
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical radical-mediated selective C(sp3)–S bond activation†
Yongli
Li‡
a,
Huamin
Wang‡
a,
Zhuning
Wang
a,
Hesham
Alhumade
bc,
Zhiliang
Huang
*a and
Aiwen
Lei
 *ab
*ab
aCollege of Chemistry and Molecular Sciences, The Institute for Advanced Studies (IAS), Wuhan University, Wuhan, 430072, Hubei, P. R. China. E-mail: aiwenlei@whu.edu.cn; zlhuang@whu.edu.cn
bChemical and Materials Engineering Department, Faculty of Engineering, King Abdulaziz University, Jeddah 21589, Saudi Arabia
cK. A. CARE Energy Research and Innovation Center, King Abdulaziz University, Jeddah 21589, Saudi Arabia
First published on 6th December 2022
Abstract
Selective C(sp3)–S bond breaking and transformation remains a particularly important, yet challenging goal in synthetic chemistry. Over the past few decades, transition metal-catalyzed cross-coupling reactions through the cleavage of C(sp3)–S bonds provided a powerful platform for the construction of target molecules. In contrast, the selective activation of widespread C(sp3)–S bonds is rarely studied and remains underdeveloped, even under relatively harsh conditions. Herein, a radical-mediated electrochemical strategy capable of selectively activating C(sp3)–S bonds is disclosed, offering an unprecedented method for the synthesis of valuable disulfides from widespread thioethers. Importantly, compared with conventional transition-metal catalyzed C–S bond breaking protocols, this method features mild, catalyst- and oxidant-free reaction conditions, as well excellent chemoselectivity towards C(sp3)–S bonds. Preliminary mechanistic studies reveal that sulfur radical species are involved in the reaction pathway and play an essential role in controlling the site-selectivity.
Sulfur-containing compounds are not only widely found in natural products, pharmaceuticals, fragrances and agrochemicals, but are also important precursors and intermediates in synthetic chemistry.1 Therefore, the chemical reactions of these valuable compounds through carbon–sulfur (C–S) bond activation would have a broadly beneficial impact on both synthetic and medicinal chemistry. Thus, the past several decades have witnessed the study of an array of transition-metal catalysts for C–S bond activation and transformation via oxidative addition (Scheme 1a).2,3 Although numerous significant advances have been made in this research area, there are still some limitations:2d (1) the oxidative addition to C–S bonds generally requires elevated temperature; (2) the resulting metal–sulfur bond is stable, and therefore, a compatible nucleophilic reagent should be used to facilitate the following transmetalation step and establish a catalytic cycle; (3) since oxidative addition of the C(sp2)-S bond is often more kinetically favorable than that of the C(sp3)–S bond, transition-metal catalyzed C–S bond transformation reactions are normally chemoselective to the C(sp2)-S bonds. In contrast, the selective activation of widespread C(sp3)-S bonds is rarely studied and remains underdeveloped, even under relatively harsh conditions. In particular, precise activation and transformation of one of the two C(sp3)-S bonds of unsymmetrical dialkyl thioethers is greatly challenging, as both C(sp3)–S bonds are very similar.2d Hence, it is significant and meaningful to develop novel methods for selective C(sp3)–S bond cleavage and transformation, especially under mild reaction conditions.
 | ||
| Scheme 1 (a) Transition-metal catalyzed C–S bond activation; (b) Schematic showing a radical-mediated strategy for C–S bond activation; (c) electrochemical selective C(sp3)–S bond activation in this work. | ||
Radical reactions have attracted extensive attention in the last few decades, particularly, along with the development of numerous modern techniques, including photocatalysis4 and electrocatalysis,5 that enable the generation of radicals in a controlled fashion. More often than not, radicals are highly reactive species, which can induce the cleavage of chemical bonds via radical hydrogen abstraction, radical substitution reactions and so on.6 Meanwhile, as unstable intermediates, pairs of radicals also have a tendency to react with each other. Given these well-known properties of radicals, we envisaged a radical-mediated formal C(sp3)–S bond activation strategy for the chemoselective conversion of unsymmetrical aryl alkyl or dialkyl thioethers (Scheme 1b). First, the starting thioether undergoes one-electron oxidation to form a transient radical cation. According to the persistent radical effect,7 the resulting sulfur-centered radical cation is able to couple with a persistent radical (˙R) and form the key intermediate I. Thereafter, the original C(sp3)–S bond of thioether is activated, which could finally be cleaved through a heterolytic process.
Organic electrochemistry provides powerful tools to address many challenges in synthetic chemistry.5 Notably, it is one of the most straightforward and practical means to produce radical species by using electrons as traceless ‘‘reagents’’ in a sustainable manner. Herein, the electrochemical technique was employed to investigate our proposal of radical-mediated formal C(sp3)–S bond activation. In this transformation, thiyl radicals were found to be effective to mediate the C–S bond activation of thioether to afford valuable unsymmetrical disulfides (Scheme 1c). Various C(sp3)–S bonds could be smoothly cleaved in a highly chemoselective manner under mild conditions: (1) As for the aryl alkyl thioethers, C(sp3)–S bonds rather than C(sp2)-S bonds were selectively cleaved; (2) With regard to dialkyl thioethers, C(primary)–S bonds took precedence over C(secondary)–S bonds for the desired activation, while C(secondary)–S bonds were more favorable than C(tertiary)–S bonds during the cleavage process.
On the basis of the above proposal, thiol was employed as a persistent radical precursor, since the resulting thiyl radical could be reversibly converted to disulfide under electrochemical conditions.8 In this regard, sec-butyl methyl thioether (1a) and 4-chlorothiophenol (2a) were initially used as reaction partners to examine the electrochemical protocol. Following extensive screening, we were pleased to discover that the desired product (3g) was produced in 79% yield with the use of CH3COOH as an additive, nBu4NPF6 as the electrolyte, carbon rode as the anode, platinum as the cathode, at 25 mA and in an air atmosphere (Table 1, entry 1). 4-Chlorothiophenol (2a) was almost completely consumed in the reaction system (Table 1, entry 1). Changing electrolytes as well as their amounts had little effect on the yields (Table 1, entries 2–4). Similarly, the yield had a slight fluctuation when we altered the amount of CH3COOH (Table 1, entries 5–7). Further screening demonstrated that the transformation took place smoothly with several electrode materials, producing the desired product in similar yields (Table 1, entries 8–10). A high yield was obtained when the reaction was carried out under a N2 atmosphere (Table 1, entry 11). Moreover, slightly lower yields were observed at 25 °C and 50 °C (Table 1, entries 12 and 13). Note that unsymmetrical disulfides are important compounds across many fields of chemistry, which encourages the development of numerous methods for their synthesis.9
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yields (%) |
| a Isolated yield in parentheses. | ||
| 1 | None | 79 (70)a |
| 2 | n Bu4NBF4(20%) | 62 |
| 3 | n Bu4NClO4(20%) | 74 |
| 4 | n Bu4NBF4(40%) | 69 |
| 5 | Without CH3COOH | 40 |
| 6 | CH3COOH(2 equiv.) | 63 |
| 7 | CH3COOH(1.6 equiv.) | 73 |
| 8 | C(+)|C(−) | 57 |
| 9 | Pt(+)|Pt(−) | 78 |
| 10 | C(+)|Ni(−) | 56 |
| 11 | N2 instead of air | 78 |
| 12 | 25 °C | 71 |
| 13 | 50 °C | 69 |
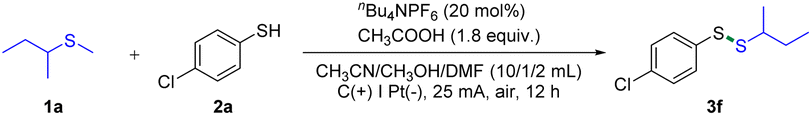
With the optimum conditions in hand, we evaluated the scope of this electrochemical C(sp3)–S bond cleavage. As can be seen in Scheme 2, this electrochemical protocol was able to chemoselectively cleave one of the two similar C(sp3)–S bonds involved in unsymmetrical dialkyl thioethers. Moreover, we found that the C(primary)–S bond is more reactive than C(secondary)–S bonds. For example, the S–Me bonds of methyl alkyl thioethers were precisely cleaved to form the target products (3a–3d). In addition, highly selective cleavage of C(primary)–S bonds rather than C(tertiary)–S bonds was observed using this protocol (3e–3i). It is worth mentioning that unprotected alcohol was well tolerated in this transformation (3g). Dialkyl thioethers containing levulinic acid and ibuprofen moieties reacted smoothly with sec-butyl methyl thioether (1a), providing desired products in 61% yields (3h and 3i). Similarly, the C(primary)-S bond instead of the C(quaternary)–S bond was chopped, when tert-butyl methyl thioether was employed as the substrate (3j). Furthermore, product 3k was obtained in high selectivity, demonstrating that the cleavage of the C(secondary)–S bond is more favored than the cleavage of the C(tertiary)–S bond. Additionally, halogen and CF3 groups were also investigated, offering various disulfides with moderate to good yields (3m–3p and 3r). Since electron-rich thiophenols are easily oxidized and decomposed at the anode, lower yields were observed (3l and 3q). Unfortunately, amine and nitro groups were not compatible with this protocol (For details, see the ESI†). Herein, this electrochemical strategy offers high chemoselectivity for C(sp3)–S bond activation of unsymmetrical dialkyl thioethers, which is difficult to be achieved by the previous methods.
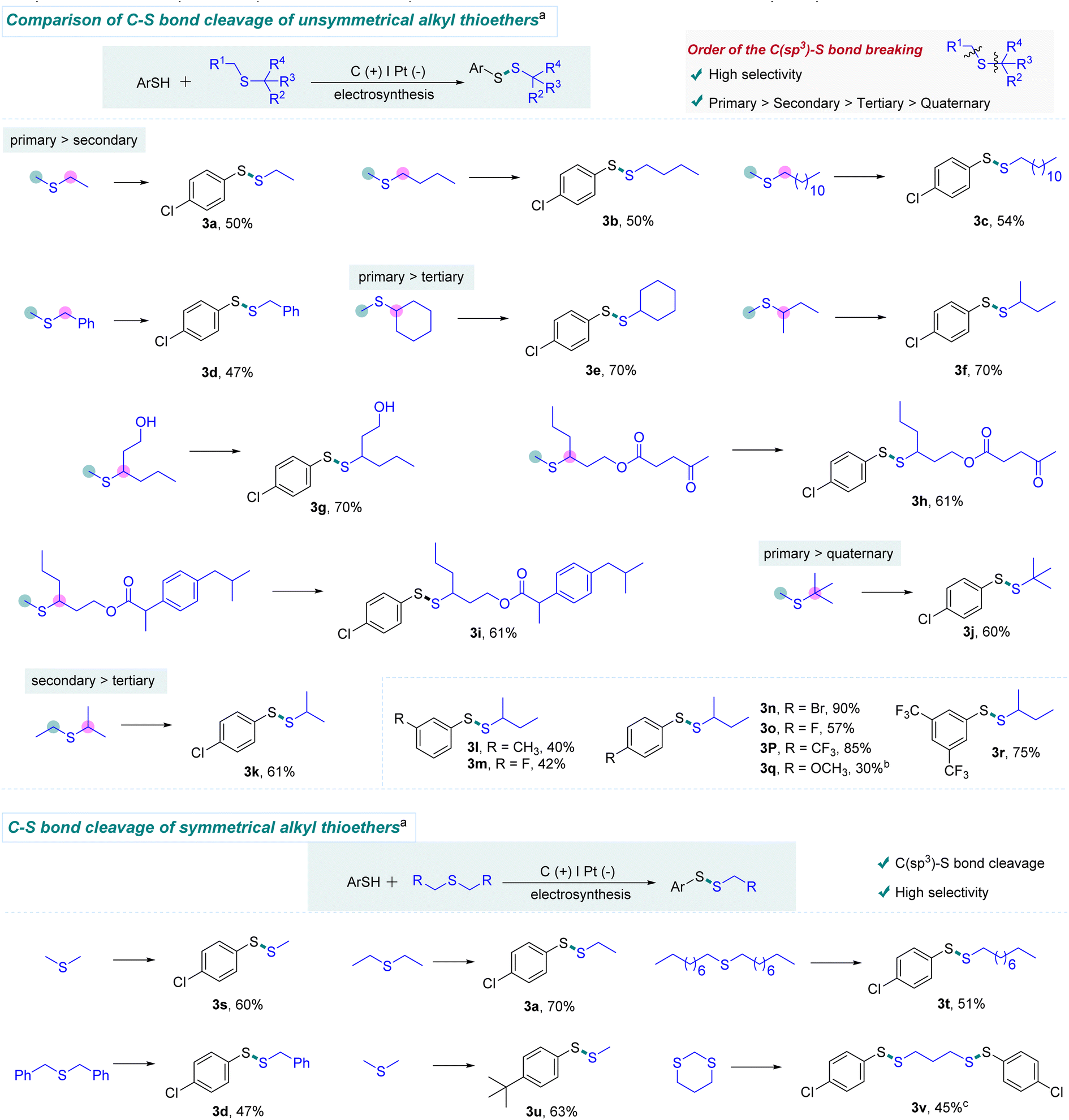 | ||
| Scheme 2 Substrate scope of chemoselective electrochemical C–S bond activation. a Reaction conditions: Carbon rod anode, Pt plate (15 mm × 15 mm × 0.3 mm) cathode, constant current (25 mA), thiols (0.6 mmol), thioethers (3 equiv.), nBu4NPF6 (0.2 equiv.), CH3COOH (1.8 equiv.), and 37 °C, in CH3CN/CH3OH/DMF (10/1/2, 13 mL) in air for 12 h, and in an undivided cell, isolated yields; b 4-methoxydiphenyl disulfide as the substrate; c 1,3-dithiane (1.8 mmol) and 2a (1.2 mmol). | ||
We next turned our attention to symmetric alkyl-sulfur-alkyl compounds (Scheme 2). Delightfully, thiomethylation could be smoothly achieved with dimethyl disulfide as the substrate under the standard conditions, offering compound 3s in 60% yield. Thus, this electrochemical protocol provides an efficient route for thiomethylation, which is regarded as a challenging issue in organic synthesis.9a Additionally, other symmetrical chain alkyl thioethers were tested. Dipropyl and dinonyl thioethers were also well tolerated (3a and 3t). It is worth mentioning that dibenzyl thioether was amenable to this protocol, offering the desired product through the cleavage of the C(benzyl)–S bond (3d). Interestingly, 1,3-dithiane could be smoothly converted to the ring-opening product 3v through the cleavage of two C–S bonds.
In order to investigate the utility of this electrochemical method, thioethers involving both C(sp2)–S and C(sp3)–S bonds were evaluated. Contrary to transition metal catalysis, C(sp3)–S bonds instead of C(sp2)–S bonds were selectively cleaved, as shown in Scheme 3. A series of alkyl mercaptans worked well with phenylmethyl thioether, providing the corresponding products in moderate yields (3w-3ab). In addition, a variety of aryl thiophenols and heterocyclic thiophenol were suitable to react with aryl methyl thioethers, selectively offering unsymmetrical aryl disulfides in moderate yields (3ac-3af). The thioether containing a hexafluorobutyl group could also generate the target product (3ah) in 56% yield. Pleasingly, other kinds of C (alkyl)–S bonds were also examined, selectively forming the corresponding product 3ai through the cleavage of C(sp3)–S bonds. These results further reveal the good chemoselectivity of this strategy. Notably, thiomethylation reactions are challenging in synthetic chemistry.9a By using our protocol, methylthiomethyl acetate (1b) was used as a substrate to examine thiomethylation. As shown in Scheme 3, a series of alkyl mercaptans reacted smoothly with 1b to generate the thiomethylation products (3aj-3an). Encouragingly, cysteine derivative could be transformed into target product (3am) in 58% yield.
 | ||
| Scheme 3 Standard conditions: Carbon rod anode, nickel plate (15 mm × 15 mm × 1 mm) or Pt (15 mm × 15 mm × 0.3 mm) cathode, constant current (25 mA), thiols (0.6 mmol), thioethers (3 equiv.), nBu4NPF6 (0.2 equiv.), CH3COOH (1.8 equiv.), and 37 °C, in CH3CN/CH3OH/DMF (10/1/2, 13 mL) in air for 12 h, and in an undivided cell, isolated yields. a 60 °C, without DMF; b Carbon rod anode, nickel plate (15 mm × 15 mm × 1 mm) cathode, constant current (15 mA), thiols (0.6 mmol), (methylthio)methyl acetate (3 equiv.), nBu4NPF6 (0.2 equiv.), and 60 °C, in CH3CN/CH3OH (10/0.1, 10.1 mL) in air for 8 h; c (ethylthio)methyl acetate (3 equiv.). | ||
To provide mechanistic detail, cyclic voltammograms were recorded and the results are illustrated in Scheme 4a. An oxidation peak of 4-chlorothiophenol (2a) in the mixed solvent (DMF/CH3CN/CH3OH) was detected at 1.27 V and 1.87 V. Additionally, oxidation peaks of sec-butyl methyl thioether (1a) could also be observed at about 1.73 V. These results revealed that 2a was first oxidized to bis(4-chlorophenyl) disulfide (2aa) at the anode. Further investigation found that bis(4-chlorophenyl) disulfide could be oxidized at 1.82 V. And it could be reduced at the cathode, and its reduction peak was detected at −1.52 V. Moreover, the radical trapping experiment was carried out. A sulfur radical from 4-chlorothiophenol (2a) was trapped by TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) and the reaction was completely suppressed (Scheme 4b). Additionally, this electrochemical process was monitored by F19 NMR experiments (Scheme 4c). The results suggest that disulfide formed from thiol is likely to be the intermediate of this protocol (Scheme 4c). To further probe details of this transformation, intermediate experiments were also performed. As shown in Scheme 4d, bis(4-chlorophenyl) disulfide (2aa) reacted smoothly with dimethyl thioether (1c) to offer the desired product (3s) in 40% yield, which also suggested that 2aa might be the intermediate. Furthermore, 1b could be observed during the reaction by GC-MS. And according to our previous work,1c this compound (1b) is likely to be an intermediate in this system. Hence, 1b was used as a substrate to react with 2a, which offered compound (3s) in 45% yield and this result supported our hypothesis (Scheme 4d). Subsequently, a control experiment was performed and detected by GC-MS (Scheme 4e). In addition to the desired product (3d), another compound (1e) was also detected by GC-MS after the reaction, which was a by-product of the departing part (Scheme 4e, for details, see the ESI†).
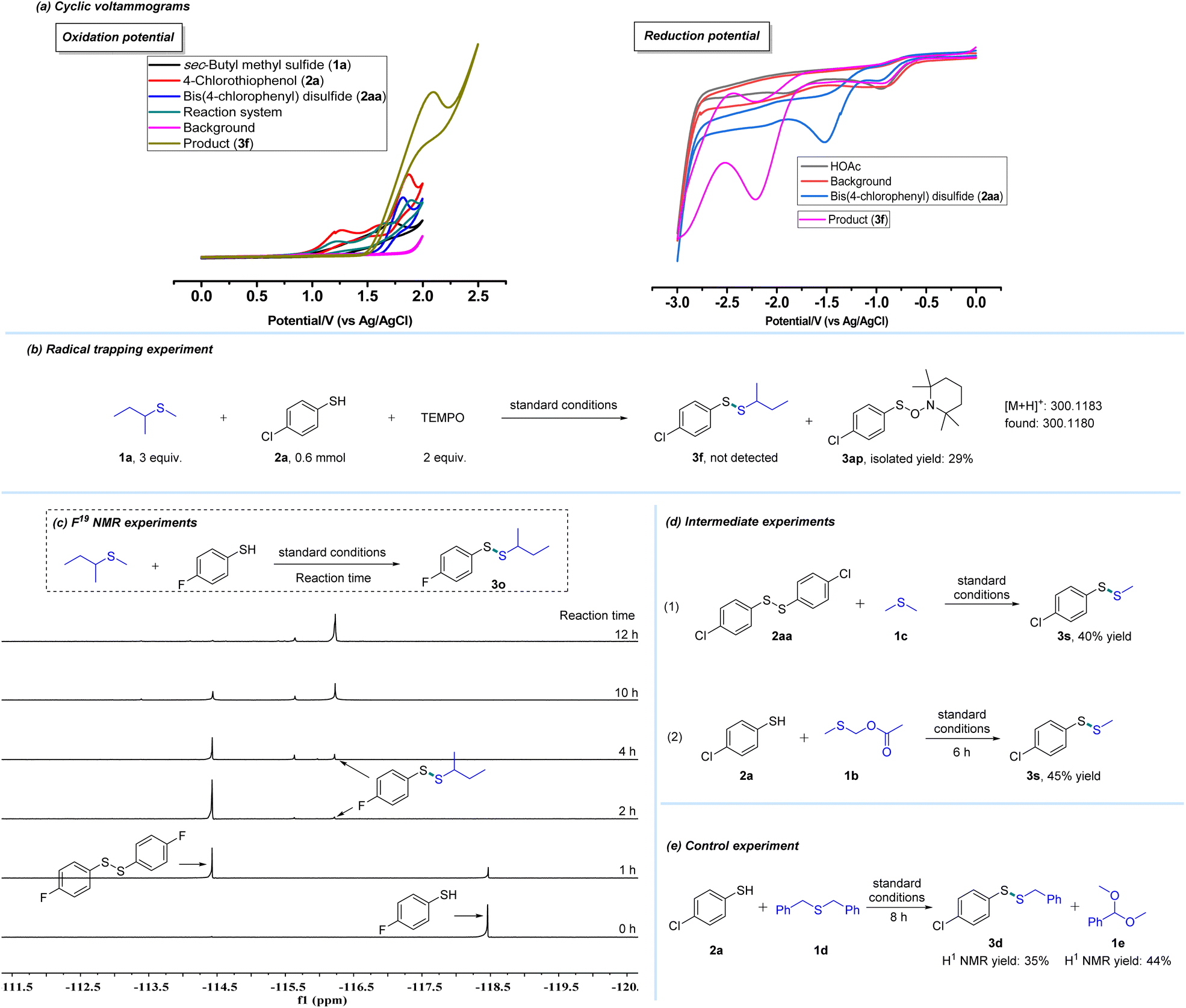 | ||
| Scheme 4 Mechanistic studies. (a) Cyclic voltammograms of related compounds (0.1 mmol) in CH3CN/DMF/CH3OH (10/2/1, 13 mL), using a glass carbon working electrode, Pt wire as the counter electrode and Ag/AgCl as the reference electrode at a 50 mV s−1 scan rate. (b) Radical trapping experiment. (c) F19 NMR experiments. (d) Intermediate experiments. (e) Control experiment. | ||
Based on the mechanistic studies, we proposed a possible mechanism in Scheme 5. Thioether (1) was oxidized at the anode to form a sulfur radical cation which could be attacked by ROH (2) to construct α-oxy thioether (3).1c Then, further oxidation of compound 3 offered radical cation (4). At the same time, symmetric disulfide (6) was generated through the homo-coupling of thiol (5) at the anode. Electron transfer to disulfide (6) from the cathode provided the radical anion (7), which released a thiol radical (9) and a thiol anion (8). Compound (10) was formed through the reaction of (4) with the thiol radical (9). Finally, the desired product (11) could be formed through a further substitution reaction.
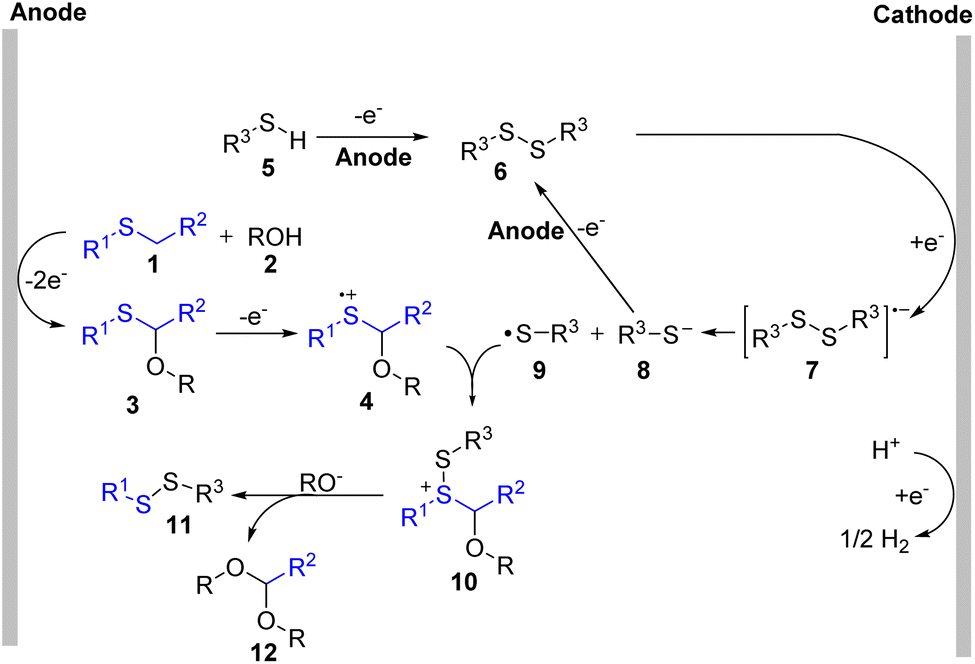 | ||
| Scheme 5 Proposed mechanism for selective C–S bond breaking. | ||
Conclusions
In summary, an unprecedented electrochemical radical-mediated approach to selectively cleave C(sp3)–S bonds is described. Compared with conventional transition-metal catalyzed C–S bond activation protocols, this method features mild, catalyst- and oxidant-free reaction conditions, as well as excellent chemoselectivity towards C(sp3)–S bond cleavage. In particular, this electrochemical protocol is able to precisely cleave one of the two similar C(sp3)–S bonds involved in unsymmetrical dialkyl thioethers, which is an important yet challenging goal in synthetic chemistry. Notably, the challenging thiomethylation could be achieved as well, by this strategy. Preliminary mechanistic studies revealed that radical species are involved in the reaction pathway and play an essential role in the site-selective activation of C(sp3)–S bonds. We anticipate that this catalyst-free electrochemical strategy will enable a broad variety of novel cross-coupling reactions through the cleavage of C(sp3)–S bonds.Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI.†Author contributions
Y. L., H. W. and Z. W. performed and analyzed experiments. A. L., Y. L., H. W. and Z. H. conceived the project and designed the experiments. A. L., Y. L., H. W. and Z. H. wrote the manuscript. All the authors (Y. L., H. W., Z. W., H. A., Z. H., A. L.) checked the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China no. 2021YFA1500100, National Natural Science Foundation of China (22031008) and Science Foundation of Wuhan (2020010601012192).Notes and references
- (a) N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 RSC; (b) S. Song, Y. Zhang, A. Yeerlan, B. Zhu, J. Liu and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 2487–2491 CrossRef CAS PubMed; (c) H. Wang, M. He, Y. Li, H. Zhang, D. Yang, M. Nagasaka, Z. Lv, Z. Guan, Y. Cao, F. Gong, Z. Zhou, J. Zhu, S. Samanta, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2021, 143, 3628–3637 CrossRef CAS PubMed; (d) T. Fuchigami and S. Inagi, Acc. Chem. Res., 2020, 53, 322–334 CrossRef CAS PubMed; (e) Y. Liu, X.-Y. Yu, J.-R. Chen, M.-M. Qiao, X. Qi, D.-Q. Shi and W.-J. Xiao, Angew. Chem., Int. Ed., 2017, 56, 9527–9531 CrossRef CAS PubMed.
- (a) L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260–11261 CrossRef CAS; (b) Y. Wang, L.-F. Deng, X. Zhang, Z.-D. Mou and D. Niu, Angew. Chem., Int. Ed., 2021, 60, 2155–2159 CrossRef CAS PubMed; (c) T. Delcaillau, P. Boehm and B. Morandi, J. Am. Chem. Soc., 2021, 143, 3723–3728 CrossRef CAS PubMed; (d) J. Lou, Q. Wang, P. Wu, H. Wang, Y.-G. Zhou and Z. Yu, Chem. Soc. Rev., 2020, 49, 4307–4359 RSC; (e) H. Prokopcová and C. O. Kappe, Angew. Chem., Int. Ed., 2008, 47, 3674–3676 CrossRef PubMed; (f) J. M. Villalobos, J. Srogl and L. S. Liebeskind, J. Am. Chem. Soc., 2007, 129, 15734–15735 CrossRef CAS PubMed; (g) W. K. Haug, E. R. Wolfson, B. T. Morman, C. M. Thomas and P. L. McGrier, J. Am. Chem. Soc., 2020, 142, 5521–5525 CrossRef CAS PubMed.
- (a) Z. Lian, B. N. Bhawal, P. Yu and B. Morandi, Science, 2017, 356, 1059–1063 CrossRef CAS PubMed; (b) X.-F. Jiang, H. Huang, Y.-F. Chai, T. L. Lohr, S.-Y. Yu, W. Lai, Y. Pan, M. Delferro and T. J. Marks, Nat. Chem., 2017, 9, 188–193 CrossRef CAS PubMed; (c) T. Sugahara, K. Murakami, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 9329–9333 CrossRef CAS PubMed.
- For selective reviews of visible light-mediated photoredox catalysis in the radical synthesis field, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) J. W. Beatty and C. R. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed; (c) J. Chen, X. Hu, L. Lu and W. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC; (d) S.-L. Meng, X.-B. Li, C.-H. Tung and L.-Z. Wu, Chem, 2021, 7, 1431–1450 CrossRef CAS; (e) F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CrossRef CAS.
- (a) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed; (b) J. B. Xiang, M. Shang, Y. Kawamata, H. Lundberg, S. H. Reisberg, M. Chen, P. Mykhailiuk, G. Beutner, M. R. Collins, A. Davies, M. D. Bel, G. M. Gallego, J. E. Spangler, J. Starr, S. L. Yang, D. G. Blackmond and P. S. Baran, Nature, 2019, 573, 398–402 CrossRef CAS PubMed; (c) D. A. Frey, S. H. K. Reddy and K. D. Moeller, J. Org. Chem., 1999, 64, 2805–2813 CrossRef CAS PubMed; (d) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed; (e) Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293–3298 CrossRef CAS PubMed; (f) P. Xiong, H.-H. Xu and H.-C. Xu, J. Am. Chem. Soc., 2017, 139, 2956–2959 CrossRef CAS PubMed; (g) C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS PubMed; (h) J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed; (i) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed; (j) N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef CAS PubMed; (k) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed; (l) S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed; (m) O. Hammerich and B. Speiser, 5th edition of Organic electrochemistry. Boca Raton, FL, CRC press, 2016 Search PubMed; (n) R. Hayash, A. Shimizu and J.-I. Yoshida, J. Am. Chem. Soc., 2016, 138, 8400–8403 CrossRef PubMed; (o) J. Li, W. Huang, J. Chen, L. He, X. Cheng and G. Li, Angew. Chem., Int. Ed., 2018, 57, 5695–5698 CrossRef CAS PubMed; (p) L.-H. Jie, B. Guo, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2022, 144, 2343–2350 CAS; (q) C. Ma, P. Fang, D. Liu, K.-J. Jiao, P.-S. Gao, H. Qiu and T.-S. Mei, Chem. Sci., 2021, 12, 12866–12873 RSC; (r) J.-H. Qin, M.-J. Luo, D.-L. An and J.-H. Li, Angew. Chem., Int. Ed., 2021, 60, 1861–1868 CrossRef CAS PubMed.
- (a) Radicals in Organic Synthesis, ed., P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, Germany, 2001 Search PubMed; (b) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (c) P. Sivaguru, Z. Wang, G. Zanoni and X. Bi, Chem. Soc. Rev., 2019, 48, 2615–2656 RSC; (d) X. Qiu, Y. Sang, H. Wu, X.-S. Xue, Z. Yan, Y. Wang, Z. Cheng, X. Wang, H. Tan, S. Song, G. Zhang, X. Zhang, K. N. Houk and N. Jiao, Nature, 2021, 597, 64–69 CrossRef CAS PubMed; (e) F. Dénès, C. H. Schiesser and P. Renaud, Chem. Soc. Rev., 2013, 42, 7900–7942 RSC; (f) X. Wu, Z. Ma, T. Feng and C. Zhu, Chem. Soc. Rev., 2021, 50, 11577–11613 RSC; (g) A. Bhunia and A. Studer, Chem, 2021, 7, 2060–2100 CrossRef CAS; (h) F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS PubMed; (i) W. Li, W. Li, D. K. Leonard, J. Rabeah, K. Junge, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 10693–10697 CrossRef CAS PubMed.
- (a) D. Griller and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13–19 CrossRef CAS; (b) H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed.
- F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed.
- (a) X. Xiao, M. Feng and X. Jiang, Angew. Chem., Int. Ed., 2016, 55, 14121–14125 CrossRef CAS PubMed; (b) Z. Wu and D. A. Pratt, J. Am. Chem. Soc., 2020, 142, 10284–10290 CrossRef CAS PubMed; (c) X.-B. Li, Z.-J. Li, Y.-J. Gao, Q.-Y. Meng, S. Yu, R. G. Weiss, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2014, 53, 2085–2089 CrossRef CAS PubMed; (d) F. Wang, Y. Chen, W. Rao, L. Ackermann and S.-Y. Wang, Nat. Commun., 2022, 13, 2588–2598 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05507d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |