DOI:
10.1039/D2SC05313F
(Edge Article)
Chem. Sci., 2023,
14, 251-256
Photocatalytic enantioselective Minisci reaction of β-carbolines and application to natural product synthesis†
Received
23rd September 2022
, Accepted 23rd November 2022
First published on 24th November 2022
Abstract
A highly efficient enantioselective direct C–H functionalization of β-carbolines via a Minisci-type radical process under a photo-redox and chiral phosphoric acid cooperative catalytic system has been disclosed. Through this protocol, a wide range of C1 aminoalkylated β-carbolines were constructed directly with high levels of enantioselectivities from readily available β-carbolines and alanine-derived redox-active esters. This transformation allows straightforward access to highly valuable enantioenriched β-carbolines, which are an intriguing structural motif in valuable natural products and synthetic bio-active compounds. This protocol has been utilized as a highly efficient synthetic strategy for the concise asymmetric total synthesis of marine alkaloids eudistomin X, (+)-eudistomidin B and (+)-eudistomidin I.
Introduction
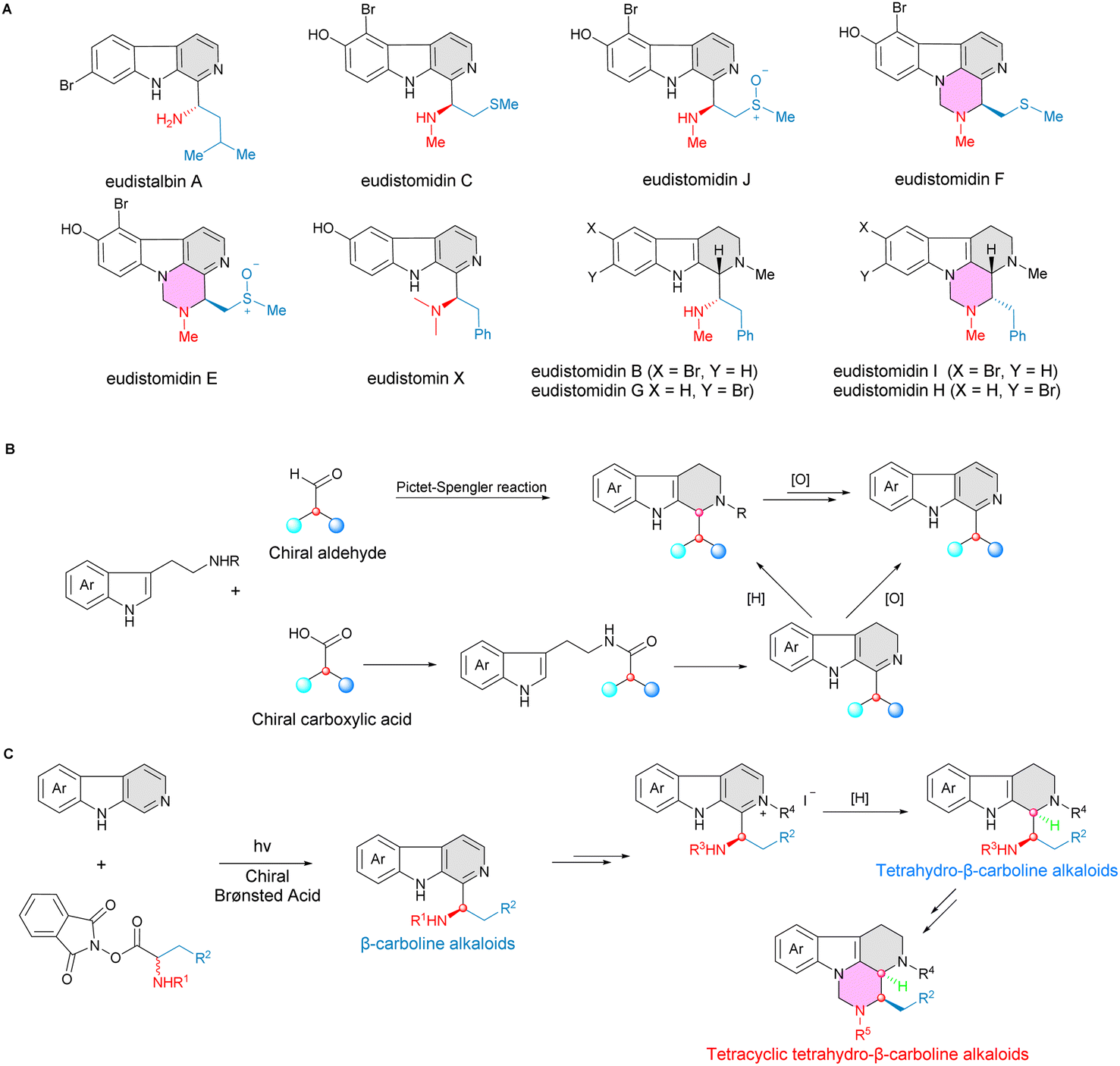
Radical chemistry has exhibited enormous potential in direct C–H functionalization of N-heteroarenes to construct complex molecules.1–4 However, the corresponding catalytic asymmetric radical transformations remain a significant challenge due to the lack of compatible effective enantioselective catalytic systems and the high reactivity of radical species.4,5 Liu6 and Zhang7,8 have presented copper-catalyzed enantioselective C(sp2)–H alkylation of electron-deficient azoles via alkyl radical intermediates, forming valuable chiral azoles with good to excellent regio- and enantioselectivities. With the rapid development of photochemistry, it has been shown that the cooperative photo-catalysis strategy plays an essential role in catalytic asymmetric radical transformations.9–11 Jiang,12 Phipps,13–17 and Fu18 have successfully developed catalytic asymmetric Minisci-type reactions of N-heteroarenes to achieve direct enantioselective C–H functionalization of electron-deficient N-heteroarenes with high level of enantiocontrol using chiral phosphoric acids as the catalysts under photoredox-catalytic conditions. Studer and co-workers reported an enantioselective cascade radical reaction via a Minisci-type process to form heterocyclic γ-amino acids and 1,2-diamine derivatives under a photoredox and chiral