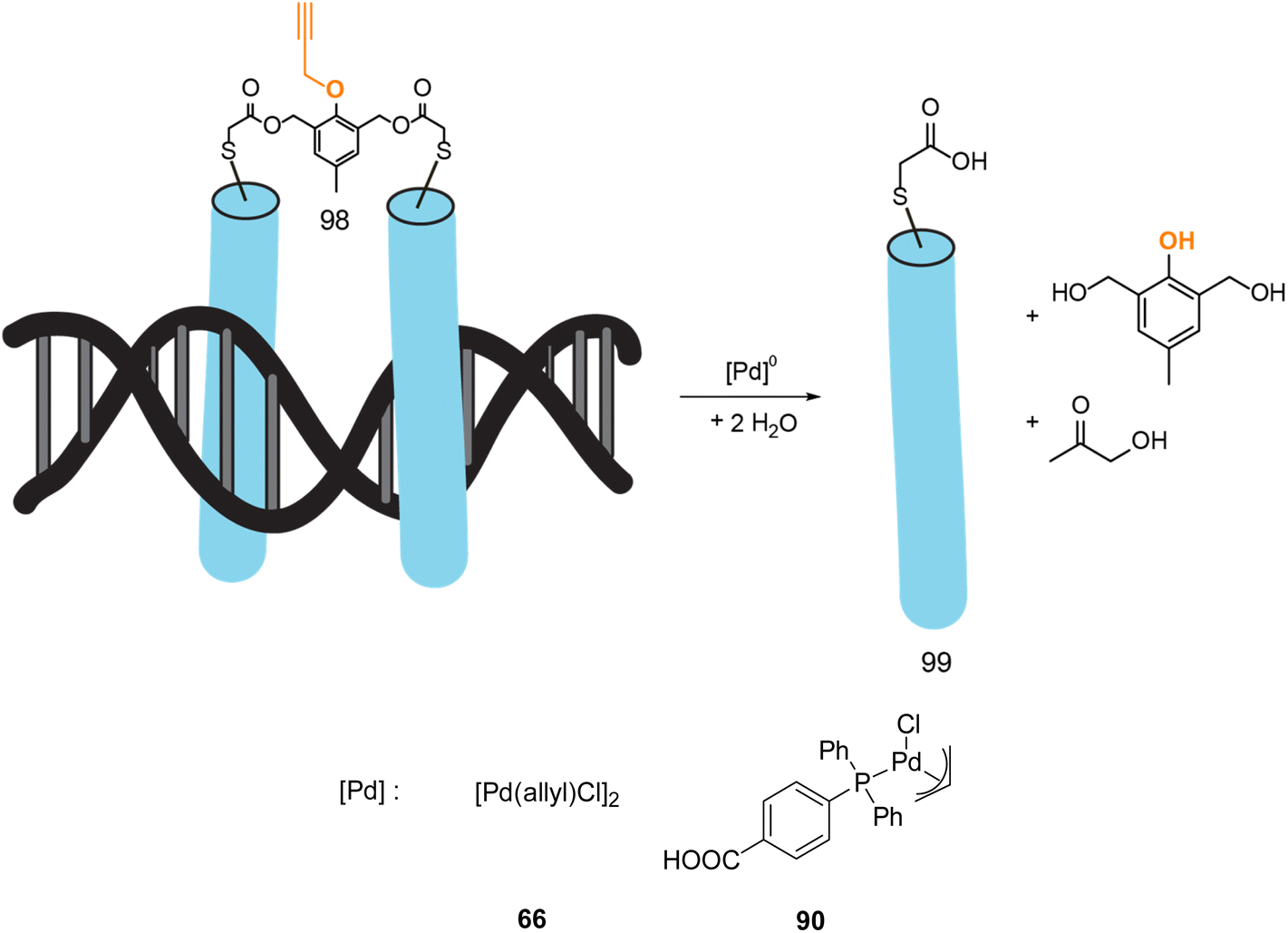 | ||
| Fig. 32 Control of DNA binding based on a palladium-mediated uncaging process under mild and physiological conditions reported by Mascareñas et al.75 | ||
The use of palladium complexes has greatly expanded the range of transformations that can be achieved in cellulo, making possible C–C cross-coupling reactions for instance. Considering that all the potential of the Pd-catalysed reactions has not been reached yet, considerable breakthroughs can be expected in the future. Within ten years, noticeable improvements have already been brought about regarding the catalyst loading, which was drastically reduced. However, it is still often difficult to unambiguously identify the active species (that could be palladium(0) nanoparticles) present in the cells.
5. Platinum
Platinum complexes have been widely used in organic chemistry but their application in cellulo as a catalyst is rare and limited by the toxicity of some PtII complexes. However, PtIV complexes are less toxic and can be designed bearing a photosensitizer (Riboflavin or Pyropheophorbide) able to perform photoreduction of PtIV in PtII within cells inducing the targeted release of cisplatin (CisPt) or analogs.76,77 Recently, Bernardes and coworkers reported a bond cleavage reaction of pentynoyl amides and N-propargyl groups by using platinum catalysts (K2PtCl4 and CisPt) to release secondary amines from stable tertiary amides (Fig. 33).78 This reaction was used to release the anticancer agents monomethyl auristatin E (MMAE) and 5-pFU from the protected precursors 100 and 93, respectively, to induce cytotoxicity in mammalian cell cultures. Unfortunately, it turned out that CisPt was too toxic at the concentration required for catalysis (C = 2.5 μM). In contrast, K2PtCl4 was found not to affect HeLa cell viability at concentrations below 50 mM, where catalyst loading is still efficient. The incubation of HeLa cells with K2PtCl4 and the prodrugs induces a two-fold increase in toxicity compared to control experiments with a non-decaging derivative or with 5-pFU 93 alone. In a colorectal Zebrafish xenograft model, this system induced a decrease of tumor sizes. Note that the presence of a nucleophile (such as glutathione) was found to lower the reaction rate even if the catalyst remained active. To overcome these issues, the team suggested the use of platinum nanoparticles or the optimisation of the complexes with new organic ligands to limit the deactivation.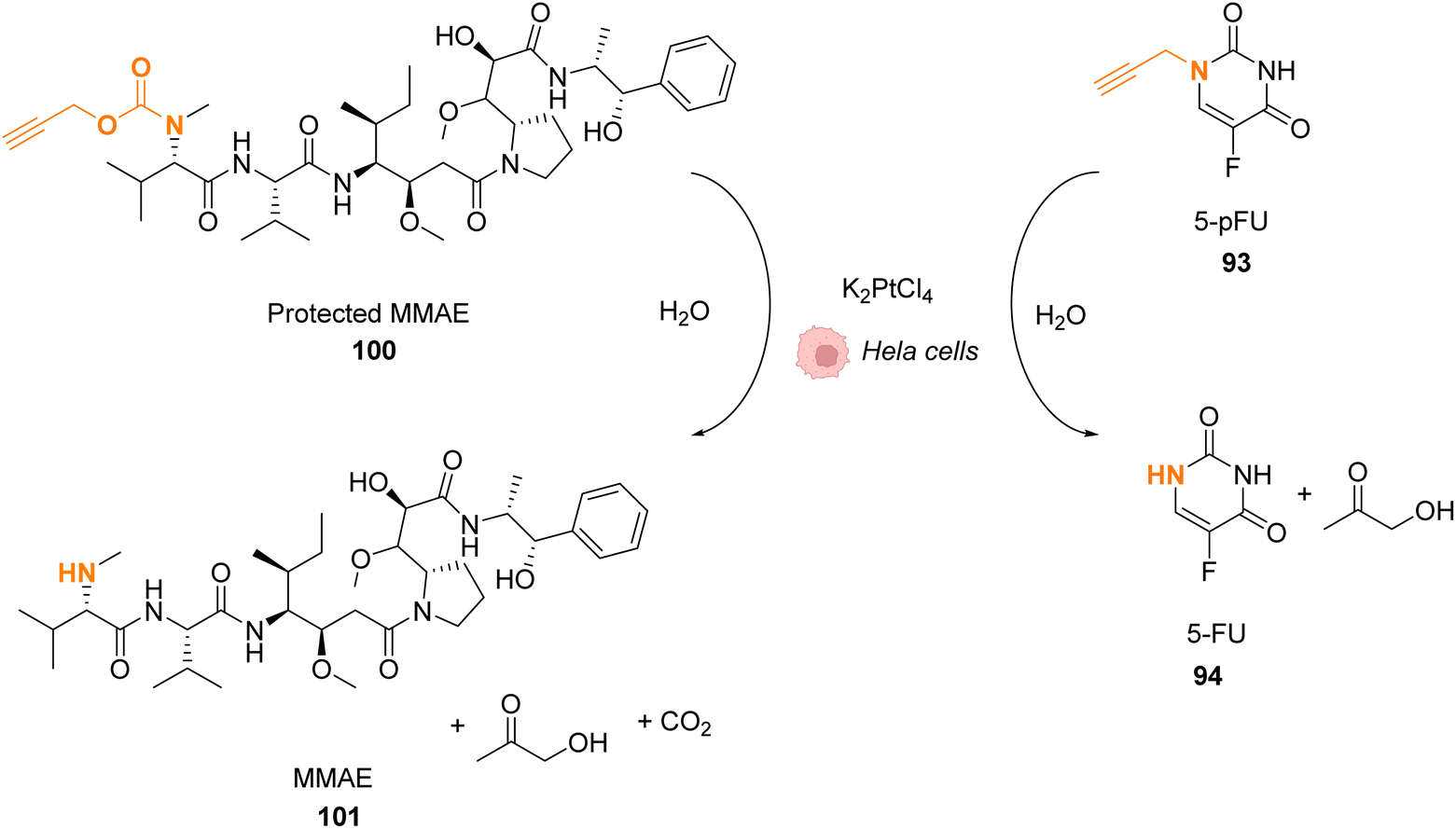 | ||
| Fig. 33 Pentynoyl and propargyl amine deprotection of prodrugs by platinum(II) complexes inside HeLa cells.76 | ||
In 2020, Huang and coworkers proposed a solution to overcome the platinum toxicity issue by developing a PtIV catalyst 102 covalently linked to an O2-propargyl diazeniumdiolate moiety, which is non-toxic toward normal cells but is reduced in cancer cells by cytoplasmic reductants to form a PtII species.79 This reduction releases CisPt and O2-propargyl-caged diazeniumdiolate 103 (Fig. 34). Unlike PtIV, PtII catalyses the cleavage of O2-propargyl, leading to the release of NO donor 104 and finally NO. NO donors were previously studied for their antiproliferative properties against cancer cells and were found to have potential as cancer therapeutic agents. The PtIV complex 102 was shown to induce O/N propargyl bond cleavage in various molecules, under biocompatible conditions, in good yields and with high efficiency. The simultaneous release of CisPt and NO induces selective and synergistic anticancer activity in vivo.80–83 PtIV complex 102 enters the cells and has a higher toxicity against human ovarian cancer A2780 cells compared to normal IOSE80 epithelial human cells. This strategy is flexible and could be used to decage other molecules of interest for several biological applications.
