
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium-catalyzed enantioselective C–H alkynylation of sulfoxides in diverse patterns: desymmetrization, kinetic resolution, and parallel kinetic resolution†
Lingheng
Kong
 a,
Yun
Zou
a,
Xiao-Xi
Li
b,
Xue-Peng
Zhang
a and
Xingwei
Li
*ab
a,
Yun
Zou
a,
Xiao-Xi
Li
b,
Xue-Peng
Zhang
a and
Xingwei
Li
*ab
aSchool of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710062, China. E-mail: lixw@snnu.edu.cn
bInstitute of Molecular Science and Engineering, Institute of Frontier and Interdisciplinary Sciences, Shandong University, Qingdao 266237, China
First published on 4th December 2022
Abstract
Rhodium-catalyzed enantioselective C–H alkynylation of achiral and racemic sulfoxides is disclosed with alkynyl bromide as the alkynylating reagent. A wide range of chiral sulfoxides have been constructed in good yield and excellent enantioselectivity (up to 99% ee, s-factor up to > 500) via desymmetrization, kinetic resolution, and parallel kinetic resolution under mild reaction conditions. The high enantioselectivity was rendered by the chiral cyclopentadienyl rhodium(III) catalyst paired with a chiral carboxamide additive. The interactions between the chiral catalyst, the sulfoxide, and the chiral carboxylic amide during the C–H bond cleavage offer the asymmetric induction, which is validated by DFT calculations. The chiral carboxamide functions as a base to promote C–H activation and offers an additional chiral environment during the C–H cleavage.
Introduction
Metal-catalyzed enantioselective C–H functionalization has emerged as a powerful strategy to synthesize complex chiral organic scaffolds owing to the atom- and step-economy of the reaction system and availability of the C–H substrates (arenes).1 A large array of transition metal catalysts have been successfully employed in enantioselective C–H bond activation. Among the chiral ligands employed, chiral phosphines are predominant, as in palladium, rhodium(I), iridium(I), and nickel(0) catalysts.2 In addition, great achievements have been made in palladium-catalyzed C–H bond activation using monoprotected amino acids (MPAA) as privileged chiral ligands.3 Chiral NHC ligands also allowed the development of intriguing C–H activation systems.4 Recently, with the plethora of racemic/achiral C–H activation systems enabled by trivalent metals via a typical concerted-metalation-deprotonation mechanism, group IX complexes stabilized by chiral cyclopentadienyl ligands (CpXM(III)) have proven as efficient and versatile catalysts in asymmetric C–H activation of diverse arenes,5 and elegant systems have been reported by the groups of Cramer,6 Rovis,7 You,8 Wang,9 Li,10 Waldmann,11 and others.12 Chelation-assistance remains a predominant strategy in C–H bond activation, and strong coordination groups such as amide,6a,d,e,7,8c,e,9,10e,11,12a,c imine,6b,10c pyridyl,8a,b,8d pyrimidyl,10a,f nitrone,10b,d and thioether10a in the arene are typically necessary to ensure catalytic reactivity (Scheme 1a). The employment of arenes bearing a weak directing group have been less explored in this context, with the exception of biarylphosphine oxides and sulfoxonium ylides as substrates.13 Thus, development of asymmetric C–H activation systems with weak and functionalizable chelating groups is under great demand.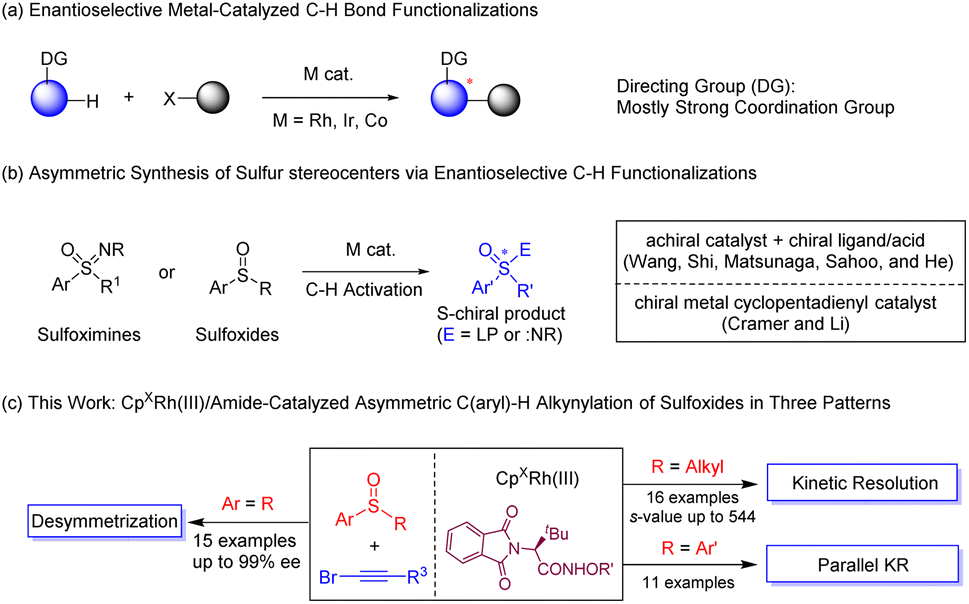 | ||
| Scheme 1 Transition-metal-catalyzed enantioselective C–H activation. | ||
