
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of the solvent in the self-assembly and binding properties of [1 + 1] tetra-imine bis-calix[4]pyrrole cages†
Chiara F. M.
Mirabella
 ab,
Gemma
Aragay
a and
Pablo
Ballester
*ac
ab,
Gemma
Aragay
a and
Pablo
Ballester
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology (BIST), Avgda. Països Catalans, 16, 43007 Tarragona, Spain. E-mail: pballester@iciq.es
bUniversitat Rovira i Virgili, Departament de Química Analítica i Química Orgànica, c/Marcel·lí Domingo,1, 43007 Tarragona, Spain
cICREA, Passeig Lluís Companys, 23, 08010 Barcelona, Spain
First published on 23rd November 2022
Abstract
We report the self-assembly of shape-persistent [1 + 1] tetra-imine cages 1 based on two different tetra-α aryl-extended calix[4]pyrrole scaffolds in chlorinated solvents and in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CDCl3:CD3CN solvent mixture. We show that the use of a bis-N-oxide 4 (4,4′-dipyridyl-N,N′-dioxide) as template is not mandatory to induce the emergence of the cages but has a positive effect on the reaction yield. We use 1H NMR spectroscopy to investigate and characterize the binding properties (kinetic and thermodynamic) of the self-assembled tetra-imine cages 1 with pyridine N-oxide derivatives. The cages form kinetically and thermodynamically stable inclusion complexes with the N-oxides. For the bis-N-oxide 4, we observe the exclusive formation of 1:1 complexes independently of the solvent used. In contrast, the pyridine-N-oxide 5 (mono-topic guest) produces inclusion complexes displaying solvent dependent stoichiometry. The bis-N-oxide 4 is too short to bridge the gap between the two endohedral polar binding sites of 1 by establishing eight ideal hydrogen bonding interactions. Nevertheless, the bimolecular 4⊂1 complex results as energetically favored compared to the 52⊂1 ternary counterpart. The inclusion of the N-oxides, 4 and 5, in the tetra-imine cages 1 is significantly faster in chlorinated solvents (minutes) than in the 9:1 CDCl3:CD3CN solvent mixture (hours). We provide an explanation for the similar energy barriers calculated for the formation of the 4⊂1 complex using the two different ternary counterparts 52⊂1 and (CD3CN)2⊂1 as precursors. We propose a mechanism for the in–out guest exchange processes experienced by the tetra-imine cages 1.
:1 CDCl3:CD3CN solvent mixture. We show that the use of a bis-N-oxide 4 (4,4′-dipyridyl-N,N′-dioxide) as template is not mandatory to induce the emergence of the cages but has a positive effect on the reaction yield. We use 1H NMR spectroscopy to investigate and characterize the binding properties (kinetic and thermodynamic) of the self-assembled tetra-imine cages 1 with pyridine N-oxide derivatives. The cages form kinetically and thermodynamically stable inclusion complexes with the N-oxides. For the bis-N-oxide 4, we observe the exclusive formation of 1:1 complexes independently of the solvent used. In contrast, the pyridine-N-oxide 5 (mono-topic guest) produces inclusion complexes displaying solvent dependent stoichiometry. The bis-N-oxide 4 is too short to bridge the gap between the two endohedral polar binding sites of 1 by establishing eight ideal hydrogen bonding interactions. Nevertheless, the bimolecular 4⊂1 complex results as energetically favored compared to the 52⊂1 ternary counterpart. The inclusion of the N-oxides, 4 and 5, in the tetra-imine cages 1 is significantly faster in chlorinated solvents (minutes) than in the 9:1 CDCl3:CD3CN solvent mixture (hours). We provide an explanation for the similar energy barriers calculated for the formation of the 4⊂1 complex using the two different ternary counterparts 52⊂1 and (CD3CN)2⊂1 as precursors. We propose a mechanism for the in–out guest exchange processes experienced by the tetra-imine cages 1.
Introduction
Molecular cages possess three-dimensional cavities with multiple portals that are capable of including sizable guests. Included guests reside in the “inner phase”, whose properties are different from those of the outer phase i.e. the bulk solution.1,2 The portals connecting the two phases and their sizes control the in and out exchange of the guests. These singular features make cage containers attractive molecular designs for different applications such as molecular recognition,3–5 stabilization of reactive species6 and mediation of chemical reactivity,7,8 among others.9–12The first examples of molecular cages date back to the middle 80s’. In pioneering works, Cram13 and Collet14 reported the preparation of molecular containers using exclusively irreversible covalent bonds. The large kinetic stability of the purely covalent cages contrasted with its high synthetic cost and the low yield in which they were produced. Dynamic covalent chemistry (DCC) constitutes an efficient synthetic strategy for the modular self-assembly of three-dimensional molecular containers in high yields.15,16 DCC combines the strength of covalent bonds with the error-checking nature of reversible interactions. The formation of imine bonds is a well-established reversible chemical transformation suitable for DCC. In fact, many shape-persistent organic cages, a.k.a. porous organic cages (POCs), were self-assembled from reversible condensation reactions between simple aldehyde and amine building blocks.17,18 The resulting poly-imine POCs displayed a well-defined interior cavity that did not collapse into more dense or twisted structures owing to the conformational restrictions imposed by the imine bonds.19 The first Schiff base molecular container was described in 1991 by Quan and Cram, via the condensation reaction between two molecules of a tetra-aldehyde resorcin[4]arene cavitand and four molecules of 1,3-diaminobenzene.20 The derived octa-imine cage was able to include a series of guests forming kinetically stable hemicarceplexes owing to constrictive binding.2 The ground state of the cage complexes had twisted portals that were too small to allow the free passage of the guests. The guest exchange was associated with the untwisting of the structure of the host, the loss of many close contacts and the maximization of the cross section of at least one portal. Thus, the calculated free energy barriers for the guest exchange were significantly larger (>20 kcal mol−1) than the absolute value of the free binding energy (>−3 kcal mol−1).
The binding affinity and selectivity displayed by synthetic molecular cages tends to be outperformed by those of biological receptors.21,22 For example, the lack of converging polar groups in the inner cavities of most synthetic molecular containers restricts their binding selectivity to non-polar guests being size and shape complementary. On the other hand, the extensive use of aromatic panels to shape the inner cavity of the synthetic molecular containers makes the incorporation of polar converging groups synthetically challenging. This serves as explanation for the reduced number of endohedrally functionalized synthetic molecular cages described in literature. Nevertheless, in recent times, several approaches have been disclosed in trying to overcome this limitation.23 For example, pyrrole units were incorporated in imine-based dynamic covalent cages to endow their cavities with polar hydrogen bond donor groups (Fig. 1).24,25
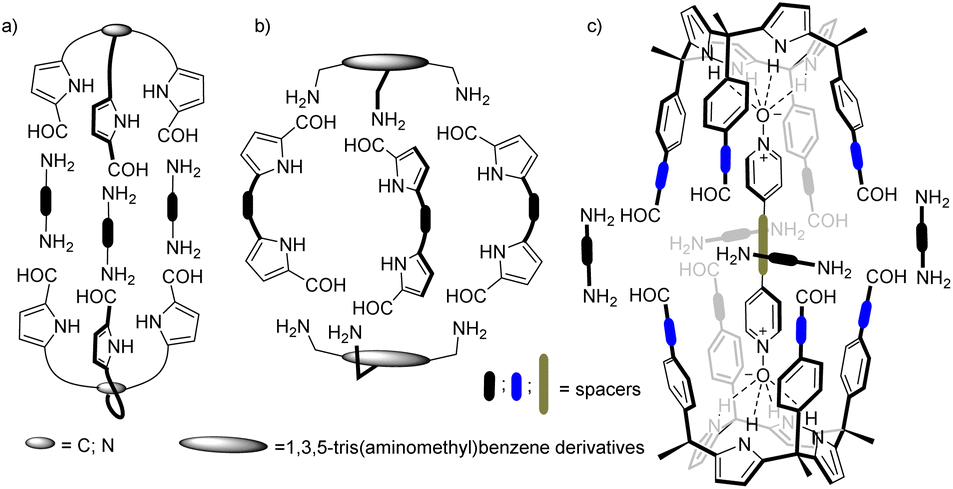 | ||
| Fig. 1 Schematic representation of pyrrole-based cages formed by Schiff base condensation of polyamines and poly-formyl building blocks. | ||
Beer and co-workers described the synthesis of bis(tripyrrolyl) cryptand-like hexa-imine cages via a [2 + 3] Schiff base condensation of triformyl-alkyl-tripyrrolyls and α,ω-alkyl diamines (Fig. 1a).26 The X-ray structures of the hexa-imine cryptands revealed the inclusion of one molecule of the reactant diamine establishing multiple hydrogen bonding interactions with the converging polar functions inwardly directed with respect to the container's cavity. The authors suggested that the formation of the cages was templated by the diamines.
In the same vein, Roelens and co-workers, reported the quantitative self-assembly of a tris-pyrrolic hexa-imine macro-bicyclic cage via [2 + 3] condensation of a 1,3,5-tris(aminomethyl)benzene derivative and pyrrole-2,5-dicarboxyaldehyde.27 A one pot reduction of the hexa-imine yielded the corresponding hexa-amine cage that selectively bound β-D-monosaccharides of the gluco series.
More recently, Sessler and co-workers also prepared a series of hexa-imine cages via [2 + 3] condensation reaction between 1,3,5-(tris-aminomethyl)benzene derivatives and diformyldipyrrolyl linkers having pyridine and naphthyridine spacers (Fig. 1b). The cage systems acted as shape-persistent POCs for the selective adsorption of CO2.28–30
Our group introduced the use of α,α,α,α-aryl-extended calix[4]pyrrole scaffolds for the construction of hydrogen-bonded31 and mechanically-bonded32 molecular capsules with polar interiors. We also described the template-directed self-assembly of octa-imine cages having polar interiors via the [2 + 4] Schiff base condensation reaction of a tetra-formyl aryl-extended calix[4]pyrrole and 1,2-diamines (Fig. 1c).33 We used 4-(4-pyridinylethynyl)pyridine-N,N′-dioxide as ditopic template. The terminal pyridyl-N-oxides knobs of the template bound two α,α,α,α-aryl-extended calix[4]pyrrole tetra-formyl units in cone conformation placing them in close proximity. The orientation of the formyl groups was also suitable for the stitching of the dimeric cage through imine bonds using 1,2-diamine linkers. Unfortunately, the template became tightly bound (locked) in the cage interior rendering the characterization of its binding properties inaccessible.
Herein, we describe the self-assembly of related tetra-imine cages 1 using bis-pyridyl-N,N′-dioxide 4 as template (Fig. 2). We also report the self-assembly of the tetra-imine cages 1 in the absence of template 4 using a 9:1 CDCl3:CD3CN solvent mixture and pure CDCl3 or CD2Cl2 solvents. These results allowed the study and characterization of the binding properties of the tetra-imine cages 1 towards bis-N-oxide 4 and pyridine-N-oxide 5. In the case of pyridine-N-oxide 5, we detected the formation of 1:1 and 1:2 complexes. Despite 4 was too short to establish an ideal ditopic interaction with the two polar hemispheres of 1, the 4⊂1 inclusion complex was thermodynamically more stable than the 52⊂1 counterpart. We investigated the kinetics for the formation of the inclusion complexes and the guests' exchange using 1H NMR spectroscopy. In the 9:1 CDCl3:CD3CN solvent mixture the inclusion of 4 in cage 1 was quite slow in reaching equilibrium (days). On the contrary, the uptakes of the guests in pure CDCl3 solution were fast (min). To our surprise, the energy barrier of the guests' exchange in pure CDCl3 (52⊂1 + 4 ⇌ 4⊂1 + 2 × 5) coincided with that of the uptake of 4 by the capsular solvate (CD3CN)2⊂1 assembled in the 9:1 CDCl3:CD3CN solvent mixture.
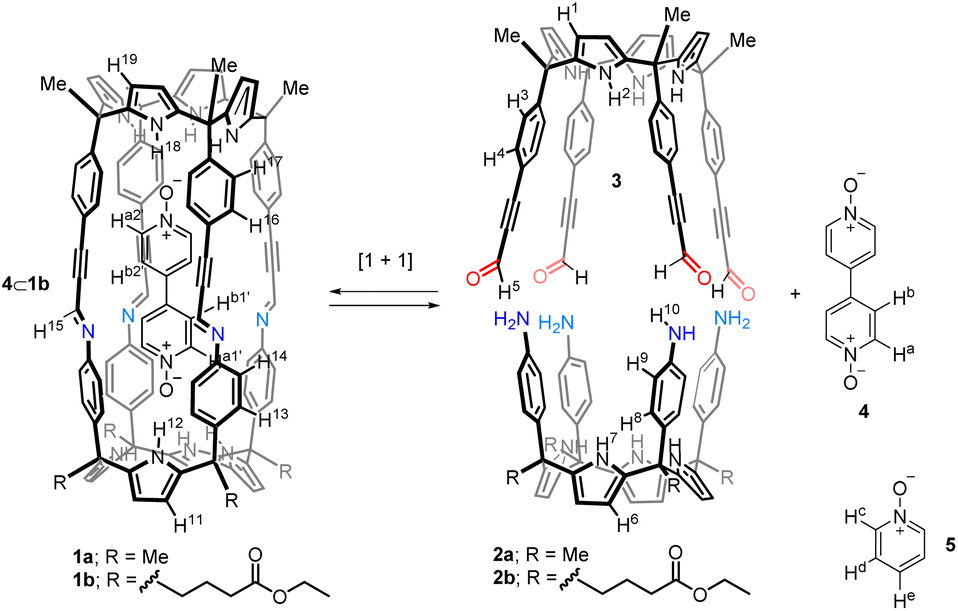 | ||
| Fig. 2 Equilibrium of the [1 + 1] Schiff base condensation reaction between the “four wall” tetra-amine calix[4]pyrroles 2 and tetra-formyl counterpart 3 assisted by 4,4′-bispyridyl N,N′-dioxide 4 as template. Line drawing structure of pyridine N-oxide 5 is also shown. | ||
Results and discussion
Synthesis of precursors
Tetra-amine “four wall” calix[4]pyrrole 2a and its lipophilic tetra-ester analogue 2b were synthesized using reported methodologies.34,35 Tetra-formyl calix[4]pyrrole 3 (Fig. 2) was also prepared uneventfully using literature procedures.33Template-assisted self-assembly of tetra-imine cages 1
Firstly, we investigated the self-assembly of cage 1a using equimolar amounts of meso-tetramethyl tetra(4-amino)phenyl calix[4]pyrrole 2a, tetra-formyl calix[4]pyrrole 3 and bis-pyridyl-N-oxide 4 in CDCl3 solution. Based on our previous results on the assembly of octa-imine dynamic covalent cages,33 we assumed that the inclusion of the ditopic template was mandatory for the emergence of the tetra-imine cage 1a. Molecular modelling (MM3) studies showed the existence of a good fit (size, shape and function complementarity) for the bis-pyridyl N-oxide 4 included in the polar cavity of 1a (Fig. S11b†).We monitored the self-assembly of 1a by 1H NMR spectroscopy and used 1,3,5-trimethoxybenzene as internal standard (i.s.). Owing to the reduced solubility of the meso-tetramethyl tetra(4-amino)phenyl calix[4]pyrrole 2a in CDCl3, the equimolar mixture of the components used in the self-assembly of the 4⊂1a cage complex produced a suspension. This had a negative impact on the yield and reaction time needed to reach the equilibrium for the self-assembly of the tetra-imine cage 1a compared to its lipophilic version 1b. Cage 1b contained meso-tetra(4-ethylbutanoate) substituents in the hemisphere deriving from the tetra-amine calix[4]pyrrole (see ESI† for details). For the sake of brevity and similarity of results, we will concentrate in describing those obtained with the tetra-imine cage 1b. The 2 mM equimolar mixture of tetra-formyl calix[4]pyrrole 3, meso-alkyl substituted tetra-amine 2b and bis-N-oxide 4 in CDCl3 produced a solution. The 1H NMR spectrum acquired approximately 3 h following the preparation of the solution mixture showed sharp and well-defined proton signals (Fig. 3c).‡ Notably, none of the proton signals of the calix[4]pyrrole precursors 3 and 2b were detected. These findings suggested the presence of a predominant and structurally well-defined species in solution.
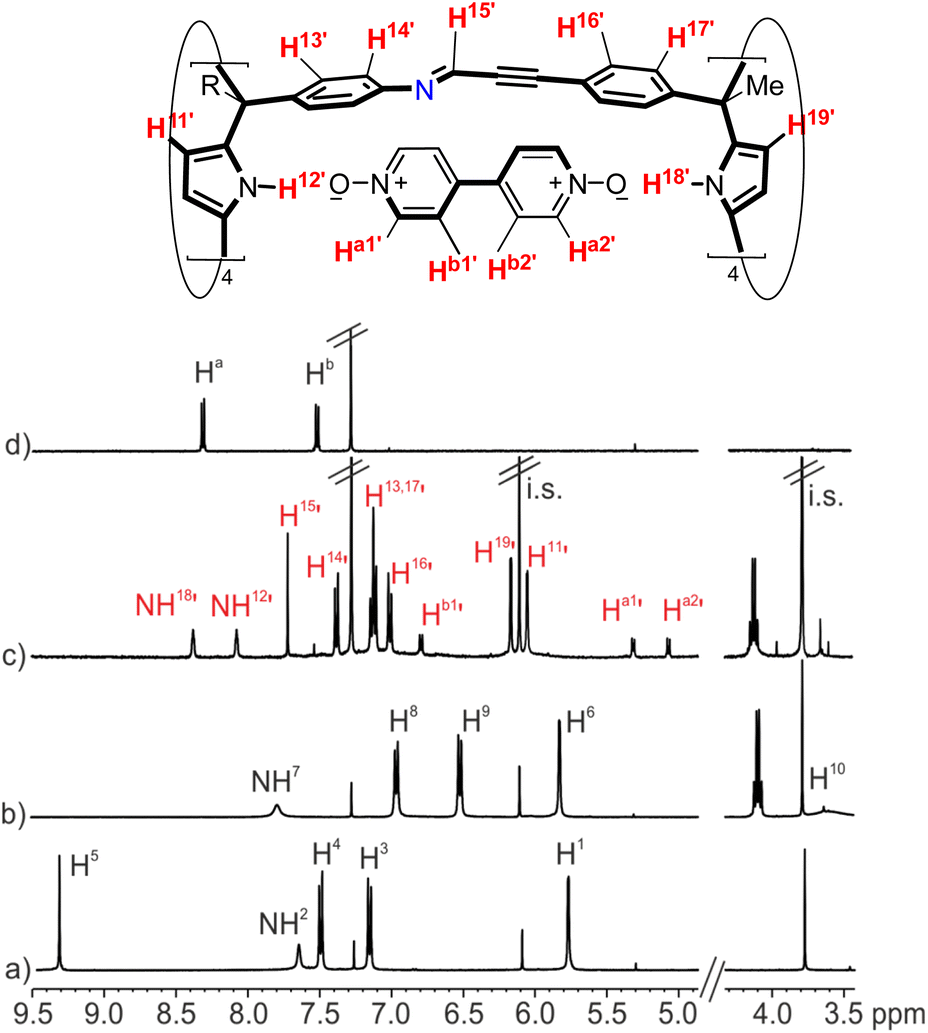 | ||
| Fig. 3 (Top) Line drawing structure of the 4⊂1b complex with the corresponding proton assignment. (Bottom) Selected regions of the 1H NMR spectra (400 MHz, at 298 K) of CDCl3 solutions of (a) tetra-formyl calix[4]pyrrole 3, (b) tetra-amine tetra-ester calix[4]pyrrole 2b, (c) tetra-imine cage complex 4⊂1b and (d) bis-pyridyl N-oxide 4. i.s.: internal standard. See Fig. 2 for proton assignment of 2b and 3. Red and primed letters correspond to protons of the 4⊂1b complex. | ||
We detected two signals for the pyrrole NHs (NH12’ and NH18’) resonating as broad singlets at δ = 8.1 and 8.4 ppm, respectively. The downfield shifts experienced by these signals compared to those in the free calix[4]pyrroles 3 and 2b were indicative of their involvement in hydrogen bonding interactions with the oxygen atoms of the pyridyl-N-oxide ends of template 4. The aromatic proton signals of template 4 appeared as four upfield shifted (Δδ = 3.2–0.4 ppm) doublets (Ha1’, Ha2’, Hb1’ and Hb2’). On the one hand, this result indicated that the bound bis-N-oxide 4 was no longer D2 symmetric. On the other hand, it suggested that the pyridyl-N-oxide ends of 4 were included in chemically different calix[4]pyrrole aromatic cavities and experienced the strong shielding effect provoked by the four meso-phenyl groups. In addition, the aromatic protons of these meso-phenyl substituents appeared as two sets of doublets. The β-pyrrole protons moved downfield and appeared as two separate singlets. Finally, we detected a sharp singlet centered at δ = 7.7 ppm, which combined with the disappearance of the formyl proton of calix[4]pyrrole 3, hinted for its assignment to an imine proton (H15’). Taken together, these results supported the predominant formation of the tetra-imine cage complex 4⊂1b in solution displaying an averaged C4v symmetry. Using integral values of selected proton signals of the 4⊂1b complex and those of the i.s., we determined that the yield for the assembly of the complex was ∼65%. We hypothesized that the remaining calix[4]pyrroles units might be involved in the formation of large polymeric aggregates whose proton signals broadened beyond detection. It is worthy to note that the downfield shifts experienced by the pyrrole NHs in the 4⊂1b complex were smaller than the ones observed in inclusion complexes of pyridine-N-oxide 5 with the modular calix[4]pyrroles 2b and 3.33 This observation suggested that the hydrogen bonds present in the 4⊂1b cage complex were longer and that 4 was too short to adequately cover the gap between the two calix[4]pyrrole binding sites of 1b. In short, the ditopic hydrogen-bonding interaction in 4⊂1b was not optimal.
Cage complex 4⊂1b was fully characterized by a complete set of high-resolution spectra (NMR and Mass Spectrometry, see ESI† for details, Fig. S5–S9 and S39†). A 1H DOSY NMR experiment36 assigned identical diffusion coefficient to the two components of the cage complex, D = 3.58 × 10−10 m2 s−1, evidencing their involvement in the same species (Fig. S10†).37 Using as reference the larger diffusion constant determined for the tetra-formyl calix[4]pyrrole 3 (D = 5.68 × 10−10 m2 s−1), we assigned a 1.6 fold increase in the hydrodynamic radius of the 4⊂1b complex diffusing as an hypothetically spherical object. Notably, the calculated spherical hydrodynamic radii for 3 and 4⊂1b were in good agreement with those of the spheres including their energy minimized structures (Fig. S11†).
Solvent-assisted self-assembly of tetra-imine cage 1b
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 CDCl3:CD3CN solvent mixture.
Aiming at investigating the binding properties of the tetra-imine cage 1b, we decided to explore its self-assembly in the absence of template 4. In previous works, we described that one molecule of acetonitrile was nicely accommodated in the polar cavity of the cone conformer of α,α,α,α-aryl-extended calix[4]pyrroles. The nitrogen atom of the bound acetonitrile molecule established four convergent hydrogen-bonds with the pyrrole NHs locking the receptor in the cone conformation.38 We hypothesized that the preorganization of the modular components, 2b and 3, of tetra-imine cage 1b in cone conformation might facilitate its self-assembly.
:1 CDCl3:CD3CN solvent mixture.
Aiming at investigating the binding properties of the tetra-imine cage 1b, we decided to explore its self-assembly in the absence of template 4. In previous works, we described that one molecule of acetonitrile was nicely accommodated in the polar cavity of the cone conformer of α,α,α,α-aryl-extended calix[4]pyrroles. The nitrogen atom of the bound acetonitrile molecule established four convergent hydrogen-bonds with the pyrrole NHs locking the receptor in the cone conformation.38 We hypothesized that the preorganization of the modular components, 2b and 3, of tetra-imine cage 1b in cone conformation might facilitate its self-assembly.
Thus, we prepared a 2 mM equimolar solution of the tetra-amine calix[4]pyrrole 2b and the tetra-formyl counterpart 3 in a 9:1 CDCl3:CD3CN solvent mixture. To our delight, after 3 h, the 1H NMR spectrum of the solution mainly showed the proton signals diagnostic of the self-assembly of the tetra-imine cage 1b (Fig. 4a).§ Most likely, two molecules of acetonitrile were included and hydrogen-bonded to the two polar binding sites of the cavity of cage 1b, resulting in the solvate inclusion complex (CD3CN)2⊂1b. Using the integral values of selected proton signals of 1b and those of the i.s., we determined that the self-assembly took place to an extent close to 65%. Notably, the yield of the self-assembly process of 1b in a 9:1 CDCl3:CD3CN solvent mixture was very similar to the one obtained for the template-assisted cage complex 4⊂1b.
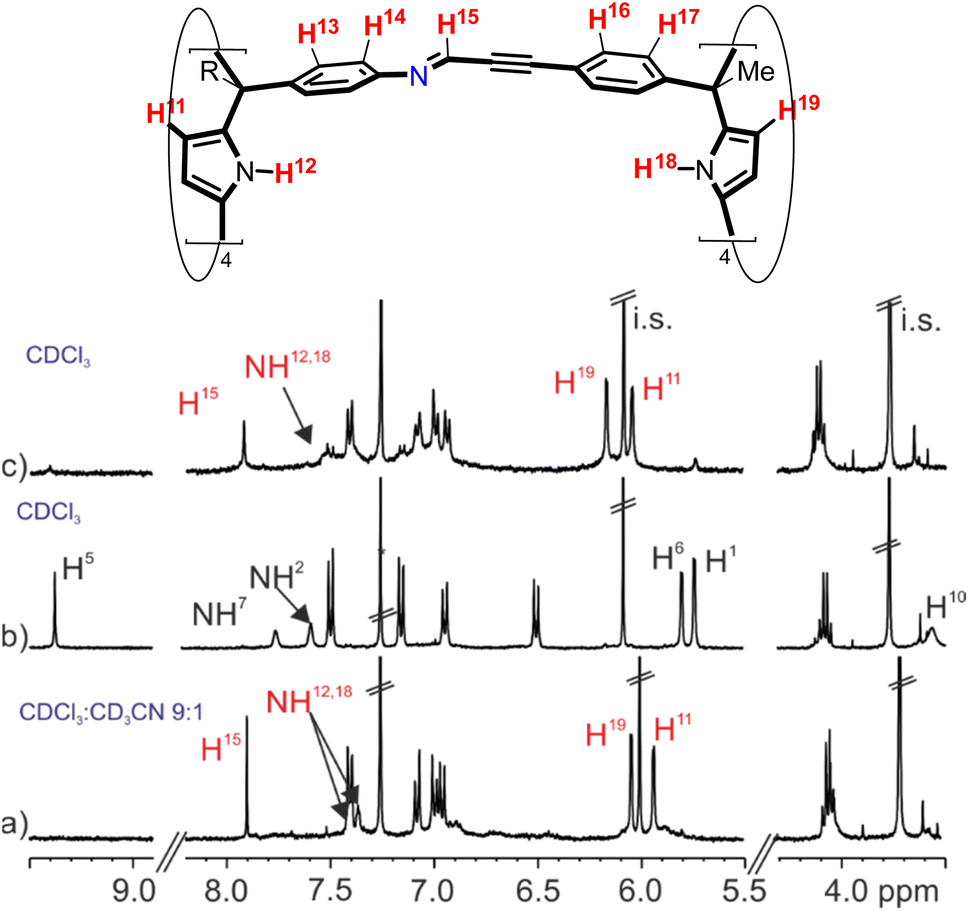 | ||
| Fig. 4 (Top) Line drawing structure of tetra-imine cage 1b with the corresponding proton assignment. (Bottom) Selected regions of the 1H NMR spectra (400 MHz, at 298 K) of an equimolar solution of tetra-formyl calix[4]pyrrole 3 and tetra-amine tetra-ester calix[4]pyrrole 2b in (a) 9:1 CDCl3:CD3CN solvent mixture after 3 h; (b) CDCl3 following its preparation and (c) CDCl3 after heating the solution at 308 K for 24 h. i.s.: internal standard. See Fig. 2 for proton assignments of 2b and 3. Red letters correspond to protons of the tetra-imine cage 1b. | ||
Luckily, single crystals suitable for X-ray diffraction grew from the sequential slow diffusion of dihalogenated benzene derivatives into a 9:1 CDCl3:CD3CN solvent mixture containing the tetra-imine cage 1b.¶ The analysis of the diffraction data revealed the solid-state structure of 1b. The four imine bonds displayed E-configuration. The imine CHs were outwardly directed with respect to internal cavity. They converged in pairs generating two differently sized portals of tetra-imine cage 1b (Portal1: average dCim1⋯Cim2 = 7.2 Å; Portal2: average dCim1⋯Cim4 = 4.3 Å) (Fig. 5).
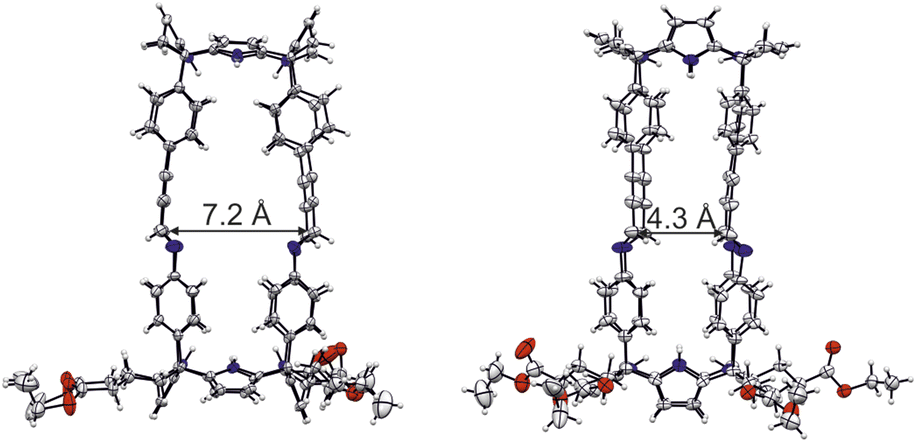 | ||
| Fig. 5 Side views of the X-ray crystal structure of tetra-imine cage 1b. The sizes of the two different portals are displayed. The structure is shown in ORTEP view with thermal ellipsoids set at 50% probability. Hydrogen atoms are shown as fixed-size spheres of 0.3 Å radius. Included guest and solvent molecules are omitted for clarity. | ||
:1 CDCl3:CD3CN solvent mixture, we decided to study the self-assembly of cage 1b in pure chlorinated solvents, such as CDCl3 and CD2Cl2.
The 1H NMR spectrum of a 2 mM equimolar mixture of tetra-formyl 3 and tetra-amine 2b in chlorinated solvents (either CDCl3 or CD2Cl2), acquired following its preparation (<15 min), mainly showed the proton signals of the two modular components. The proton signals of tetra-imine cage 1b showed a reduced intensity (Fig. 4b). Heating the solutions of the chlorinated solvents (48 h, 308 K for CDCl3 and 72 h, 303 K CD2Cl2) produced the tetra-imine cage 1b as the main species in solution. We calculated a yield of ∼50% for the self-assembly of 1b in the chlorinated solvents (Fig. 4c). The diffusion coefficient of 1b determined in CDCl3 (Fig. S23†) was almost coincident with the one measured above for the 4⊂1b cage complex in the same solvent.
It is worthy to note that the 1H NMR spectrum of cage 1b in CDCl3 solution showed broader proton signals than in a 9:1 CDCl3:CD3CN solvent mixture (Fig. 4c and a, respectively). This observation, together with the reduction in yield of the self-assembly process suggested that in pure chlorinated solvents the tetra-imine cage 1b was thermodynamically less stable and conformationally more flexible than the (CD3CN)2⊂1b analogue. The large equilibration time, the use of heat, and the observation of intermediate species supported the thermodynamic nature of the tetra-imine cage 1b.
Molecular modelling studies (MM3) of the tetra-imine cage 1b showed a nice fit of two chlorinated solvent molecules (CHCl3 or CH2Cl2) in its cavity providing sensible packing coefficient (PC)39 values 43–57% (see Fig. S29 and Table S1 in the ESI†).||
To sum up, in pure chlorinated solvents, the Schiff base condensation reaction producing 1b proceeds with lower yields (50%). It required longer time and temperature (24–48 h, >300 K) to finish (reach equilibrium) than the template assisted methodology (65%, 3 h r.t.) or the use of the 9:1 CDCl3:CD3CN solvent mixture (65%, 3 h r.t.). These results evidenced that the preorganization of the modular components of 1b in cone conformation was clearly beneficial for its self-assembly.
Binding studies in chloroform
The self-assembly of tetra-imine cage 1b, putatively including two solvent molecules (CDCl3 or CD3CN) in its cavity, opened the possibility to study its binding properties using pyridyl-N-oxides 4 and 5 as guests. All thermodynamic and kinetic studies of the inclusion complexes of 1b were performed using an excess of guest with respect to the cage concentration. We considered that the oligomeric aggregates of 2b and 3 that might be present in solution, but not visible in the 1H NMR spectrum, would also bind the pyridyl-N-oxide guests. For this reason, in the following binding studies, we used 2 equiv. of the bis-N-oxide 4 and 4 equiv. of mono-N-oxide 5 with respect to the initial concentration of the modular components (3 and 2b) used for the self-assembly of 1b.We concluded that the weakly bound CDCl3 molecules occupying the interior of cage 1b were readily replaced (low energy barrier) by the incoming N-oxide forming the thermodynamically and kinetically highly stable 4⊂1b cage complex.
:1 cage complex 52⊂1b (Fig. S32†). One pyridine-N-oxide 5 was included in each one of the two hemispheres of cage 1b. Because the two hemispheres are chemically non-equivalent, the aromatic protons of the pyridine-N-oxides resonated as six diastereotopic and separated upfield shifted signals (Hc1′, Hc2′, Hd1′, Hd2′, He1′ and He2′).
The downfield shift experienced by the pyrrole NHs in the 52⊂1b complex (i.e. Δδ = 0.86) were larger than those observed for the 4⊂1b counterpart. This result evidenced that the hydrogen bonds in the former cage complex were shorter than in the latter. DFT theoretical calculations, at the BP8641D3-def2-TZVP42 level of theory using Turbomole v7.0,43,44 of the two cage complexes also supported this conclusion: 4⊂1b (average d(H–N⋯O–N) = 3.29 ± 0.07 Å) and 52⊂1b (average d(H–N⋯O–N) = 2.88 ± 0.02 Å). For comparison, the X-ray crystal structures of aryl-extended calix[4]pyrrole complexes with pyridyl-N-oxide guests previously reported in the literature revealed an average hydrogen bonding distances (d(H–N⋯O–N)) on the range of 3.0 to 2.8 Å.45 Taken together, these results reinforced our hypothesis that the length of the bis-N-oxide 4 was too short to establish eight simultaneous optimal hydrogen bonding interactions with the two hemispheres of cage 1b (vide supra).
Considering the fast kinetics observed for the inclusion of 4 and 5 in the cavity of cage 1b in this solvent, we envisaged that it did not require the partial cleavage of imine bonds. We hypothesized that the guest uptake mechanisms involved a gating process through the calix[4]pyrrole meso-phenyl substituents. Because we did not observe significant differences in the uptake kinetics of the two guests, we propose a similar uptake mechanism for both of them. That is, the incoming guests are squeezed on passing through the enlarged portals of cage 1b resulting from a “French doors” mechanism of the meso-phenyl substituents.46,47 The inclusion of 4 in 1b can be considered as an elementary process. In contrast, the formation of the 52⊂1b complex is stepwise and requires separate “french doors” mechanisms for the two hemispheres.
We estimated in the previous section that the energy barrier for the uptake of bis-N-oxide 4 by cage 1b should be of the order of 15 kcal mol−1. Most likely, 5 is included in the cavity of 1b with a lower or similar energy barrier. The energy barrier corresponded to: (a) the concerted rotation of at least four meso-substituent in one of the capsule's hemisphere; (b) the breaking of the interactions with the released CDCl3 molecule per s; (c) the squeezing of the incoming planar N-oxides and the expelled CDCl3 molecule through opposed and wider portals of 1b available in the transition state of the guest uptake.
The 1H NMR spectrum acquired immediately after the addition of the ditopic guest 4 showed exclusively the proton signals corresponding to the 52⊂1b cage complex and those of the free bis-pyridyl N-oxides 4 and excess of 5 (Fig. 6a). After 12 hours, the signals of the protons of the 4⊂1b complex became visible. With time, they grew at the expenses of those of the 52⊂1b counterpart (Fig. 6b). It took more than 1 month for the ditopic N-oxide 4 to completely replace the two copies of pyridine-N-oxide 5 initially included in the cavity of 1b (Fig. 6c).
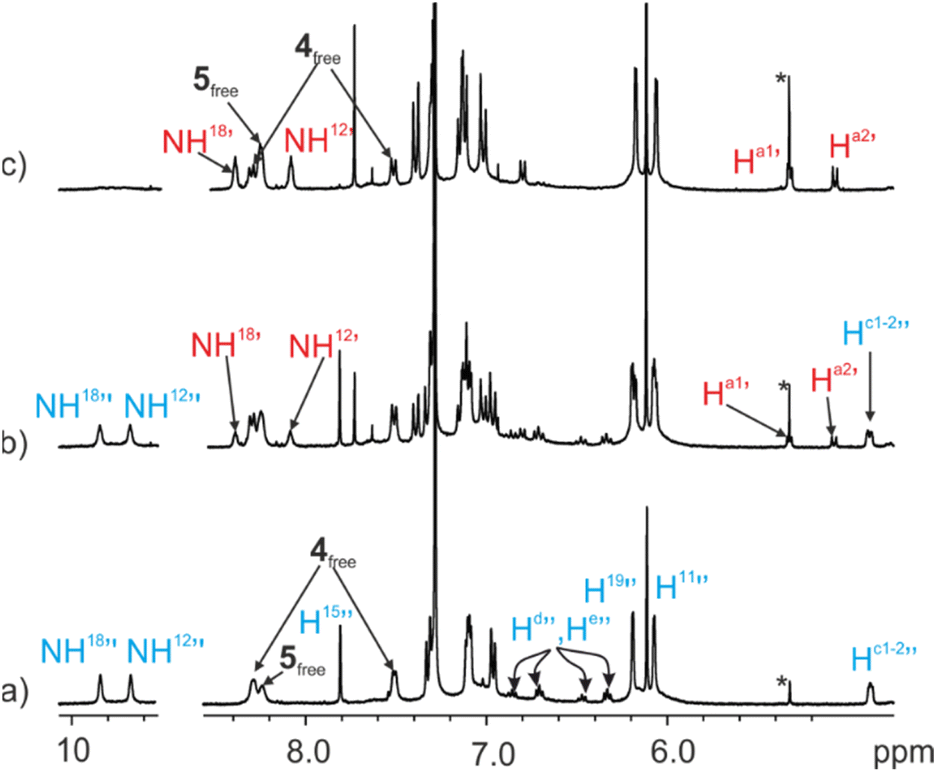 | ||
| Fig. 6 Selected regions of the 1H NMR spectra (400 MHz, at 298 K) of a CDCl3 solution of imine cage complex 52⊂1b (1 mM) in the presence of excess of bis-N-oxide 4: (a) immediately after the addition; (b) 4 days after the addition; (c) 1 month after the addition. i.s.: internal standard. See Fig. 2 for proton assignment. Primed letters indicate the proton signals of the 4⊂1b (red) and 52⊂1b (blue) complexes. Note that the polymeric aggregates of 2b and 3 present in solution also bind the N-oxides based on the integrals of the proton signals detected for the free species. However, the proton signals of the aggregates are not detected in the 1H NMR spectra due to extensive broadening. | ||
We fit the experimental kinetic data of the guests' exchange experiment (Fig. S36†) to the theoretical kinetic model of an irreversible bimolecular reaction (A + B → C). Using the parameter estimation module of the COPASI Software Version 4.2548 we obtained a good fit returning a rate constant value of kexch = 8.7 × 10−4 M−1 s−1. This value was translated into a free energy barrier of ca. ΔG‡ = 21.6 kcal mol−1 for the guests' exchange reaction (298 K). Notably, the free energy barrier calculated for the exchange reaction 4 + 52⊂1b ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) 2 5 + 4⊂1b using kinetic 1H NMR experiments was in line with our lower estimate for the exchange barrier of 4 (>20 kcal mol−1), determined for the reaction 4 + 4⊂1b 4 + 4⊂1b, owing to the lack of exchange cross-peaks in the corresponding EXSY experiment (see section describing the results of the inclusion of 4 in 1b).
2 5 + 4⊂1b using kinetic 1H NMR experiments was in line with our lower estimate for the exchange barrier of 4 (>20 kcal mol−1), determined for the reaction 4 + 4⊂1b 4 + 4⊂1b, owing to the lack of exchange cross-peaks in the corresponding EXSY experiment (see section describing the results of the inclusion of 4 in 1b).
We reasoned that both exchange processes should occur via structurally related transition states involving the breaking of a similar number and type of intermolecular interactions. As could be expected, the guests' exchange experiment starting from the 4⊂1b complex and adding guest 5 in excess did not produce, after more than a month, noticeable changes in the 1H NMR spectrum of the mixture. The results above assigned a larger thermodynamic stability to the 4⊂1b complex (ΔΔG > 3 kcal mol−1) compared to the three particles 52⊂1b counterpart. It also supported the use of the theoretical kinetic model for an irreversible bimolecular reaction in the fit of the guests' exchange kinetic data.
Binding studies in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 CDCl3:CD3CN solvent mixture
:1 CDCl3:CD3CN solvent mixture
Having kinetically and thermodynamically characterized the inclusion complexes of cage 1b with the pyridine-N-oxides derivatives 4 and 5 in CDCl3 solution, we became interested in their characterization in 9:1 CDCl3:CD3CN solvent mixture.
:1 CDCl3:CD3CN solvent mixture produced, after 10 min, a 1H NMR spectrum displaying exclusively the proton signals of the starting (CH3CN)2⊂1b cage complex and the free bis-pyridyl N-oxide 4 (Fig. 7b). The solution mixture was left standing at r.t. for 24 h and reanalyzed using 1H NMR spectroscopy. After the elapsed time, we observed the emergence of a new set of proton signals corresponding to the 4⊂1b complex. Notably, (CH3CN)2⊂1b and free 4 were still the major species in solution. The 4⊂1b complex was present to a 20% extent. This result was in contrast to the quantitative formation in just few minutes of the 4⊂1b complex from 1b in CDCl3 solution. During the course of several weeks, we observed that the intensity of the proton signals of the 4⊂1b complex increased at the expenses of those of (CH3CN)2⊂1b and 4 (Fig. 7c). After one month the 4⊂1b complex was quantitatively formed in solution.
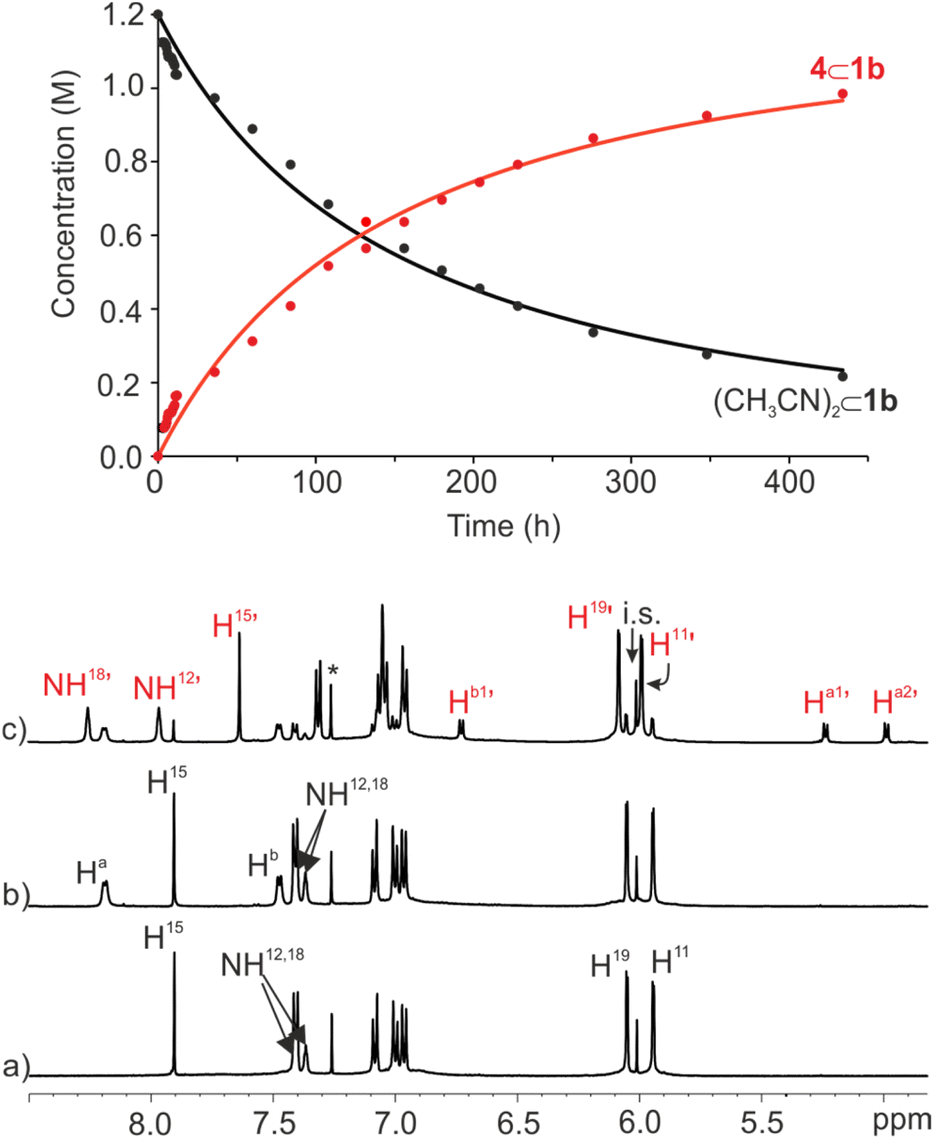 | ||
| Fig. 7 (Top) Changes in the concentration of (CD3CN)2⊂1b (black) and 4⊂1b (red) versus time (h) following the addition of 2 equiv. of bis-pyridyl N-oxide 4 (initial concentrations: [1b] = 1 mM and [4] = 2 mM). Solid lines represent the fit of the kinetic data to the rate law for a second order irreversible reaction using the parameter estimation module of COPASI Software Version 4.25. (Bottom) Selected regions of the 1H NMR spectra (500 MHz, at 298 K) of 1 mM solution of imine cage 1b in CDCl3:CD3CN 9:1 mixture: (a) before, (b) following the addition of bis-pyridyl N-oxide 4, and (c) after standing at r.t. for 18 days. i.s.: internal standard. See Fig. 2 for proton assignment. Red primed letters indicate the proton signals of the 4⊂1b cage complex. | ||
We also employed the bimolecular irreversible kinetic model (A + B → C) to mathematically analyze the time dependent concentration changes experienced by (CD3CN)2⊂1b and 4⊂1b throughout the irreversible inclusion/exchange process.48 The fit of the data returned the rate constant value for the formation of the 4⊂1b complex (inclusion of 4 in 1b) as kon = 1.0 × 10−3 M−1 s−1, which corresponded to a free energy barrier of ΔG‡ ∼21.6 kcal mol−1 (Fig. 7 top). This result indicated that the energy barrier for the formation of the 4⊂1b cage complex was almost identical starting from 52⊂1b in CDCl3 solution or (CD3CN)2⊂1b in 9:1 CDCl3:CD3CN solvent mixture. In contrast, the energy barrier for the inclusion of 4 dropped to ∼15 kcal mol−1 when the (CDCl3)2⊂1b cage complex was used as starting material in CDCl3 solution (vide supra). In short, the displacement/exchange of two polar guests (5 and CD3CN) from the cavity of 1b caused by the inclusion of 4 was associated to an increase in the free energy barrier of > 6 kcal mol−1 compared to the replacement of the less polar CDCl3 molecules. We assumed that this increase was mainly associated with the breaking of polar intermolecular interactions (hydrogen bonds, CH–π and π–π). Nevertheless, owing to the superior binding properties of 5 for the aryl-extended calix[4]pyrrole receptors, we were surprised to find out that the energy barriers of the exchange of the pyridine-N-oxide 5 and CD3CN were almost identical.
For this reason, we were keen in determining the relative thermodynamic stability of the 52⊂1b and (CD3CN)2⊂1b cage complexes in the 9:1 CDCl3:CD3CN solvent mixture.
:1 CDCl3:CD3CN solvent mixture. The 1H NMR spectrum of the mixture acquired immediately after the addition (<10 min) displayed three set of signals for the protons of 1b. The less intense set of signals corresponded to the intact (CD3CN)2⊂1b cage solvate. The other two sets of proton signals were assigned to the (CD3CN·5)⊂1b heterocomplex and the 52⊂1b homocomplex. After 1.5 h, the two inclusion complexes were exclusively present in solution in a 60:40 molar ratio that did not change over time (24 h) (Fig. 8c). These results assigned lower energy barriers to the stepwise inclusion of pyridine-N-oxide 5 in the cavity of the (CD3CN)2⊂1b cage complex than the 21.6 kcal mol−1 calculated for the bis-N-oxide 4. Moreover, they also showed that working under stoichiometric control for the formation of the 52⊂1b homocomplex, the 2:1 species was not the most favored complex in solution. Instead, the 1:1 heterocomplex (CD3CN·5)⊂1b was present to a larger extent. The (CD3CN·5)⊂1b complex was quantitatively formed in the presence of equimolar amounts of 1b and 5 (Ka[(CD3CN·5)⊂1b] > 104 M−1, see ESI for details, Fig. S37†). However, the quantitative formation of the 52⊂1b analogue required a large excess of 5 (>4 equiv.). Consequently, we conclude that the formation of the (5)2⊂1b complex occurred through a stepwise binding process featuring negative cooperativity (Ka[(CD3CN·5)⊂1b] > 4 × Ka[52⊂1b]). That is, the 52⊂1b complex is relatively higher in energy than its immediate precursor (CD3CN·5)⊂1b. Probably, the existence of steric clashes between the two pyridine-N-oxides occupying the rather limited volume of the cavity of the 52⊂1b complex was responsible for this energy increase. All together, these findings might explain the unexpected similar values calculated for the energy barriers of the guests exchange reactions leading to the 4⊂1b complex, either starting from 4 and 52⊂1b in pure CDCl3 solution or 4 and (CD3CN)2⊂1b in 9:1 CDCl3:CD3CN solvent mixture.
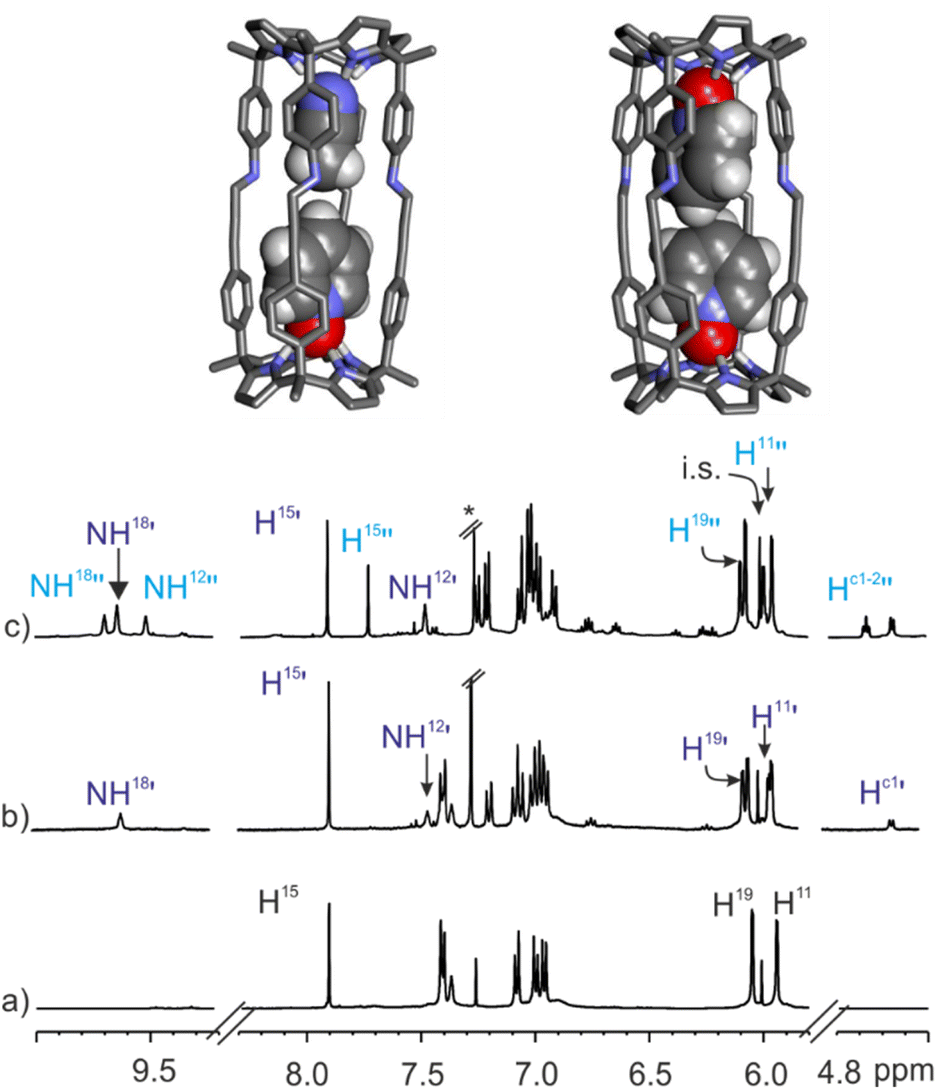 | ||
| Fig. 8 (Top) Energy minimized structures (BP86-D3-def2-TZVP DFT) of (CD3CN·5)⊂1b (main isomer observed in solution) and 52⊂1b complexes. (Bottom) Selected regions of the 1H NMR spectra (500 MHz, at 298 K) of 1 mM solution of imine cage 1b in CDCl3:CD3CN 9:1 mixture before (a), after the addition of pyridyl N-oxide 5 (0.3 mM) (b) and an excess of guest (2 mM) (c). i.s.: internal standard. See Fig. 2 for proton assignment. Dark blue primed letters indicate the signals of the major isomer of (CD3CN·5)⊂1b 1:1 complex and light blue doubled primed letters those of the 2:1 complex 52⊂1b. | ||
It is worthy to note, that in CDCl3 solution and using 1H NMR spectroscopy, we did not detect the formation of the heterocomplex (CDCl3·5)⊂1b. The exclusive formation of the 52⊂1b complex in the presence of 3 equiv. of 5 implied a cooperative binding process in CDCl3 solution. This result is in striking contrast with the negative cooperativity observed in the 9:1 CDCl3:CD3CN solvent mixture. We hypothesized that in CDCl3 the heterocomplex (CDCl3·5)⊂1b was energetically less favored than the homo counterpart 52⊂1b despite the steric clashes between the included N-oxides.
The careful analysis of the 1H NMR spectrum of the (CD3CN·5)⊂1b cage complex formed in the 9:1 CDCl3:CD3CN solvent mixture allowed us to identify two separate signals for the pyrrole NHs resonating at δ = 9.6 and 7.4 ppm (Fig. 8b). This observation was indicative of the formation of a single isomer, in which the pyridine-N-oxide 5 must be preferentially located in one of the two hemispheres of 1b.** A 2D ROESY experiment located the included N-oxide 5 in the hemisphere having the meso-p-phenyl alkynyl substituents. The co-inclusion of one molecule of acetonitrile in the opposite hemisphere produced a packing coefficient of 53% for the heterocomplex (CD3CN·5)⊂1b in comparison to the 57% calculated for the 52⊂1b (Fig. 8 top).
Conclusions
In summary, we described the self-assembly of tetra-imine shape-persistent cages 1 based on two different “four wall” tetra-α aryl-extended calix[4]pyrrole scaffolds. We showed that the presence of a bis-N-oxide template is not mandatory for the emergence of the cages. The tetra-imine cages 1 self-assembled in chlorinated solvents and in a 9:1 CDCl3:CD3CN mixture of solvents.
We demonstrated that the use of a templating guest produced a moderate increase in the yield of the self-assembly process (50% to 65%). Interestingly, the use of bound acetonitrile molecules pre-organizing the calix[4]pyrrole modular components of 1 in cone conformation had a similar effect (∼65%).
The X-ray crystal structure of the tetra-imine cage 1b revealed that the imine bonds were exclusively present as E-isomers. The imine hydrogen atoms converged in pairs defining two different sized-portals of 1b.
The tetra-imine cages 1 formed kinetically and thermodynamically stable complexes with the bis-N-oxide 4 and pyridine-N-oxide 5. On the one hand, bis-N-oxide 4 produced exclusively 1:1 complexes. On the other hand, the stoichiometry of the complexes of 1 with the mono-N-oxide 5 was shown to be solvent dependent.
The kinetics of the inclusion processes of the guests in 1 were also solvent dependent. In chlorinated solvents, the inclusion of both guests (4 and 5) was quantitative and required minutes. In contrast, in the 9:1 CDCl3:CD3CN solvent mixture and working under identical stoichiometric control, the 4⊂1b complex was exclusively formed after several weeks, while 5 rendered an equilibrium mixture of 1:1 and 2:1 complexes after several hours.
We ascribed the different kinetics (i.e. transition state energies) observed for the formation of the inclusion complexes to the nature of the solvent molecules (CDCl3 or CD3CN) that must be displaced from the solvated cages' interior, as well as, to the dissimilar sizes of the incoming guests.
Notably, in CDCl3 solution, the displacement reaction of the pyridine-N-oxide molecules in the 52⊂1 complex by the bis-N-oxide 4 was also quantitative but required more than a month. The calculated energy barriers (ΔG‡) for the above displacement reaction and that of the formation of the inclusion complex 4⊂1b from the acetonitrile solvate (CD3CN)2⊂1b, assembled in a 9:1 CDCl3:CD3CN solvent mixture, almost coincided.
Owing to the different polarities and sizes of the displaced guests (5 or CD3CN), we surmised that the 52⊂1 complex must be energetically disfavored due to steric clashes between the included guest molecules. Indeed, the study of the inclusion process of 5 in the tetra-imine 1b, performed in the 9:1 CDCl3:CD3CN solvent mixture, supported our hypothesis. That is, the quantitative formation of the 52⊂1 complex required the use of 5 in a large stoichiometric excess (>4 equiv.).
Conflicts of interest
There are no conflicts to declare.Data availability
All of the related experimental data are provided in the ESI.†Author contributions
Conceptualization, P. B.; methodology, C. M.; formal analysis, P. B.; G. A.; writing—original draft preparation, P. B., C. M. and G. A.; writing—review and editing, P. B., G. A. and C. M.; supervision, P. B.; All authors have read and agreed to the published version of the manuscript.Acknowledgements
This research was funded by Gobierno de España MICINN/AEI/FEDER (PID2020-114020GB-I00 and CEX2019-000925-S), the European Union (NOAH project H2020-MSCA-ITN project ref. 765297), the CERCA Programme/Generalitat de Catalunya, and AGAUR (2017 SGR 1123). We also thank Dr Eduardo C. Escudero-Adán for X-ray crystallography data and Prof. Dr Christoph A. Schalley and Daniel Stares from Freie Universität Berlin for their help on the mass spectrometry studies.Notes and references
- F. Hof, S. L. Craig, C. Nuckolls and J. Rebek, Angew. Chem., Int. Ed., 2002, 41, 1488–1508 CrossRef CAS.
- Container Molecules and Their Guests, ed. D. J. Cram and J. M. Cram, The Royal Society of Chemistry, Cambridge, England, 1997 Search PubMed.
- L. Tapia, I. Alfonso and J. Solà, Org. Biomol. Chem., 2021, 19, 9527–9540 RSC.
- G. Montà-González, F. Sancenón, R. Martínez-Máñez and V. Martí-Centelles, Chem. Rev., 2022, 122, 13636–13708 CrossRef.
- A. Platzek, S. Juber, C. Yurtseven, S. Hasegawa, L. Schneider, C. Drechsler, K. E. Ebbert, R. Rudolf, Q.-Q. Yan, J. J. Holstein, L. V. Schäfer and G. H. Clever, Angew. Chem., Int. Ed., 2022, e202209305 CAS.
- A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC.
- R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS.
- G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, Chem. Soc. Rev., 2021, 50, 7681–7724 RSC.
- D. Ajami and J. Rebek, Acc. Chem. Res., 2013, 46, 990–999 CrossRef CAS PubMed.
- S. La Cognata, R. Mobili, C. Milanese, M. Boiocchi, M. Gaboardi, D. Armentano, J. C. Jansen, M. Monteleone, A. R. Antonangelo, M. Carta and V. Amendola, Chem.–Eur. J., 2022, 28, e202201631 CAS.
- W. Liu and J. F. Stoddart, Chem, 2021, 7, 919–947 CAS.
- S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- D. J. Cram, S. Karbach, Y. H. Kim, L. Baczynskyj and G. W. Kallemeyn, J. Am. Chem. Soc., 1985, 107, 2575–2576 CrossRef CAS.
- J. Gabard and A. Collet, J. Chem. Soc., Chem. Commun., 1981, 1137–1139 RSC.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- J. Lindsey, New J. Chem., 1991, 15, 153–180 CAS.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC.
- M. L. C. Quan and D. J. Cram, J. Am. Chem. Soc., 1991, 113, 2754–2755 CrossRef CAS.
- L.-P. Yang, X. Wang, H. Yao and W. Jiang, Acc. Chem. Res., 2020, 53, 198–208 CrossRef CAS PubMed.
- R. A. Tromans, T. S. Carter, L. Chabanne, M. P. Crump, H. Li, J. V. Matlock, M. G. Orchard and A. P. Davis, Nat. Chem., 2019, 11, 52–56 CrossRef CAS PubMed.
- For recent examples of endohedrally functionalized molecular containers see ref. 5, 12 and 21 and references therein.
- F. Wang, C. Bucher, Q. He, A. Jana and J. L. Sessler, Acc. Chem. Res., 2022, 55, 1646–1658 CrossRef CAS.
- T. Guchhait, P. Pradhan, L. Panda and M. S. K. Rao, ChemistrySelect, 2022, 7, e202202671 CrossRef CAS.
- O. D. Fox, T. D. Rolls, M. G. B. Drew and P. D. Beer, Chem. Commun., 2001, 1632–1633 RSC.
- O. Francesconi, A. Ienco, G. Moneti, C. Nativi and S. Roelens, Angew. Chem., Int. Ed., 2006, 45, 6693–6696 CrossRef CAS.
- H. J. Han, J. H. Oh, J. L. Sessler and S. K. Kim, Chem. Commun., 2019, 55, 10876–10879 RSC.
- F. Wang, E. Sikma, Z. Duan, C. Lei, Z. Zhang, S. M. Humphrey and J. L. Sessler, J. Porphyrins Phthalocyanines, 2020, 24, 424–431 CrossRef CAS.
- F. Wang, E. Sikma, Z. Duan, T. Sarma, C. Lei, Z. Zhang, S. M. Humphrey and J. L. Sessler, Chem. Commun., 2019, 55, 6185–6188 RSC.
- P. Ferreira, G. Moncelsi, G. Aragay and P. Ballester, Chem.–Eur. J., 2021, 27, 12675–12685 CrossRef CAS.
- J. R. Romero, G. Aragay and P. Ballester, Chem. Sci., 2017, 8, 491–498 RSC.
- A. Galán, E. C. Escudero-Adán and P. Ballester, Chem. Sci., 2017, 8, 7746–7750 RSC.
- P. Ballester and G. Gil-Ramírez, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10455–10459 CrossRef CAS.
- L. Escobar, F. A. Arroyave and P. Ballester, Eur. J. Org. Chem., 2018, 2018, 1097–1106 CrossRef CAS.
- L. Avram and Y. Cohen, Chem. Soc. Rev., 2015, 44, 586–602 RSC.
- D. Sinnaeve, Concepts Magn. Reson., Part A, 2012, 40A, 39–65 CrossRef CAS.
- D. Hernández-Alonso, S. Zankowski, L. Adriaenssens and P. Ballester, Org. Biomol. Chem., 2015, 13, 1022–1029 RSC.
- S. Mecozzi, J. Rebek and Julius, Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- A. Pastor and E. Martínez-Viviente, Coord. Chem. Rev., 2008, 252, 2314–2345 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- TURBOMOLE V7.0 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B, 2016, 72, 171–179 CrossRef CAS.
- L. Escobar, E. C. Escudero-Adán and P. Ballester, Angew. Chem., Int. Ed., 2019, 58, 16105–16109 CrossRef CAS.
- L. Escobar, D. Villarón, E. C. Escudero-Adán and P. Ballester, Chem. Commun., 2019, 55, 604–607 RSC.
- S. Hoops, S. Sahle, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes and U. Kummer, Bioinformatics, 2006, 22, 3067–3074 CrossRef CAS PubMed.
- J. Stonehouse, P. Adell, J. Keeler and A. J. Shaka, J. Am. Chem. Soc., 1994, 116, 6037–6038 CrossRef CAS.
- M. Chas and P. Ballester, Chem. Sci., 2012, 3, 186–191 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data, additional binding experiments and characterization in the gas phase. CCDC 2208504. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05311j |
| ‡ Analogous reaction conditions using tetra-methyl tetra-amino calix[4]pyrrole 2a initially produced broad and ill-defined signals. Remarkably, a white solid precipitated out from this mixture. The suspension was left overnight at room temperature. After 12 h the solid was completely dissolved to yield a yellowish clear solution. The 1H NMR spectrum of this solution showed sharp and well-defined signals that were assigned to the formation of the tetra-imine cage complex 4⊂1a. |
| § The reaction performed at more diluted conditions (1 mM) produced similar yields. However, larger reaction times were needed to observe the complete consumption of the starting material. |
| ¶ Deposition number CCDC 2208504 contains the supplementary crystallographic data for this paper. |
| || 1D GOESY experiments,49 performed at different temperatures (323–238 K) using a 95:5 CHCl3:CDCl3 solution of cage 1b and by selectively exciting the CHCl3 signal did not help in revealing the proton signals of the putative CHCl3 included in the cage. Most likely, the chemical exchange between the free and bound CHCl3 was fast on the chemical shift timescale.50 |
| ** At the BP86-D3-def2-TZVP DFT level of theory using Turbomole v7.0, we computed an energy difference of 1.5 kcal mol−1 for the two isomers of the (CDCl3·5)⊂1b in favor to the one experimentally observed. |
| This journal is © The Royal Society of Chemistry 2023 |