Brønsted acid cooperative catalytic system.19 Recently, Xiao and coworkers presented a photo-promoted asymmetric Minisci reaction of 5-arylpyrimidines with α-amino-acid derived redox-active esters to construct axially and centrally chiral heterobiaryls with high levels of diastereo-, and enatioselectivities.20 In spite of this impressive progress on various asymmetric C–H radical functionalizations of electron-deficient N-heteroarenes,21 such as quinolines, isoquinolines, pyridines, and pyrimidines, the catalytic direct C–H enantioselective functionalization of β-carbolines via a radial process has not been touched.22–24 The reason for this might originate from the less electron-deficient nature of the β-carboline structure, or from its reduced reactivity due to the participation of the nitrogen lone pair of the indole moiety, or from other regioselectivity-related problems.2
Notably, chiral β-carbolines have been shown to be attractive and privileged scaffolds in a variety of natural products, bioactive agents and approved drugs, displaying diverse potent biological activities.25–29 For instance, eudistomin marine alkaloids, which bears a chiral aminoalkylated β-carboline core motif, exhibited extremely potent antiviral and antitumor activities (Fig. 1A).30–32 Compared with the well-developed catalytic asymmetric synthesis of tetrahydro-β-carbolines,33,34 the highly effective catalytic enantioselective construction of β-carbolines has been less explored (Fig. 1B).
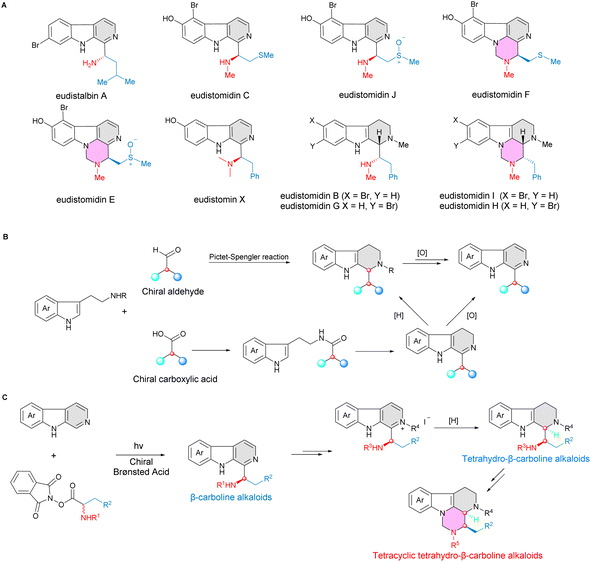 |
| | Fig. 1 Stereoselective synthesis of β-carboline and tetrahydro-β-carboline alkaloids. (A) Representative examples of naturally occurring C1-aminoalkylated bioactive β-carbolines and tetrahydro-β-carbolines. (B) Construction of β-carboline and tetrahydro-β-carboline skeletons by Pictet–Spengler reaction (well studied). (C) A new strategy for efficient construction of β-carboline and tetrahydro-β-carboline cores by photocatalytic Minisci reaction (this work). | |
Herein, we report an enantioselective Minisci reaction of β-carbolines via catalytic asymmetric C–H radical functionalization process under a photoredox and chiral phosphoric acid cooperative catalytic system to construct enantioenriched β-carbolines. This strategy has been applied as a key step in the efficient and concise asymmetric synthesis of biologically important eudistomin marine alkaloids (Fig. 1C).
Results and discussion
Reaction optimization
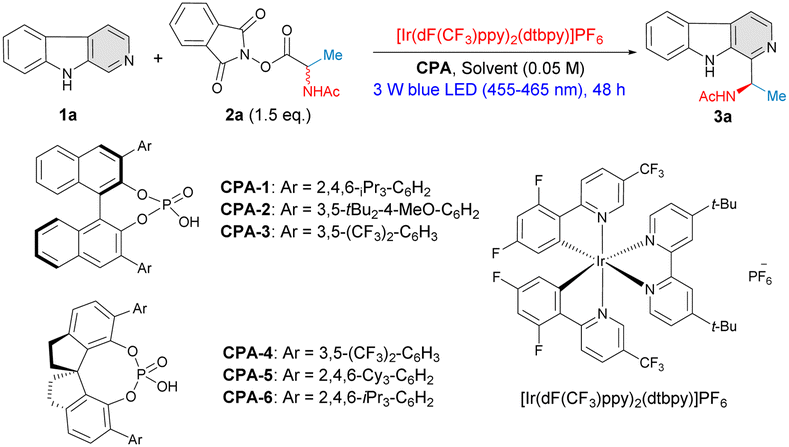
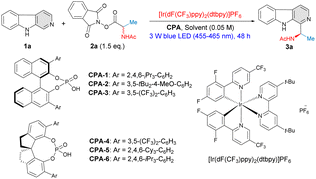
Inspired by the work of Jiang12 and Phipps,13 our study began with the reaction between β-carboline (1a) and racemic N-acetylalanine-derived redox-active ester (RAE) 2a in the presence of 5 mol% of chiral phosphoric acid catalyst CPA-1 and 1 mol% of photocatalyst [Ir(dF(CF3)ppy)2(dtbpy)]PF6 in 1,4-dioxane; the system was irradiated with blue LEDs (455–465 nm) for 48 h at 25 °C. To our delight, the reaction proceeded with excellent regioselectivity and only C1 C–H aminoalkylated product 3a was obtained in an excellent yield, although without any enantiocontrol (92% yield, 0% ee) (Table 1, entry 1). In order to obtain high levels of asymmetric induction, different types of chiral phosphoric acid catalysts were evaluated (Table 1, entries 2–6). We were pleased to find that the 3,5-(CF3)2C6H3– substituted catalyst CPA-3 delivered 3a in 83% yield with fair enantioselectivity (41% ee) (Table 1, entry 3). Notably, chiral phosphoric acids equipped with a chiral SPINOL backbone displayed a highly favorable catalytic performance, and CPA-6 (R)-STRIP gave a remarkable improvement on enantio-induction, affording 3a in 83% yield and 68% ee (Table 1, entry 6). Further screening of different solvents demonstrated that the solvent effect significantly influenced the enantioselectivity of this process (Table 1, entries 7–12). A notable improvement on both catalytic efficacy and enantioselectivity (90% yield, 80% ee) was observed when the reaction was carried out in THF (Table 1, entry 12), while other solvents showed inferior results (Table 1, entries 7–11). Lowering the reaction temperature revealed further increased enantioselectivities (Table 1, entries 13–14). Very good yield and excellent enantioselectivity (86% yield, 94% ee) was obtained when the reaction was conducted at −40 °C employing 10 mol% of CPA-6 (Table 1, entry 15). Thus, the optimal reaction conditions were established as 10 mol% of CPA-6, 1 mol% of photocatalyst [Ir(dF(CF3)ppy)2(dtbpy)]PF6 in THF (0.05 M), irradiated by 3 W blue LEDs at −40 °C for 72 hours. The influence of N-protecting groups of the RAEs ranging from formyl, acetyl, tert-butyloxycarbonyl (-Boc), benzoyl (-Bz), to benzyloxycarbonyl (-Cbz) groups were also evaluated, indicating the N-acetylalanine-derived RAE was the most suitable substrate in terms of yield and ee (for more details, see ESI†). The effects of photocatalyst and light intensity of the reaction were also investigated (for more details, see ESI†).
Table 1 Optimization of reaction conditionsa
|
|
| Entry |
Solvent |
Cat. (mol%) |
T (°C) |
Yieldb (%) |
eec (%) |
|
Reaction conditions: 1a (0.1 mmol), 2a (0.15 mmol), [Ir(dF(CF3)ppy)2(dtbpy)]PF6 (1 mol%), solvent (2.0 mL), 48 h.
Yield of isolated product.
Determined by HPLC analysis on a chiral stationary phase.
Reaction time: 72 h.
|
| 1 |
1,4-Dioxane |
CPA-1 (5) |
25 |
92 |
0 |
| 2 |
1,4-Dioxane |
CPA-2 (5) |
25 |
58 |
19 |
| 3 |
1,4-Dioxane |
CPA-3 (5) |
25 |
83 |
41 |
| 4 |
1,4-Dioxane |
CPA-4 (5) |
25 |
81 |
46 |
| 5 |
1,4-Dioxane |
CPA-5 (5) |
25 |
82 |
58 |
| 6 |
1,4-Dioxane |
CPA-6 (5) |
25 |
83 |
68 |
| 7 |
MeCN |
CPA-6 (5) |
25 |
67 |
44 |
| 8 |
Toluene |
CPA-6 (5) |
25 |
91 |
56 |
| 9 |
CH2Cl2 |
CPA-6 (5) |
25 |
68 |
59 |
| 10 |
DME |
CPA-6 (5) |
25 |
92 |
59 |
| 11 |
EtOAc |
CPA-6 (5) |
25 |
74 |
73 |
| 12 |
THF |
CPA-6 (5) |
25 |
90 |
80 |
| 13 |
THF |
CPA-6 (5) |
−20 |
88 |
88 |
| 14d |
THF |
CPA-6 (5) |
−40 |
85 |
91 |
| 15d |
THF |
CPA-6 (10) |
−40 |
86 |
94 |
Substrate scopes
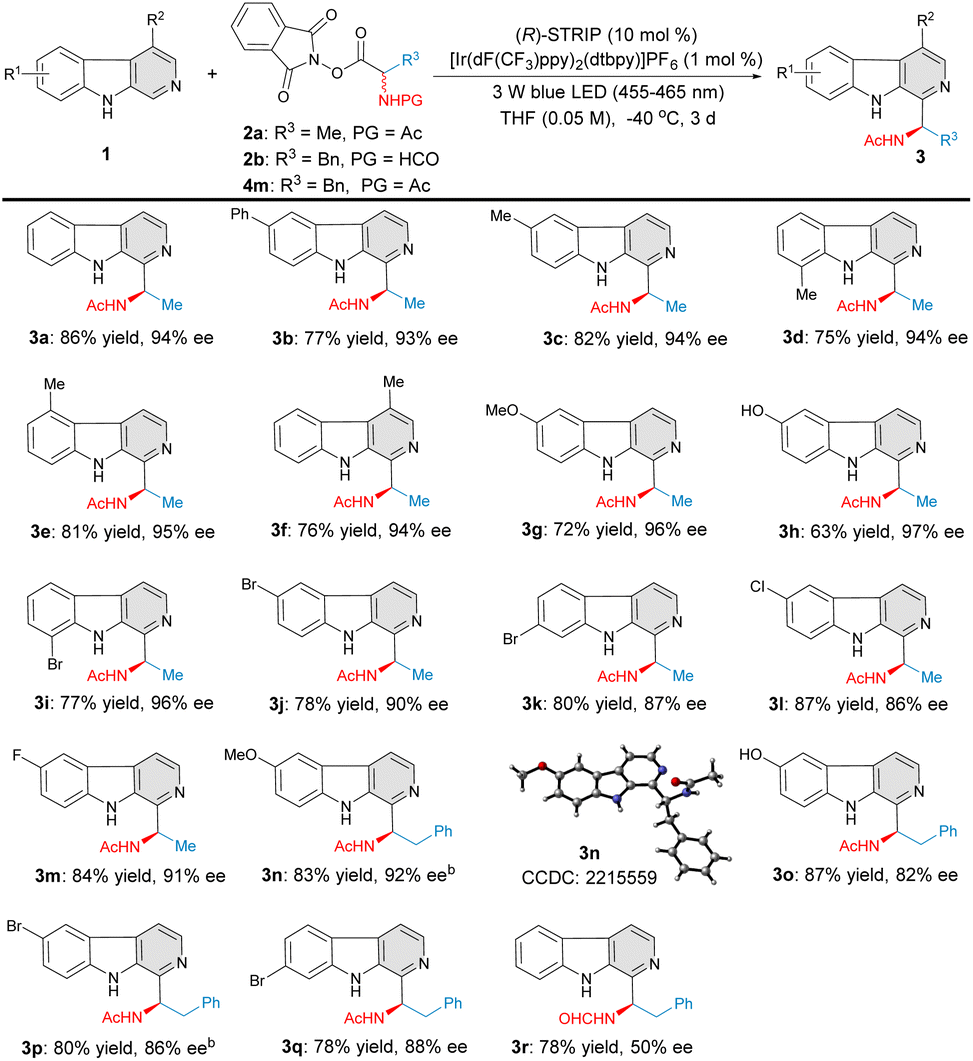
With the established reaction conditions in hand, the scope and generality of this protocol was investigated (Fig. 2). Firstly, a variety of β-carboline substrates were evaluated with N-acetylalanine-derived RAE 2a under the optimal reaction conditions. Gratifyingly, variations in the structural and electronic properties of β-carboline substrates were all tolerated well. Different types of β-carbolines bearing either electron-rich (2a–2h, 2n–2o) or electron-poor (2i–2m, 2p–2q) substituents were smoothly transformed to the desired C–H aminoalkyl functionalized products with high yields and excellent levels of enantioselectivitiy. 6-Methyl-, 8-methyl-, 5-methyl-, 4-methyl-, and 6-methoxy substituted β-carbolines were all proven to be suitable substrates under this catalytic system, affording the corresponding products 3c–3g in 72–82% yield with 94–96% ee. β-Carboline 1h bearing a free hydroxyl group was well compatible in terms of both reactivity and asymmetric induction, giving the target product 3h with 63% yield and 97% ee. Electron-withdrawing groups, such as Cl–, Br– and F–, at different positions on the β-carboline scaffold were demonstrated to have little influence on yield and enantioselectivity (3i–3m, 77–87% yield and 86–96% ee). Considering the fact that various eudistomin marine alkaloids exhibit a phenylalanyl moiety, N-acetylphenylalanine-derived RAE 4m was also examined in this transformation. Various C–H phenylalanyl-substituted β-carbolines 3n–3q were obtained with high level of yields and enantioselectivities (78–87% yield, 82–92% ee). Interestingly, product 3r was obtained from the N-formylphenylalanine-derived RAE 2b in good yield but moderate enantioselectivity, possibly due to the steric hindrance effect.
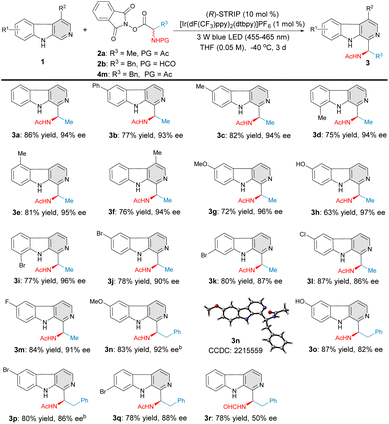 |
| | Fig. 2 Substrate scope of β-carbolines. aReaction conditions: 1 (0.2 mmol), 2a, 2b, or 4m (0.3 mmol), [Ir(dF(CF3)ppy)2(dtbpy)]PF6 (1 mol%), (R)-STRIP (10 mol%), THF (0.05 M), 3 W Blue LEDs (455–465 nm), −40 °C, 3 d. bTHF (0.025 M). | |
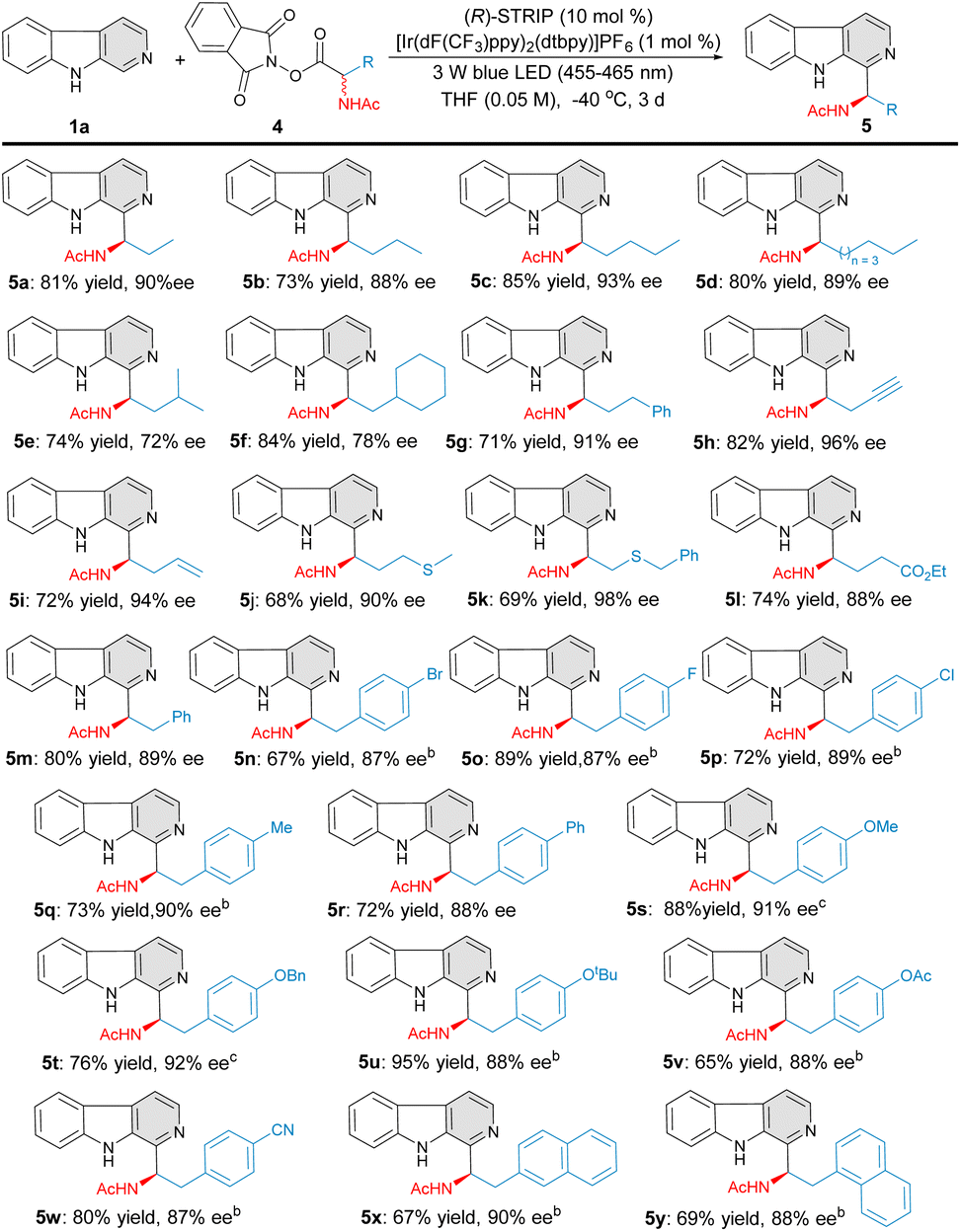
Encouraged by the above results, we further evaluated a wide range of amino-acid derived RAEs (Fig. 3). Alanine homologue substrates with different carbon chain length were first examined. Clearly, the carbon chain length of the amino-acid derived RAEs exhibits little influence on the yields and enantioselectivities, and the corresponding C–H aminoalkyl functionalization products 5a–5d were effectively constructed (73–85% yield, 88–93% ee). Sterically more hindered amino-acid derived RAEs were also tolerated in this transformation, albeit with slightly decreased enantioselectivities (5e–5f, 72–78% ee). Various functional groups, such as phenyl (5g), thioethers (5j–5k), and esters (5l), were well tolerated, providing the desired products with excellent asymmetric induction (5g–5l, 88–98% ee). To our delight, terminal alkenyl- and alkynyl-containing RAEs 4h–4i transformed efficaciously under this reaction conditions to furnish the products 5h and 5i with excellent yields and enantioselectivities. A series of 4-substituted phenylalanine-derived RAEs were further evaluated, affording the products 5m–5w in good yields and excellent level of enantiocontrol (65–95% yield, 87–92% ee). 1-Naphthyl and 2-naphthyl-substituted amino-acids were also tolerated in this catalytic system, affording the target products 5x–5y smoothly in good yields and enantioselectivities (67–69% yield, 88–90% ee).
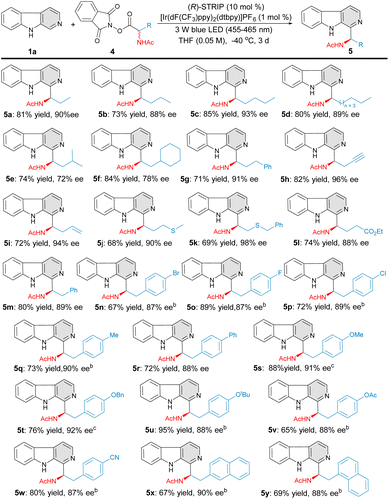 |
| | Fig. 3 Substrate scope of redox active esters (RAEs). aReaction conditions: 1a (0.2 mmol), 4 (0.3 mmol), [Ir(dF(CF3)ppy)2(dtbpy)]PF6 (1 mol%), (R)-STRIP (10 mol%), THF (0.05 M), 3 W Blue LEDs (455–465 nm), −40 °C, 3 d. bTHF (0.025 M). cTHF (0.04 M). | |
Total synthesis of eudistomin marine alkaloids
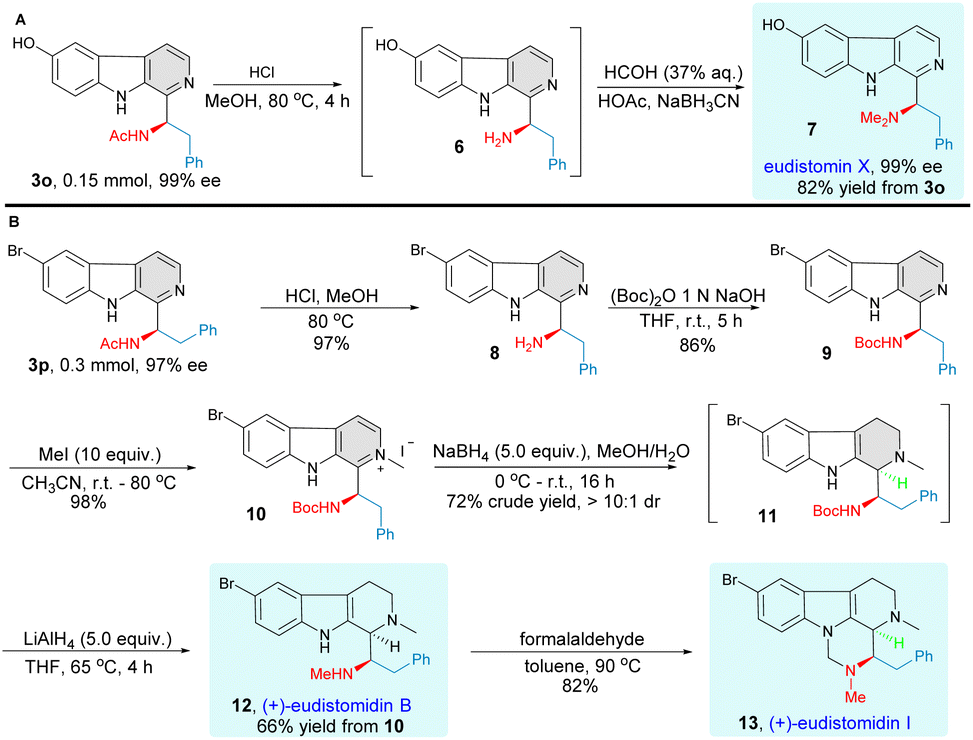
The synthetic potential and utility of this methodology were demonstrated in the total synthesis of eudistomin marine alkaloids (Fig. 4). Firstly, we introduced this enantioselective C–H bond aminoalkyl functionalization as a straightforward synthetic strategy for the concise synthesis of marine alkaloid eudistomin X.35,36 Simple treatment of the optically pure C–H functionalization product 3o, obtained after a simple recrystallization, with deacetylation and aminomethylation conditions afforded the target natural product eudistomin X directly with 82% yield without any loss of enantiomeric purity. We then focused our attention on applying this asymmetric Minisci reaction for the highly efficient synthesis of tetrahydro-β-carboline natural products eudistomidin B30,37 and eudistomidin I.32,38 To our great delight, compound 3p underwent acetyl group cleavage and subsequent N-Boc protection successfully to furnish the desired product 9. The transformation of compound 9 to the natural product (+)-eudistomidin B proceeded smoothly in a highly diastereoselective manner (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) by a three-step sequence, which included the formation of β-carboline methiodide 10, selective reduction of pyridine methiodide with NaBH4,39 and reduction of N-Boc to N-Me. Then, treatment of (+)-eudistomidin B with formaldehyde via a cyclocondensation process readily afforded the desired (+)-eudistomidin I. The absolute configuration of optically pure compound 3n was established as R by an X-ray crystallographic analysis and the structure of compounds 7, 12 and 13 were confirmed by comparing the reported spectroscopic data and optical rotation of natural eudistomin X,35,36 eudistomin B30,37 and eudistomin I32,38 (for more details, see ESI†).
:1 dr) by a three-step sequence, which included the formation of β-carboline methiodide 10, selective reduction of pyridine methiodide with NaBH4,39 and reduction of N-Boc to N-Me. Then, treatment of (+)-eudistomidin B with formaldehyde via a cyclocondensation process readily afforded the desired (+)-eudistomidin I. The absolute configuration of optically pure compound 3n was established as R by an X-ray crystallographic analysis and the structure of compounds 7, 12 and 13 were confirmed by comparing the reported spectroscopic data and optical rotation of natural eudistomin X,35,36 eudistomin B30,37 and eudistomin I32,38 (for more details, see ESI†).
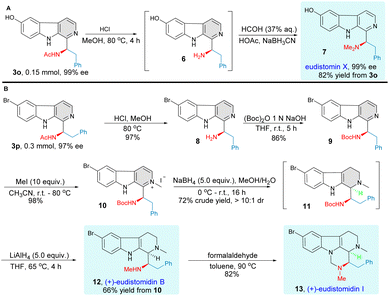 |
| | Fig. 4 Total synthesis of β-carboline marine alkaloids. (A) Total synthesis of eudistomin X. (B) Total synthesis of (+)-eudistomidin B and (+)-eudistomidin I. | |
Proposed mechanism
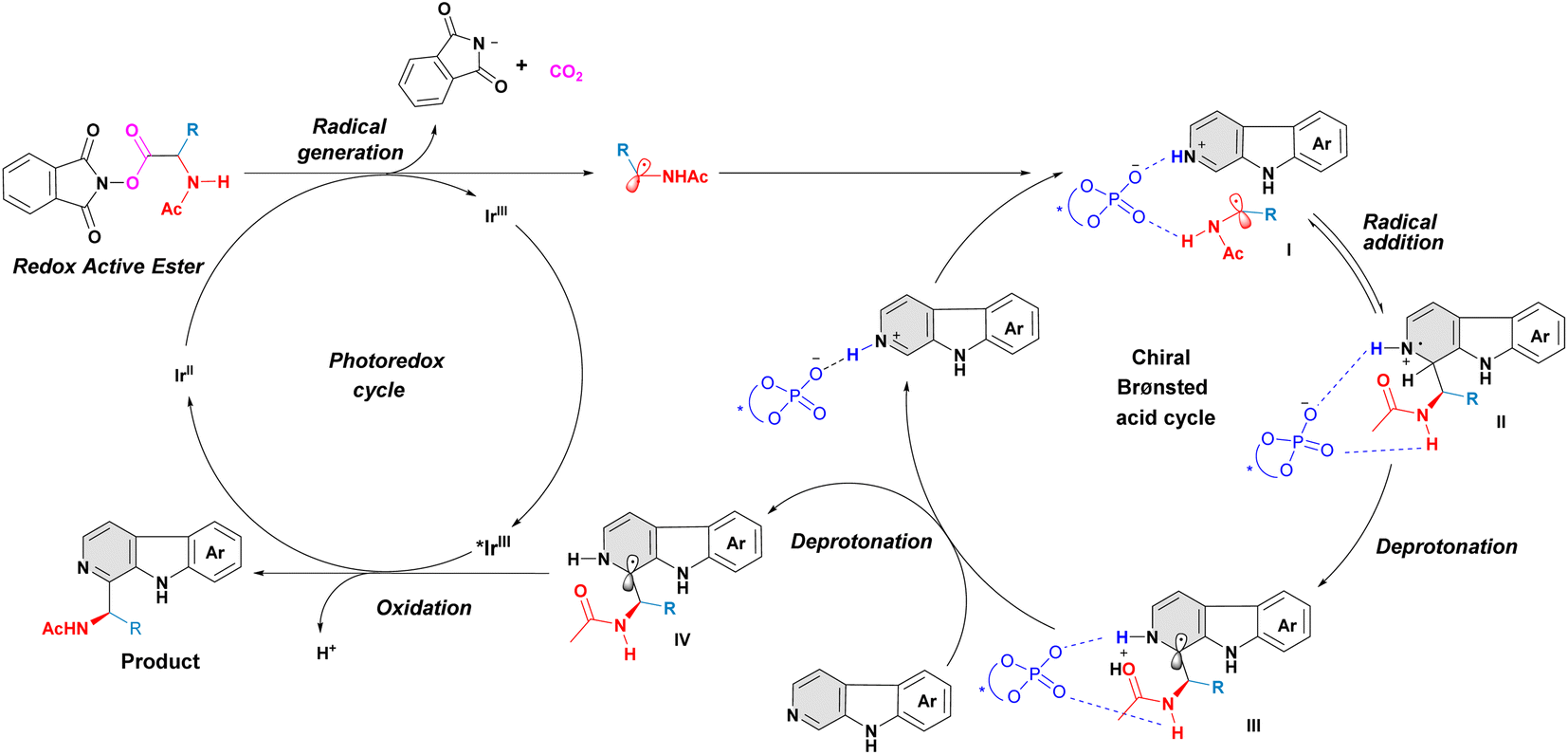
A plausible mechanism for this asymmetric Minisci reaction of β-carbolines is shown in Fig. 5.12–18 Firstly, the α-aminoalkyl radical, generated from α-amino-acid derived redox-active ester, interacts with β-carboline, aided by the bifunctional phosphoric acid catalyst, affording indeterminate I. A subsequent reversible radical addition results in a radical cation species II. The intermediate II then undergoes an internal proton abstraction promoted by the carbonyl oxygen of the N-acetyl group,15 giving intermediate III. Subsequently, the deprotonation of intermediate III occurs with the help of an external β-carboline and forms the neutral radical intermediate IV. This deprotonation is considered as an irreversible and enantiodetermining step in this transformation.15 Finally, the dissociated radical intermediate IV transforms into the desired C–H aminoalkyl-functionalized product via a single-electron oxidation process.
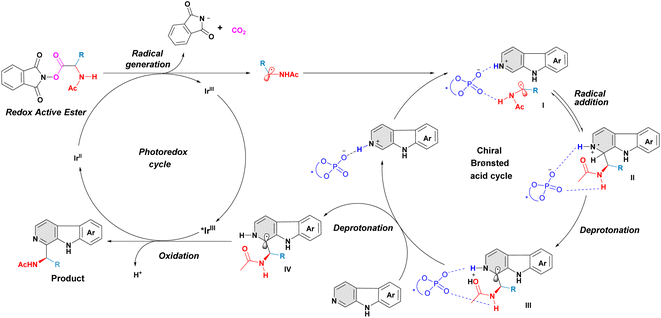 |
| | Fig. 5 Proposed catalytic cycle. | |
Conclusions
In summary, we present a photocatalytic direct C–H enantioselective functionalization of β-carbolines via a Minisci-type radial process using a chiral phosphoric acid as the chiral catalyst. This transformation allows straightforward access to a wide range of highly functionalized β-carbolines from simple and readily available starting materials with high level of enantioselectivity. This protocol has been applied as a highly efficient synthetic strategy for the concise asymmetric total synthesis of marine alkaloids. Further explorations on other catalytic asymmetric radical C–H functionalizations of N-heteroarenes in this field are on the way.
Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.† Crystallographic data for compound 3n has been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC2215559.
Author contributions
M. P. performed the most experiments, collected and analyzed the data and wrote the draft; Y. J. assisted in some experiments and helped in synthesis of substrates; S. G. conceived the idea, guided this project and revised the manuscript.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (22001260); Shenzhen Science and Technology Innovation Committee (JCYJ20210324101810028); Guangdong Basic and Applied Basic Research Foundation (2021A1515010016); the Shenzhen Key Laboratory of Computer Aided Drug Discovery (ZDSYS20201230165400001); China Postdoctoral Science Foundation (2019M663172) and the Guangdong Provincial Key Laboratory of Catalysis (2020B121201002).
Notes and references
- M. A. J. Duncton, MedChemComm, 2011, 2, 1135–1161 RSC.
- F. O'Hara, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2013, 135, 12122–12134 CrossRef PubMed.
- K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302–9332 CrossRef CAS.
- R. S. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- X.-L. Liu, L.-B. Jiang, M.-P. Luo, Z. Ren and S.-G. Wang, Org. Chem. Front., 2022, 9, 265–280 RSC.
- X. L. Su, L. Ye, J. J. Chen, X. D. Liu, S. P. Jiang, F. L. Wang, L. Liu, C. J. Yang, X. Y. Chang and Z. L. Li, Angew. Chem., Int. Ed., 2021, 60, 380–384 CrossRef CAS PubMed.
- C. Li, B. Chen, X. Ma, X. Mo and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2130–2134 CrossRef CAS.
- X. Ma and G. Zhang, ACS Catal., 2021, 11, 5108–5118 CrossRef CAS.
- C. Prentice, J. Morrisson, A. D. Smith and E. Zysman-Colman, Beilstein J. Org. Chem., 2020, 16, 2363–2441 CrossRef.
- M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chem. Rev., 2021, 122, 1654–1716 CrossRef PubMed.
- W. Yao, E. A. Bazan-Bergamino and M. Y. Ngai, ChemCatChem, 2022, 14, e202101292 CAS.
- X. Liu, Y. Liu, G. Chai, B. Qiao, X. Zhao and Z. Jiang, Org. Lett., 2018, 20, 6298–6301 CrossRef CAS PubMed.
- R. S. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419–422 CrossRef CAS.
- J. P. Reid, R. S. Proctor, M. S. Sigman and R. J. Phipps, J. Am. Chem. Soc., 2019, 141, 19178–19185 CrossRef CAS.
- K. Ermanis, A. C. Colgan, R. S. Proctor, B. W. Hadrys, R. J. Phipps and J. M. Goodman, J. Am. Chem. Soc., 2020, 142, 21091–21101 CrossRef CAS PubMed.
- R. S. Proctor, P. Chuentragool, A. C. Colgan and R. J. Phipps, J. Am. Chem. Soc., 2021, 143, 4928–4934 CrossRef CAS PubMed.
- A. C. Colgan, R. S. Proctor, D. C. Gibson, P. Chuentragool, A. S. Lahdenperä, K. Ermanis and R. J. Phipps, Angew. Chem., Int. Ed., 2022, e202200266 CAS.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed.
- D. Zheng and A. Studer, Angew. Chem., Int. Ed., 2019, 131, 15950–15954 CrossRef.
- D. Liang, J.-R. Chen, L.-P. Tan, Z.-W. He and W.-J. Xiao, J. Am. Chem. Soc., 2022, 144, 6040–6049 CrossRef CAS.
- Y. Yin, X. Zhao and Z. Jiang, Chinese J. Org. Chem., 2022, 42, 1609–1625 CrossRef.
- G. Lin, Y. Wang, Q. Zhou, W. Tang, J. Wang and T. Lu, Synth. Commun., 2011, 41, 3541–3550 CrossRef CAS.
- K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 125, 2136–2140 CrossRef.
- M. E. Zhidkov and V. A. Kaminskii, Tetrahedron Lett., 2013, 54, 3530–3532 CrossRef CAS.
- R. Cao, W. Peng, Z. Wang and A. Xu, Curr. Med. Chem., 2007, 14, 479–500 CrossRef CAS PubMed.
- J. Dai, W. Dan, U. Schneider and J. Wang, Eur. J. Med. Chem., 2018, 157, 622–656 CrossRef CAS PubMed.
- S. Aaghaz, K. Sharma, R. Jain and A. Kamal, Eur. J. Med. Chem., 2021, 216, 113321 CrossRef PubMed.
- A. Beato, A. Gori, B. Boucherle, M. Peuchmaur and R. Haudecoeur, J. Med. Chem., 2021, 64, 1392–1422 CrossRef CAS.
- B. Luo and X. Song, Eur. J. Med. Chem., 2021, 224, 113688 CrossRef CAS PubMed.
- J. Kobayashi, J. F. Cheng, T. Ohta, S. Nozoe, Y. Ohizumi and T. Sasaki, J. Org. Chem., 1990, 55, 3666–3670 CrossRef CAS.
- M. A. Rashid, K. R. Gustafson and M. R. Boyd, J. Nat. Prod., 2001, 64, 1454–1456 CrossRef CAS.
- T. Suzuki, T. Kubota and J. i. Kobayashi, Bioorg. Med. Chem. Lett., 2011, 21, 4220–4223 CrossRef CAS PubMed.
- A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS PubMed.
- J. Stöckigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 CrossRef.
- R. Finlayson, A. Brackovic, A. Simon-Levert, B. Banaigs, R. F. O'Toole, C. H. Miller and B. R. Copp, Tetrahedron Lett., 2011, 52, 837–840 CrossRef CAS.
- P. Schupp, T. Poehner, R. Edrada, R. Ebel, A. Berg, V. Wray and P. Proksch, J. Nat. Prod., 2003, 66, 272–275 CrossRef CAS.
- Y. Takahashi, H. Ishiyama, T. Kubota and J. i. Kobayashi, Bioorg. Med. Chem. Lett., 2010, 20, 4100–4103 CrossRef CAS PubMed.
- H. Ishiyama, K. Yoshizawa and J. i. Kobayashi, Tetrahedron, 2012, 68, 6186–6192 CrossRef CAS.
- I. W. Still and J. McNulty, Tetrahedron Lett., 1995, 36, 7965–7966 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yi-Jie
Gu
a and
Shou-Guo
Wang
a,
Yi-Jie
Gu
a and
Shou-Guo
Wang