Enantioenriched sulfoxides are ubiquitous as pharmaceuticals and bioactive compounds, and they have also been utilized as chiral ligands in asymmetric catalysis.14 Traditional protocols in synthesis of chiral sulfoxides such as resolution, nucleophilic substitution, and biocatalytic reactions generally suffer from stoichiometric amounts of chiral reagents, poor efficiency, and/or limited substrate scope.15 The C–H activation strategy was not introduced to asymmetric synthesis of chiral sulfoxides until very recently, likely due to inhibitive binding of the sulfur atom.16 In 2018, Wang and coworkers reported Pd(II)-catalyzed enantioselective C–H olefination of diaryl sulfoxides through parallel kinetic resolution (PKR) and desymmetrization utilizing monoprotected amino acid (Ac-Leu-OH) as the ligand.17 Recently, the He group reported Cp*Ir(III)-catalyzed enantioselective C–H amidation of dibenzyl sulfoxides through desymmetrization and parallel kinetic resolution also using a MPAA ligand, with the sulfur atom serves being a directing group (Scheme 1b).18 Shi and coworkers achieved Pd-catalyzed C–H alkynylation-kinetic resolution of a special class of sulfoxides with 2-pyridyl as a directing group.16b Despite the progress and the powerful role of chiral CpXRh(III) catalysts, no asymmetric catalytic system had been established in CpXRh(III)-catalyzed C–H activation using sulfoxides as arene substrates. On the other hand, alkynes as reactive organic intermediates have been employed in a wide variety of catalytic transformations, especially annulation reactions. Meanwhile, direct alkynylation of C(sp2)-H bonds in high stereoselectivity and activity is highly desirable owing to the significance of the alkynyl functionality and the chiral platform in the product. Of note, hypervalent iodine-based alkynes and alkynyl bromides have been extensively studied in Rh/Ir-catalyzed C–H activation.19 Nevertheless, enantioselective C–H alkynylation remains largely limited. Herein, we report rhodium-catalyzed enantioselective sulfoxide-directed C–H alkynylation utilizing both achiral and racemic sulfoxides, delivering chiral sulfoxides in high stereoselectivity via diverse patterns such as desymmetrization, kinetic resolution, and parallel kinetic resolution (Scheme 1c).
Results and discussion
We initially investigated the feasibility of the alkynylation of diphenyl sulfoxide (1a) with 1-bromo-2-(triisopropylsilyl)acetylene (2a) in the presence of a chiral rhodium cyclopentadienyl catalyst ((R)-Rh1-Rh4, Table 1). The desired alkynylated product 3aa was indeed obtained in 32% yield and 25% ee when catalyzed by (R)-Rh1 (4 mol%)/AgSbF6 (16 mol%) in the presence of LiOAc (20 mol%) and Ag2CO3 (1.0 equiv.) in DCE at 30 °C for 48 h (entry 1). Switching the LiOAc additive to a tert-butyl-substituted chiral carboxylic acid improved the enantioselectivity to 51% ee (entries 2–4). Catalyst screening revealed that (R)-Rh3 was superior to others (entries 5–7). To our delight, tert-Leucine-derived chiral amide20A4 significantly improved the enantioselectivity to 86% ee (entry 8). A brief evaluation of the silver additive indicated that AgOTf performed best, affording 3aa in 65% yield and 90% ee (entries 9–11). Further investigation of the chiral amides (A5–A9) indicated that employment of A8 bearing an N-OiPr group afforded the product in the highest ee (91%, entries 12–16). The enantioselectivity remained even the temperature was increased to 55 °C (entry 17). Finally, the alkynylated product was obtained in an improved yield (72%) and enantioselectivity (91% ee) when Ag2O (1.2 equiv.) was used as a base in the presence of 4 Å molecular sieves (entry 19). Lower yield was obtained when the catalyst loading was decreased to 4 mol% (entry 20). The employment of a chiral catalyst was necessary since switching to an achiral catalyst-(S)-A8 only afforded poor enantioselectivity (entry 21).|
|
|||||
|---|---|---|---|---|---|
| Entry | [Rh] | Ag salt | Additive | Yield (%) | ee(%)b |
| a Reaction conditions: rac-1 (0.05 mmol), 2 (0.06 mmol), (R)-Rh (4 mol%), Ag salt (16 mol%), additive (20 mol%), and Ag2CO3 (1.0 equiv.) in DCE (1.0 mL) at 30 °C for 48 h under N2 in a sealed reaction tube, isolated yield. b The ee value was determined by chiral HPLC. c At 55 °C. d Ag2O (1.2 equiv.) at 55 °C. e Ag2O (1.2 equiv.) and 4 Å MS (25 mg) at 55 °C. f 0.1 mmol. g (R)-Rh (2 mol%). | |||||
| 1 | Rh1 | AgSbF6 | LiOAc | 32 | 25 |
| 2 | Rh1 | AgSbF6 | (S)-A1 | 25 | 37 |
| 3 | Rh1 | AgSbF6 | (S)-A2 | 30 | 46 |
| 4 | Rh1 | AgSbF6 | (S)-A3 | 56 | 51 |
| 5 | Rh2 | AgSbF6 | (S)-A3 | 48 | 37 |
| 6 | Rh3 | AgSbF6 | (S)-A3 | 51 | 59 |
| 7 | Rh4 | AgSbF6 | (S)-A3 | 57 | −40 |
| 8 | Rh3 | AgSbF6 | (S)-A4 | 60 | 86 |
| 9 | Rh3 | AgOTf | (S)-A4 | 65 | 90 |
| 10 | Rh3 | AgBF4 | (S)-A4 | 62 | 89 |
| 11 | Rh3 | AgNTf2 | (S)-A4 | 65 | 89 |
| 12 | Rh3 | AgOTf | (S)-A5 | 54 | 79 |
| 13 | Rh3 | AgOTf | (S)-A6 | — | — |
| 14 | Rh3 | AgOTf | (S)-A7 | 60 | 90 |
| 15 | Rh3 | AgOTf | (S)-A8 | 66 | 91 |
| 16 | Rh3 | AgOTf | (S)-A9 | 55 | 56 |
| 17c | Rh3 | AgOTf | (S)-A8 | 54 | 90 |
| 18d | Rh3 | AgOTf | (S)-A8 | 68 | 92 |
| 19e | Rh3 | AgOTf | (S)-A8 | 72 (70)f | 91 |
| 20e,g | Rh3 | AgOTf | (S)-A8 | 46 | 94 |
| 21 | Rh5 | AgOTf | (S)-A8 | 42 | 5 |

With the optimal conditions in hand (Table 1, entry 19), we next examined the scope of this enantioselective C–H alkynylation-desymmetrization (Scheme 2). Achiral diarylsulfoxides bearing electron-withdrawing (COOMe), donating (Me and OMe), and halogen groups at the para or ortho position of the benzene ring, all coupled effectively with alkyne 2a, affording the desired products in 48–97% yield and 91–99% ee (3aa-3ja). Compatibility of diverse ortho substituents highlighted tolerance of steric effect of the benzene ring. In addition, the reaction could also be carried out on a 1 mmol scale, and the product 3fa was still isolated in 96% ee. Diarylsulfoxides with meta-Me and F substituents reacted selectively at the less hindered ortho site, and the desired products were obtained in 89–90% ee (3ka and 3la). Replacement of the TIPS group in the alkyne by a TBS, TES, or a siloxyl-based tertiary alkyl group afforded the product in moderate yields and excellent 90–95% ee (3ab, 3ac, and 3fd).
 | ||
| Scheme 2 Substrate scope of desymmetrization reactions. a reaction conditions: achiral sulfoxide 1 (0.1 mmol), 2 (0.12 mmol), (R)-Rh3 (4 mol%), AgOTf (16 mol%), (S)-A8 (20 mol%), Ag2O (1.2 equiv.), 4 Å MS (50 mg) in DCE (2.0 mL) at 55 °C for 48 h under N2 in a sealed reaction tube, isolated yield. bThe ee was determined by chiral HPLC. cAt 45 °C. | ||
To better explore the reaction patterns, kinetic resolution of biased racemic sulfoxides was also successfully realized under our standard reaction conditions (Scheme 3). The reaction of (iso-propylsulfinyl)benzene was first examined using alkyne 2a as the coupling partner. Racemic (iso-propylsulfinyl)benzenes containing electron-donating groups (Me), withdrawing groups (CF3), as well as halogen substituents (F and Br) at the para and the meta position of the benzene ring were well tolerated, furnishing the desired products 5aa-5ga in consistently excellent enantioselectivities (89–99% ee), and the recovered sulfoxide substrate was obtained in 54–98% ee, which corresponds to a s-factor ranging from 67 to 544. As expected, the presence of an ortho Me, Cl, Br, and CO2Et group was also well compatible with this protocol, delivering the alkynylation products in high yields and enantioselectivities (5ha-5ka, 91–99% ee), and the recovered substrates 4h–4k were isolated in 41–51% yields with 74–98% ee. The arene ring has been extended to a fused one, and the alkynylation of 1-naphthoyl sulfoxide (4l) afforded the product in 98% ee (s = 224). Extension of the S-alkyl group to a methyl led to diminished reactivity, and the product 5ma was isolated in 93% ee (s = 34). To our delight, mono- and disubstituted products (5na, and 5na′) were obtained in excellent enantioselecttivity when a benzyl-substituted sulfoxide 4n was used. Excellent yield and enantioselectivity were also realized for a TBS (5ib) or a propargyl silyl ether-substituted (5dd) alkynyl bromide. The (R) configuration of a recovered sulfoxide substrate 4m was determined based on a previous report.21
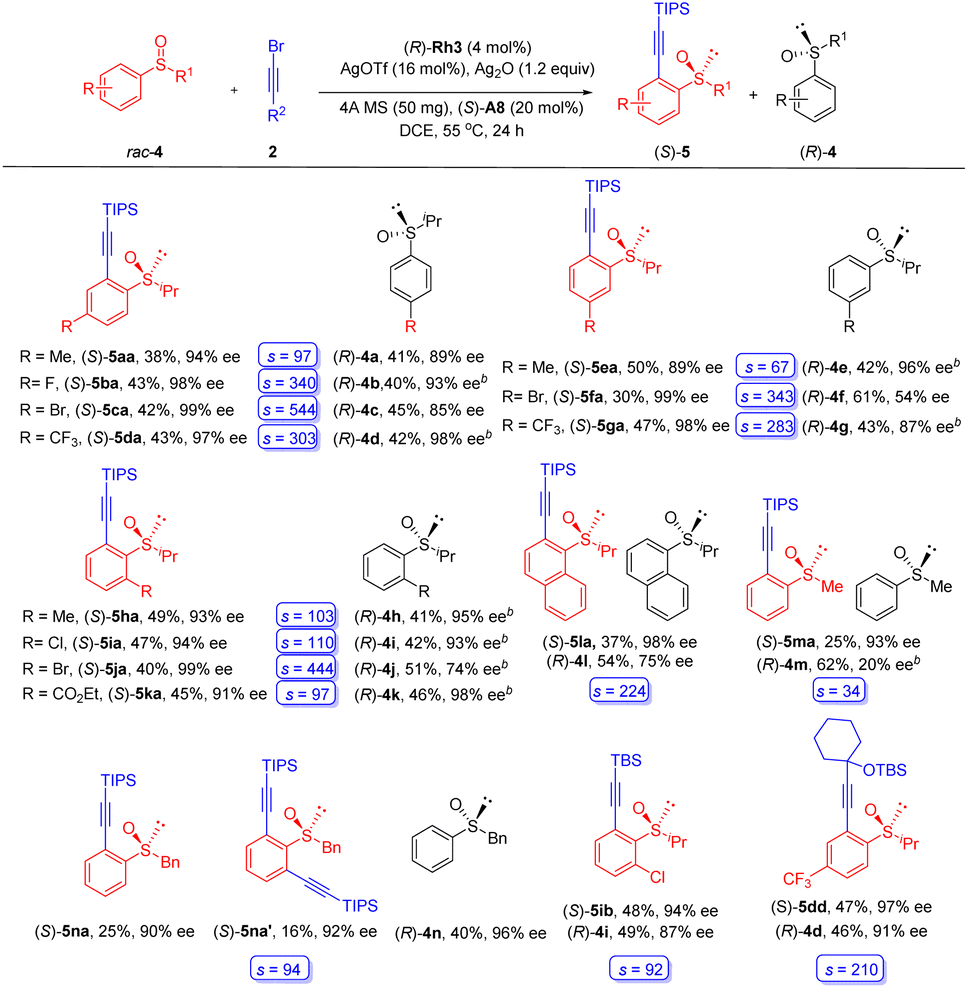 | ||
| Scheme 3 Substrate scope of kinetic resolution reactions. aReaction conditions: (rac)-4 (0.2 mmol), 2 (0.3 mmol), (R)-Rh3 (4 mol%), AgOTf (16 mol%), (S)-A8 (20 mol%), Ag2O (1.2 equiv.), 4 Å MS (50 mg) in DCE (2.0 mL) at 55 °C for 24 h under N2 in a sealed reaction tube, isolated yield. b At 35 °C. c Calculated according to s = ln[(1 − C)(1 − ee2a)]/ln[(1 − C)(1 + ee2a)]. | ||
To further broaden the substrate scope and the asymmetric reaction patterns, biased diaryl sulfoxides were then examined toward dynamic kinetic resolution since both arene rings may undergo competitive alkynylation (Scheme 4). Indeed, the alkynylation occurred at both arene rings under the standard reaction conditions, furnishing products 7 and 8 in a parallel kinetic resolution (PKR) fashion. The scope of this reaction seems broad. Thus, racemic sulfoxides 6 with two different ortho substituents in the benzene rings were applicable, giving the two alkynylated products in good yields and high ee values (7aa-7fa, 8aa-8fa, 71–97% ee). Slightly lower enantioselectivity was observed when two different meta substituents were introduced (7ga, 87% ee; 8ga, 86% ee), likely because these substituents are distal to the reaction site or the directing group. A dialkynylated product was obtained in 97% ee (7ha) and the other single alkynylated product was isolated in 48% yield and 95% ee (8ha) when a para substituted sulfoxide 1-methoxy-2-(p-tolylsulfinyl)benzene was employed. A mixture of products (7ia + 8ia) were isolated using 1-chloro-4-(p-tolylsulfinyl)benzene as an arene. For the alkynyl bromide substrates, different alkynyl terminus such as TBS, TES, and propargyl silyl ether were compatible, giving the corresponding products 7ab-7ad and 8ab-8ad in high yields and enantioselectivity under slightly modified conditions using a different base (silver carbonate). The absolute configuration of ethynyl sulfoxide derived from product 7aa was determined to be (S) by X-ray crystallography (CCDC 2207905).
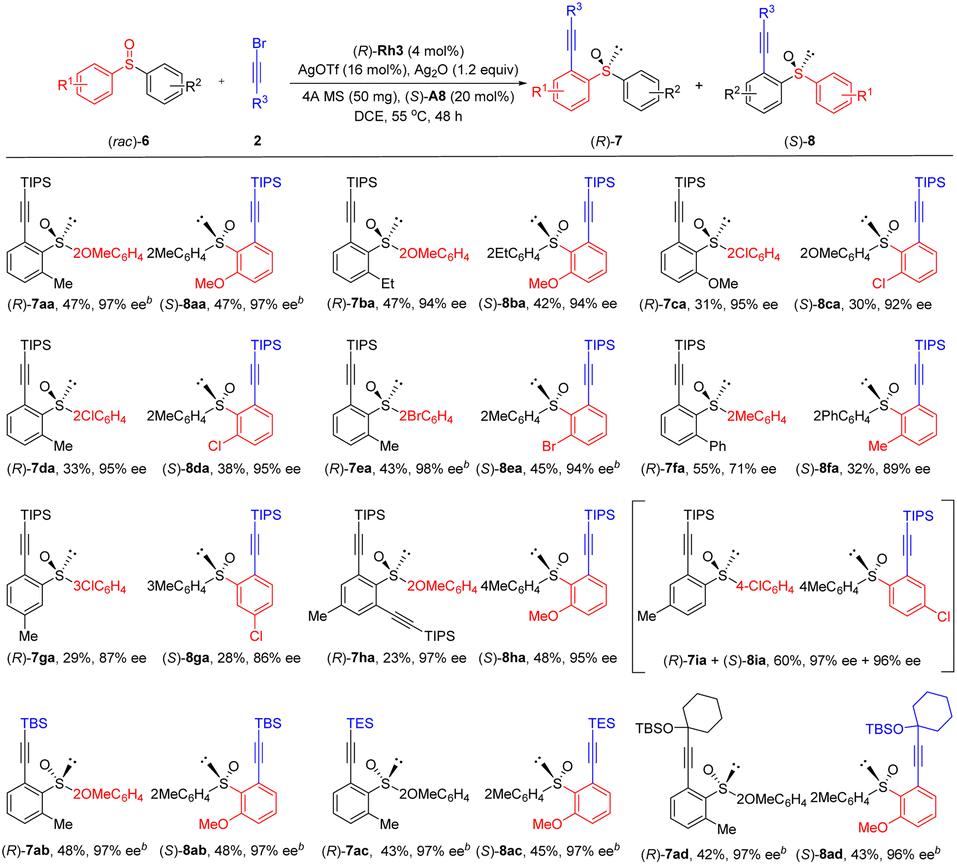 | ||
| Scheme 4 Substrate scope of parallel kinetic resolution. aReaction conditions: rac-6 (0.1 mmol), 2 (0.15 mmol), (R)-Rh3 (4 mol%), AgOTf (16 mol%), (S)-A1 (20 mol%), Ag2O (1.2 equiv.), 4 Å MS (50 mg) in DCE (2.0 mL) at 55 °C for 48 h under N2 in a sealed reaction tube, isolated yield. bAg2CO3 (1.0 equiv.) was used at 45 °C. | ||
To demonstrate the synthetic utility of the chiral products, synthetic applications have been briefly demonstrated (Scheme 5). The triisopropylsilyl (TIPS) group could be readily removed to give the ethynyl sulfoxide 9 in 97% yield and 89% ee. Meanwhile, the (S) configuration of product 9 was determined by X-ray crystallography (CCDC 2207903) and was extrapolated to the other products. Chiral ethynyl sulfoxide 9 was further transformed to a triazole 10 in attenuated enantiopurity via a cycloaddition reaction.
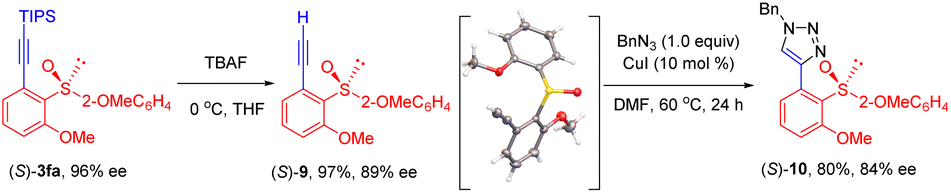 | ||
| Scheme 5 Synthetic applications. | ||
Several experiments have been conducted to probe the reaction mechanism (Scheme 6). H/D exchange experiments in the presence of 2a was conducted under the standard conditions and afforded product 3aa-dn with deuteration at the ortho position (Scheme 6a), indicating reversibility of the C–H activation. Isotope effect was observed in side-by side reaction (KIE = 2.4), thus indicating C–H cleavage is likely involved in the turnover-limiting step (Scheme 6b). Lower yield and enantioselectivity (53% yield and 47% ee) were obtained when the oppositely configured (R)-A8 was designated as additive, indicating that the match between the catalyst and chiral amide was crucial (Scheme 6c), as has been disclosed in other stereo-determining C–H activation processes.20,22 In another experiment, enantio-enriched sulfoxide substrate (S)-6h was synthesized and was subjected to the reaction conditions in order to evaluate the parallel kinetic resolution. The (S)-6h reacted selectively at the o-OMe-attached benzene, leading to the corresponding product 8ha in 96% ee, suggesting that regiodivergent parallel kinetic resolution (PKR) was involved in this transformation (Scheme 6d).
 | ||
| Scheme 6 Mechanistic studies. | ||
A series of DFT23 calculations were then conducted at the B3LYP/Def2TZVPP//Def2SVP level24,25 to understand the origin of the enantioselectivity. Only the C–H activation event was explored because the turnover-limiting feature has been revealed by the KIE experiment. The reaction follows a concerted metalation-deprotonation pathway, and the chiral amide functions as a base to bridge the rhodium and the reactive C–H proton. Due to prochirality of the diaryl sulfoxide and the two possible binding modes of the chiral amide group, eight possible deprotonation pathways have been investigated for the concerted metalation-deprotonation process (Scheme 6e). Interestingly, the amide oxygen rather than nitrogen is found to serve as the effective proton acceptor. Our preliminary studies revealed that the lowest (S)-forming transition state S,ivTS (15.8 kcal mol−1) is 8.3 kcal mol−1 lower in energy than the lowest (R)- forming transition state R,iiiTS (24.1 kcal mol−1) due to the loss of the hydrogen-bonding interaction between amide additive and substrate in the latter case (see Scheme S1†), which is consistent with our experimental studies. By passing this lowest (S)-forming transition state, the cyclometallation process is nearly thermoneutral. Thus, the chiral amide offers additional chiral environment in the second coordination sphere of the catalyst to assist the C–H activation.
Conclusions
In summary, we have developed a rhodium(III)-catalyzed enantioselective C–H alkynylation of sulfoxides with alkynyl bromides as the alkylating reagent. Diverse asymmetric reaction patterns such as desymmetrization, kinetic resolution, and parallel kinetic resolution have been realized for the sulfoxide substrates. The chiral catalyst and the chiral amide additive collectively played an important role during the C–H bond activation. A broad range of chiral sulfoxides were constructed in good yields and consistently excellent enantioselectivities (up to 99% ee). Further studies on selective and catalytic synthesis of sulfur- or phosphorus-stereogenic products are in progress in our laboratories.Data availability
Further details of the experimental procedure, 1H, and 13C NMR, HPLC spectra, and X-ray crystallographic data for 7aa and 9 are available in the ESI.†Author contributions
X. L. conceived the ideas and directed the project. L. K. and Y. Z. performed the experiments. X.-X. L. and X.-P. Z. conducted the computational studies. X. L. and L. K. wrote the manuscript with feedback from all authors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Financial support from the China Postdoctoral Science Foundation (2019M653530) and the SNNU are gratefully acknowledged.Notes and references
- (a) S. J. O'Malley, K. L. Tan, A. Watzke, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2005, 127, 13496–13497 CrossRef PubMed; (b) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (c) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (d) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (e) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588–5598 RSC; (f) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (g) Z. Shi, D. C. Koester, M. Boultadakis-Arapinis and F. Glorius, J. Am. Chem. Soc., 2013, 135, 12204–12207 CrossRef CAS PubMed; (h) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173–6214 RSC; (i) C. Sambiagio, D. Schonbauer, R. Blieck, T. DaoHuy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnurch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- (a) Phosphorus Ligands in Asymmetric Catalysis: Synthesis and Applications, ed, A. Bçrner, Wiley-VCH, Wein-heim, 2008, Vol. 1–3 Search PubMed; (b) Privileged Chiral Ligands and Catalysts, ed, Q.-L. Zhou, Wiley-VCH, Weinheim, 2011 Search PubMed; (c) J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010 Search PubMed.
- For selected reviews, see (a) X. Yu, Z.-Z. Zhang, J.-L. Niu and B.-F. Shi, Org. Chem. Front., 2022, 9, 1458–1484 RSC; (b) T. G. SaintDenis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed; (c) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed For selected examples, see; (d) B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 CrossRef CAS; (e) B.-F. Shi, Y.-H. Zhang, J. K. Lam, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 460–461 CrossRef CAS PubMed; (f) Q.-F. Wu, P.-X. Shen, J. He, X.-B. Wang, F. Zhang, Q. Shao, R.-Y. Zhu, C. Mapelli, J. X. Qiao, M. A. Poss and J.-Q. Yu, Science, 2017, 355, 499–502 CrossRef CAS PubMed.
- (a) P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2015, 127, 633–637 CrossRef; (b) J.-B. Ma, X. Zhao, D. Zhang and S.-L. Shi, J. Am. Chem. Soc., 2022, 144, 13643–13651 CrossRef CAS PubMed; (c) M. Chen and J. Montgomery, ACS Catal., 2022, 12, 11015–11023 CrossRef CAS.
- (a) C. Davies, S. Shaaban and H. Waldmann, Trends in Chem., 2022, 4, 318–330 CrossRef CAS; (b) C. Pan, S.-Y. Yin, Q. Gu and S.-L. You, Org. Biomol. Chem., 2021, 19, 7264–7275 RSC; (c) J. Mas-Roselló, A. G. Herraiz, B. Audic, A. Laverny and N. Cramer, Angew. Chem., Int. Ed., 2021, 60, 13198–13224 CrossRef PubMed; (d) C.-X. Liu, W.-W. Zhang, S.-Y. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 14025–14040 CrossRef CAS; (e) T. K. Achar, S. Maiti, S. Jana and D. Maiti, ACS Catal., 2020, 10, 13748–13793 CrossRef CAS; (f) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed; (g) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed.
- (a) S.-G. Wang, Y. Liu and N. Cramer, Angew. Chem., Int. Ed., 2019, 58, 18136–18140 CrossRef CAS; (b) M. Brauns and N. Cramer, Angew. Chem., Int. Ed., 2019, 58, 8902–8906 CrossRef CAS PubMed; (c) C. Duchemin and N. Cramer, Chem. Sci., 2019, 10, 2773–2777 RSC; (d) B. Ye, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 507–511 CrossRef CAS PubMed; (e) B. Ye and N. Cramer, Science, 2012, 338, 504–506 CrossRef CAS PubMed.
- (a) I. S. Hassan, J. T. Fuller, V. N. Dippon, A. N. Ta, M. W. Danneman, B. R. McNaughton, A. N. Alexandrova and T. Rovis, Chem. Sci., 2022, 13, 9220–9224 RSC; (b) I. S. Hassan, A. N. Ta, M. W. Danneman, N. Semakul, M. Burns, C. H. Basch, V. N. Dippon, B. R. McNaughton and T. Rovis, J. Am. Chem. Soc., 2019, 141, 4815–4819 CrossRef CAS PubMed; (c) T. K. Hyster, L. Knorr, T. R. Ward and T. Rovis, Science, 2012, 338, 500–503 CrossRef CAS PubMed.
- (a) C. Pan, S.-Y. Yin, S.-B. Wang, Q. Gu and S.-L. You, Angew. Chem., Int. Ed., 2021, 60, 15510–15516 CrossRef CAS; (b) Q. Wang, W.-W. Zhang, C. Zheng, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 114–120 CrossRef CAS PubMed; (c) W.-J. Cui, Z.-J. Wu, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2020, 142, 7379–7385 CrossRef CAS PubMed; (d) J. Zheng, W.-J. Cui, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5242–5245 CrossRef CAS PubMed; (e) J. Zheng, S.-B. Wang, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 4540–4544 CrossRef CAS PubMed.
- (a) T. Li, C. Zhou, X. Yan and J. Wang, Angew. Chem., Int. Ed., 2018, 57, 4048–4052 CrossRef CAS; (b) H. Li, X. Yan, J. Zhang, W. Guo, J. Jiang and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 6732–6736 CrossRef CAS PubMed; (c) G. Li, X. Yan, J. Jiang, H. Liang, C. Zhou and J. Wang, Angew. Chem., Int. Ed., 2020, 59, 22436–22440 CrossRef CAS PubMed; (d) H. Liang, W. Guo, J. Li, J. Jiang and J. Wang, Angew. Chem., Int. Ed., 2022, 61 DOI:10.1002/anie.202204926.
- (a) Y. Zou, P. Wang, L. Kong and X. Li, Org. Lett., 2022, 24, 3189–3193 CrossRef CAS PubMed; (b) F. Wang, J. Jing, Y. Zhao, X. Zhu, X.-P. Zhang, L. Zhao, P. Hu, W.-Q. Deng and X. Li, Angew. Chem., Int. Ed., 2021, 60, 16628–16633 CrossRef CAS; (c) B. Liu, P. Xie, J. Zhao, J. Wang, M. Wang, Y. Jiang, J. Chang and X. Li, Angew. Chem., Int. Ed., 2021, 60, 8396–8400 CrossRef CAS PubMed; (d) L. Kong, X. Han, S. Liu, Y. Zou, Y. Lan and X. Li, Angew. Chem., Int. Ed., 2020, 59, 7188–7192 CrossRef CAS PubMed; (e) R. Mi, G. Zheng, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2019, 58, 17666–17670 CrossRef CAS PubMed; (f) M. Tian, D. Bai, G. Zheng, J. Chang and X. Li, J. Am. Chem. Soc., 2019, 141, 9527–9532 CrossRef CAS PubMed; (g) B. Shen, B. Wan and X. Li, Angew. Chem., Int. Ed., 2018, 57, 15534–15538 CrossRef CAS PubMed.
- (a) H. Li, R. Gontla, J. Flegel, C. Merten, S. Ziegler, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2019, 58, 307–311 CrossRef CAS PubMed; (b) Z.-J. Jia, C. Merten, R. Gontla, C. G. Daniliuc, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2017, 56, 2429–2434 CrossRef CAS PubMed.
- (a) E. A. Trifonova, N. M. Ankudinov, A. A. Mikhaylov, D. A. Chusov, Y. V. Nelyubina and D. S. Perekalin, Angew. Chem., Int. Ed., 2018, 57, 7714–7718 CrossRef CAS PubMed; (b) C. M. B. Farr, A. M. Kazerouni, B. Park, C. D. Poff, J. Won, K. R. Sharp, M.-H. Baik and S. B. Blakey, J. Am. Chem. Soc., 2020, 142, 13996–14004 CrossRef CAS PubMed; (c) X. Chen, S. Yang, H. Li, B. Wang and G. Song, ACS Catal., 2017, 7, 2392–2396 CrossRef CAS; (d) S. Maity, T. J. Potter and J. A. Ellman, Nat. Catal., 2019, 2, 756–762 CrossRef CAS PubMed.
- (a) Y.-S. Jang, Ł. W. niak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901–12905 CrossRef CAS PubMed; (b) P. Wang, Y. Huang, J. Jing, F. Wang and X. Li, Org. Lett., 2022, 24, 2531–2535 CrossRef CAS PubMed.
- For reviews, see: (a) R. Bentley, Chem. Soc. Rev., 2005, 34, 609–624 RSC; (b) G. Sipos, E. E. Drinkel and R. Dorta, Chem. Soc. Rev., 2015, 44, 3834–3860 RSC; (c) S. Otocka, M. Kwiatkowska, L. Madalinska and P. Kielbasinski, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS PubMed; (d) T. Jia, M. Wang and J. Liao, Top. Curr. Chem., 2019, 377, 1–29 CrossRef CAS PubMed; (e) J. Diesel and N. Cramer, ACS Catal., 2019, 9, 9164–9177 CrossRef CAS . For examples, see:; (f) A. Osorio-Lozada, T. Prisinzano and H. F. Olivo, Tetrahedron: Asymmetry, 2004, 15, 3811–3815 CrossRef CAS; (g) J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347, 19–31 CrossRef CAS; (h) T. Jia, P. Cao, B. Wang, Y. Lou, X. Yin, M. Wang and J. Liao, J. Am. Chem. Soc., 2015, 137, 13760–13763 CrossRef CAS PubMed.
- (a) E. Wojaczynska and J. Wojaczynski, Chem. Rev., 2010, 110, 4303–4356 CrossRef CAS PubMed; (b) I. Fernandez and N. Khiar, Chem. Rev., 2003, 103, 3651–3705 CrossRef CAS PubMed.
- For a recent review, see: (a) W. Liu, J. Ke and C. He, Chem. Sci., 2021, 12, 10972–10984 RSC For examples, see:; (b) T. Zhou, M.-X. Jiang, P.-F. Qian, Q.-J. Yao, X.-T. Xu, K. Zhang and B.-F. Shi, Org. Lett., 2021, 23, 7910–7915 CrossRef CAS PubMed; (c) T. Zhou, P.-F. Qian, J.-Y. Li, Y.-B. Zhou, H.-C. Li, H.-Y. Chen and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 6810–6816 CrossRef CAS PubMed; (d) K. Mukherjee, N. Grimblat, S. Sau, K. Ghosh, M. Shankar, V. Gandon and A. K. Sahoo, Chem. Sci., 2021, 12, 14863–14870 RSC . While sulfoxide had been previously introduced as a chiral directing group in C–H activation, enantioenriched substrates were employed. See:; (e) C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871–13875 CrossRef CAS PubMed.
- Y.-C. Zhu, Y. Li, B.-C. Zhang, F.-X. Zhang, Y.-N. Yang and X.-S. Wang, Angew. Chem., Int. Ed., 2018, 57, 5129–5133 CrossRef CAS PubMed.
- W. Liu, W. Yang, J. Zhu, Y. Guo, N. Wang, J. Ke, P. Yu and C. He, ACS Catal., 2020, 10, 7207–7215 CrossRef CAS.
- (a) Y. Li, J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 6743 CrossRef CAS PubMed; (b) F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780–4787 CrossRef CAS PubMed; (c) C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53(a), 2722–2726 CrossRef CAS PubMed; (d) Z. Ruan, N. Sauermann, E. Manoni and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 3172–3176 CrossRef CAS PubMed; (e) E. Tan, M. Zanini and A. M. Echavarren, Angew. Chem., Int. Ed., 2020, 59, 10470–10473 CrossRef CAS PubMed.
- C.-W. Zhang, X.-Q. Hu, Y.-H. Dai, P. Yin, C. Wang and W.-L. Duan, ACS Catal., 2022, 12, 193–199 CrossRef CAS.
- X. Kan, J.-C. Wang, Z. Chen, J.-Q. Du, J.-L. Kan, W.-Y. Li and Y.-B. Dong, J. Am. Chem. Soc., 2022, 144, 6681–6686 CrossRef CAS PubMed.
- Y.-S. Jang, M. Dieckmann and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 15088 CrossRef CAS PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (d) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- (a) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2207905 and 2207903. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05310a |
| This journal is © The Royal Society of Chemistry 2023 |