
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable and practical formation of carbon–carbon and carbon–heteroatom bonds employing organo-alkali metal reagents
Lu-Qiong
Huo
a,
Xin-Hao
Wang
a,
Zhenguo
Zhang
c,
Zhenhua
Jia
 *c,
Xiao-Shui
Peng
*ab and
Henry N. C.
Wong
*ab
*c,
Xiao-Shui
Peng
*ab and
Henry N. C.
Wong
*ab
aSchool of Science and Engineering, Shenzhen Key Laboratory of Innovative Drug Synthesis, The Chinese University of Hong Kong (Shenzhen), Longgang District, Shenzhen, China. E-mail: xspeng@cuhk.edu.cn; hncwong@cuhk.edu.hk
bDepartment of Chemistry, and State Key Laboratory of Synthetic Chemistry, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong SAR, China
cInstitute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing, 211816, China. E-mail: iaszhjia@njtech.edu.cn
First published on 21st December 2022
Abstract
Metal-catalysed cross-coupling reactions are amongst the most widely used methods to directly construct new bonds. In this connection, sustainable and practical protocols, especially transition metal-catalysed cross-coupling reactions, have become the focus in many aspects of synthetic chemistry due to their high efficiency and atom economy. This review summarises recent advances from 2012 to 2022 in the formation of carbon–carbon bonds and carbon–heteroatom bonds by employing organo-alkali metal reagents.
1 Introduction
Organometallic reagents are frequently used to afford new organic molecules since they facilitate the catalysed process of organic reactions as well as the formation of specific bonds. Some efficient, economical and green catalytic processes involving organometallic reagents feature a wide range of applications, such as the synthesis of natural products and drugs, chemical biology and materials science, etc.1 Arising from the emergence of metal-catalysed cross-coupling reactions, the versatile molecules can be directly obtained using various of available organometallic reagents, such as organozinc reagents, organomagnesium reagents, organoboron reagents, organosilicon reagents and organotin reagents.2 Alkaline metals and s-group metal complexes are abundant in nature and easily prepared.3 However, organo-alkali reagents have not gained much attention in the field of cross-coupling reaction, presumably due to their natively high reactivities.Although the Suzuki–Miyaura, Stille, Kumada and Negishi cross-coupling reactions have matured in the last several decades for the formation of Csp2–Csp2 and Csp2–Csp3 bonds,4 the direct employment of organolithium reagents in cross-coupling reactions has been neglected for a long time. In addition, the participation of organo-alkali reagents in cross-coupling processes might generate toxic wastes, which is a vital and detrimental factor in the pharmaceutical industry. It is, therefore, expected that the development of new strategies considering the economical, environment-friendly and earth-abundant themes would become a research hot spot in organic synthesis. This review is not only a summary of recent progress but also an inspiration to the community for the expansion of related fields to further develop more environment-friendly and economic processes.
2 Organo-alkali metal reagents in the formation of carbon–carbon and carbon–heteroatom bonds
2.1 Organo-alkali metal reagents acting coupling partners
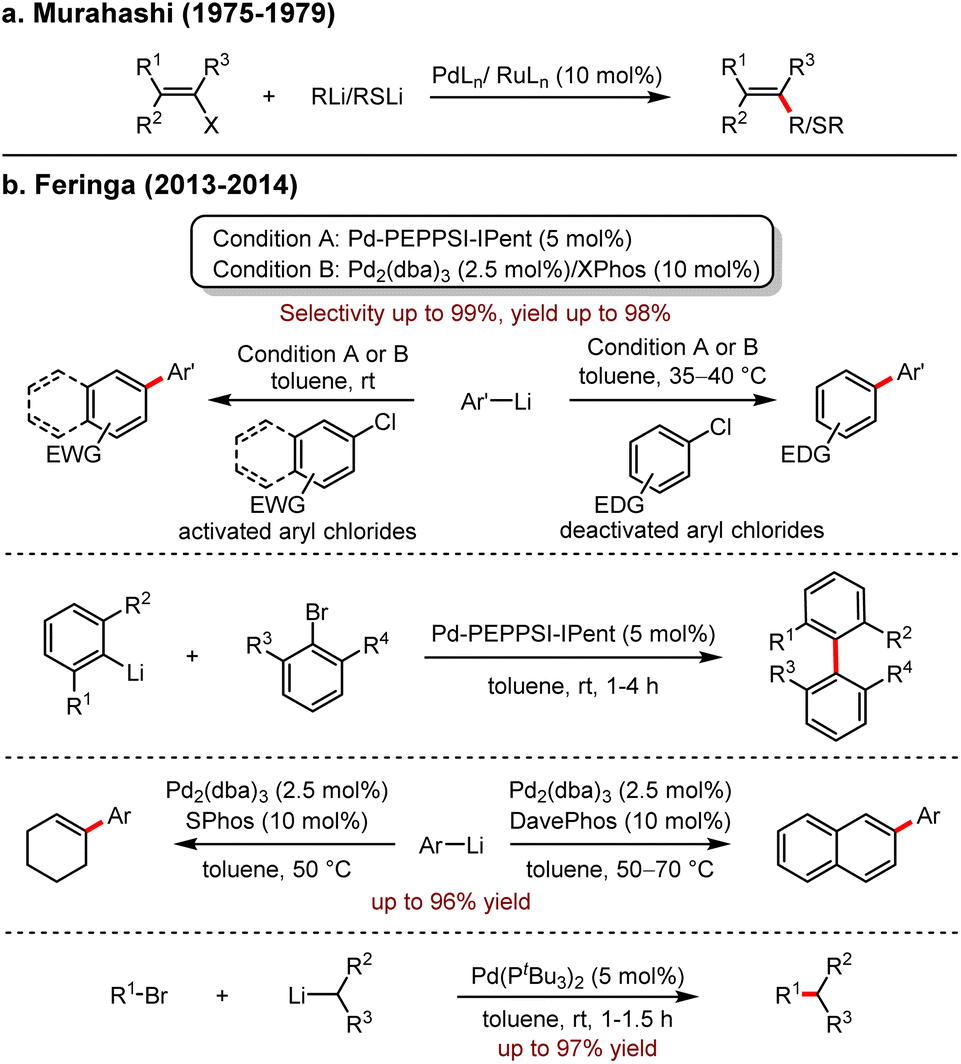 | ||
| Scheme 1 (a) Stereoselective synthesis of alkenes and alkenyl sulfides from alkenyl halides using Pd and Ru catalysts. (b) Feringa's work: Pd-catalysed cross-coupling with organolithiums. | ||
In 2015, Feringa revealed an efficient Pd-catalysed cross-coupling to introduce CH2 moiety using Me3SiCH2Li, which is a useful synthetic building block in the preparation of ArCH2SiMe3 (Scheme 2a).8 The reaction proceeded smoothly at room temperature and tolerated well with both electron-rich and electron-deficient groups, even with sterically hindered substrates. It is noteworthy that the pyridine group was also suitable for this process with high selectivity. Moreover, the unprotected hydroxyl group was also tolerated, and the corresponding product was isolated in 65% yield. In the presence of excess organolithium reagents, aryl bromides bearing multiple functional groups could be coupled with Me3SiCH2Li to afford the products in 66–82% yields.
 | ||
| Scheme 2 (a) Pd-catalysed cross-coupling of (hetero)aryl chlorides with Me3SiCH2Li. (b) Pd-catalysed cross-coupling of aryl halides with organolithium reagents under neat conditions. (c) Pd-catalysed fast cross-coupling of (hetero)aryl bromides with organolithium reagents. | ||
Subsequently, an efficient cross-coupling reaction of organohalides with organolithium reagents under neat conditions was developed also by the same group (Scheme 2b).9 This protocol avoided many undesired manipulations, such as inert atmosphere, low temperature, dilution and slow addition of substrate. Other notable features are the low catalyst loading down to 0.1 mol%, the scalability of up to 120 mmol and the short reaction time in 10 minutes. The organolithium reagents are commercially available or could be prepared following general procedures such as lithium–halogen exchange or ortho-directed lithiation. In all cases, only a small amount of ether was used to enhance the solubility. Importantly, no dehalogenation occurred, indicating that the competitive β-hydride elimination/olefin dissociation did not proceed. Moreover, the coupling of sterically hindered bromides with organolithium reagents to afford biaryls was also achieved successfully at room temperature in a short reaction time (10 minutes). The protocol exhibited high efficiencies and selectivities with SiMe3CH2Li and aryllithiums. While nHexLi and nBuLi reacted with solid substrates, dehalogenated by-products were produced.
Inspired by Schoenebeck's Pd-catalysed reactions in which Grignard reagents or organozinc reagents coupled very rapidly with aryl halides,10 Feringa and co-workers investigated the possibility of fast cross-coupling with organolithiums to aryl halides. In 2017, an ultrafast coupling reaction between aryl- or alkyl-lithium reagents and aryl bromides in 5 s to 5 min with the participation of 2–3 nm Pd nanoparticles was uncovered.11 In contrast to the inert atmosphere, the presence of molecular oxygen is crucial for the formation of this powerful catalyst, and the protocol exhibited a broad range of substrate scope, including naphthalene derivatives in 61–95% yields, anthracene in 97% yield, unprotected phenol in 83% yield, and the substrate bearing an epoxide functional group afforded 1 in 58% yield. Surprisingly, this protocol provided an alternative approach to access ferrocene derivatives, albeit in relatively lower yields, for example, 2 in 20% yield and 3 in 40% yield, respectively (Scheme 2c).
There are several methods to access unsymmetrical ketones, including Friedel–Crafts acylation, oxidation of secondary alcohols, nucleophilic addition of carboxylic acid derivatives with organometallic reagents, etc. However, the challenge remains for the modification of unprotected ketones with organolithium reagents, because the highly reactive organolithium reagents likely lead to 1,2-addition to the carbonyl group.
In the same year, an efficient protocol involving 1,2-addition and Pd-catalysed cross-coupling was disclosed by Feringa and co-workers.12 In this work, the organolithium compound R1Li underwent 1,2-addition to Weinreb amide to form a stable tetrahedral intermediate, which simultaneously masked the ketone, and was immediately followed by a one-pot Pd-catalysed cross-coupling with the second organolithium reagent R2Li to produce unsymmetric ketones (Scheme 3a). To avoid the competing Buchwald–Hartwig coupling, they employed a masked keto-tetrahedral intermediate to undergo the desired transformation. Subsequently, a general one-pot process involving nucleophilic addition of organolithium reagents to amides was developed in 2019, which formed aminolithium compounds in situ following a direct Buchwald–Hartwig amination to provide aminoaryl ketones up to 89% yield (Scheme 3b).13 A wide range of aryllithium nucleophiles and amides was tolerated, leading to the corresponding aminodiphenylketone derivatives. The successful coupling with cyclic or acyclic amines included morpholine, methylpiperazine, dihydroindole and tetrahydroisoquinoline moieties.
 | ||
| Scheme 3 (a) One-pot 1,2-addition/Pd-catalysed cross-coupling of Weinreb amides. (b) Pd-catalysed one-pot approach to functionalised ketones via nucleophilic addition/Buchwald–Hartwig amination strategy. (c) Pd-catalysed one-pot sequential coupling with alkyl- or aryllithium reagents with functionalised nucleophiles. | ||
Biaryls, especially sterically hindered tri- and tetra-ortho-substituted biaryls are the fundamental skeleton of functional molecules, which are widely found in natural products, organic materials, pharmaceuticals and ligands.14 Although biaryls could be obtained via the use of organoboron compounds or organozinc reagents, these methods usually featured long reaction times and high temperatures. In 2019, Organ, Feringa and co-workers15 reported a one-pot method for the rapid construction of biaryls (Scheme 3c). It is noteworthy that this strategy avoided the requirement of slowly adding the alkyllithium reagents, which were added directly in a one-shot manner. Moreover, the authors also disclosed an efficient way to selectively couple with multiple nucleophilic reagents by controlling the temperature, which enabled the divergent synthesis of various complex molecules.
Feringa and co-workers developed a series of Murahashi coupling reactions for the direct coupling of alkyl-, alkenyl-, and (hetero)aryl-lithium reagents with various aryl or alkenyl halides as electrophilic partners.5–9,11–13 To circumvent lithium–halogen exchange and homo-coupling side reactions, their strategies include a slow dropwise addition (usually over one hour) of extremely dilute organolithium in a nonpolar solvent (e.g. toluene) or in neat and the use of electron-rich ligands such as PtBu3.
In the same year, Capriati and co-workers16 reported a novel Pd-catalysed cross-coupling reaction of aryl bromides or chlorides with organolithium reagents on water and found that sodium chloride was treated as an efficient additive to provide the corresponding products (Scheme 4). The advantages of this method are illustrated as (a) the simple manipulation in air, avoiding any anhydrous cautions, (b) the addition of organolithium reagents without the use of a syringe pump or in a slow dropwise manner and (c) the ultra-fast rate of the reaction (20 s). The versatility of this protocol was also demonstrated to tolerate electron-withdrawing and electron-donating groups, albeit, with lithium phenylethynyl, the yield was lower to afford the alkyne 4 in 17% yield.
 | ||
| Scheme 4 Pd-catalysed cross-coupling of (hetero)aryl halides with organolithium reagents on water at room temperature and under air. | ||
Despite the remarkable progress in metal-catalysed Csp2–Csp2 coupling with aryllithiums, the Csp2–Csp3 coupling reaction remains rare. In particular, the combination of aryl chloride with alkyllithium has not been reported. In 2020, Gessner and co-workers17 disclosed the employment of palladium catalysts for the coupling of aryl chlorides with alkyllithium reagents (Scheme 5). This method tolerated electron-rich (59–88% yields) and electron-deficient (56–96% yields) substrates. Moreover, the ratio of branch and linear products 5 was over 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, when isopropyl- or isobutyl-lithium reagent was utilised.
:1, when isopropyl- or isobutyl-lithium reagent was utilised.
 | ||
| Scheme 5 Pd-catalysed direct coupling of aryl chlorides with alkyllithium reagents. | ||
Biaryl compounds with an aza-heterocyclic core are excellent pharmacodynamic groups, which are widely found in small-molecule drugs.18 In 2020, a continuous flow strategy to prepare the organolithium reagents for the synthesis of heteroaryl sulfoxides in gram scale was reported by Lima, Sedelmeier and co-workers.19 As shown in Scheme 6, lithium heteroaryl sulfites were generated in a continuous flow process and simply separated, which was then subjected to a direct Pd-catalysed cross-coupling with aryl halides, affording bis-heteroaryls in moderate to good yields.
 | ||
| Scheme 6 Pd-catalysed desulfinative cross-couplings with heteroaromatic lithium sulfinates towards the synthesis of bis-heteroaryls. | ||
Allenes, featuring the two consecutive orthogonal double bonds with an sp-hybridised carbon atom as its center, has a wide range of applications in materials, ligands and bioactive compounds.20 In 2020, Feringa and co-workers reported a palladium-catalysed cross-coupling of propadienyl- or propargyl-lithium reagents with aryl bromides to furnish highly functionalised propadiene derivatives in 46–93% yields.21 This protocol provided a fast and efficient method for the selective synthesis of trisubstituted or tetrasubstituted propadiene, avoiding the formation of propargyl by-products (Scheme 7a). In the same year, a Sonogashira type reaction in the combination of lithium acetylide reagents with aryl bromides was also developed.22 Most examples proceeded under mild conditions to give the corresponding arylacetylene derivatives in 45 minutes. It is noted that the broad substrate scope of this method with excellent selectivities was demonstrated by its significant functional group tolerance, including electron-deficient, electron-rich substrates, compounds, and polybromo-containing substrates. Importantly, for 6-bromocoumarin 6, the undesired nucleophilic addition by organolithium reagents was inhibited and the corresponding coupling product was obtained in 21% yield (Scheme 7b).