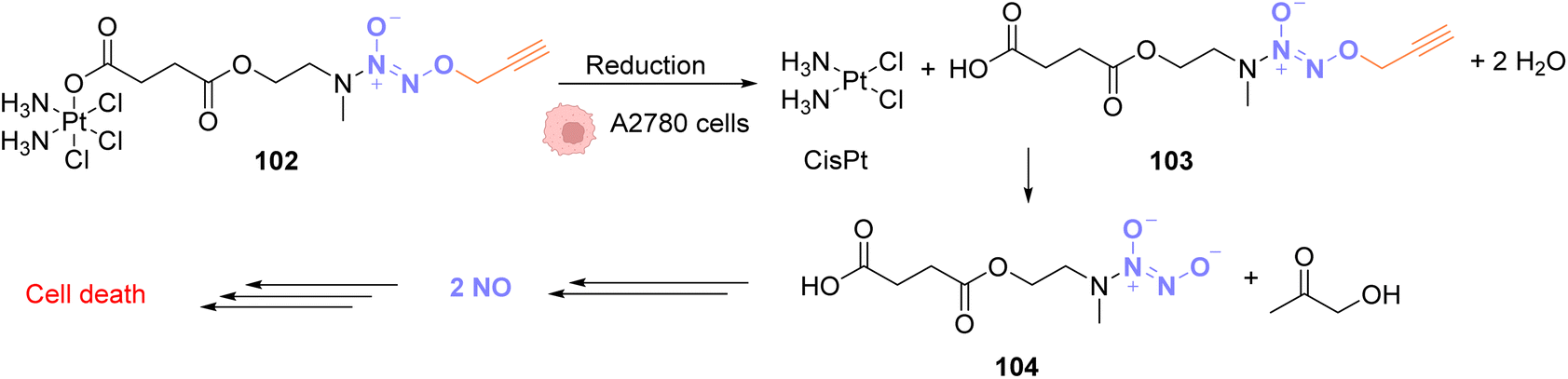 | ||
| Fig. 34 O 2-propargyl cleavage and NO release inside A2780 cells using a platinum(IV) precatalyst of low toxicity.79 | ||
While platinum salts are part of the WHO Essential Medicine List, or maybe because of this, their in vivo applications as catalysts remain rather scarce. This can also be due to the harsh conditions (such as prolonged heating) that are usually required for platinum catalysis to be efficient, which makes it, in most cases, inapplicable for biological applications.
6. Gold
While metallic gold is biocompatible, gold salts are well-known to be cytotoxic, and therefore the development of ligands to prevent their toxicity is essential. Nevertheless, AuI and AuIII salts are also notoriously alkynophilic and triple bonds are very useful biorthogonal functions. Therefore, using AuI or AuIII-based catalysts to react with alkynes inside cell is a very attractive strategy.6.1 Catalysis
As AuIII is cytotoxic,4 the first work involving this salt in an in cellulo catalysis was aimed to detect traces of AuIII through a reaction that it would specifically catalyse. A series of systems were studied based on the same principle: the generation of a fluorescent probe upon reaction with AuIII. For instance, rhodamine derivative 105 was synthesised and incubated in HaCaT cells. Without gold(III) salts, no fluorescence was observed, while upon addition of AuCl3 or HAuCl4, the products rearranged into a fluorescent molecule 106 (Fig. 35).84,85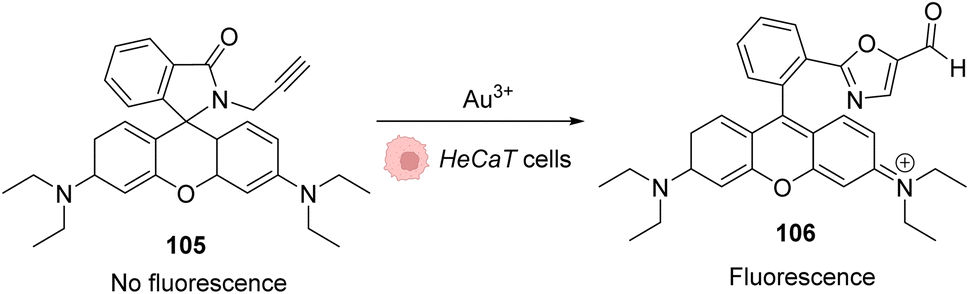 | ||
| Fig. 35 Generation of fluorescence inside HeCaT cells by the generation of 106 after incubation with AuIII.84,85 | ||
The same principle has been used with other fluorophores like Bodipy86 or coumarins. Hence, Kim and co-workers developed a system based on an apo-coumarin 107 that is not fluorescent but upon a hydroarylation reaction selectively catalysed by AuCl3 the fluorescence is turned-on with the formation of coumarin 108.87 To determine the probe sensitivity, the emission spectra of a solution of the apo-coumarin 107 at several concentrations of AuIII were investigated. The limit of detection of the probe was determined to be 64 ppb (parts per billion), which meant that the probe was relevant for biological applications since 40 ppm of AuIII are necessary to be strongly toxic (90% cell death) towards K562 human cells. Fluorescence imaging was successfully performed in HaCaT cells, with 10 μM of AuIII and 50 μM of the coumarin precursor 107 (Fig. 36). A blank experiment also confirmed that the presence of both the AuIII and molecule 107 was necessary to obtain fluorescence. Interestingly, in this case, the AuI complex, Ph3PAuCl, did not induce the turn-on of the fluorescence.
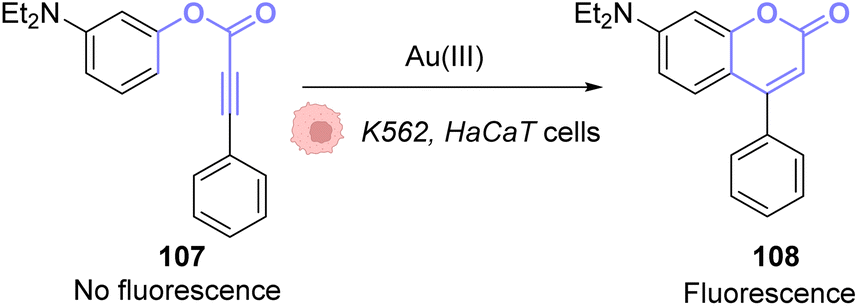 | ||
| Fig. 36 Gold(III)-catalysed hydroarylation of non-fluorescent 107 into a fluorescent coumarin.87 | ||
To perform the same reaction using AuI complexes, Mascareñas and co-workers used AuCl complex 109 bearing a water-soluble ligand.88 The cytotoxicity of the AuI catalyst 109 as well as the colourless compound 111 and the green/blue product 113 were tested in HeLa cells. None of them showed significant toxicity individually. Reactivity tests were carried out in HeLa cells incubated with 50 μM of the AuCl complex 109 and 100 μM of the colourless substrate 111. Fluorescence developed in the cells incubated with both compounds. The authors also tested the capacity of different gold catalysts to enter cells. The cellular uptake of the complexes varied greatly depending on the ligands. AuIII complexes without the cage-like ligand of 109 have a much higher concentration inside cells but a lower catalytic activity, while the complex 109 has a considerably better catalytic performance. They also showed that this reaction could be achieved orthogonally compared to other metal-based catalysis, in particular ruthenium promoted allyl cleavage (Fig. 37). Cellular tests carried out in the presence of substrate 111 and 112 as well as the AuCl catalysts 109 and the ruthenium catalyst 110 revealed infrared fluorescence coming from product 114 and green and blue staining coming from product 113. Cross experiments also revealed that the ruthenium catalyst 110 could not promote the hydroarylation of 111 and the gold complex could not induce decaging of 112. This is an example of two biorthogonal reactions that are successful and compatible inside living cells.
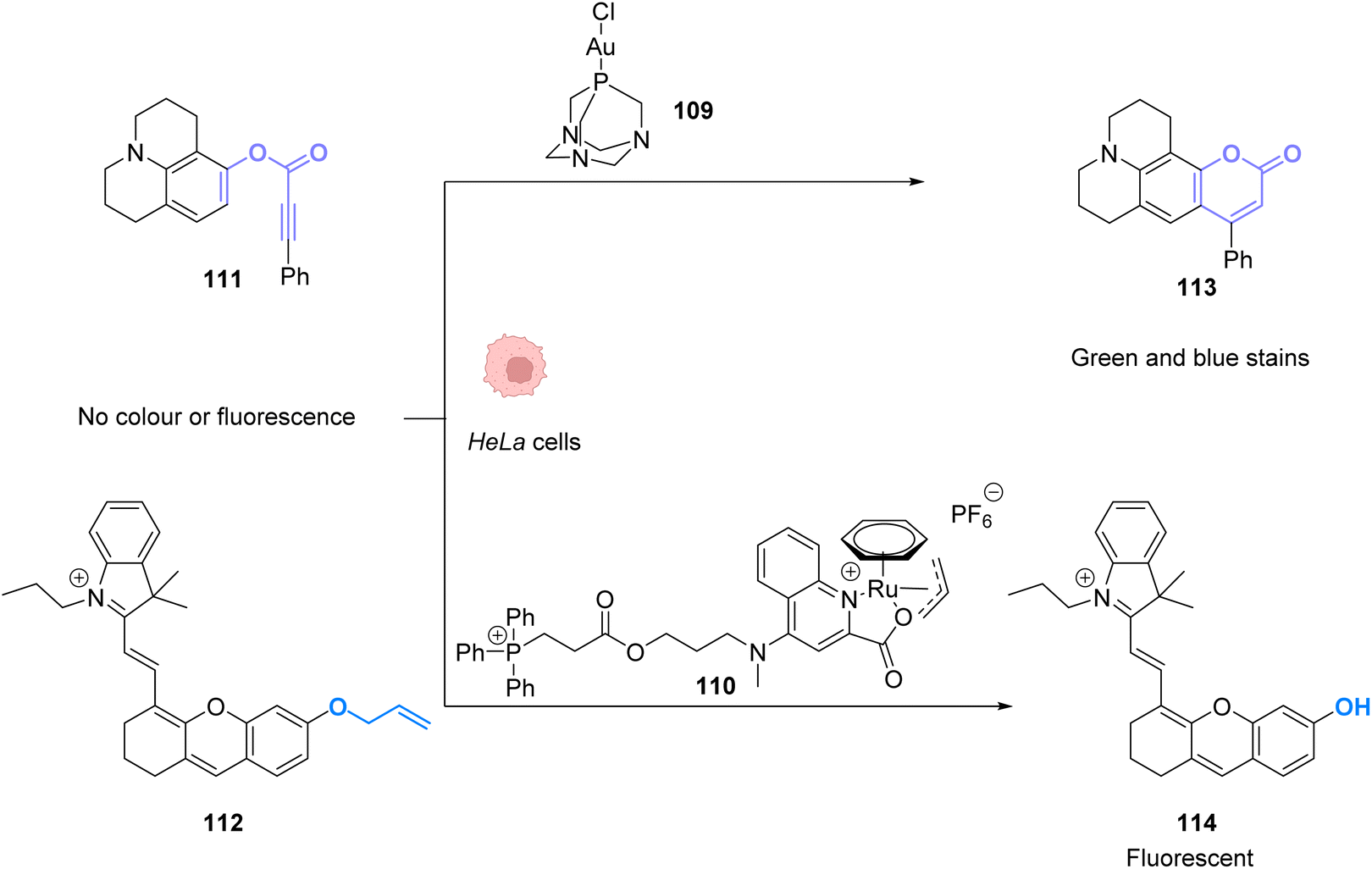 | ||
| Fig. 37 Simultaneous AuI and RuII catalysis inside HeLa cells. Gold-catalysed hydroarylation and allyl-deprotection catalysed by Ru.88 | ||
To avoid the cytotoxicity of AuCl, Mascareñas used a specific phosphine water soluble ligand. To use other ligands, such as NHCs, whose complexes with AuI are known to be cytotoxic, Zou and co-workers developed an in situ activation strategy for NHC–AuI complexes.89 The idea was to use NHC–AuI–alkyne complexes such as 115, which are known to be unreactive with thiols and therefore non-cytotoxic. The authors confirmed their stability with bio-relevant thiols. They next studied a NHC–gold–alkyne complex as a trans-metalation agent on a Pd complex. A series of PdII complexes were used and appeared to undergo transmetalation both in organic and aqueous solvents. A mixture of NHC–gold complex 115 and PdII salts in a buffer was also shown to induce the hydroarylation, already used before, to afford a fluorescent coumarin. For instance, NHC-Au complex 115 induced transmetallation on Pd(OAc)2 to eventually form Pd0 and NHC-AuOAc 116, which in turn catalysed the hydroarylation of 111 into fluorescent 113 (Fig. 38). Finally, this mixture was used to induce fluorescence inside A549 cancer cells and in zebrafish. The authors showed that only the mixture of the three components of the reaction, the NHC–Au 115 Pd complex and the coumarin precursor, induced a significant increase in fluorescence, demonstrating that the transmetallation/hydroarylation sequence was at play in cellulo and in vivo. It is worth noting that if the gold complex seems non-cytotoxic in a short period of time necessary for the reaction, longer incubation times induce cytotoxicity and angiogenesis perturbation in zebrafish. Therefore, this bimetallic strategy was also used to activate the cytotoxicity of the gold complex in targeted cells. It is also clear from this example that ligand design is essential to obtain gold catalysts, which are both reactive and non-toxic.
 | ||
| Fig. 38 PdII-triggered transmetalation activates AuI compound 115 and induces hydroarylation of the coumarin precursor to form a fluorescent coumarin in A549 cancer cells and zebrafish.89 | ||
To apply gold catalysis within fully developed living animals, Tanaka and co-workers developed a gold complex conjugated to a protein capable of catalysing an amide bond formation.90 Gold is used here to activate a propargyl ester to convert it into an amide. The authors showed that the presence of the gold catalyst 117 was necessary for the coupling of ester 118 with amine 119 (Fig. 39A). To target specific organs, the gold complex was conjugated to a coumarin that binds strongly to albumin forming complex 120. The albumin used here is heavily N-glycosylated with two types of oligosaccharides for liver targeting (Fig. 39B). The complex N-glycoalbumin/coumarin-gold was then studied. Two different compounds were tested, each having a different targeting glycan, as described in the team's previous work:91 Glyco-Au (Sia) with α(2-6)-disialoglycans targets the liver and galactose-terminated glycans Glyco-Au (Gal) targets the intestines. In vivo tests were carried out on live nude mice. After injecting Gluco-Au systems in the mice, fluorescent propargyl ester probes were administered (Fig. 39C). The glycoalbumin system provided a targeted delivery of the AuIII complexes within the first 30 min. A selective labelling was revealed by the amide bond formation between the amines present at the surface of the targeted organs and the fluorescent ester probes. This system is very promising as it functions on living animals, but the cytotoxicity of the gold complex is not discussed here and should probably be investigated before further applications.
 | ||
| Fig. 39 (A) Cyclometalated AuIII complex 117 structure and reactivity. (B) Preparation of glycoalbumins as “transition-metal carriers” to produce glyco-Au complexes. (C) In vivo fluorescence labelling by 120-catalysed amide bond formation between propargyl ester-based imaging probes and surface.90 | ||
Compared to platinum, gold catalysis generally requires milder conditions, making it more adequate for biological applications. In general, the reactions involve the π-activation of a triple bond, which grants them high potential for bio-orthogonal transformations and many developments are bound to follow these seminal studies. It is worth reminding that a gold complex (Auranofin) has been an approved drug, for rheumatoid arthritis, since 1985.
6.2 Photocatalysis
There has been no in vivo photocatalysis involving gold described in the literature so far. In 2020, Zou et al. described photoactivatable gold drugs,92 where they take advantage of the inactivity of Au-hydride complexes and their capacity to bind to thiols under irradiation. As the dissociation of the hydride ligands only occurs when the compounds are irradiated by visible light, they exhibit a promising photo-activated cytotoxic activity. However, no catalytic reaction is involved in the process.7. Copper
Copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) has gained tremendous attraction for biological applications. This reaction connects a terminal alkyne and an azide to form a triazole and serves as a standard example of click chemistry.93 It is an improved version of Huisgen cycloaddition that requires high temperatures and pressures. Sharpless and Meldal found that by using CuI as a catalyst it was possible to achieve much better reaction rates under milder and more achievable reaction conditions (Fig. 40).94,95 Its applications are extremely varied and include chemical biology, in particular by Bertozzi.96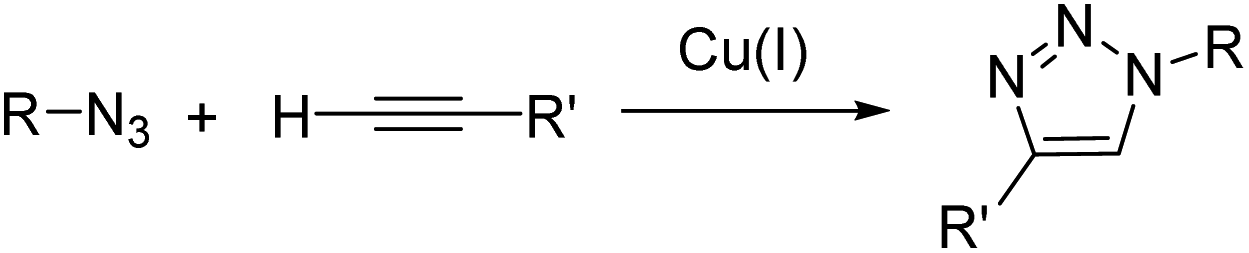 | ||
| Fig. 40 CuAAC reaction. | ||
Yet, the classic CuAAC has some major disadvantages in terms of biorthogonality. It is generally incompatible with thiols which are abundant inside cells. The CuAAC catalysts are usually deactivated by common cellular thiols. The major limitation is the reactivity and high toxicity of CuI ions. To overcome these drawbacks, researchers developed CuII catalysts that can be introduced into living organisms at a high concentration and then reduced to CuI. However, this is not a perfect solution, as many reducing agents are often not biocompatible either. In 2011, Sletten and Bertozzi described one of the most elegant solutions: a copper-free click reaction.97 They found that while linear alkynes were unreactive at physiological temperatures, cyclooctynes easily reacted with azide-labeled proteins or glycans. They successfully improved the reaction rates by adapting the cyclooctyne structure. In this metal-centered review, we focus on how other approaches were developed to implement biorthogonal CuAAC reactions with interesting biological applications.
7.1 Catalysis
Cai and co-workers investigated in 2017 the effect of tris(triazolylmethyl)amine-based ligands on the CuAAC reaction performed inside living cells, using a biotin-coumarin-azide derivative as a probe (Fig. 41A). The alkyne partner consisted of proteins that were metabolically modified to incorporate homopropargyl glycine (HPG).98 The coumarin moiety plays the role of a fluorogenic indicator, whilst the biotin moiety is an additional handle for other type of labelling (for example with fluorescein) via biotin-avidin interactions. Cell penetrating peptide Tat was attached to the ligand to enhance the efficiency, while avoiding the cytotoxicity of copper to make the CuI catalyst 121, produced in situ by copper sulphate and sodium ascorbate. The authors indeed observed that cell viability was higher in the presence of the ligands compared to the cells that were incubated with only CuI, even though the proliferation was slower than that of the control cells, without a ligand or CuI. In cellulo reactions were performed on HPG-incorporating HUVEC and OVCAR5 cell lines and the best results were obtained for a ligand modified with the Tat peptide that facilitates cellular uptake. The cell viability remained above 75% and the efficiency of the reaction varied between the cytosol and the membrane. Yields were measured using a fluorogenic reaction assay. The standard for the 100% yield was measured by dissolving the membrane proteins with SDS, meaning that all alkyl groups of the cell were converted to an HPG-containing group. However, for live cell tests, only the HPG groups present at the cell membrane were available for the reaction. Therefore, the yields measured for reactions taking place on the membrane surface are considered to be underestimated. On the membrane surface, the yield could reach up to 18%, while in the cytosol only 0.8% could be achieved. This was attributed to the presence of thiols such as GSH inside the cytosol, which hindered the reaction.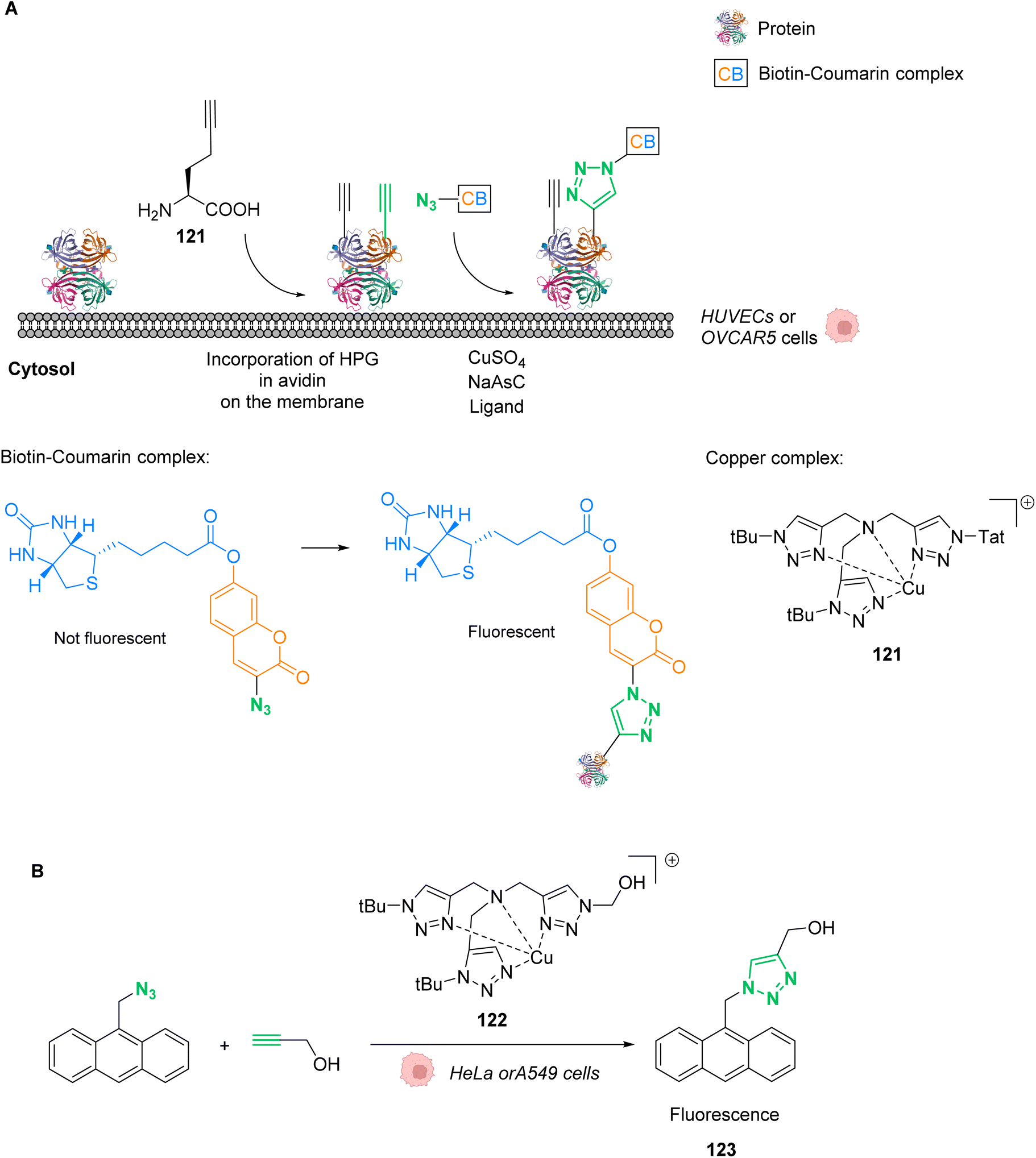 | ||
| Fig. 41 (A) In cellulo CuAAC reaction between coumarin/biotin azides and HPG-incorporating proteins.98 (B) In cellulo CuAAC reaction that produces a fluorescent probe without the use of sodium ascorbate.99 | ||
One year later, Mascareñas and co-workers developed a discrete copper(I) catalyst 122 using the same tris(triazolylmethyl)amine-based ligand BTTE (Fig. 41B).99 In this study, the compound is capable of catalysing intracellular CuAAC annulations between two exogenous and freely spreading substrates without the need for sodium ascorbate. The reaction was performed between 9-(azidomethyl)anthracene, that has almost no fluorescence, and an alkyl, to produce the fluorescent triazole 123. Tests were performed on HeLa and A549 cells, incubated with 50 μM of the copper catalyst 122, 100 μM of the azide and 200 μM of the alkyne. For this specific compound, no sodium ascorbate was necessary to assist with the reactivity. After 60 minutes, the researchers could observe blue fluorescence across the cytoplasm and unaltered morphology of the cells.
Later, in 2021, Mascareñas and co-workers performed a metal carbene transfer reaction inside mammalian cells. They demonstrated how CuII catalysts could promote the intracellular cyclisation of alpha-keto diazocarbenes 124 with ortho-amino arylamines via an N–H carbene insertion.100 They chose to study the reaction with diaminonaphthalene 123, which can form easy-to-monitor fluorescent benzoquinoxaline products 125–126. After testing different metals, it was concluded that the Cu(OAc)2 catalyst gave the best yield, even at a 1 mol% loading. Preliminary tests of the reaction were carried out under different biologically relevant conditions to confirm its biorthogonality. Curiously, the authors noticed that when performed in PBS, the reaction tolerates certain biomolecules better such as glutathione. They suggested that the buffer accelerated the targeted reaction due to its hydrophobic effects and disfavoured the formation of inactive Cu complexes. Finally, the reaction was carried out in HeLa mammalian cells. The authors confirmed that the diamine 123 presented a very low emission when excited at 385 nm while product 125 displayed high fluorescence (Fig. 42). After 1.5 h of incubation, fluorescence was observed in the cytosol, due to the production of the product 125, as confirmed by mass spectrometry. To broaden biological applications, the authors investigated the synthesis of the toxic compound 126 in HeLa cells. This compound, also referred to as Tyrphostin AG1385, promoted a decrease in the cell viability of 40% after 12 h of incubation at 50 μM. Compound 126 is known to provoke mitochondrial fragmentation, which suggests that the decrease in cell viability is a consequence of the reaction having been successfully carried out inside the cells, resulting in cell death (Fig. 42).
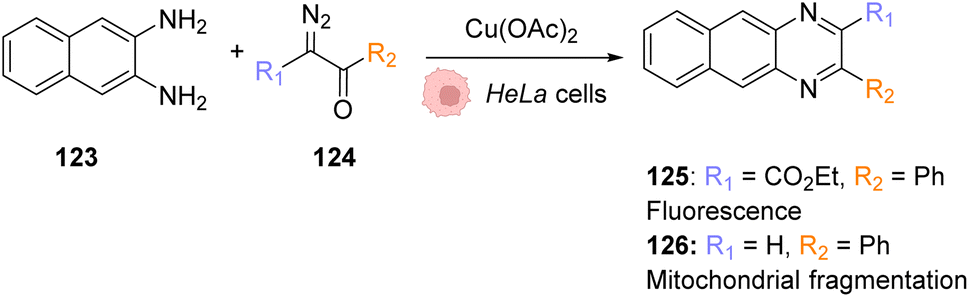 | ||
| Fig. 42 Copper-catalysed metal carbene transfer reaction in HeLa cells for the assembly of quinoxalines such as the fluorescent compound 125 and the toxic compound 126, capable of inducing mitochondrial fragmentation.100 | ||
More work has been performed with in vivo copper catalysis focused on nanoparticles, which fall out of the scope of the present review, but that should nonetheless be cited for completeness. In 2016, Zimmerman et al. developed CuI-containing nanoparticles that can catalyse the CuAAC click reaction in bacteria and mammalian cells.101 The CuI catalyst is then generated in situ with sodium ascorbate acting as a CuII reducer. That same year, Bradley and co-workers described the use of entrapped copper nanoparticles (E-Cu-NPs) imbedded in a polymeric support as a biocompatible catalyst for the same reaction.102 They demonstrated that these E-Cu-NPs could be used as catalysts for the cycloaddition of two benign components to make a Combretastatin A4 analogue in situ, a potent anticancer drug.
Copper is an abundant first-row transition metal, which is much cheaper than previously mentioned metals. With copper catalysis, new click and carbene insertion reactions were developed and reported. However, copper can easily access different oxidation states, making unwanted toxicity difficult to control. Moreover, the shift between oxidation states can also disturb the catalysis and requires sterically hindering ligands to protect and stabilize the copper centre.
7.2 Photocatalysis
The only in vivo photocatalysis described involving copper investigates the use of mesoporous carbon nanospheres, which also fall out of the scope of the present review. Ren and Qu described the use of near infra-red light and photothermal conversion to promote an in vivo CuI catalysed azide–alkyne 1,3-cycloaddition (CuAAC) reaction.1038. Iron
Examples of in vivo application of artificial iron-based catalysts are scarce. Meggers and co-workers identified in 2012 an iron(III) [Fe(TPP)Cl] complex 127 as an efficient catalyst for in cellulo reduction of aromatic azides to amines (Fig. 43A).104 Aromatic amines are useful functional groups in drugs and thus the in situ reduction of azides has shown some promise as a prodrug activation process.105,106 The authors first studied the reaction under biologically relevant conditions and in living mammalian cells. Compound 127 was administered to HeLa cells and its azide-reduction capabilities were followed by fluorescence microscopy, using a profluorophore of rhodamine 110 128. More specifically, cellular fluorescence was monitored by live-cell imaging with a confocal fluorescence microscope. HeLa cells were pre-incubated with 100 μL of the non-fluorescent rhodamine azide 128 for 25 min and subsequently treated with 10 μL of [Fe(TPP)Cl] (Fig. 43B). The production of rhodamine 110 3 inside cells was demonstrated within the first 10 minutes, with a 28-fold fluorescence increase observed. The authors also showed that the catalyst 127 successfully reduced the inactive compound 129 to form the anticancer drug candidate MS-275 130 (Fig. 43C). However, in vivo experiments in C. elegans nematodes and D. rerio zebrafish did not prove so successful, as rhodamine 110 3 fluorescence was detected prior to catalyst [Fe(TPP)Cl] addition. In both cases, strong fluorescence was observed after 20 to 30 minutes of incubation, the catalyst addition being redundant.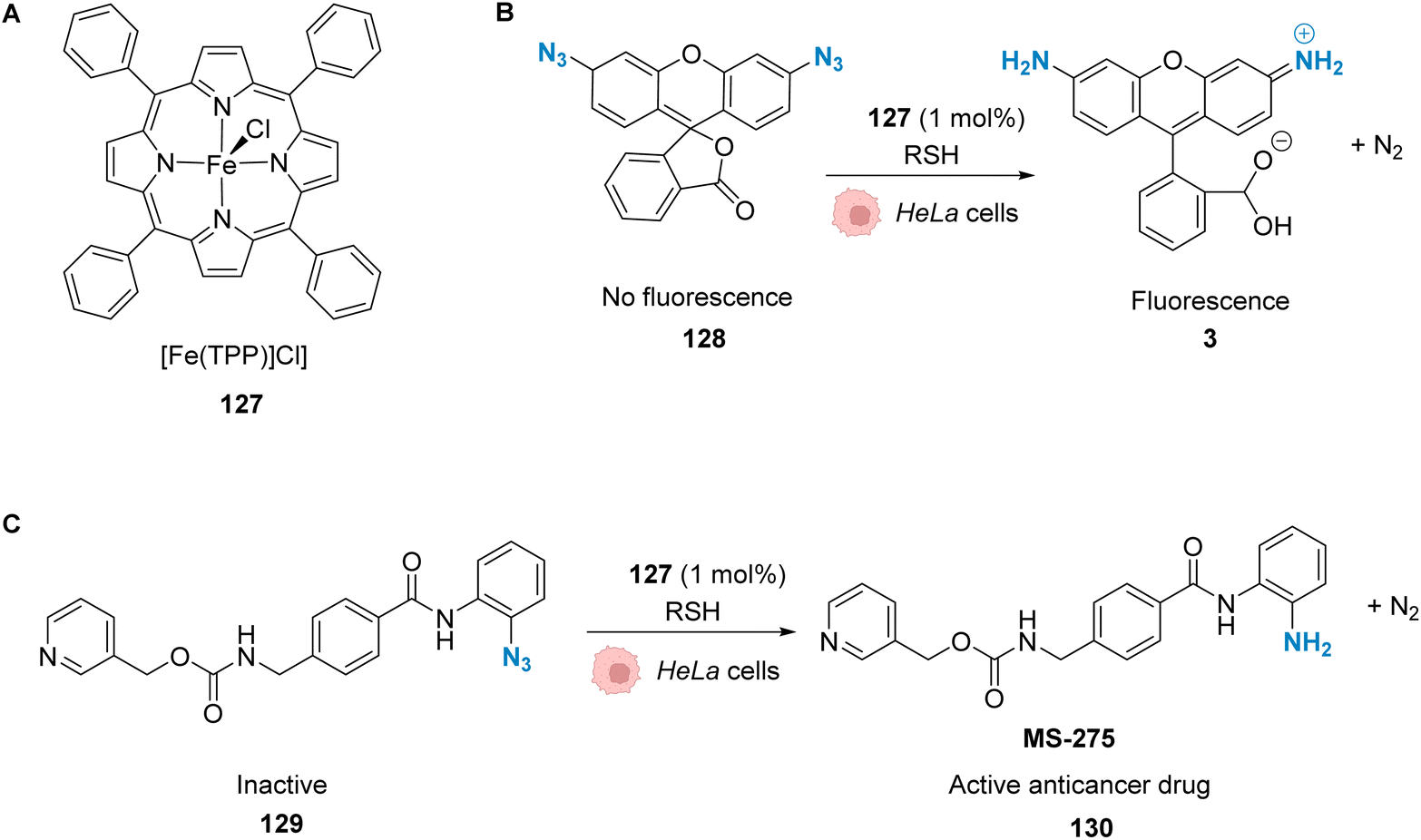 | ||
| Fig. 43 (A) Structure of a 5,10,15,20-tetraphenyl-21H,23H-porphine (TPP)-containing complex, [Fe(TPP)Cl]. (B) Iron-mediated reduction of a rhodamine 110 derivative to form fluorescent rhodamine 110. (C) Reduction of the azide molecule 129 to the anticancer drug candidate MS-275.104 | ||
Even though iron is as abundant and cheap as copper, it is not as toxic, which makes it even more interesting for biological purposes. However, most of the reactions that have been reported with iron complexes rely on their Lewis acidity, which makes it difficult to transpose them in a biological environment. Yet, as shown above, it is possible to achieve in vivo iron catalysis and we believe that this could certainly be a promising field in the future.
9. Conclusion and prospects
Metal-promoted catalysis within cells with a discrete metal complex and an exogenous substrate is a challenge due to the presence of high concentrations of diverse nucleophilic species such as thiols or amines, which can deactivate the catalyst. In addition, the catalyst must be active even at diluted concentrations and have good biocompatibility and cellular uptake. Despite these significant challenges, important breakthroughs in the field have been achieved over the past few years, as described in this perspective. These advances have mainly revolved around improving the biocompatibility of the catalysts, the turnover, the catalytic loading and finally the reaction rates to avoid catalyst deactivation. Improvements to the catalyst were mainly achieved by caging the catalyst and having an in situ release of the metal complex, by activation of the complex with other components present only in the target organisms or even by targeted delivery to the membrane by using cofactors and their affinities. The use of light to activate the compounds allows having a locally specific reaction. The possibility to tune the wavelength of absorption by changing the metal ligands is another tool for depth specificity. However, the use of light can be limited by the localisation of the target area. Deep tissues are less accessible to light-induced activation. Moreover light itself can be toxic towards tissues, if it is close to UV wavelengths.107Heavy noble metals such as ruthenium, iridium, platinum and palladium are a scarce natural resource and complicated to extract. In contrast, first row metals such as zinc, copper, cobalt and manganese are essential for life and already present in the human body.108 This is a reassuring factor for patients or companies when it comes to the development of potential treatments and medication based on metals. We are thus confident that within the next few years there will be more studies performed on first-row transition metals for in vivo catalysis. Particularly, there are a lot of innovation opportunities regarding their use for metal-based photocatalysis.109 As mentioned before, photocatalysis can reduce the side effects of treatments through local drug activation. This suggests that more catalysts will have the possibility to reach clinical trials, which will improve the visibility of this field of research. The development of new catalysts will also expand the range of reactions which are compatible with cells. Compared to the ever-increasing panel of reactions that can be performed and of the metals that can be used in catalysis, only very few are currently applied in cellulo. In addition, the use of nanoparticles has many advantages and seems very promising for in vivo metal catalysis. There are already several examples of metal-based nanoparticles being used for catalysis101,102 and photocatalysis103 afore-mentioned in this review. Another factor that could boost this research field is the improvement of the current tools used to study intracellular catalysis. Do, Nguyen and Nguyen tackled this issue in their recent review, where they analysed the four major techniques mentioned in our perspective:24 fluorescence microscopy, flow cytometry, mass spectrometry and biological assays. Clearly the next steps will be to bring these innovative strategies to the clinic to develop valuable therapeutic and/or diagnostic tools and the tremendous advances that have recently been achieved, and that are the focus of this perspective, have definitively set the stage to address these future challenges.
Author contributions
H. M. and F. F. wrote the manuscript. K. C., S. R., M. S. and G. G. revised and supervised the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Abbreviations
| Alloc | Allyloxycarbonyl |
| ArM | Artificial metalloprotein |
| Asc | Ascorbic acid/ascorbate |
| CuAAC | Copper-catalysed azide–alkyne cycloaddition |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| DHA | Dehydroascorbic acid |
| DMEM | Dulbecco's modified eagle medium |
| DNA | Deoxyribonucleic acid |
| EGFR | Estimated glomerular filtration rate |
| EPR | Enhanced permeability and retention |
| GFP | Green fluorescent protein |
| GSH | Glutathione |
| HEPES | (2-Hydroxyethyl)-1-piperazineethanesulfonic acid |
| HER2 | ERBB2 gene. ERBB is abbreviated from erythroblastic oncogene B |
| IC50 | Inhibitory concentration |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| LED | Light-emitting diode |
| MCNs | Mesoporous carbon nanospheres |
| MONP | Metal–organic nanoparticle |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NAD(P)H | Nicotinamide adenine dinucleotide phosphate |
| NADH/NAD+ | Nicotinamide adenine dinucleotide |
| NIR | Near infra-red |
| NO | Nitric oxide |
| NPs | Nanoparticles |
| PBS buffer | Phosphate-buffered saline |
| p-Erk | Phosphorylated extracellular signal-regulated kinases |
| PDT | Photo-dynamic therapy |
| PNA | Peptide nucleic acid |
| Rho | Rhodamine |
| ROS | Reactive oxygen species |
| RuAtAC | Ruthenium-catalysed azide–thioalkyne cycloadditions |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SET | Singlet electron transfer |
| TONs | Turn-over number |
| TPP | Thiamine pyrophosphate |
| 5-FU | Fluorouracil |
Acknowledgements
We are grateful for financial support from the ANR (ANR-20-CE07-0035), the ERC Consolidator Grant PhotoMedMet to G. G. (GA 681679), and the program “Investissements d’ Avenir” launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G. G.).References
- J. Chen, J. Wang, K. Li, Y. Wang, M. Gruebele, A. L. Ferguson and S. C. Zimmerman, Polymeric “Clickase” Accelerates the Copper Click Reaction of Small Molecules, Proteins, and Cells, J. Am. Chem. Soc., 2019, 141(24), 9693–9700, DOI:10.1021/jacs.9b04181.
- D. C. Luther, R. Huang, T. Jeon, X. Zhang, Y.-W. Lee, H. Nagaraj and V. M. Rotello, Delivery of Drugs, Proteins, and Nucleic Acids Using Inorganic Nanoparticles, Adv. Drug Deliv. Rev., 2020, 156, 188–213, DOI:10.1016/j.addr.2020.06.020.
- Y. Liu and Y. Bai, Design and Engineering of Metal Catalysts for Bio-Orthogonal Catalysis in Living Systems, ACS Appl. Bio Mater., 2020, 3(8), 4717–4746, DOI:10.1021/acsabm.0c00581.
- T. W. Hambley, Developing New Metal-Based Therapeutics: Challenges and Opportunities, Dalton Trans., 2007, 43, 4929, 10.1039/b706075k.
- M. Patra and G. Gasser, Organometallic Compounds: An Opportunity for Chemical Biology?, ChemBioChem, 2012, 13(9), 1232–1252, DOI:10.1002/cbic.201200159.
- S. J. Lippard, The Inorganic Side of Chemical Biology, Nat. Chem. Biol., 2006, 2(10), 504–507, DOI:10.1038/nchembio1006-504.
- C. R. Corso and A. Acco, Glutathione System in Animal Model of Solid Tumors: From Regulation to Therapeutic Target, Crit. Rev. Oncol./Hematol., 2018, 128, 43–57, DOI:10.1016/j.critrevonc.2018.05.014.
- R. A. Goyer, Nutrition and Metal Toxicity, Am. J. Clin. Nutr., 1995, 61(3), 646S–650S, DOI:10.1093/ajcn/61.3.646S.
- D. P. Nguyen, H. T. H. Nguyen and L. H. Do, Tools and Methods for Investigating Synthetic Metal-Catalyzed Reactions in Living Cells, ACS Catal., 2021, 11(9), 5148–5165, DOI:10.1021/acscatal.1c00438.
- W. Liu, E. E. Watson and N. Winssinger, Photocatalysis in Chemical Biology: Extending the Scope of Optochemical Control and Towards New Frontiers in Semisynthetic Bioconjugates and Biocatalysis, Helv. Chim. Acta, 2021, 104(12), e2100179, DOI:10.1002/hlca.202100179.
- B. Lozhkin and T. R. Ward, Bioorthogonal Strategies for the in vivo Synthesis or Release of Drugs, Bioorg. Med. Chem., 2021, 45, 116310, DOI:10.1016/j.bmc.2021.116310.
- J. J. Soldevila-Barreda and N. Metzler-Nolte, Intracellular Catalysis with Selected Metal Complexes and Metallic Nanoparticles: Advances toward the Development of Catalytic Metallodrugs, Chem. Rev., 2019, 119(2), 829–869, DOI:10.1021/acs.chemrev.8b00493.
- A. Seoane and J. L. Mascareñas, Exporting Homogeneous Transition Metal Catalysts to Biological Habitats, Eur. J. Org. Chem., 2022, 2022(32), e202200118, DOI:10.1002/ejoc.202200118.
- S. Gutiérrez, M. Tomás-Gamasa and J. L. Mascareñas, Organometallic Catalysis in Aqueous and Biological Environments: Harnessing the Power of Metal Carbenes, Chem. Sci., 2022, 13(22), 6478–6495, 10.1039/D2SC00721E.
- S. Alonso-de Castro, A. Terenzi, J. Gurruchaga-Pereda and L. Salassa, Catalysis Concepts in Medicinal Inorganic Chemistry, Chem.–Eur. J., 2019, 25(27), 6651–6660, DOI:10.1002/chem.201806341.
- J. Tu, M. Xu and R. M. Franzini, Dissociative Bioorthogonal Reactions, ChemBioChem, 2019, 20(13), 1615–1627, DOI:10.1002/cbic.201800810.
- M. O. N. van de L'Isle, M. C. Ortega-Liebana and A. Unciti-Broceta, Transition Metal Catalysts for the Bioorthogonal Synthesis of Bioactive Agents, Curr. Opin. Chem. Biol., 2021, 61, 32–42, DOI:10.1016/j.cbpa.2020.10.001.
- W. Wang, X. Zhang, R. Huang, C.-M. Hirschbiegel, H. Wang, Y. Ding and V. M. Rotello, In Situ Activation of Therapeutics through Bioorthogonal Catalysis, Adv. Drug Delivery Rev., 2021, 176, 113893, DOI:10.1016/j.addr.2021.113893.
- M. Martínez-Calvo and J. L. Mascareñas, Organometallic Catalysis in Biological Media and Living Settings, Coord. Chem. Rev., 2018, 359, 57–79, DOI:10.1016/j.ccr.2018.01.011.
- J. G. Rebelein and T. R. Ward, In Vivo Catalyzed New-to-Nature Reactions, Curr. Top. Biotechnol., 2018, 53, 106–114, DOI:10.1016/j.copbio.2017.12.008.
- T.-C. Chang and K. Tanaka, In Vivo Organic Synthesis by Metal Catalysts, Bioorg. Med. Chem., 2021, 46, 116353, DOI:10.1016/j.bmc.2021.116353.
- J. Wang, X. Wang, X. Fan and P. R. Chen, Unleashing the Power of Bond Cleavage Chemistry in Living Systems, ACS Cent. Sci., 2021, 7(6), 929–943, DOI:10.1021/acscentsci.1c00124.
- J. J. Soldevila-Barreda, I. Romero-Canelón, A. Habtemariam and P. J. Sadler, Transfer Hydrogenation Catalysis in Cells as a New Approach to Anticancer Drug Design, Nat. Commun., 2015, 6(1), 6582, DOI:10.1038/ncomms7582.
- D. P. Nguyen, H. T. H. Nguyen and L. H. Do, Tools and Methods for Investigating Synthetic Metal-Catalyzed Reactions in Living Cells, ACS Catal., 2021, 11(9), 5148–5165, DOI:10.1021/acscatal.1c00438.
- P. Destito, C. Vidal, F. López and J. L. Mascareñas, Transition Metal-Promoted Reactions in Aqueous Media and Biological Settings, Chem.–Eur. J., 2021, 27(15), 4789–4816, DOI:10.1002/chem.202003927.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Ruthenium(II)-Catalyzed C–H Bond Activation and Functionalization, Chem. Rev., 2012, 112(11), 5879–5918, DOI:10.1021/cr300153j.
- G. C. Vougioukalakis and R. H. Grubbs, Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts, Chem. Rev., 2010, 110(3), 1746–1787, DOI:10.1021/cr9002424.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113(7), 5322–5363, DOI:10.1021/cr300503r.
- J. Xuan and W.-J. Xiao, Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2012, 51(28), 6828–6838, DOI:10.1002/anie.201200223.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, Photoredox Catalysis in Organic Chemistry, J. Org. Chem., 2016, 81(16), 6898–6926, DOI:10.1021/acs.joc.6b01449.
- W. Han Ang and P. J. Dyson, Classical and Non-Classical Ruthenium-Based Anticancer Drugs: Towards Targeted Chemotherapy, Eur. J. Inorg. Chem., 2006, 2006(20), 4003–4018, DOI:10.1002/ejic.200600723.
- P.-S. Kuhn, V. Pichler, A. Roller, M. Hejl, M. A. Jakupec, W. Kandioller and B. K. Keppler, Improved Reaction Conditions for the Synthesis of New NKP-1339 Derivatives and Preliminary Investigations on Their Anticancer Potential, Dalton Trans, 2015, 44(2), 659–668, 10.1039/C4DT01645A.
- S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. van Werkhoven, E. Alessio, G. Sava, J. H. Beijnen and J. H. M. Schellens, Phase I/II Study with Ruthenium Compound NAMI-A and Gemcitabine in Patients with Non-Small Cell Lung Cancer after First Line Therapy, Invest. New Drugs, 2015, 33(1), 201–214, DOI:10.1007/s10637-014-0179-1.
- F. Lentz, A. Drescher, A. Lindauer, M. Henke, R. A. Hilger, C. G. Hartinger, M. E. Scheulen, C. Dittrich, B. K. Keppler and U. Jaehde, Pharmacokinetics of a Novel Anticancer Ruthenium Complex (KP1019, FFC14A) in a Phase I Dose-Escalation Study, Anti-Cancer Drugs, 2009, 20(2), 97–103, DOI:10.1097/CAD.0b013e328322fbc5.
- K. Lin, Z.-Z. Zhao, H.-B. Bo, X.-J. Hao and J.-Q. Wang, Applications of Ruthenium Complex in Tumor Diagnosis and Therapy, Front. Pharmacol., 2018, 9, 1323, DOI:10.3389/fphar.2018.01323.
- C. Streu and E. Meggers, Ruthenium-Induced Allylcarbamate Cleavage in Living Cells, Angew. Chem., Int. Ed., 2006, 45(34), 5645–5648, DOI:10.1002/anie.200601752.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Progress towards Bioorthogonal Catalysis with Organometallic Compounds, Angew. Chem., Int. Ed., 2014, 53(39), 10536–10540, DOI:10.1002/anie.201404547.
- M. I. Sánchez, C. Penas, M. E. Vázquez and J. L. Mascareñas, Metal-Catalyzed Uncaging of DNA-Binding Agents in Living Cells, Chem. Sci., 2014, 5(5), 1901–1907, 10.1039/C3SC53317D.
- M. Tomás-Gamasa, M. Martínez-Calvo, J. R. Couceiro and J. L. Mascareñas, Transition Metal Catalysis in the Mitochondria of Living Cells, Nat. Commun., 2016, 7(1), 12538, DOI:10.1038/ncomms12538.
- Y. Okamoto, R. Kojima, F. Schwizer, E. Bartolami, T. Heinisch, S. Matile, M. Fussenegger and T. R. Ward, A Cell-Penetrating Artificial Metalloenzyme Regulates a Gene Switch in a Designer Mammalian Cell, Nat. Commun., 2018, 9(1), 1943, DOI:10.1038/s41467-018-04440-0.
- Z. Zhao, X. Tao, Y. Xie, Q. Lai, W. Lin, K. Lu, J. Wang, W. Xia and Z. Mao, In Situ Prodrug Activation by an Affibody-Ruthenium Catalyst Hybrid for HER2-Targeted Chemotherapy, Angew. Chem., Int. Ed., 2022, 61(26), e202202855, DOI:10.1002/anie.202202855.
- C. Vidal, M. Tomás-Gamasa, A. Gutiérrez-González and J. L. Mascareñas, Ruthenium-Catalyzed Redox Isomerizations inside Living Cells, J. Am. Chem. Soc., 2019, 141(13), 5125–5129, DOI:10.1021/jacs.9b00837.
- J. Miguel-Ávila, M. Tomás-Gamasa and J. L. Mascareñas, Intracellular Ruthenium-Promoted (2+2+2) Cycloadditions, Angew. Chem., Int. Ed., 2020, 59(40), 17628–17633, DOI:10.1002/anie.202006689.
- C. Weng, L. Shen, J. W. Teo, Z. C. Lim, B. S. Loh and W. H. Ang, Targeted Antibacterial Strategy Based on Reactive Oxygen Species Generated from Dioxygen Reduction Using an Organoruthenium Complex, JACS Au, 2021, 1(9), 1348–1354, DOI:10.1021/jacsau.1c00262.
- S. Angerani and N. Winssinger, Visible Light Photoredox Catalysis Using Ruthenium Complexes in Chemical Biology, Chem.–Eur. J., 2019, 25(27), 6661–6672, DOI:10.1002/chem.201806024.
- P. K. Sasmal, S. Carregal-Romero, W. J. Parak and E. Meggers, Light-Triggered Ruthenium-Catalyzed Allylcarbamate Cleavage in Biological Environments, Organometallics, 2012, 31(16), 5968–5970, DOI:10.1021/om3001668.
- K. K. Sadhu, T. Eierhoff, W. Römer and N. Winssinger, Photoreductive Uncaging of Fluorophore in Response to Protein Oligomers by Templated Reaction in Vitro and in Cellulo, J. Am. Chem. Soc., 2012, 134(49), 20013–20016, DOI:10.1021/ja310171s.
- Y. Chen, A. S. Kamlet, J. B. Steinman and D. R. Liu, A Biomolecule-Compatible Visible-Light-Induced Azide Reduction from a DNA-Encoded Reaction-Discovery System, Nat. Chem., 2011, 3(2), 146–153, DOI:10.1038/nchem.932.
- K. K. Sadhu and N. Winssinger, Detection of MiRNA in Live Cells by Using Templated Ru II -Catalyzed Unmasking of a Fluorophore, Chem.–Eur. J., 2013, 19(25), 8182–8189, DOI:10.1002/chem.201300060.
- K. K. Sadhu, E. Lindberg and N. Winssinger, In Cellulo Protein Labelling with Ru-Conjugate for Luminescence Imaging and Bioorthogonal Photocatalysis, Chem. Commun., 2015, 51(93), 16664–16666, 10.1039/C5CC05405B.
- S. Sato and H. Nakamura, Ligand-Directed Selective Protein Modification Based on Local Single-Electron-Transfer Catalysis, Angew. Chem., Int. Ed., 2013, 52(33), 8681–8684, DOI:10.1002/anie.201303831.
- S. Sato, K. Morita and H. Nakamura, Regulation of Target Protein Knockdown and Labeling Using Ligand-Directed Ru(Bpy) 3 Photocatalyst, Bioconjugate Chem., 2015, 26(2), 250–256, DOI:10.1021/bc500518t.
- A. Gutiérrez-González, P. Destito, J. R. Couceiro, C. Pérez-González, F. López and J. L. Mascareñas, Bioorthogonal Azide–Thioalkyne Cycloaddition Catalyzed by Photoactivatable Ruthenium(II) Complexes, Angew. Chem., Int. Ed., 2021, 60(29), 16059–16066, DOI:10.1002/anie.202103645.
- A. Zamora, G. Vigueras, V. Rodríguez, M. D. Santana and J. Ruiz, Cyclometalated Iridium(III) Luminescent Complexes in Therapy and Phototherapy, Coord. Chem. Rev., 2018, 360, 34–76, DOI:10.1016/j.ccr.2018.01.010.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Recent Progress in Photosensitizers for Overcoming the Challenges of Photodynamic Therapy: From Molecular Design to Application, Chem. Soc. Rev., 2021, 50(6), 4185–4219, 10.1039/D0CS00173B.
- H. Huang, S. Banerjee and P. J. Sadler, Recent Advances in the Design of Targeted Iridium(III) Photosensitizers for Photodynamic Therapy, ChemBioChem, 2018, 19(15), 1574–1589, DOI:10.1002/cbic.201800182.
- L. Zhang and D. Ding, Recent Advances of Transition Ir(III) Complexes as Photosensitizers for Improved Photodynamic Therapy, VIEW, 2021, 2(6), 20200179, DOI:10.1002/VIW.20200179.
- Z. Liu, I. Romero-Canelón, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, The Potent Oxidant Anticancer Activity of Organoiridium Catalysts, Angew. Chem., Int. Ed., 2014, 53(15), 3941–3946, DOI:10.1002/anie.201311161.
- S. Bose, A. H. Ngo and L. H. Do, Intracellular Transfer Hydrogenation Mediated by Unprotected Organoiridium Catalysts, J. Am. Chem. Soc., 2017, 139(26), 8792–8795, DOI:10.1021/jacs.7b03872.
- N. Singh, A. Gupta, P. Prasad, P. Mahawar, S. Gupta and P. K. Sasmal, Iridium-Triggered Allylcarbamate Uncaging in Living Cells, Inorg. Chem., 2021, 60(17), 12644–12650, DOI:10.1021/acs.inorgchem.1c01790.
- H. Huang, S. Banerjee, K. Qiu, P. Zhang, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stavros, G. Gasser, H. Chao and P. J. Sadler, Targeted Photoredox Catalysis in Cancer Cells, Nat. Chem., 2019, 11(11), 1041–1048, DOI:10.1038/s41557-019-0328-4.
- C. Huang, C. Liang, T. Sadhukhan, S. Banerjee, Z. Fan, T. Li, Z. Zhu, P. Zhang, K. Raghavachari and H. Huang, In-vitro and In-vivo Photocatalytic Cancer Therapy with Biocompatible Iridium(III) Photocatalysts, Angew. Chem., Int. Ed., 2021, 60(17), 9474–9479, DOI:10.1002/anie.202015671.
- J. Li and P. R. Chen, Moving Pd-Mediated Protein Cross Coupling to Living Systems, ChemBioChem, 2012, 13(12), 1728–1731, DOI:10.1002/cbic.201200353.
- C. D. Spicer, T. Triemer and B. G. Davis, Palladium-Mediated Cell-Surface Labeling, J. Am. Chem. Soc., 2012, 134(2), 800–803, DOI:10.1021/ja209352s.
- N. Li, R. K. V. Lim, S. Edwardraja and Q. Lin, Copper-Free Sonogashira Cross-Coupling for Functionalization of Alkyne-Encoded Proteins in Aqueous Medium and in Bacterial Cells, J. Am. Chem. Soc., 2011, 133(39), 15316–15319, DOI:10.1021/ja2066913.
- N. Li, C. P. Ramil, R. K. V. Lim and Q. Lin, A Genetically Encoded Alkyne Directs Palladium-Mediated Protein Labeling on Live Mammalian Cell Surface, ACS Chem. Biol., 2015, 10(2), 379–384, DOI:10.1021/cb500649q.
- J. Li, S. Lin, J. Wang, S. Jia, M. Yang, Z. Hao, X. Zhang and P. R. Chen, Ligand-Free Palladium-Mediated Site-Specific Protein Labeling Inside Gram-Negative Bacterial Pathogens, J. Am. Chem. Soc., 2013, 135(19), 7330–7338, DOI:10.1021/ja402424j.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Palladium-Triggered Deprotection Chemistry for Protein Activation in Living Cells, Nat. Chem., 2014, 6(4), 352–361, DOI:10.1038/nchem.1887.
- J. Wang, S. Zheng, Y. Liu, Z. Zhang, Z. Lin, J. Li, G. Zhang, X. Wang, J. Li and P. R. Chen, Palladium-Triggered Chemical Rescue of Intracellular Proteins via Genetically Encoded Allene-Caged Tyrosine, J. Am. Chem. Soc., 2016, 138(46), 15118–15121, DOI:10.1021/jacs.6b08933.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Megia-Fernandez, A. Lilienkampf and M. Bradley, Intracellular Delivery of a Catalytic Organometallic Complex, Chem. Commun., 2017, 53(50), 6712–6715, 10.1039/C7CC02988H.
- T. Lv, J. Wu, F. Kang, T. Wang, B. Wan, J.-J. Lu, Y. Zhang and Z. Huang, Synthesis and Evaluation of O2 -Derived Diazeniumdiolates Activatable via Bioorthogonal Chemistry Reactions in Living Cells, Org. Lett., 2018, 20(8), 2164–2167, DOI:10.1021/acs.orglett.8b00423.
- M. Martínez-Calvo, J. R. Couceiro, P. Destito, J. Rodríguez, J. Mosquera and J. L. Mascareñas, Intracellular Deprotection Reactions Mediated by Palladium Complexes Equipped with Designed Phosphine Ligands, ACS Catal., 2018, 8(7), 6055–6061, DOI:10.1021/acscatal.8b01606.
- D. Cherukaraveedu, P. T. Cowling, G. P. Birch, M. Bradley and A. Lilienkampf, Solid-Phase Synthesis of Biocompatible N-Heterocyclic Carbene–Pd Catalysts Using a Sub-Monomer Approach, Org. Biomol. Chem., 2019, 17(22), 5533–5537, 10.1039/C9OB00716D.
- J. Konč, V. Sabatino, E. Jiménez-Moreno, E. Latocheski, L. R. Pérez, J. Day, J. B. Domingos and G. J. L. Bernardes, Controlled In-Cell Generation of Active Palladium(0) Species for Bioorthogonal Decaging, Angew. Chem., Int. Ed., 2022, 61(8), e202113519, DOI:10.1002/anie.202113519.
- J. Rodríguez, C. Pérez-González, M. Martínez-Calvo, J. Mosquera and J. L. Mascareñas, Deactivation of a Dimeric DNA-Binding Peptide through a Palladium-Mediated Self-Immolative Cleavage, RSC Adv., 2022, 12(6), 3500–3504, 10.1039/D1RA09180H.
- S. Alonso-de Castro, E. Ruggiero, A. Ruiz-de-Angulo, E. Rezabal, J. C. Mareque-Rivas, X. Lopez, F. López-Gallego and L. Salassa, Riboflavin as a Bioorthogonal Photocatalyst for the Activation of a Pt IV Prodrug, Chem. Sci., 2017, 8(6), 4619–4625, 10.1039/C7SC01109A.
- Z. Wang, N. Wang, S.-C. Cheng, K. Xu, Z. Deng, S. Chen, Z. Xu, K. Xie, M.-K. Tse, P. Shi, H. Hirao, C.-C. Ko and G. Zhu, Phorbiplatin, a Highly Potent Pt(IV) Antitumor Prodrug That Can Be Controllably Activated by Red Light, Chem, 2019, 5(12), 3151–3165, DOI:10.1016/j.chempr.2019.08.021.
- B. L. Oliveira, B. J. Stenton, V. B. Unnikrishnan, C. R. de Almeida, J. Conde, M. Negrão, F. S. S. Schneider, C. Cordeiro, M. G. Ferreira, G. F. Caramori, J. B. Domingos, R. Fior and G. J. L. Bernardes, Platinum-Triggered Bond-Cleavage of Pentynoyl Amide and N -Propargyl Handles for Drug-Activation, J. Am. Chem. Soc., 2020, 142(24), 10869–10880, DOI:10.1021/jacs.0c01622.
- T. Sun, T. Lv, J. Wu, M. Zhu, Y. Fei, J. Zhu, Y. Zhang and Z. Huang, General Strategy for Integrated Bioorthogonal Prodrugs: Pt(II)-Triggered Depropargylation Enables Controllable Drug Activation In Vivo, J. Med. Chem., 2020, 63(22), 13899–13912, DOI:10.1021/acs.jmedchem.0c01435.
- B. Azizzadeh, H. T. Yip, K. E. Blackwell, S. Horvath, T. C. Calcaterra, G. M. Buga, L. J. Ignarro and M. B. Wang, Nitric Oxide Improves Cisplatin Cytotoxicity in Head and Neck Squamous Cell Carcinoma, Laryngoscope, 2001, 111(11), 1896–1900, DOI:10.1097/00005537-200111000-00004.
- M. Kielbik, I. Szulc-Kielbik, M. Nowak, Z. Sulowska and M. Klink, Evaluation of Nitric Oxide Donors Impact on Cisplatin Resistance in Various Ovarian Cancer Cell Lines, Toxicol. In Vitro, 2016, 36, 26–37, DOI:10.1016/j.tiv.2016.07.005.
- D. A. Wink, J. A. Cook, D. Christodoulou, M. C. Krishna, R. Pacelli, S. Kim, W. DeGraff, J. Gamson, Y. Vodovotz, A. Russo and J. B. Mitchell, Nitric Oxide and Some Nitric Oxide Donor Compounds Enhance the Cytotoxicity of Cisplatin, Nitric Oxide, 1997, 1(1), 88–94, DOI:10.1006/niox.1996.0108.
- J. Zhao, S. Gou, Y. Sun, R. Yin and Z. Wang, Nitric Oxide Donor-Based Platinum Complexes as Potential Anticancer Agents, Chem.–Eur. J., 2012, 18(45), 14276–14281, DOI:10.1002/chem.201201605.
- M. Jung Jou, X. Chen, K. M. K. Swamy, H. Na Kim, H.-J. Kim, S. Lee and J. Yoon, Highly Selective Fluorescent Probe for Au3+ Based on Cyclization of Propargylamide, Chem. Commun., 2009, 46, 7218, 10.1039/b917832e.
- Y.-K. Yang, S. Lee and J. Tae, A Gold(III) Ion-Selective Fluorescent Probe and Its Application to Bioimagings, Org. Lett., 2009, 11(24), 5610–5613, DOI:10.1021/ol902325u.
- J.-B. Wang, Q.-Q. Wu, Y.-Z. Min, Y.-Z. Liu and Q.-H. Song, A Novel Fluorescent Probe for Au(iii)/Au(i) Ions Based on an Intramolecular Hydroamination of a Bodipy Derivative and Its Application to Bioimaging, Chem. Commun., 2012, 48(5), 744–746, 10.1039/C1CC16128H.
- J. H. Do, H. N. Kim, J. Yoon, J. S. Kim and H.-J. Kim, A Rationally Designed Fluorescence Turn-On Probe for the Gold(III) Ion, Org. Lett., 2010, 12(5), 932–934, DOI:10.1021/ol902860f.
- C. Vidal, M. Tomás-Gamasa, P. Destito, F. López and J. L. Mascareñas, Concurrent and Orthogonal Gold(I) and Ruthenium(II) Catalysis inside Living Cells, Nat. Commun., 2018, 9(1), 1913, DOI:10.1038/s41467-018-04314-5.
- Y. Long, B. Cao, X. Xiong, A. S. C. Chan, R. W. Sun and T. Zou, Bioorthogonal Activation of Dual Catalytic and Anti-Cancer Activities of Organogold(I) Complexes in Living Systems, Angew. Chem., Int. Ed., 2021, 60(8), 4133–4141, DOI:10.1002/anie.202013366.
- K. Tsubokura, K. K. H. Vong, A. R. Pradipta, A. Ogura, S. Urano, T. Tahara, S. Nozaki, H. Onoe, Y. Nakao, R. Sibgatullina, A. Kurbangalieva, Y. Watanabe and K. Tanaka, In Vivo Gold Complex Catalysis within Live Mice, Angew. Chem., Int. Ed., 2017, 56(13), 3579–3584, DOI:10.1002/anie.201610273.
- K. Tanaka, Chemically Synthesized Glycoconjugates on Proteins: Effects of Multivalency and Glycoform in Vivo, Org. Biomol. Chem., 2016, 14(32), 7610–7621, 10.1039/C6OB00788K.
- H. Luo, B. Cao, A. S. C. Chan, R. W. Sun and T. Zou, Cyclometalated Gold(III)-Hydride Complexes Exhibit Visible Light-Induced Thiol Reactivity and Act as Potent Photo-Activated Anti-Cancer Agents, Angew. Chem., Int. Ed., 2020, 59(27), 11046–11052, DOI:10.1002/anie.202000528.
- N. K. Devaraj and M. G. Finn, Introduction: Click Chemistry, Chem. Rev., 2021, 121(12), 6697–6698, DOI:10.1021/acs.chemrev.1c00469.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41(14), 2596–2599, DOI:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4.
- C. W. Tornøe, C. Christensen and M. Meldal, Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides, J. Org. Chem., 2002, 67(9), 3057–3064, DOI:10.1021/jo011148j.
- J. A. Prescher and C. R. Bertozzi, Chemistry in Living Systems, Nat. Chem. Biol., 2005, 1(1), 13–21, DOI:10.1038/nchembio0605-13.
- E. M. Sletten and C. R. Bertozzi, From Mechanism to Mouse: A Tale of Two Bioorthogonal Reactions, Acc. Chem. Res., 2011, 44(9), 666–676, DOI:10.1021/ar200148z.
- S. Li, L. Wang, F. Yu, Z. Zhu, D. Shobaki, H. Chen, M. Wang, J. Wang, G. Qin, U. J. Erasquin, L. Ren, Y. Wang and C. Cai, Copper-Catalyzed Click Reaction on/in Live Cells, Chem. Sci., 2017, 8(3), 2107–2114, 10.1039/C6SC02297A.
- J. Miguel-Ávila, M. Tomás-Gamasa, A. Olmos, P. J. Pérez and J. L. Mascareñas, Discrete Cu(i) Complexes for Azide–Alkyne Annulations of Small Molecules inside Mammalian Cells, Chem. Sci., 2018, 9(7), 1947–1952, 10.1039/C7SC04643J.
- S. Gutiérrez, M. Tomás-Gamasa and J. L. Mascareñas, Exporting Metal-Carbene Chemistry to Live Mammalian Cells: Copper-Catalyzed Intracellular Synthesis of Quinoxalines Enabled by N−H Carbene Insertions, Angew. Chem., Int. Ed., 2021, 60(40), 22017–22025, DOI:10.1002/anie.202108899.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, A Highly Efficient Single-Chain Metal–Organic Nanoparticle Catalyst for Alkyne–Azide “Click” Reactions in Water and in Cells, J. Am. Chem. Soc., 2016, 138(35), 11077–11080, DOI:10.1021/jacs.6b04477.
- J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J. Mullins and M. Bradley, Copper Catalysis in Living Systems and In Situ Drug Synthesis, Angew. Chem., 2016, 128(50), 15891–15895, DOI:10.1002/ange.201609837.
- Y. You, F. Cao, Y. Zhao, Q. Deng, Y. Sang, Y. Li, K. Dong, J. Ren and X. Qu, Near-Infrared Light Dual-Promoted Heterogeneous Copper Nanocatalyst for Highly Efficient Bioorthogonal Chemistry in Vivo, ACS Nano, 2020, 14(4), 4178–4187, DOI:10.1021/acsnano.9b08949.
- P. K. Sasmal, S. Carregal-Romero, A. A. Han, C. N. Streu, Z. Lin, K. Namikawa, S. L. Elliott, R. W. Köster, W. J. Parak and E. Meggers, Catalytic Azide Reduction in Biological Environments, ChemBioChem, 2012, 13(8), 1116–1120, DOI:10.1002/cbic.201100719.
- Z. Pianowski, K. Gorska, L. Oswald, C. A. Merten and N. Winssinger, Imaging of MRNA in Live Cells Using Nucleic Acid-Templated Reduction of Azidorhodamine Probes, J. Am. Chem. Soc., 2009, 131(18), 6492–6497, DOI:10.1021/ja809656k.
- A. R. Lippert, E. J. New and C. J. Chang, Reaction-Based Fluorescent Probes for Selective Imaging of Hydrogen Sulfide in Living Cells, J. Am. Chem. Soc., 2011, 133(26), 10078–10080, DOI:10.1021/ja203661j.
- I. Plattfaut, E. Demir, P. C. Fuchs, J. L. Schiefer, E. K. Stürmer, A. K. E. Brüning and C. Opländer, Characterization of Blue Light Treatment for Infected Wounds: Antibacterial Efficacy of 420, 455, and 480 Nm Light-Emitting Diode Arrays Against Common Skin Pathogens Versus Blue Light-Induced Skin Cell Toxicity, Photobiomodulation, Photomed., Laser Surg., 2021, 39(5), 339–348, DOI:10.1089/photob.2020.4932.
- M. A. Zoroddu, J. Aaseth, G. Crisponi, S. Medici, M. Peana and V. M. Nurchi, The Essential Metals for Humans: A Brief Overview, J. Inorg. Biochem., 2019, 195, 120–129, DOI:10.1016/j.jinorgbio.2019.03.013.
- C. B. Larsen and O. S. Wenger, Photoredox Catalysis with Metal Complexes Made from Earth-Abundant Elements, Chem.–Eur. J., 2018, 24(9), 2039–2058, DOI:10.1002/chem.201703602.
Footnote |
| † These authors have contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2023 |