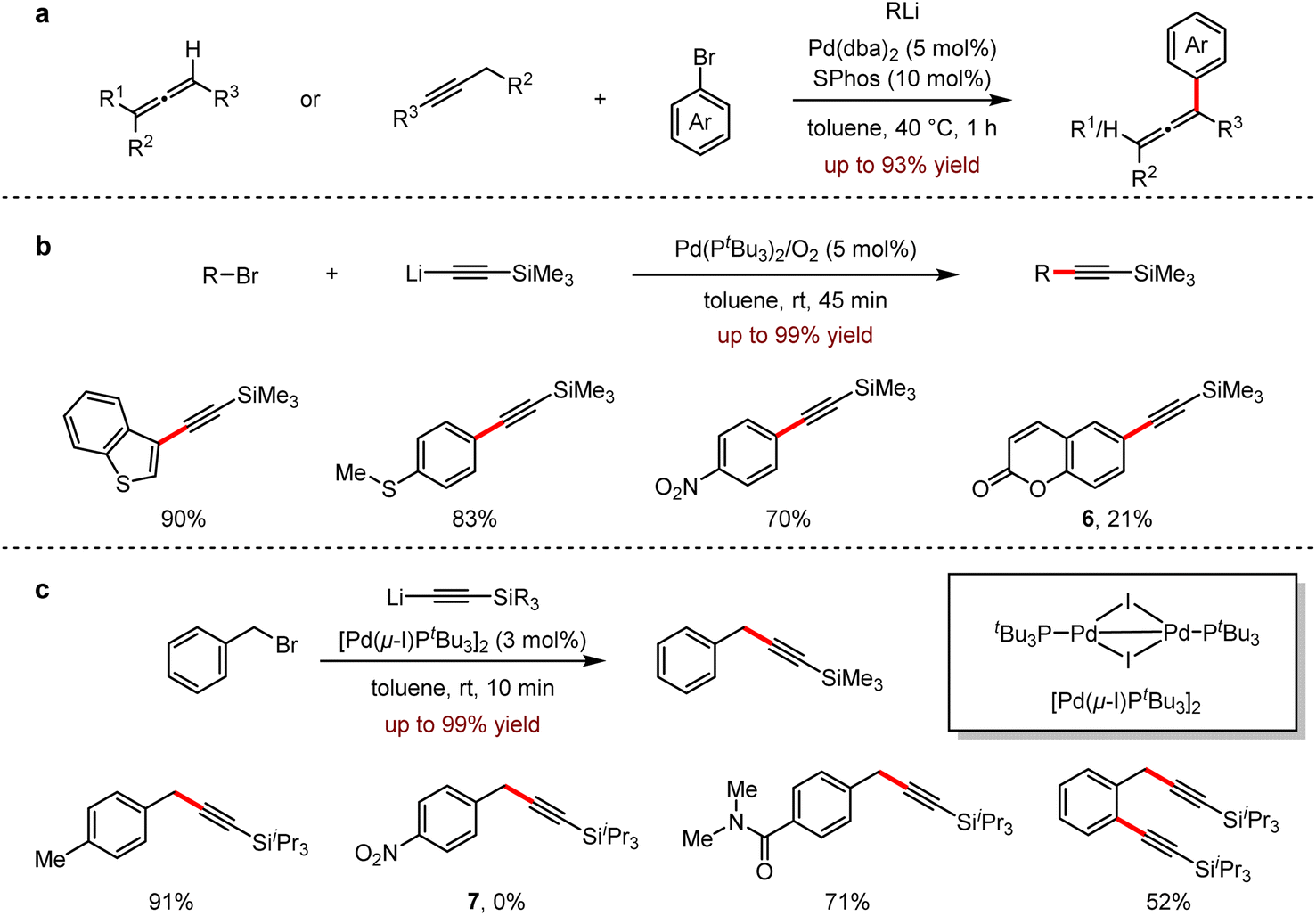 | ||
| Scheme 7 (a) Synthesis and functionalisation of allenes by Pd-catalysed cross-coupling with organolithium. (b) Pd-catalysed cross-coupling with lithium acetylides and aryl bromides. (c) Pd-catalysed cross-coupling of benzyl bromides with lithium acetylides. | ||
In 2021, Feringa and coworkers subsequently reported the rapid coupling of organolithium reagents with aromatic (pseudo) halides.23 This efficient protocol of cross-coupling reaction with benzyl bromide and lithium acetylenes allowed the presence of organolithium-sensitive functional groups such as esters, nitrile, amides and boronic esters. The generality of this methodology was also presented with its excellent functional group tolerance. However, there's no desired product 7 detected for the coupling between triisopropylsilyllithium and 1-(bromomethyl)-4-nitrobenzene (Scheme 7c).
Very recently, the efficient remote Csp3–H activation reactions involving the direct Pd-catalysed cross-coupling of aryllithium reagents with terminal alkynes were disclosed by Wong, Peng and co-workers (Scheme 8).24 This strategy showed a broad functional group tolerance with excellent regioselectivities, affording the desired products up to 98% yield. It should be noted that the sterically hindered alkynes proceeded smoothly under optimal conditions. The late modification of the natural product (+)-δ-tocopherol further demonstrated the synthetic feasibility of the methodology.
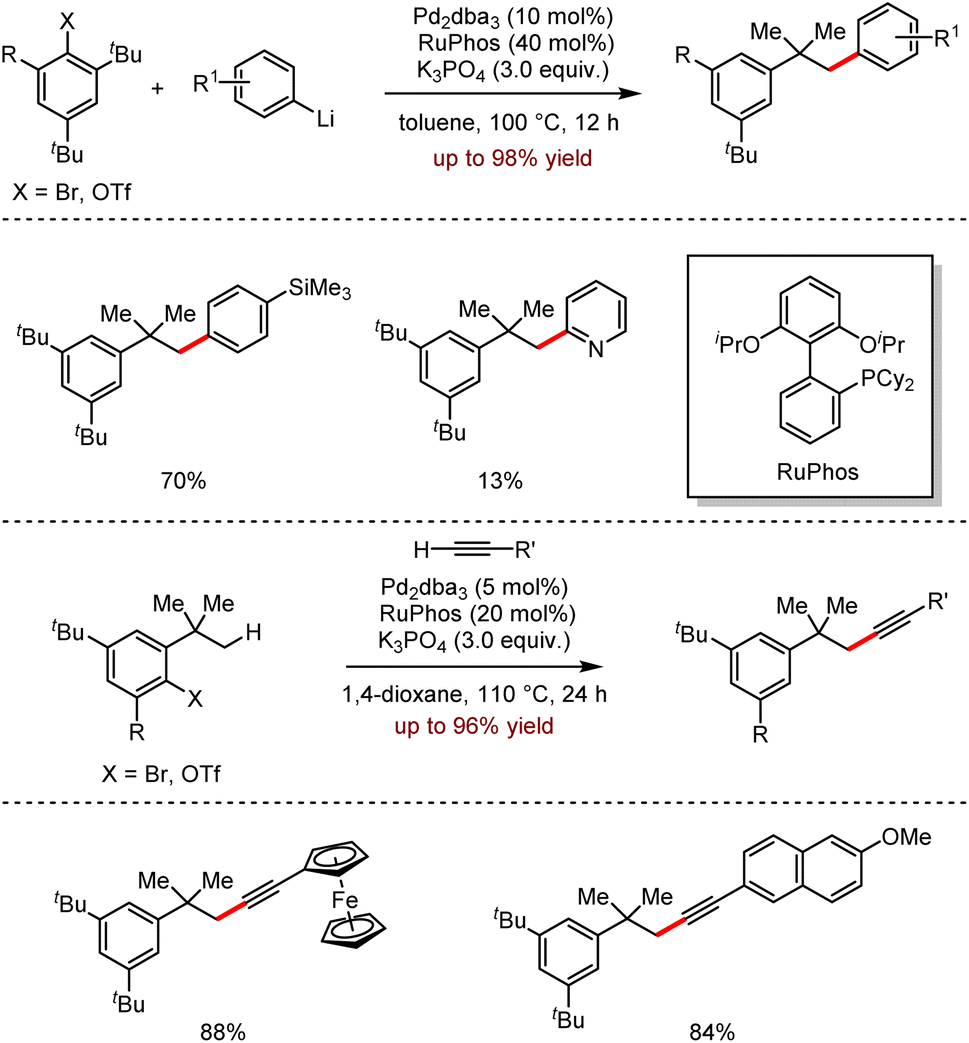 | ||
| Scheme 8 Pd-catalysed intermolecular arylation and alkynylation with organolithiums and terminal alkynes. | ||
In 2014, Rueping and co-workers recorded a novel synthetic pathway which involved the direct demethoxylation with organolithium reagent LiCH2SiMe3via nickel catalysis.26 (Scheme 9). The corresponding cross-coupling products were produced in 61–99% yields. Especially, naphthalene derivatives bearing two methoxy groups at diverse positions could selectively deliver the mono-substituted product 8 in 63% yield by treatment with an excess of 1,7-dimethoxynaphthalene, or the di-substituted product 9 in 97% yield. Nitrogen-containing heterocyclic derivatives such as indole, pyrrole and tetrahydroquinoline are also suitable for this demethoxylation reaction. It is worth mentioning that the natural product dimethoxy-β-estradiol was directly modified to afford its derivative 10 in 87% yield.
 | ||
| Scheme 9 Ni-catalysed dealkoxylative Caryl–Csp3 cross-coupling reaction. | ||
The successful conversion of the existing structurally diverse allyl silyl ethers and oxygenated heterocycles to allylsilanes provides powerful synthetic tools for the construction of functional molecular scaffolds and allows for a variety of subsequent structural modifications. In 2015, Rueping and co-workers reported a novel nickel-catalysed dealkylation cross-coupling reaction that converted allyl silyl ethers to the corresponding allylsilane and alcohol derivatives with high stereoselectivities in moderate to excellent yields.27 Moreover, the reaction underwent smoothly for diverse monosubstituted and disubstituted alkene substrates. The coupling between enol ethers and oxygen-, nitrogen- and sulfur-containing heterocycles also proceeded well under optimal conditions.
To demonstrate the potential application of this protocol, a scale-up experiment with 1 mol% catalyst loading was carried out and the corresponding product trimethyl(2-(naphthalen-2-yl)allyl)silane was obtained in 98% yield. When benzofurans, containing a natural Csp2–O bond, were treated as the substrates, the Csp2–O bonds were cleavaged to give the ring-opening products selectively in the presence of Ni(COD)2 and SIPr·HCl (Scheme 10).
 | ||
| Scheme 10 Ni-catalysed dealkoxylation of enol ether silylation. | ||
In 2016, Feringa and co-workers disclosed an alternative method of Ni-catalysed cross-coupling with easy-to-prepare organolithium reagents as coupling partners (Scheme 11).28 Both nickel(II) complex C1 and [NiCl2(depe)] (depe = bis(diethylphosphino)ethane) were efficient catalysts to catalyse the cross-coupling reactions with organometallic lithium.
 | ||
| Scheme 11 Ni-catalysed cross-coupling of (hetero)aryl electrophiles with organolithium reagents. | ||
When heteroaryllithium was coupled with the aromatic precursors, the corresponding polyaromatic products were generated in 25–89% yields. For instance, trifluoromethyl-substituted aromatic compounds reacted efficiently with phenyllithium; moreover, 1-fluoro-4-(trifluoromethyl)benzene was less reactive than 1-bromo-4-(trifluoromethyl)benzene or 1-chloro-4-(trifluoromethyl)benzene. However, the expected cross-coupling reaction between 3-bromo-N,N-dimethylaniline and phenyllithium was not observed without the detection of the corresponding product, instead of the dehalogenated by-product.
In 2016, Uchiyama and co-workers reported Ni-catalysed cross-coupling between organolithium and ether or ammonium salts, featuring the cleavage of carbon–oxygen and carbon–nitrogen bonds simultaneously to construct the carbon–carbon bonds (Scheme 12).29
 | ||
| Scheme 12 Ni-catalysed cross-coupling of ethers or aryl ammonium salts with organolithiums via C–O or C–N bond cleavage. | ||
They found π-rich heterocyclic aromatic lithium reagents are well tolerated, while π-deficient heterocyclic aromatic lithium reagents, such as thienyllithium and furanyllithium, exhibited rather disappointing reactivities. In most examples, alkenyl- and alkyl-lithium worked well, but the cross-coupling between acyclic alkenyllithium with 2-methoxynaphthalene was insufficient to give 12 in 35% yield. For chiral aryl methyl ether substrates, the absolute configuration of biaryl products (R)-13 and (S)-13 was maintained without any racemisation, especially at the acidic benzylic position.
Late transition metal-catalysed cross-couplings have been widely employed to install various functional groups into unsaturated substrates30 The probable mechanism has been also revealed by simulating these catalytic systems, involving the exchange of transition metals with isoelectronic properties to provide useful mechanistic insights. In the above studies, the anionic Nickel–ate complexes are more efficient in oxidative addition with aryl halides or alkenyl halides as compared to the use of nickel salts.31 In particular, the anionic Nickel–ate species contribute to the cleavage of carbon–halogen bonds due to a strong nucleophilicity.32 Despite the prevalence of reports related to the anionic cross-coupling reactions, relevant studies on the electron-rich Nickel–ate complexes remain relatively rare.
Kambe et al. found that Lithioalkenyl sulfoximines bearing a chiral nucleofuge, failed to undergo nucleophilic substitution with organolithium or Fritsch–Buttenberg–Wiechell (FBW) rearrangement at 0 °C.33 When metalloalkenyl sulfoximines were treated by organolithium agents, alkenyllithiums were generated via the transformation between an anionic Ni–ate and Ni–vinylidene. Therefore, the Ni0–vinylidene was obtained by releasing PhS(O)N(Li)Me, indicating that the electron-rich nickel complex is necessary for the following process (Scheme 13a). Numerous studies on metal–vinylidene complexes have shown that the rearrangement of acetylene to vinylidene is a thermodynamically favorable process.34 Accordingly, a highly reduced nickel center was employed to generate Ni–vinylidenes.
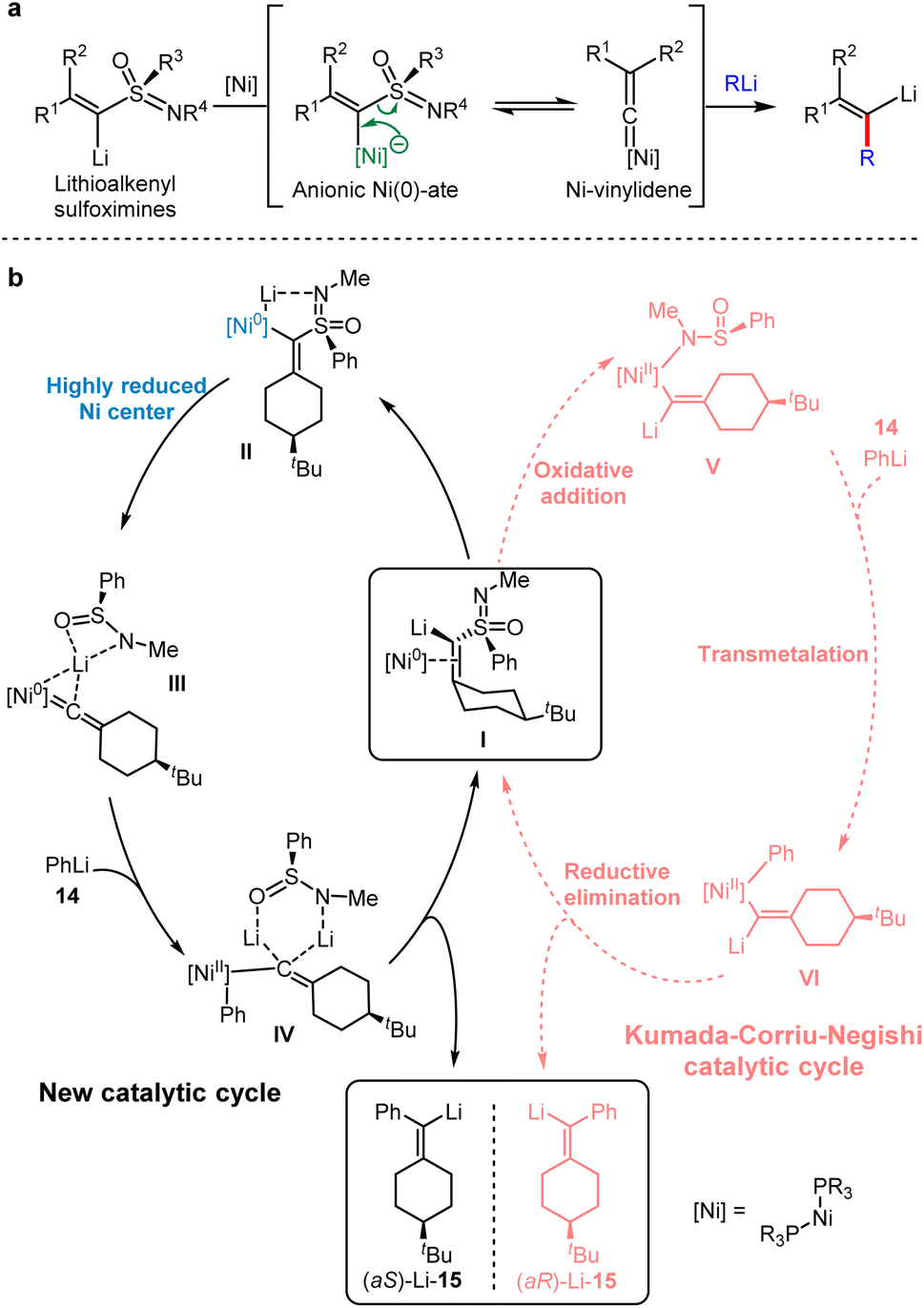 | ||
| Scheme 13 Ni-catalysed anionic cross-coupling reaction of lithium sulfonimidoyl alkylidene carbenoids with organolithiums. (a) Ni-catalysed anionic cross-coupling reaction (ACCR). (b) Proposed mechanism for ACCR (Solid lines depicted the proposed reaction pathway of the Ni-catalysed ACCR. Dashed lines depicted the oxidative addition pathway, which induced the opposite stereoselectivity). | ||
In 2020, Gais and co-workers combined experimental and computational results to record a novel catalytic cycle involving the conversion of Ni–ate induced by organolithium with Ni–vinylidene intermediates via α-elimination.35 The detailed mechanism of the anionic cross-coupling reactions (ACCR) of Ni-catalysed metalloalkenyl sulfoximines was elaborated.
As shown in Scheme 13b at the right side in the dashed line, a typical Kumada–Corriu–Negishi (KCN) cross-coupling pathway initiated from the oxidative addition to provide Ni(II) complexes. Following the transmetallation with phenyllithium and the reductive elimination, streptavidin lithium was generated. Alternatively, at the left side in the solid line, the neutral Ni0 complex was involved, which produced Ni–ate species by the mediation of lithium ions, and this transient intermediate underwent α-elimination to give Ni–vinylidene. DFT calculations indicated that the reaction energy barrier would be 6 kcal mol−1 higher without the participation of lithium ions, indicating the importance of lithium ions in the catalytic cycle. After the addition of PhLi 14, a Ni(II) intermediate with carbon anion IV was generated, which was probably stabilised by two lithium ions. Finally, the vinyl lithium 15 product was afforded by the reductive elimination of IV with the regeneration of I.
Ni-catalysed cross-coupling of aryl ethers is an effective method for the construction of carbon–carbon and carbon–heteroatom bonds. Moreover, the mechanism may differ from the conventional coupling reactions due to the inertness of the sp2 carbon–oxygen bond, which is thermodynamically and kinetically unfavourable for the oxidative addition of aryl ethers to nickel. Wang, Uchiyama and co-workers36 provided the theoretical support for the alternative anionic pathway through DFT calculations to identify the formation of the anionic Ni0–ate as the key in the process of C–O bond cleavage. Only a few catalytic intermediates have been confirmed; therefore, direct experimental evidence to elucidate the anionic pathway is still lacking.
In 2021, Borys and Hevia reported detailed studies to disclose the ionic pathway, in which electron-rich hetero-bimetallic Ni–ates were obtained by the addition of organometallic nucleophiles to a Ni(0) center.37 Based on their experimental investigation and computational studies, the probable mechanism is shown in Scheme 14. Initially, the addition of Ni(cod)2 (I) and PhLi(donor)n afforded lithium nickelate II (1:1) and VI (2:1), following the coordination with 2-methoxynaphthalene to generate the intermediates III or VII, respectively. The coordination pulls the substrate closer to the active nickel center and facilitates the oxidative addition to nickel. Subsequently, the cleavage of the C–O bond occurs through the presumable transition state IV to deliver the Ni(II) species V or VIII by the release of the lithium methoxide, which is consistent with the computational calculations. Furthermore, the possible transition state IV suggests that aryl ether substrates containing π-extended aromatic systems can generate more stable dearomatised intermediates. Eventually, Ni(0) catalysts (I or II) are regenerated by the reductive elimination of Ni(II) to furnish the catalytic cycles.
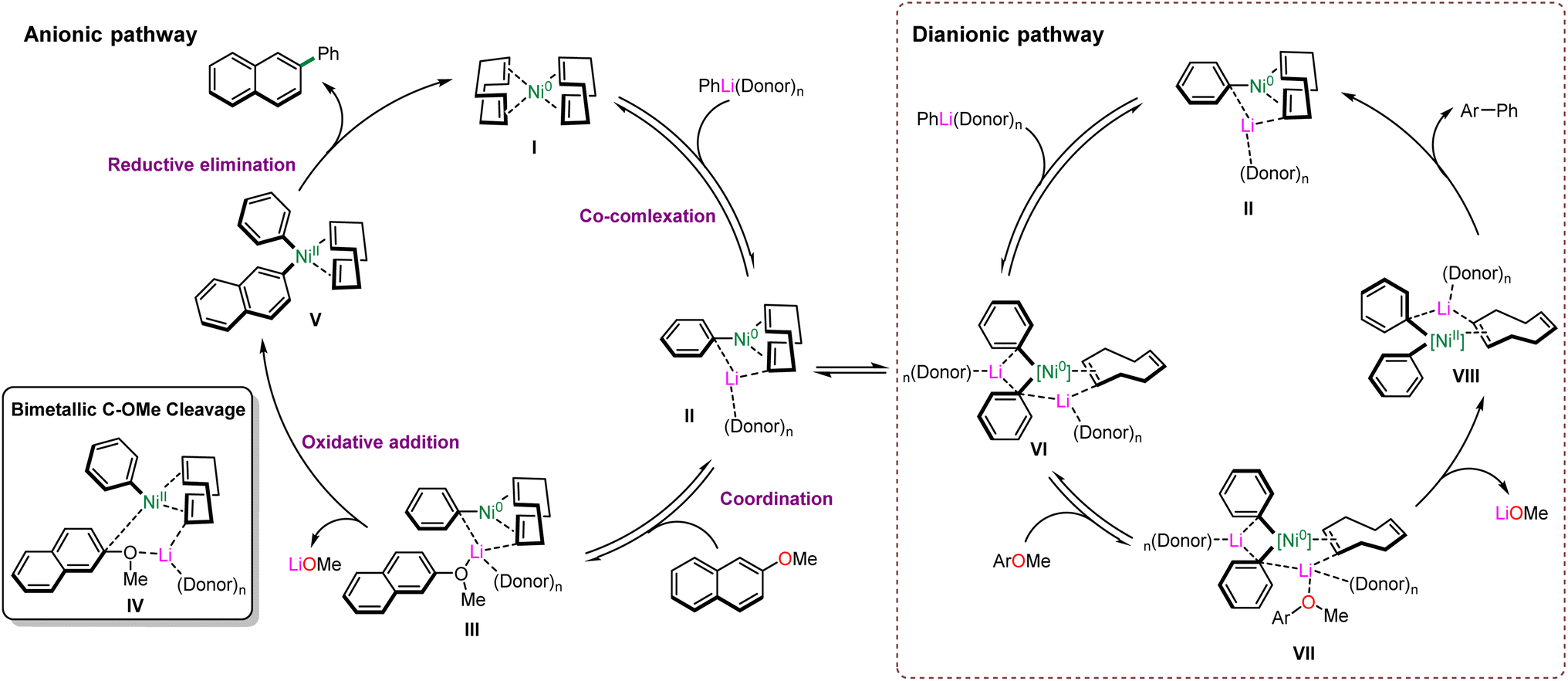 | ||
| Scheme 14 Proposed mechanism (anionic and dianionic) of Ni-catalysed cross-coupling of aryl ethers. | ||
In 2016, Wong's group developed an efficient Fe-catalysed cross-coupling of organolithium reagents with a variety of organic bromides under mild conditions to construct diverse of Csp2–Csp3 bonds and Csp3–Csp3 bonds, demonstrating that organolithium reagents can also be employed as the partners in iron-catalysed reactions.39 Shortly after, they also realised iron-catalysed cross-coupling reactions with organolithium reagents to forge Csp2–Csp2 bonds and other types of carbon–carbon bonds, which were allowed to practically prepare 1,3-butadiene and biaryls via homo-coupling using alkenyl-lithium or aryl-lithium reagents.40
In 2020, Wong and co-workers reported a strategy of Fe-catalysed homo-coupling reaction without the assistance of ligands.41 As shown in Scheme 15, this reaction exhibited broad functional group compatibility to afford various biaryls in moderate to good yields. Especially, when the bromotetraphenylene was subjected to the standard conditions, the expected product 16 was obtained in a 31% yield.
 | ||
| Scheme 15 Fe-catalysed homo-coupling with aryllithium reagents. | ||
Subsequently, they also reported a protocol of Fe-catalysed cross-coupling of organolithium compounds with haloalkenes (Scheme 16).42 In the presence of Fe(acac)2 and XPhos, the desired products were produced with good functional group tolerance. To further demonstrate the potential industrial application, a gram scale reaction was realised in this work.
 | ||
| Scheme 16 Fe-catalysed Csp2–Csp2 cross-coupling with aryllithium. | ||
In 2013, Duan and co-workers established a cooperative cross-coupling reaction catalysed by cobalt and titanium with Grignard reagents or organolithium. As shown in Scheme 17, the reactions underwent efficiently with the combination of Ti(OEt)4 as co-catalyst, CoCl2 as catalyst and PBu3 as ligand, leading to the biaryl products in moderate to excellent yields.44
 | ||
| Scheme 17 Co/Ti catalysed cooperative couplings of aryl halides with organometallics. | ||
A series of control experiments indicated that in the absence of titanate the homo-coupling dominated the reaction, suggesting the necessity for a bimetallic co-catalyst. The generality of this reaction was shown as the good functional group tolerance, such as thienyl, pyridinyl, quinolinyl, and naphthyl etc. Both Grignard reagents and organolithium reagents could be reacted with aryl bromides and chlorides under mild conditions. Moreover, aryl bromide was more active than aryl chloride, for instance, the biaryl product was obtained in 56% yield using 1-bromo-4-methoxybenzene as the starting material, while only a trace amount of product was detected with 1-chloro-4-methoxybenzene as the substrate.
In 2012, Smith et al. presented a palladium and copper catalysed cross-coupling reaction of aryl halides and an in situ formed silicon–lithium reagents.45 From the insight of the probable mechanism, this relay process involved anion relay chemistry (ARC). On the basis of this work, they designed and prepared a series of this-type reagents.46 Furthermore, the polymer-supported siloxane-transfer reagents were also produced and screened including PSTA-I20 (17), PSTA-I200′ (18), PSTA-II20 (19), PSTA-II200 (20) and PSTA-I200 (21) to enhance the reaction efficiency and their stability after several recycles (Scheme 18). Under standard conditions, the reaction exhibited excellent functional group tolerance and the yield was up to 98% yield.
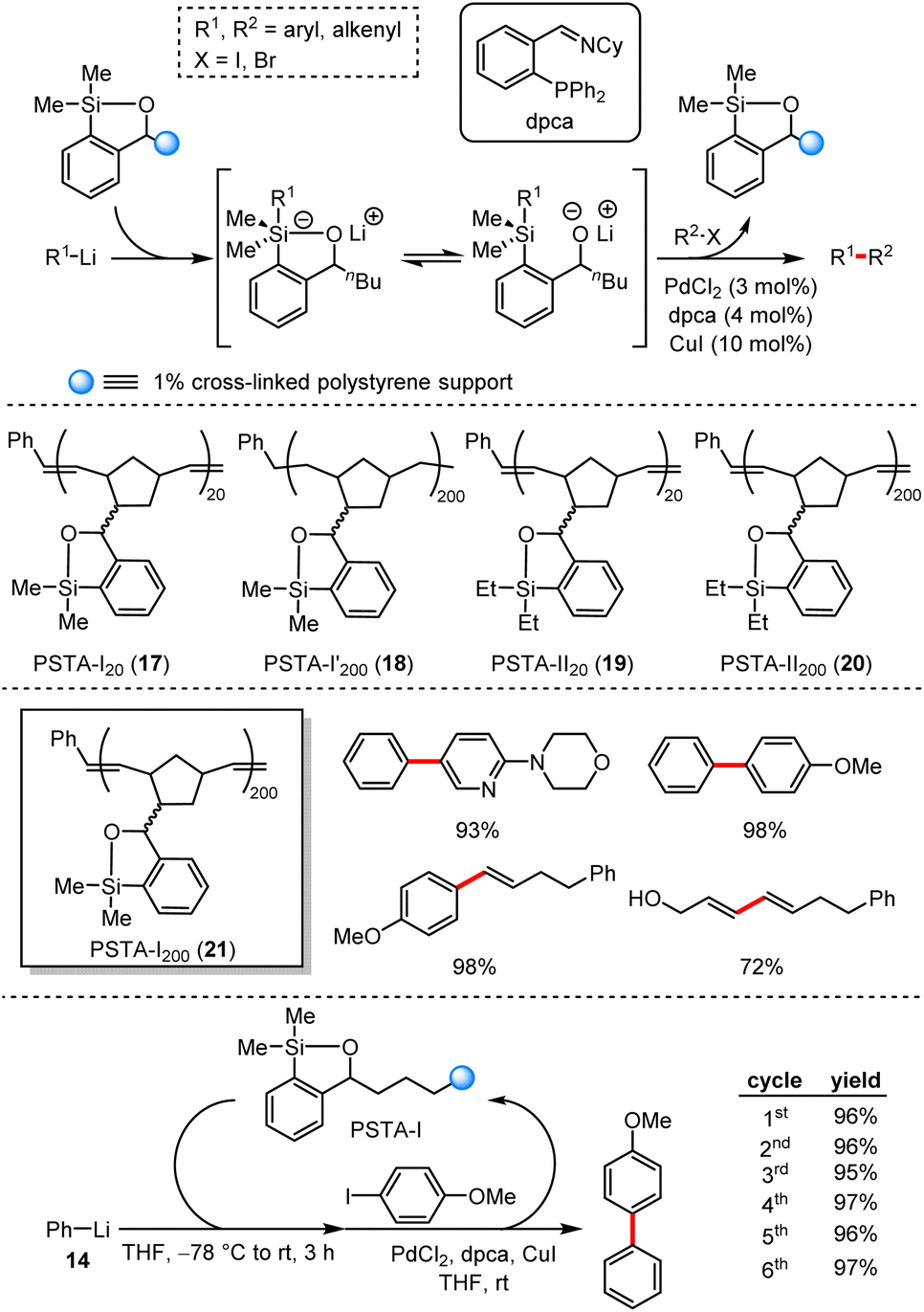 | ||
| Scheme 18 Polymer-supported silicon-transfer agents for cross-coupling reactions with organolithium reagents. | ||
Diaryl sulfone is an important motif in organic synthesis with diverse biological activities such as the inhibition of HIV-1 reverse transcriptase, antibacterial and antitumour properties.51 In 2017, Manolikakes and co-workers reported a nickel-catalysed cross-coupling reaction between sodium sulfite and (hetero)aryl halides, indicating that sodium sulfite was a promising coupling partner (Scheme 19).52a The versatility of this method was demonstrated in a wide range of aryl bromides or activated aryl chlorides with various sodium salt of aryl sulfonic acids; moreover, substrates bearing electron-rich groups gave higher yields. For the aromatic heterocyclic bromides, the corresponding sulfone products were provided in 50–95% yields.
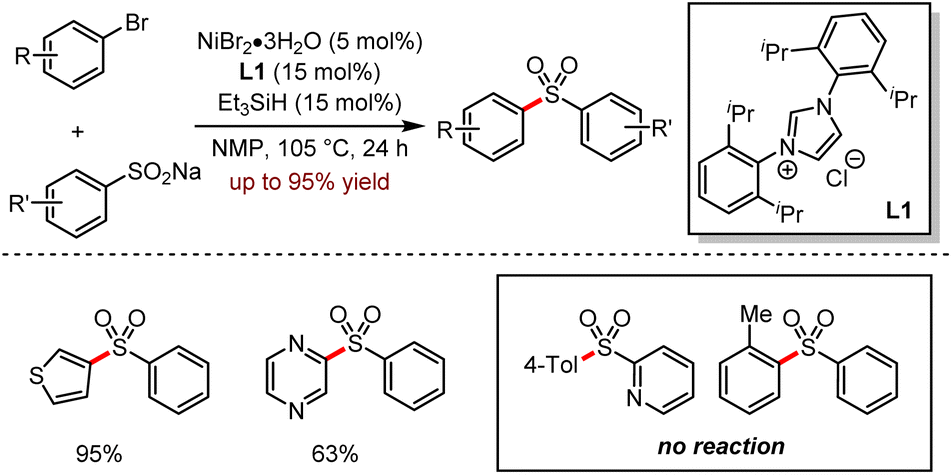 | ||
| Scheme 19 Ni-catalysed synthesis of diaryl sulfones from aryl halides and sodium sulfinates. | ||
Subsequently, they reported a manganese-mediated C–H sulfonylation via the coupling reaction between organosodium or organolithium reagents and 1,4-dimethoxybenzenes (Scheme 20).52b These organolithium and organosodium reagents could be easily obtained from sulfur dioxide as their precursors. It is worth noting that the reactions proceeded efficiently at room temperature to afford various sulfones in moderate to good yields. But this protocol was limited to the use of excess stoichiometric metal salts, considering the atom economy.
 | ||
| Scheme 20 Mn(OAc)3 mediated C–H sulfonylation of 1,4-dimethoxybenzenes with sodium and lithium sulfinates (HFIP = hexafluoroisopropanol). | ||
Furthermore, Manolikakes and coworkers also reported an effective cross-coupling of sodium sulfites with aryl iodides by photoredox/nickel dual catalysis (Scheme 21).53
 | ||
| Scheme 21 Visible-light mediated photoredox/nickel dual catalysis for the cross-coupling of aryl iodides with sulfonic acid salts. | ||
This method exhibited a board scope and tolerated diverse functional groups to afford the corresponding diaryl sulfones up to 89% yield. Specifically, aryl iodides bearing substituents at the para and meta positions or with sensitive groups such as trifluoromethyl, cyano, ester and carbonyl were suitable for this strategy. Moreover, substrates with free amino, hydroxyl and carboxyl groups achieved the corresponding products in moderate to good yields.
Transition metal-catalysed cross-coupling between two fragments or reagents has become one of the most efficient and reliable synthetic methods for the construction of structurally and functionally diverse organic molecules.54 The general methods for the preparation of organometallic reagents employed the transmetallation of organolithium reagents or Grignard reagents, which transferred the organic moieties from Mg or Li centers to Zn or B. Despite their extensive use in organic synthesis, these methods still have some limitations, for instance, the unexpensive organic chlorides are less reactive and more difficult to be utilised than organic bromides or organic iodides in cross-coupling reactions. Recently, Knochel et al. addressed this issue by using iPrMgCl·LiCl reagent to activate the substrate.55 The alternative approach to the desired coupling with aryl chlorides is the direct use of organosodium reagents as the coupling partners. In 2019, Asako and co-workers56 reported a Pd-catalysed cross-coupling of organosodium reagents with aryl chlorides to provide the corresponding biaryl products (Scheme 22).
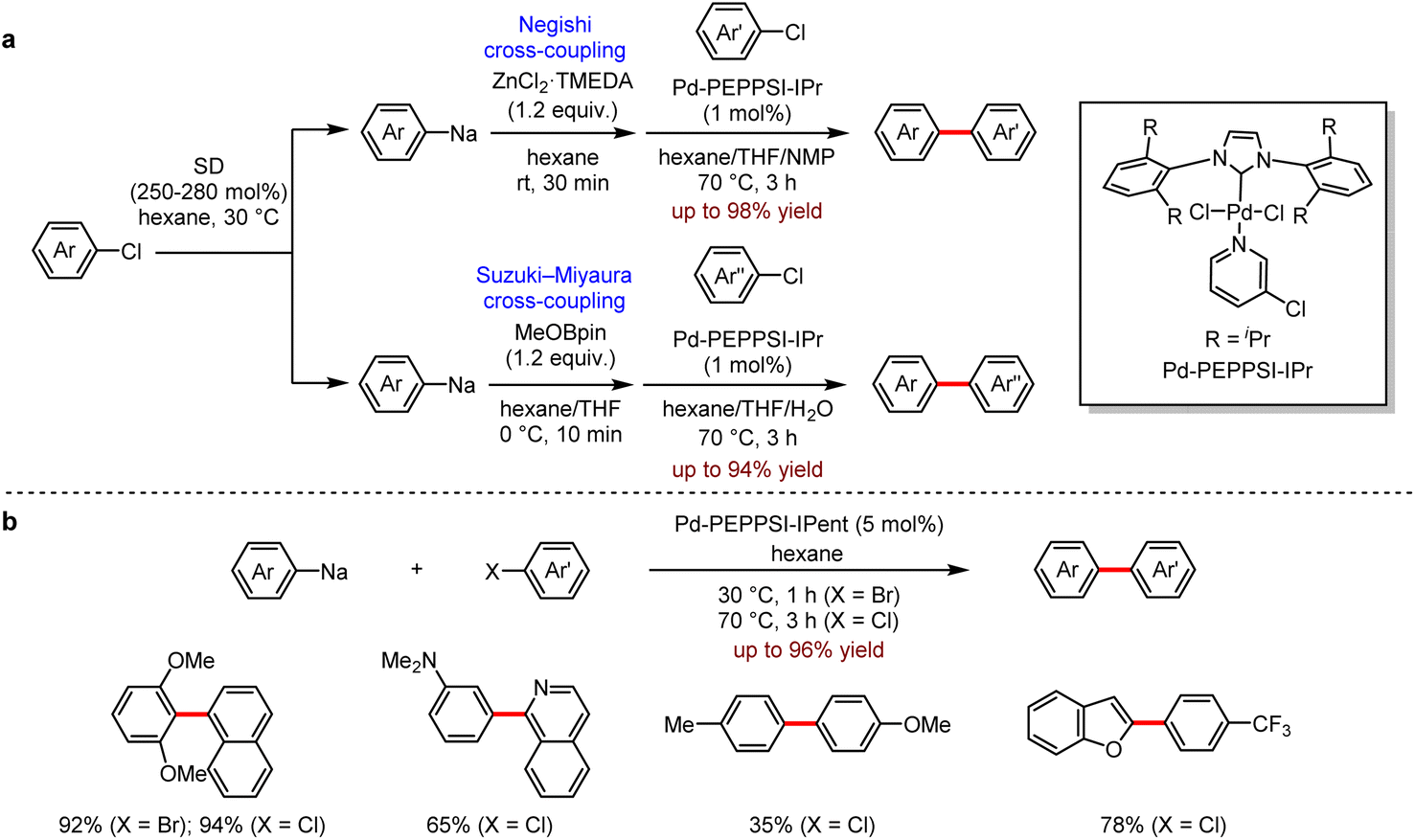 | ||
| Scheme 22 (a) Pd-catalysed Negishi or Suzuki–Miyaura cross-coupling reactions with arylsodiums. (b) Pd-catalysed cross-coupling reactions with arylsodiums directly. (c) Pd-catalysed Negishi cross-coupling reactions with arylsodiums. (d) Pd-catalysed Suzuki–Miyaura cross-coupling reactions with aryl- and alkenylsodiums. (e) Pd-catalysed cross-coupling reactions of arylsodiums and 2-chloronaphthalene directly. | ||
Firstly, they found that the transmetallation of organosodium reagents with ZnCl2–TMEDA or boronic esters generated the corresponding organozinc or organoarylboron reagents, which were subsequently applied in Pd-catalysed Negishi and Suzuki–Miyaura coupling (Scheme 22a). In addition, the organosodium reagents could be directly utilised in Pd-catalysed cross-coupling of aryl chlorides or aryl bromides to give the coupling products efficiently (Scheme 22b).
They also developed a reliable method to prepare a variety of (hetero)aryl and alkenyl sodium compounds by halogen–sodium exchange.57 A wide range of organosodium reagents were obtained (Scheme 23). The aryl-, (hetero)aryl- and alkenyl-sodium compounds were in situ produced from their corresponding bromides and neopentylsodium, which was easily prepared from neopentyl chloride and sodium disperse. Especially, 1,3,5-tris(4-bromophenyl)benzene 22 and tetrakis(4-bromophenyl)methane 23 succeeded to yield the corresponding products with neopentylsodium, which were not available by other approaches (Scheme 23a). This strategy also allowed a one-pot Negishi reaction (Scheme 23b) and Suzuki–Miyaura reaction (Scheme 23c) under mild conditions. In addition, the direct coupling of arylsodium reagents and aryl chloride proceeded successfully (Scheme 23d). It is noted that the organosodium reagent does not need to be added slowly.
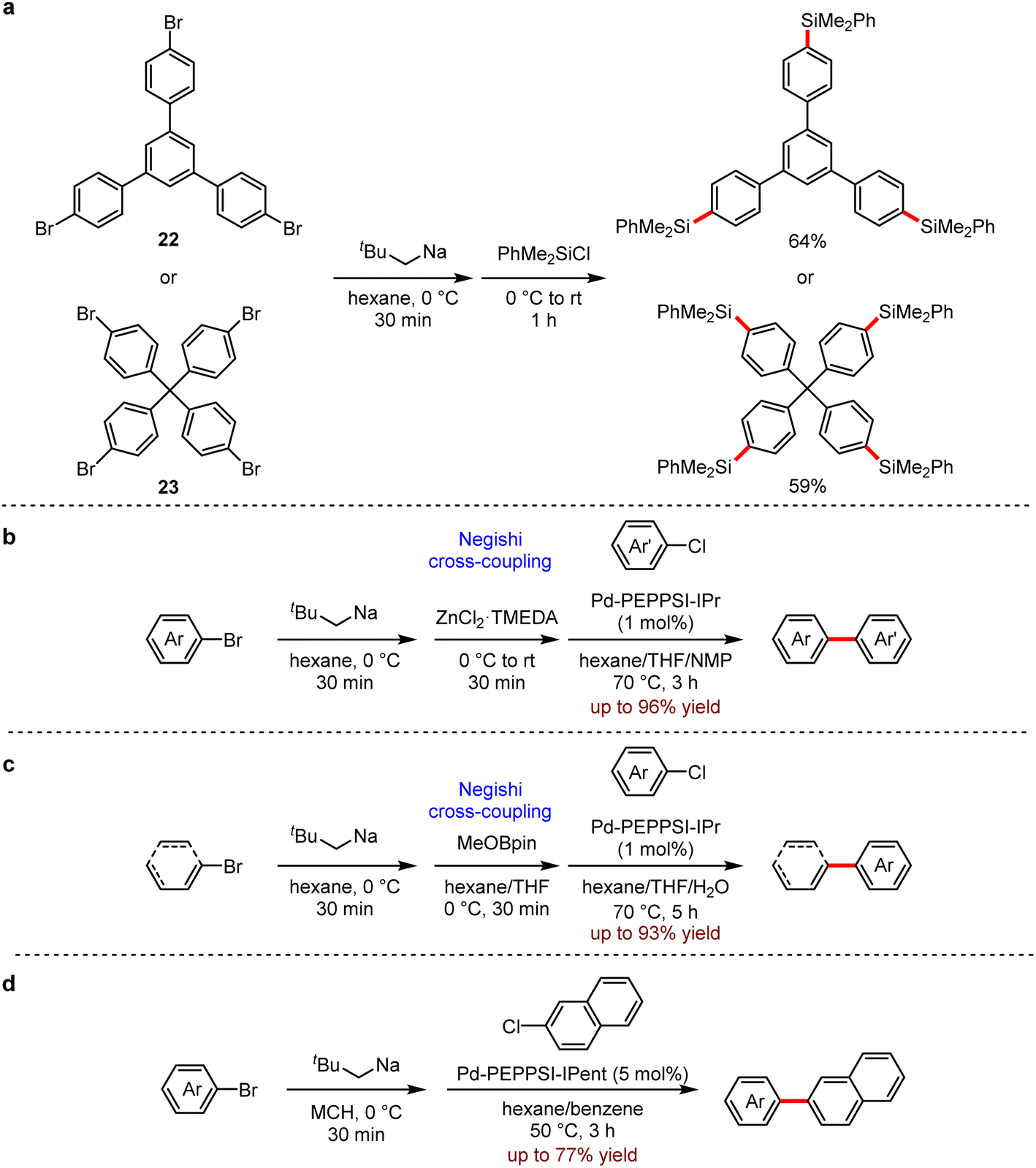 | ||
| Scheme 23 (a) Multisodiation through bromine–sodium exchange. (b) Pd-catalysed Negishi cross-coupling reactions using arylsodiums. (c) Pd-catalysed Suzuki–Miyaura cross-coupling reactions using aryl- and alkenylsodiums. (d) Pd-catalysed direct cross-coupling reactions of arylsodiums and 2-chloronaphthalene (MCH = methylcyclohexane). | ||
Although new interest and some new insights into the use of organosodium reagents have recently been disclosed, the development of versatile reactions remains a challenge due to the lack of general and reliable methods for their preparation. Considering the abundance of sodium, we expect that more and more chemists will explore new and novel chemistry in organosodium chemistry.
2.2 Organo-alkali metal reagents acting as additives
Organoalkali metal reagents, especially organolithium reagents, have also been utilised as additives to facilitate the desired reactions. In 2018, Bedford and co-workers reported a novel iron-catalysed Suzuki–Miyaura cross-coupling reaction promoted by alkyllithium-activated aryl pinacol boronic esters under mild conditions.58 As shown in Scheme 24, the key success of this transformation was the use of π-coordinating pyrrole as the directing group to achieve the coupling process efficiently. | ||
| Scheme 24 Fe-catalysed substrate-directed Suzuki–Miyaura coupling reaction. | ||
In 2016, Morken developed a conjunctive cross-coupling reaction to achieve chiral alcohols (Scheme 25).59 This approach combined three simple starting materials including organolithium reagents, organoboronic esters and electrophiles to forge two new carbon–carbon bonds with high enantioselectivity. When alkenyl- or aryl-trifluoromethanesulfonates were used as electrophiles, the reactivity was significantly improved compared with other electrophiles.
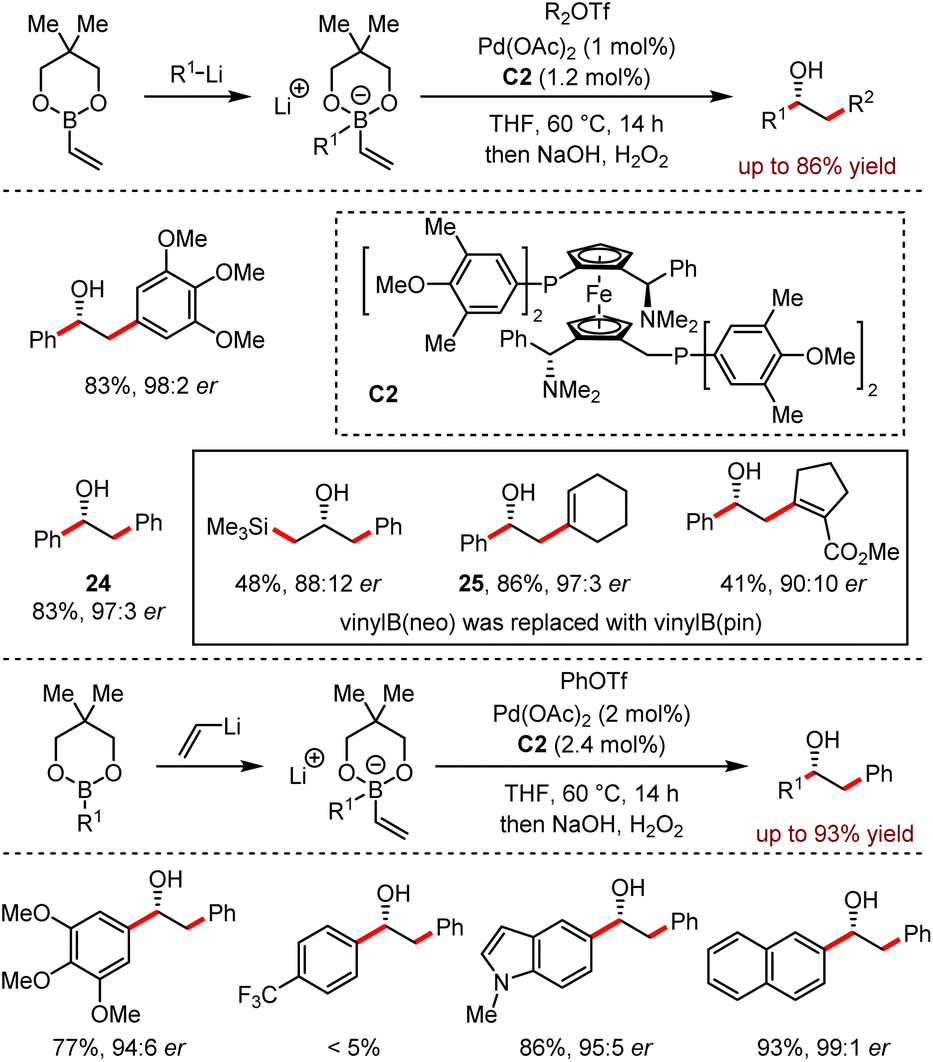 | ||
| Scheme 25 Conjunctive cross-coupling enabled by metal-induced metallate rearrangement in the combination of organoboronates and organolithium. | ||
In addition, the yields and stereoselectivities of the reaction majorly relied on the in situ formed borate complexes. For example, the neopentyl glycol-derived borate complexes provided 24 in 83% yield with 97:3 er and 25 in 86% yield with 97:3 er respectively. To analyse the by-products, both the direct Suzuki–Miyaura products and the partial consumption of organoborate esters led to low efficiency. For the substrates bearing with primary and secondary alkyl groups, the alkyl group migration also disturbed the outcome of the target product. This method exhibited a broad functional group tolerance, although the trifluoromethyl group was inert and only a trace amount of the corresponding product was detected.
Moreover, the addition of vinyllithium to organoborate could also generate the ate complexes, which underwent the subsequent Pd-catalysed coupling reaction efficiently. Control experiments showed that the presence of lithium halide salts affected the reaction negatively.
Polar organometallic reagents, such as organolithium and Grignard reagents, are widely utilised in synthetic chemistry to produce a variety of novel organometallic compounds.60 Hevia and co-workers60b–d designed a series of polar mixed-metal reagents (Scheme 26) with synergistic effects. By isolation and characterisation of key organometallic intermediates, they revealed the potential of s-block co-catalysis, including enhanced reaction performance, greater functional group tolerance and exceptional regioselectivity.
 | ||
| Scheme 26 (a) Sodium magnesiate catalysed hydroamination of isocyanates. (b) Synthesis of potassium magnesiate and donor-induced redistribution processes to form higher-order species (PMDETA = pentamethyldiethylenetriamine). | ||
The mixture of [NaMg(CH2SiMe3)3] (26) and [(THF)NaMg(NPh2)3-(THF)] (27) (Scheme 26a) could selectively catalyse the hydroamination/trimerisation of isocyanates by modulating the space of the isocyanate substituents.60b,c The well-defined dipotassium-tetrakis(alkyl)magnesates allowed direct magnesium–hydrogen exchange reactions of various (hetero)aryl substrates (Scheme 26b).60d,e Although the direct cross-coupling reactions with the mixed metal reagents have not been explored, their unique chemical properties and spatial advantages allowed for a potential application in organic synthesis.
In 2017, a novel protocol of regioselective mono- and di-ferric transformations under mild conditions, using sodium perferrate base [(dioxane)0.5NaFe{N(SiMe3)2}3] (28), paired with iron bis(amide) Fe(N(SiMe3)2)2 and Na[N(SiMe3)2] was reported by Hevia and co-workers.61 The structure of sodium tris(amido)ferrate (28) was confirmed by X-ray crystallography. The complex (29) was generated from 28 with 1,3-difluorobenzene, which was followed by the electrophilic addition with iodide to give 1,3-difluoro-2-iodobenzene (30) in 84% yield (Scheme 27a). A wide range of fluoroaromatic derivatives was obtained by the mono- or bimetallisation pathway (Scheme 27b). Moreover, the key mechanistic insight of this transformation was observed by the capture of highly reactive iron intermediate. Using 1,3,5-trifluorobenzene derivatives as the substrates, an unusual two-fold C–H/three-fold C–F activation proceeded at 80 °C with an excess 28 to give 31 in 87% yield as a yellow crystal and the biferrogenic compounds 32 in 47% yield and 33 in 46% yield, respectively (Scheme 27c).
 | ||
| Scheme 27 (a) Sodium-mediated ferration of 1,3-difluorobenzene with [(dioxane)0.5NaFe{N(SiMe3)2}3]. (b) Sodium-mediated regioselective ferration of fluoroarenes via C–H and C–F bond activation. (c) Two-fold C–H/three-fold C–F activation of 1,3,5-trifluorobenzenes. | ||
Organozinc–lithium reagents could enhance the nucleophilic ability of tetraorganozincates, exhibiting unique functional group tolerance. As shown in Scheme 28, the mixed-metal organometallic reagents were used in the C–H alkylation or arylation of heteroaromatics such as acridines or pyrazines by coordination of alkali metals adjacent to the anion activation site.62 In 2020, efficient conjugated addition to nitroolefins employing lithium tetraorganozincates was reported under transition metal-free and ligand-free conditions (Scheme 29).63 This reaction allows highly chemo-selective access to nitromethane derivatives up to 98% isolated yields.
 | ||
| Scheme 28 Selected examples of s-block bimetallic cooperation in zinc chemistry for transition metal-free C–C bond-forming processes (DDQ = dichlorodicyanoquinone). | ||
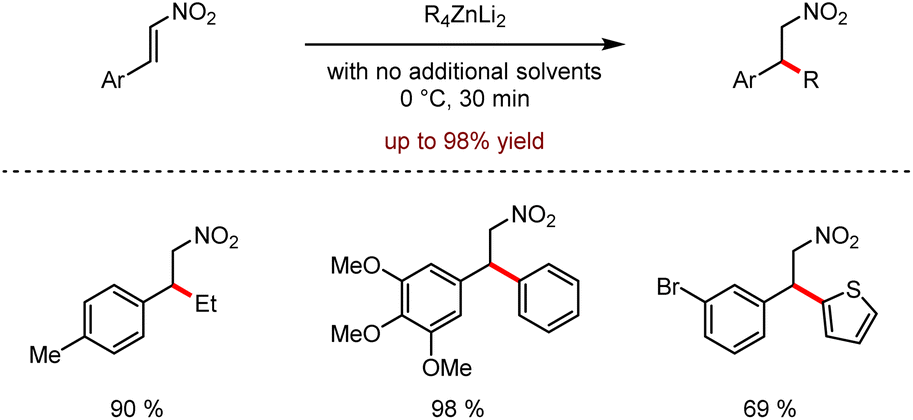 | ||
| Scheme 29 Nucleophilic addition of tetraorganozincates to nitroolefins. | ||
2.3 Organo-alkali reagents acting as nucleophiles
Organo-alkali reagents are conventionally used as nucleophiles in organic synthesis due to the convenient preparation and high activities, involving various functional group transformations. In 2012, a general method for the preparation of isoquinolines was reported (Scheme 30a).64 The Pd-catalysed cross-coupling of o-cyano arene-nonaflates and α-alkoxycarbonyl surrogates afforded o-cyanoalkene derivatives in situ, which were followed by nucleophilic addition with organolithium reagents in one-pot to provide various isoquinolines up to 99% yield.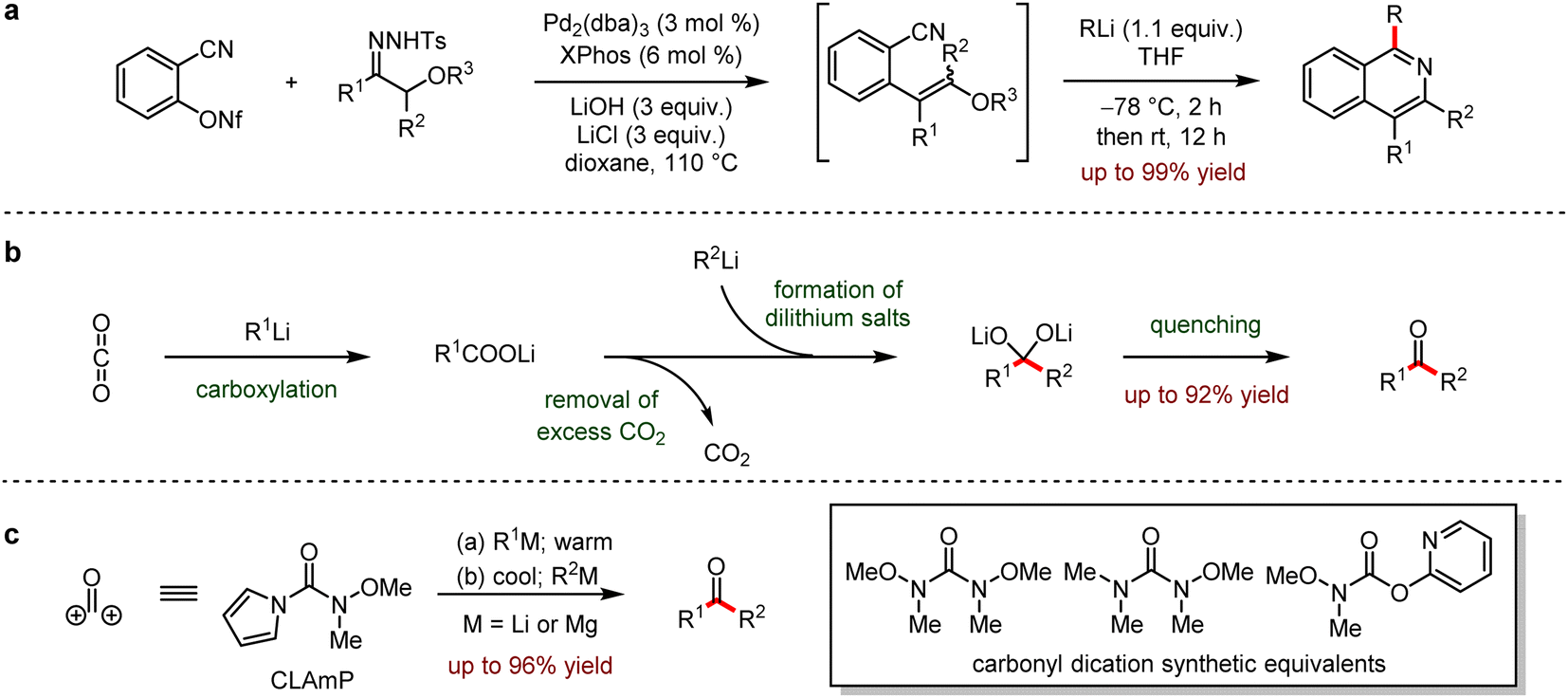 | ||
| Scheme 30 (a) Synthesis of isoquinolines via nucleophilic addition between o-cyanoalkenes and organolithiums. (b) Continuous flow process to access ketones from organolithiums and CO2. (c) Unsymmetrical synthesis of ketones with a pyrrole-bearing formal carbonyl dication linchpin reagent. | ||
In 2014, Hatton, Jamison and coworkers65 disclosed an efficient continuous flow process to access ketones by the nucleophilic addition of carbon dioxide with organolithium or Grignard reagents (Scheme 30b). This process exhibited the significant advantages to inhibit side reactions, which would yield by-products including undesired symmetrical ketone and tertiary alcohol. Moreover, this method allowed the continuous generation of organometallic reagents in a facile and scalable three-step flow process.
In 2015, Sarpong and co-workers reported a strategy to prepare the asymmetric ketones with pyrrole derivatives that were the synthetic equivalent of formyl carbonyl dication (Scheme 30c).66 Compared to other carbonyl surrogates, carbonyl linchpin N,O-dimethylhydroxylamine pyrrole enabled the electrophilic addition of less reactive or thermally unstable nucleophiles such as Grignard reagents or organolithium reagents to provide the desired ketones.
Deep eutectic solvents (DESs) are an emerging green medium that have a significantly lower melting point than pure single solvent. In addition, many of their physicochemical characteristics are also adjustable.67 The promising utilisations of DESs have been well documented recently.67,68 In 2020, an efficient and chemo-selective addition of aldehydes or epoxides with highly polarised lithium phosphides (LiPR2) was disclosed, which were generated in the recyclable eutectic mixtures of 1ChCl and 2Gly (Scheme 31a).69 This environmental-friendly protocol showed a fast reaction rate and obtained α-hydroxy-phosphine oxide or β-hydroxy-phosphine oxide, respectively, in 62–94% yields at room temperature and in air. In 2021, Capriati, and co-workers reported the nucleophilic addition of amides with organolithium reagents in a cyclopentane methyl ether environment to give ketones up to 93% yield (Scheme 31b).70 This protocol inhibited the over-addition of nucleophilic reagents and showed the potential for scale-up and recovery of the reaction medium.
 | ||
| Scheme 31 (a) In situ generation of lithium phosphides and one-pot chemoselective addition to aldehydes and epoxides. (b) Nucleophilic addition of amides by organolithium compounds in air (CPME = cyclopentyl methyl ether). | ||
3 Application of organo-alkali metal reagents in the synthesis of functional molecules
Metal-catalysed carbon–carbon bond and carbon–heteroatom bond formations are also important in medicinal chemistry, especially for the structural modification of small molecule drugs. Moreover, some well-established methods have been applied in industrial production. A timely summary of existing results would herein provide a necessary update for drug development.Feringa and co-workers employed a one-pot 1,2-addition and Pd-catalysed cross-coupling strategy to prepare cyclopropylaryl ketones, which are versatile intermediates for the synthesis of various drugs.12 They applied the strategy to obtain cyclopropylphenyl ketone, which was utilised to prepare a HIV protease inhibitor 34 (Scheme 32a) according to the precesured in references.
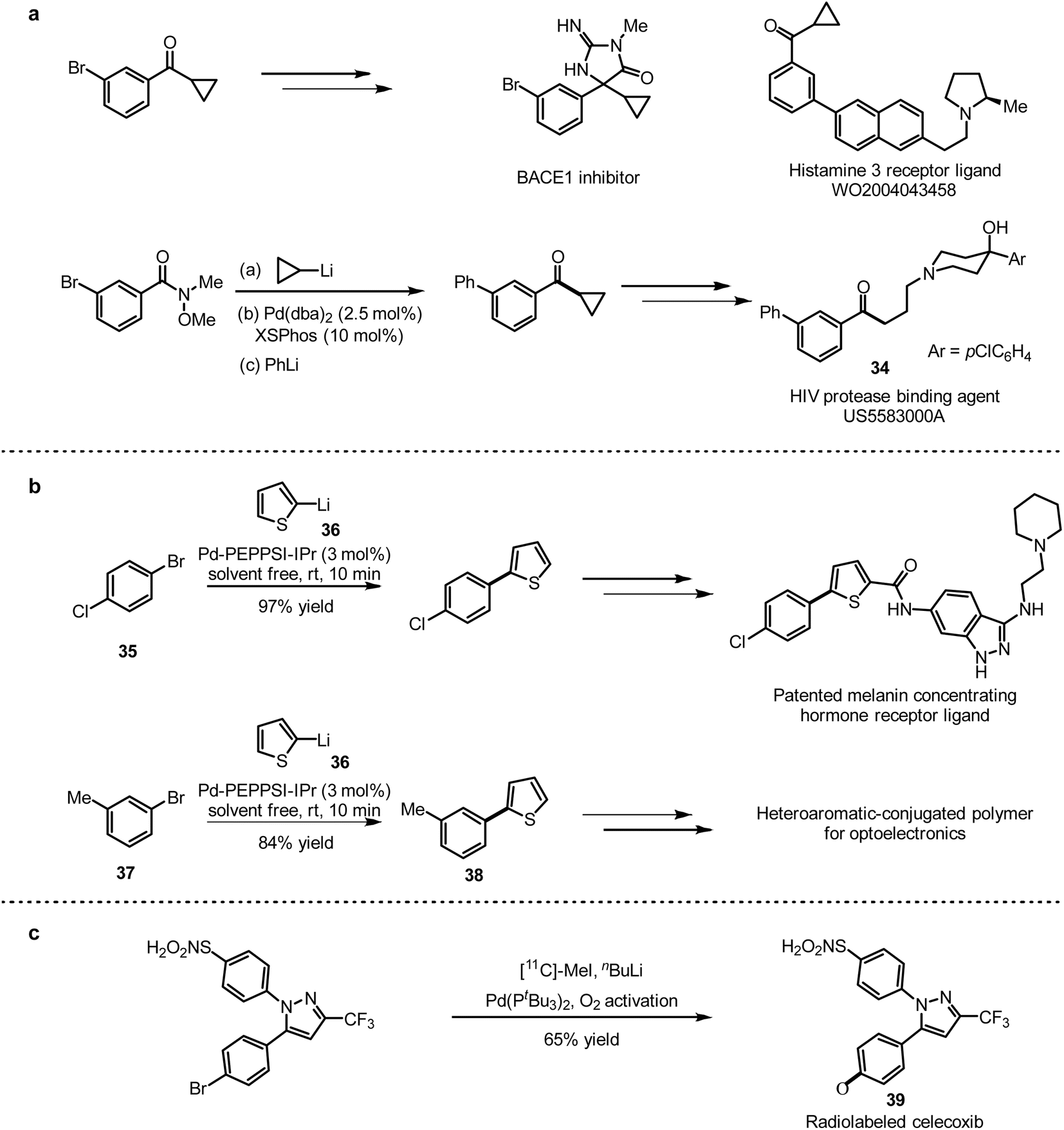 | ||
| Scheme 32 (a) Synthetic application of the Pd-catalysed one-pot 1,2 addition/cross-coupling of Weinreb amides. (b) Synthetic application of Pd-catalysed cross-coupling of aryl halides with organolithium reagents under neat conditions. (c) Synthesis of radiolabeled celecoxib via Pd-catalysed ultrafast cross-coupling with organolithium reagents. | ||
To further demonstrate the application of this method, 1-bromo-4-chlorobenzene (35) and 2-thienyllithium (36) were coupled with 3 mol% Pd-PEPPSI-IPr as a catalyst to generate the desired coupling product with high selectivity in 97% yield, which was used for the synthesis of melanin-concentrating hormone receptor ligands71 (Scheme 32b). In addition, the combination of 3-methyl-1-bromobenzene (37) and 2-thienyllithium (36) provided the coupling product 38, which was applied to prepare polymers for optoelectronic devices.72 A general method of isotopic labelling was also provided in this work, introducing CD3-labeled and short-lived 11C radioisotopes (t1/2(11C) = 20.3 min) for positron emission tomography (PET) imaging.73 This isotopic labelling protocol is an efficient complement to the conventional quenching strategy. With this method in hand, they succeed to prepare [11C]celecoxib (39) with 11C-labeled MeI (Scheme 32c).
Axial asymmetric units are widely found in functional molecules and related fields. For example, 2,2′-dihydroxy-1,1′-binaphthyl (BINOL) derivatives are commonly used as catalysts or ligands in asymmetric synthesis and also as important constituent sequences in optical materials or supramolecular structures.74a Uchiyama and co-workers74b tested the feasibility of introducing BINOL units into polymers by a condensation strategy (Scheme 33a). The results showed that dilithium reagents (39) prepared by lithium–bromine exchange could be coupled with dimethoxynaphthalene ether (41) to give the desired polymers 42, rather than the unsuccessful attempts with Grignard reagents. Considering the significant potential of sulfone groups in organic synthesis, Manolikakes and co-workers explored a dual-catalytic strategy in the late-stage modification of small molecule drugs.52 They found that the cross-coupling of sulfites (43) with heteroaryl iodides led to the nonsteroidal anti-inflammatory drug phenazone (44), and with aryl iodide (45) modified the core structure of sildenafil, respectively (Scheme 33b).
 | ||
| Scheme 33 (a) Synthesis of axially asymmetric structures by Ni-catalysed polycondensation. (b) Visible-light photoredox/nickel dual catalysis cross-coupling of sulfinic acid salts with aryl iodides for late-stage modification of small molecular drugs. (c) Pd-catalysed nucleophilic addition/Buchwald–Hartwig amination for the synthesis of AMA37. | ||
In 2019, Feringa and coworkers disclosed a general one-pot approach to afford functionalised ketones by nucleophilic addition/Buchwald–Hartwig amination.13 The application of this method was also proved for the efficient synthesis of asymmetrical ketone, which was finally transformed into AMA37 (46), an isoform-specific phosphoinositide 3-kinase inhibitor with an IC50 = 0.27 μM for DNA-dependent protein kinase (DNA-PK) inhibition (Scheme 33c).75 It is noted that the efficient conversion of organolithium reagents provided 79% reaction mass efficiency (RME) and the only waste was the lowly toxic lithium chloride, which indicated that this process was environmentally friendly.
As shown in Scheme 34, cationic photosensitisers (49) for photodynamic therapy could be obtained by Pd-catalysed cross-coupling of 4-bromo-N,N-dimethylaniline (47) with alkyllithium (48), which furnished efficiently in 45 minutes under mild reaction conditions, compared to the previous Sonogashira cross-coupling.76 Other examples include the synthesis of potential inhibitor intermediates (50) for the construction of aurora kinase A and B,77 which are enzymes overexpressed in human tumors, as well as the synthesis of 1,4-bis(4-(phenylethynyl) phenylethynyl)benzene-based films for the application in light-emitting diodes.78 The combination of ethynyllithium and fluorobenzyl alkyne 51 with 3 mol% [Pd(μ-I)PtBu3]2 as catalyst delivered 52, which is the key intermediate towards the synthesis of CRTH2 receptor antagonists 53. Employing the same strategy, the precursor of lignans could be obtained by the coupling of 5-(bromomethyl)-1,2,3-trimethoxybenzene (54) with the organolithium reagent (Scheme 35). The above applications suggest that organolithium reagents as the partners directly involved in cross-coupling reactions have great potential for the synthesis of diverse bioactive molecules, especially key pharmaceutical intermediates, as well as in polymers and functional materials.
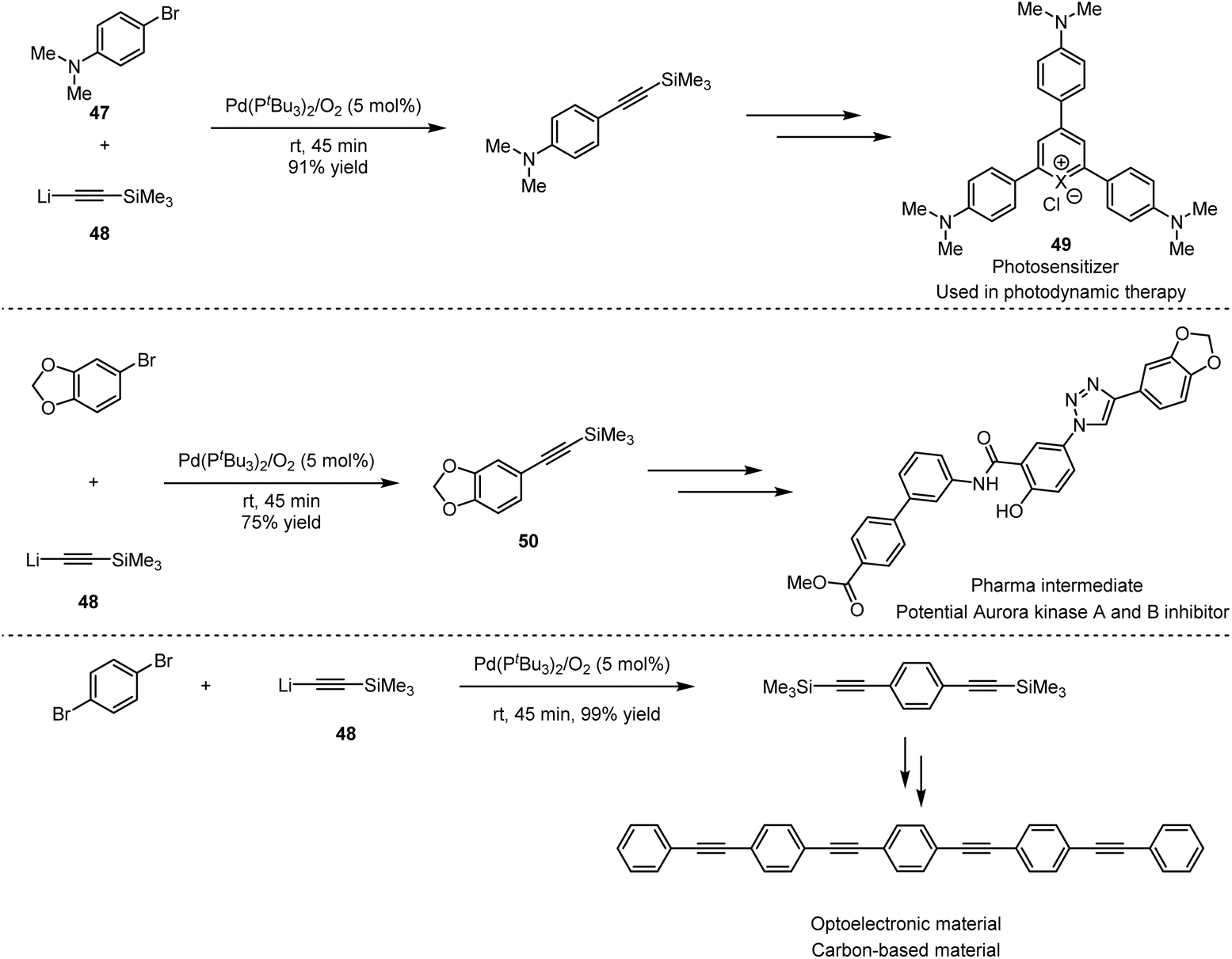 | ||
| Scheme 34 Potential applications of Pd-catalysed cross-coupling of benzyl bromides with lithium acetylides. | ||
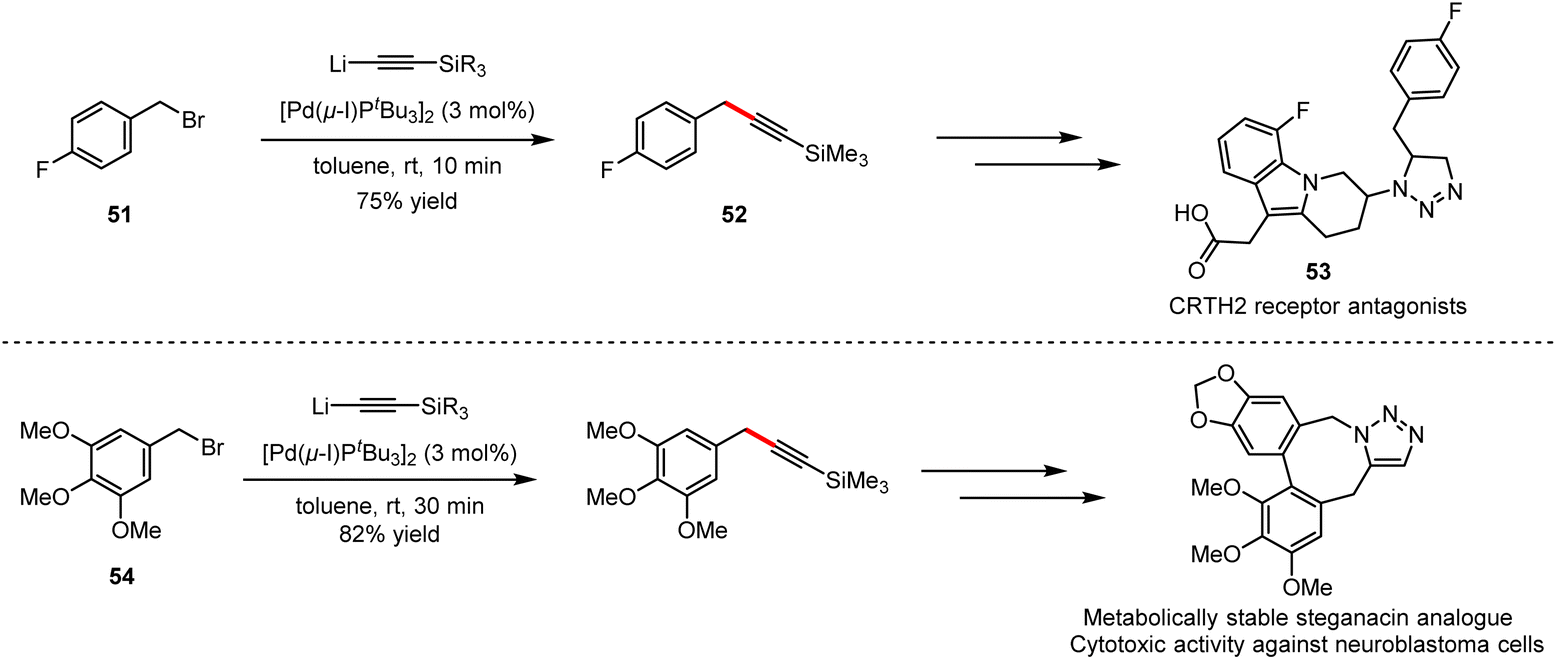 | ||
| Scheme 35 Potential applications of the Pd-catalysed cross-coupling of benzyl bromides with lithium acetylides. | ||
4 Conclusions
This review summarises the recent utilisation of organic alkali metal reagents, especially organolithium reagents, as partners in metal-catalysed cross-coupling reactions, revealing effective approaches towards the synthesis of carbon–carbon and carbon–heteroatom bonds. To achieve these desired transformations, synthetic challenges including the homo-coupling between organo-alkali metal reagents and the undesired lithium–halogen exchange must be inhibited and well overcome. Many methodologies for nickel- and iron-based catalytic protocols involving cross-coupling with organolithium reagents have emerged in recent years, suggesting that these earth-abundant metals in nature have promising prospects, they indeed have potential to replace the use of expensive and toxic transition metals. Nonetheless, there are still several major challenges in this field: (a) very few asymmetric examples have been achieved; (b) there is still lack of enough attention to organosodium reagents in this field compared to organolithium reagents; and (c) broad substrate scope of these methods needs to be further explored.Author contributions
Lu-Qiong Huo, Xin-Hao Wang, and ZhenGuo Zhang: preparing the original draft. ZhenHua Jia, Xiao-Shui Peng, and Henry N.C. Wong: writing, reviewing, editing, and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (NSFC) (22271243, 21971219), the Research Team Program and “Guangdong Leading Talent Program” for Guangdong Introducing Innovative and Entrepreneurial Teams, Shenzhen Science and Technology Innovation Committee for the Shenzhen Key Laboratory Scheme, Research Grants Council (Hong Kong) (GRF CUHK14304819), and the Innovation and Technology Commission (Hong Kong) in the form of subsidy to the State Key Laboratory of Synthetic Chemistry, and the University Development Fund Grants from The Chinese University of Hong Kong, Shenzhen.References
- For recent reviews, see: (a) C. Fricke, T. Sperger, M. Mendel and F. Schoenebeck, Angew. Chem., Int. Ed., 2021, 60, 3355–3366 CrossRef CAS PubMed; (b) K. H. Shaughnessy, Isr. J. Chem., 2020, 60, 180–194 CrossRef CAS; (c) P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497–5508 CrossRef CAS; (d) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- (a) C. C. C. Johansson Seechurn, O. M. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed; (b) A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed; (c) F. Christoffel and T. R. Ward, Catal. Lett., 2018, 148, 489–511 CrossRef CAS; (d) D. Roy and Y. Uzomi, Adv. Synth. Catal., 2018, 360, 602–605 CrossRef CAS; (e) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- (a) M. Schlosser, Organometallics in Synthesis: A Manual, Wiley, 2nd edn, 2002 Search PubMed; (b) D. Seyferth, Organometallics, 2009, 28, 2–33 CrossRef CAS; (c) J. F. Nobis, L. F. Moormeier and R. E. Robinson, Adv. Chem., 1959, 23, 63–68 CAS; (d) D. Seyferth, Organometallics, 2001, 20, 2940–2955 CrossRef CAS; (e) T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247–9262 CrossRef CAS PubMed; (f) https://www.rsc.org/periodic-table/, accessed 10 November 2022; (g) Z. Chen, H. Yang, Z. Kang, M. Driess and P. W. Menezes, Adv. Mater., 2022, 34, 2108432 CrossRef CAS PubMed.
- For recent reviews, see: (a) M. M. Heravi, V. Zadsirjan, P. Hajiabbasi and H. Habidi, Chem. Mon., 2019, 150, 535–591 CrossRef CAS; (b) Z. Qureshi, C. Toker and M. Lautens, Synthesis, 2017, 49, 1–16 CAS; (c) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed.
- (a) M. Yamamura, I. Moritani and S. Murahashi, J. Organomet. Chem., 1975, 91, 39–42 CrossRef; (b) S. Murahashi, M. Yamamura, K. Yanagisawa, N. Mita and K. Kondo, J. Org. Chem., 1979, 44, 2408–2417 CrossRef CAS.
- (a) V. Hornillos, M. Giannerini, C. Vila, M. Fañanás-Mastral and B. L. Feringa, Org. Lett., 2013, 15, 5114–5117 CrossRef CAS PubMed; (b) C. Vila, V. Hornillos, M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, Chem.–Eur. J., 2014, 20, 13078–13083 CrossRef CAS PubMed; (c) C. Vila, M. Giannerini, V. Hornillos, M. Fañanás -Mastral and B. L. Feringa, Chem. Sci., 2014, 5, 1361–1367 RSC; (d) M. Giannerini, V. Hornillos, C. Vila, M. Fañanás-Mastral and B. L. Feringa, Angew. Chem., Int. Ed., 2013, 52, 13329–13333 CrossRef CAS PubMed.
- J. D. Firth and P. O'Brien, ChemCatChem, 2015, 7, 395–397 CrossRef CAS.
- D. Heijnen, V. Hornillos, B. P. Corbet, M. Giannerini and B. L. Feringa, Org. Lett., 2015, 17, 2262–2265 CrossRef CAS PubMed.
- E. B. Pinxterhuis, M. Giannerini, V. Hornillos and B. L. Feringa, Nat. Commun., 2016, 7, 11698 CrossRef PubMed.
- (a) M. Aufiero, T. Scattolin, F. Proutiere and F. Schoenebeck, Organometallics, 2015, 34, 5191–5195 CrossRef CAS; (b) I. Kalvet, G. Magnin and F. Schoenebeck, Angew. Chem., Int. Ed., 2017, 56, 1581–1585 CrossRef CAS PubMed.
- D. Heijnen, F. Tosi, C. Vila, M. C. A. Stuart, P. H. Elsinga, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 3354–3359 CrossRef CAS PubMed.
- M. Giannerini, C. Vila, V. Hornillos and B. L. Feringa, Chem. Commun., 2016, 52, 1206–1209 RSC.
- J. de Jong, D. Heijnen, H. Helbert and B. L. Feringa, Chem. Commun., 2019, 55, 2908–2911 RSC.
- (a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed; (b) G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed.
- N. Sinha, D. Heijnen, B. L. Feringa and M. G. Organ, Chem.–Eur. J., 2019, 25, 9180–9184 CrossRef CAS PubMed.
- G. Dilauro, A. F. Quivelli, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., Int. Ed., 2019, 58, 1799–1802 CrossRef CAS PubMed.
- T. Scherpf, H. Steinert, A. Großjohann, K. Dilchert, J. Tappen, I. Rodstein and V. H. Gessner, Angew. Chem., Int. Ed., 2020, 59, 20596–20603 CrossRef CAS PubMed.
- (a) P. J. Hajduk, M. Bures, J. Praestgaard and S. W. Fesik, J. Med. Chem., 2000, 43, 3443–3447 CrossRef CAS PubMed; (b) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed; (c) J. Klekota and F. P. Roth, Bioinformatics, 2008, 24, 2518–2525 CrossRef CAS PubMed.
- F. Lima, J. André, A. Marziale, A. Greb, S. Glowienke, M. Meisenbach, B. Schenkel, B. Martin and J. Sedelmeier, Org. Lett., 2020, 22, 6082–6085 CrossRef CAS PubMed.
- (a) P. Rivera-Fuentes and F. Diederich, Angew. Chem., Int. Ed., 2012, 51, 2818–2828 CrossRef CAS PubMed; (b) X. Pu, X. Qi and J. M. Ready, J. Am. Chem. Soc., 2009, 131, 10364–10365 CrossRef CAS PubMed; (c) A. Hoffmann-Rçder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196–1216 CrossRef PubMed; (d) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS PubMed.
- J. Mateos-Gil, A. Mondal, M. C. Reis and B. L. Feringa, Angew. Chem., Int. Ed., 2020, 59, 7823–7829 CrossRef CAS PubMed.
- H. Helbert, P. Visser, J. G. H. Hermens, J. Buter and B. L. Feringa, Nat. Catal., 2020, 3, 664–671 CrossRef CAS.
- A. Mondal, P. Visser, A. M. Doze, J. Buter and B. L. Feringa, Chem. Commun., 2021, 57, 7529–7532 RSC.
- P. Chen, Z. Y. Wang, J. X. Wang, X. S. Peng and H. N. C. Wong, Org. Chem. Front., 2022, 9, 3438–3445 RSC.
- (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed; (b) T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319–6332 CrossRef CAS; (c) N. D. Schley and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 16588–16593 CrossRef CAS PubMed.
- M. Leiendecker, C. C. Hsiao, L. Guo, N. Alandini and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 12912–12915 CrossRef CAS PubMed.
- L. Guo, M. Leiendecker, C. C. Hsiao, C. Baumann and M. Rueping, Chem. Commun., 2015, 51, 1937–1940 RSC.
- D. Heijnen, J. B. Gualtierotti, V. Hornillos and B. L. Feringa, Chem.–Eur. J., 2016, 22, 3991–3995 CrossRef CAS PubMed.
- Z. K. Yang, D. Y. Wang, H. Minami, H. Ogawa, T. Ozaki, T. Saito, K. Miyamoto, C. Wang and M. Uchiyama, Chem.–Eur. J., 2016, 22, 15693–15699 CrossRef CAS PubMed.
- (a) N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem, 2017, 1, 25 CrossRef CAS PubMed; (b) A. Biffis, P. Centomo, A. D. Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed; (c) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed; (d) F. S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC.
- R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed.
- (a) J. Terao and N. Kambe, Acc. Chem. Res., 2008, 41, 1545–1554 CrossRef CAS PubMed; (b) J. Terao, H. Watanabe, A. Ikumi, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2002, 124, 4222–4223 CrossRef CAS PubMed.
- (a) L. R. Reddy, H. J. Gais, C. W. Woo and G. Raabe, J. Am. Chem. Soc., 2002, 124, 10427–10434 CrossRef CAS PubMed; (b) H. J. Gais, L. R. Reddy, G. S. Babu and G. Raabe, J. Am. Chem. Soc., 2004, 126, 4859–4864 CrossRef CAS PubMed; (c) J. Terao and N. Kambe, Acc. Chem. Res., 2008, 41, 1545–1554 CrossRef CAS PubMed; (d) J. Terao, H. Watanabe, A. Ikumi, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2002, 124, 4222–4223 CrossRef CAS PubMed.
- (a) D. G. Johnson, J. M. Lynam, N. S. Mistry, J. M. Slattery, R. J. Thatcher and A. C. Whitwood, J. Am. Chem. Soc., 2013, 135, 2222–2234 CrossRef CAS PubMed; (b) O. J. S. Pickup, I. Khazal, E. J. Smith, A. C. Whitwood, J. M. Lynam, K. Bolaky, T. C. King, B. W. Rawe and N. Fey, Organometallics, 2014, 33, 1751–1761 CrossRef CAS; (c) S. W. Roh, K. Choi and C. Lee, Chem. Rev., 2019, 119, 4293–4356 CrossRef CAS PubMed.
- I. Erdelmeier, J. Won, S. Park, J. Decker, G. Below, M. H. Baik and H. J. Gais, Chem.–Eur. J., 2020, 26, 2914–2926 CrossRef CAS PubMed.
- (a) H. Ogawa, H. Minami, T. Ozaki, S. Komagawa, C. Wang and M. Uchiyama, Chem.–Eur. J., 2015, 21, 13904–13908 CrossRef CAS PubMed; (b) K. Kojima, Z. K. Yang, C. Wang and M. Uchiyama, Chem. Pharm. Bull., 2017, 65, 862–868 CrossRef CAS PubMed.
- A. M. Borys and E. Hevia, Angew. Chem., Int. Ed., 2021, 60, 24659–24667 CrossRef CAS PubMed.
- (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed; (b) A. Correa, O. G. Mancheno and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC; (c) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS PubMed; (d) I. Bauer and H. J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed; (e) R. B. Bedford, Acc. Chem. Res., 2015, 48, 1485–1493 CrossRef CAS PubMed.
- Z. H. Jia, Q. Liu, X. S. Peng and H. N. C. Wong, Nat. Commun., 2016, 7, 10614 CrossRef PubMed.
- (a) X. L. Lu, M. Shannon, X. S. Peng and H. N. C. Wong, Org. Lett., 2019, 21, 2546–2549 CrossRef CAS PubMed; (b) Q. Liu, Z. Y. Wang, X. S. Peng and H. N. C. Wong, J. Org. Chem., 2018, 83, 6325–6333 CrossRef CAS PubMed; (c) Z. L. Zhong, Z. Y. Wang, S. F. Ni, L. Dang, H. K. Lee, X. S. Peng and H. N. C. Wong, Org. Lett., 2019, 21, 700–704 CrossRef CAS PubMed; (d) Z. L. Zhong, X. S. Peng and H. N. C. Wong, Chin. Chem. Lett., 2019, 30, 1463–1467 CrossRef CAS.
- Z. Y. Wang, X. S. Peng and H. N. C. Wong, Asian J. Org. Chem., 2020, 9, 1834–1840 CrossRef CAS.
- P. Chen, Z. Y. Wang, X. S. Peng and H. N. C. Wong, Org. Lett., 2021, 23, 4385–4390 CrossRef CAS PubMed.
- For selected reviews on dual transition metal catalysis, see: (a) J. F. Li, Y. X. Luan and M. Ye, Sci. China: Chem., 2021, 64, 1923–1937 CrossRef CAS; (b) U. B. Kim, D. J. Jung, H. J. Jeon, K. Rathwell and S. G. Lee, Chem. Rev., 2020, 120, 13382–13433 CrossRef CAS PubMed; (c) F. Romiti, J. del Pozo, P. H. S. Paioti, S. A. Gonsales, X. Li, F. W. W. Hartrampf and A. H. Hoveyda, J. Am. Chem. Soc., 2019, 141, 17952–17961 CrossRef CAS PubMed; (d) M. M. Lorion, K. Maindan, A. R. Kapdi and L. Ackermann, Chem. Soc. Rev., 2017, 46, 7399–7420 RSC; (e) D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705–1718 RSC; (f) J. Fu, X. Huo, B. Li and W. Zhang, Org. Biomol. Chem., 2017, 15, 9747–9759 RSC; (g) M. H. Pérez-Temprano, J. A. Casares and P. Espinet, Chem.–Eur. J., 2012, 18, 1864–1884 CrossRef PubMed; (h) J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931–6943 RSC.
- J. Zeng, K. M. Liu and X. F. Duan, Org. Lett., 2013, 15, 5342–5345 CrossRef CAS PubMed.
- A. B. Smith III, A. T. Hoye, D. M. Solorio, W. S. Kim and R. B. Tong, J. Am. Chem. Soc., 2012, 134, 4533–4536 CrossRef PubMed.
- (a) M. H. Nguyen and A. B. Smith III, Org. Lett., 2013, 15, 4258–4261 CrossRef CAS PubMed; (b) D. Martinez-Solorio, A. T. Hoye, M. H. Nguyen and A. B. Smith III, Org. Lett., 2013, 15, 2454–2457 CrossRef CAS PubMed; (c) M. H. Nguyen and A. B. Smith III, Org. Lett., 2014, 16, 2070–2073 CrossRef CAS PubMed; (d) M. H. Nguyen, K. T. O'Brien and A. B. Smith III, J. Org. Chem., 2017, 82, 11056–11071 CrossRef CAS PubMed.
- (a) D. Seyferth, Organometallics, 2006, 25, 2–24 CrossRef CAS; (b) D. Seyferth, Organometallics, 2009, 28, 2–33 CrossRef CAS.
- J. F. Nobis, L. F. Moormeier and R. E. Robinson, Adv. Chem. Ser., 1959, 23, 63–68 CrossRef CAS.
- H. Gilman and G. F. Wright, J. Am. Chem. Soc., 1933, 55, 2893–2896 CrossRef CAS.
- (a) R. A. Benkeser, D. J. Foster, D. M. Sauve and J. F. Nobis, Chem. Rev., 1957, 57, 867–894 CrossRef CAS; (b) M. Schlosser, Angew. Chem., Int. Ed., 1964, 3, 287–306 CrossRef; (c) M. Schlosser, Angew. Chem., Int. Ed., 1964, 3, 362–373 CrossRef.
- C. J. Dinsmore, T. M. Williams, T. J. O'Neill, D. Liu, E. Rands, J. Culberson, R. B. Lobell, K. S. Koblan, N. E. Kohl, J. B. Gibbs, A. I. Oliff, S. L. Graham and G. D. Hartman, Bioorg. Med. Chem. Lett., 1999, 9, 3301–3306 CrossRef CAS PubMed.
- (a) N. W. Liu, S. Liang, N. Margraf, S. Shaaban, V. Luciano, M. Drost and G. Manolikakes, Eur. J. Org. Chem., 2017, 2018, 1208–1210 CrossRef; (b) S. Liang, Y. Ren and G. Manolikakes, Eur. J. Org. Chem., 2017, 2017, 4117–4120 CrossRef CAS.
- N. W. Liu, K. Hofman, A. Herbert and G. Manolikakes, Org. Lett., 2018, 20, 760–763 CrossRef CAS PubMed.
- E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed.
- A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333–3336 CrossRef CAS PubMed.
- S. Asako, H. Nakajima and K. Takai, Nat. Catal., 2019, 2, 297–303 CrossRef CAS.
- S. Asako, I. Takahashi, H. Nakajima, L. Ilies and K. Takai, Chem. Commun., 2021, 4, 76 CrossRef CAS.
- H. M. O'Brien, M. Manzotti, R. D. Abrams, D. Elorriaga, H. A. Sparkes, S. A. Davis and R. B. Bedford, Nat. Catal., 2018, 1, 429–437 CrossRef.
- L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70–74 CrossRef CAS PubMed.
- (a) For a recent historical overview on organolithium chemistry see: U. Wietelmann and J. Klett, Z. Anorg. Allg. Chem., 2018, 644, 194–204 CrossRef CAS PubMed; (b) A. Hernán-Gómez, T. D. Bradley, A. R. Kennedy, Z. Livingstone, S. D. Robertson and E. Hevia, Chem. Commun., 2013, 49, 8659–8661 RSC; (c) M. De Tullio, A. Hernán-Gómez, Z. Livingstone, W. Clegg, A. R. Kennedy, R. W. Harrington, A. Antiñolo, A. Martínez, F. Carrillo-Hermosilla and E. Hevia, Chem.–Eur. J., 2016, 22, 17646–17656 CrossRef CAS PubMed; (d) L. Davin, A. Hernán-Gómez, C. McLaughlin, A. R. Kennedy, R. McLellan and E. Hevia, Dalton Trans., 2019, 48, 8122–8130 RSC; (e) P. Mastropierro, A. R. Kennedy and E. Hevia, Eur. J. Inorg. Chem., 2020, 2021, 1016–1022 CrossRef.
- L. C. H. Maddock, T. Nixon, A. R. Kennedy, M. R. Probert, W. Clegg and E. Hevia, Angew. Chem., Int. Ed., 2018, 57, 187–191 CrossRef CAS PubMed.
- (a) A. Hernán-Gómez, E. Herd, M. Uzelac, T. Cadenbach, A. R. Kennedy, I. Borilovic, G. Aromí and E. Hevia, Organometallics, 2015, 34, 2614–2623 CrossRef; (b) S. E. Baillie, V. L. Blair, D. C. Blakemore, D. Hay, A. R. Kennedy, D. C. Prydeb and E. Hevia, Chem. Commun., 2012, 48, 1985–1987 RSC; (c) E. Hevia, Chimia, 2020, 74, 681–688 CrossRef CAS PubMed.
- M. Dell'Aera, F. M. Perna, P. Vitale, A. Altomare, A. Palmieri, L. C. H. Maddock, L. J. Bole, A. R. Kennedy, E. Hevia and V. Capriati, Chem.–Eur. J., 2020, 26, 8742–8748 CrossRef PubMed.
- L. Florentino, F. Aznar and C. Valdes, Org. Lett., 2012, 14, 2323–2325 CrossRef CAS PubMed.
- J. Wu, X. Q. Yang, Z. He, X. W. Mao, T. A. Hatton and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 8416–8420 CrossRef CAS PubMed.
- S. T. Heller, J. N. Newton, T. T. Fu and R. Sarpong, Angew. Chem., Int. Ed., 2015, 54, 9839–9843 CrossRef CAS PubMed.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- Selected examples for DES: (a) S. Ghinato, G. Dilauro, F. M. Perna, V. Capriati, M. Blangetti and C. Prandi, Chem. Commun., 2019, 55, 7741–7744 RSC; (b) M. J. Rodríguez-Álvarez, J. García-Álvarez, M. Uzelac, M. Fairley, C. T. O'Hara and E. Hevia, Chem.–Eur. J., 2018, 24, 1720–1725 CrossRef PubMed; (c) L. Cicco, M. J. Rodríguez-Álvarez, F. M. Perna, J. García-Álvarez and V. Capriati, Green Chem., 2017, 19, 3069–3077 RSC; (d) L. Cicco, S. Sblendorio, R. Mansueto, F. M. Perna, A. Salomone, S. Florio and V. Capriati, Chem. Sci., 2016, 7, 1192–1199 RSC; (e) S. E. García-Garrido, A. P. Soto, E. Hevia and J. García-Álvarez, Eur. J. Inorg. Chem., 2021, 2021, 3116–3130 CrossRef.
- L. Cicco, A. Fombona-Pascual, A. Sánchez-Condado, G. A. Carriedo, F. M. Perna, V. Capriati, A. P. Soto and J. García-Álvarez, ChemSusChem, 2020, 13, 4967–4973 CrossRef CAS PubMed.
- S. Ghinato, D. Territo, A. Maranzana, V. Capriati, M. Blangetti and C. Prandi, Chem.–Eur. J., 2021, 27, 2868–2874 CrossRef CAS PubMed.
- M. Ronsheim and G. L. Araldi, WO 2008061109 (A2), 2008.
- K. Kawamoto and T. Tominaga, JP 2008306170 (A), 2008.
- (a) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed; (b) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC; (c) E. I. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed; (d) V. L. Andersen, H. D. Hansen, M. M. Herth, G. M. Knudsen and J. L. Kristensen, Bioorg. Med. Chem. Lett., 2014, 24, 2408–2411 CrossRef CAS PubMed; (e) H. G. Lee, P. J. Milner, M. S. Placzek, S. L. Buchwald and J. M. Hooker, J. Am. Chem. Soc., 2015, 137, 648–651 CrossRef CAS PubMed; (f) P. J. H. Scott, Angew. Chem., Int. Ed., 2009, 48, 6001–6004 CrossRef CAS PubMed.
- (a) L. Pu, Acc. Chem. Res., 2017, 50, 1032–1040 CrossRef CAS PubMed; (b) Z. K. Yang, N. X. Xu, R. Takita, A. Muranaka, C. Wang and M. Uchiyama, Nat. Commun., 2018, 9, 1587 CrossRef PubMed.
- M. Andrs, J. Korabecny, D. Jun, Z. Hodny, J. Bartek and K. Kuca, J. Med. Chem., 2015, 58, 41–71 CrossRef CAS PubMed.
- K. A. Leonard, M. I. Nelen, L. T. Anderson, S. L. Gibson, R. Hilf and M. R. Detty, J. Med. Chem., 1999, 42, 3942–3952 CrossRef CAS PubMed.
- Y. Jeong, J. Lee and J. S. Ryu, Bioorg. Med. Chem., 2016, 24, 2114–2124 CrossRef CAS PubMed.
- F. Larysa, S. Guang, O. Akihiro, Y. Masayuki, O. Junzo, S. Sergei and A. Chihaya, Chem. Commun., 2007, 22, 2278–2280 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |