
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-induced defluorination acyl fluoride exchange as a fluorogenic photo-click reaction for photo-affinity labeling†
Lijun
Deng
,
Cefei
Zhang
,
Baolin
Li
,
Jielin
Fu
,
Zhong
Zhang
,
Sitong
Li
,
Xiaohu
Zhao
,
Zhishan
Su
 ,
Changwei
Hu
* and
Zhipeng
Yu
*
,
Changwei
Hu
* and
Zhipeng
Yu
*
Key Laboratory of Green Chemistry & Technology of Ministry of Education, College of Chemistry, Sichuan University, 29 Wangjiang Road, Chengdu 610064, P. R. China. E-mail: changweihu@scu.edu.cn; zhipengy@scu.edu.cn
First published on 24th February 2023
Abstract
Photo-click chemistry has emerged as a powerful tool for revolutionizing bioconjugation technologies in pharmacological and various biomimetic applications. However, enriching the photo-click reactions to expand the bioconjugation toolkit remains challenging, especially when focusing on spatiotemporal control endowed by light activation. Herein, we describe a photo-induced defluorination acyl fluoride exchange (photo-DAFEx) as a novel type of photo-click reaction that is mediated through acyl fluorides produced by the photo-defluorination of m-trifluoromethylaniline to covalently conjugate with primary/secondary amines and thiols in an aqueous environment. (TD)-DFT calculations, together with experimental discovery, indicate that the m-NH2PhF2C(sp3)–F bond in the excited triplet state is cleaved by water molecules, which is key to inducing defluorination. Intriguingly, the benzoyl amide linkages built by this photo-click reaction exhibited a satisfactory fluorogenic performance, which allowed visualization of its formation in situ. Accordingly, this photo-controlled covalent strategy was exploited not only for the decoration of small molecules, peptide cyclization and functionalization of proteins in vitro, but also for designing photo-affinity probes targeting endogenous carbonic anhydrase II (hCA-II) in living cells.
Introduction
Click chemistry has become an essential strategy for bioconjugation, benefiting from characteristics such as accessibility, smart operation and reliable efficiency. Over the past decade, sulfur(VI) fluoride exchange (SuFEx) has served as an emerging click strategy and has been employed in protein engineering and drug discovery, as it enables crosslinking toward natural nucleophilic residues with excellent specificity (Scheme 1a).1–3 Therefore, ligand-directed covalent-linking strategies are flourishing by designing probes harbouring the above features, emphasizing the site-dependent reactivity of SuFEx in combination with the target affinity of the ligands.4 Moreover, the small size of sulfur(VI) fluoride and its high resistance to hydrolysis also allows its easy incorporation into biomacromolecules for profiling of protein-related interactions in a complex biological environment.4b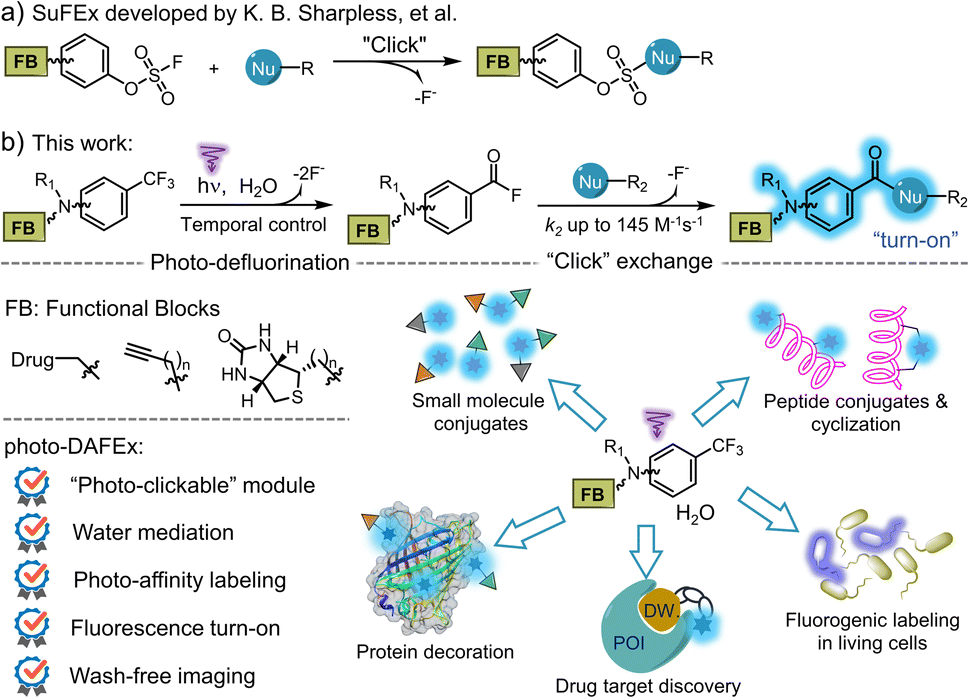 | ||
| Scheme 1 Illustration of the SuFEx and the photo-DAFEx reactions for fluorogenic functionalization of small molecules and native biomolecules. | ||
Although thermodynamic ligation approaches, including SuFEx, have enabled numerous pharmacological explorations, some inherent intractable issues are still unsolved, e.g., off-target reactivity at high dosage, off-site launching induced by the microenvironment during delivery,5 and inability to be applied to single cells.6 Light-triggered click chemistry provides alternative pathways to build covalent linkages via non-invasive manipulation.7 Scientists have successfully harnessed photo-stimulation to initiate click reactions via the in situ generation of intermediates with high reactivity and selectivity, allowing conjugation with spatiotemporal controllability.8
Most of the available photo-clickable reagents find an appropriate partner-group after photo-activation, according to their respective characteristics in various bioorthogonal scenarios. Current achievements include photo-released cycloalkyne clicking with azide;9 tetrazole or sydnone photo-cycloaddition with alkene/-yne;10,11 photo-promoted hetero-Diels–Alder (HDA)12 and IEDDA reactions;13 light-activated thiol-ene/-yne conjugations;14,15 and light-triggered phenanthrenequinone–alkene HDA reactions.16 In these scenarios, a biocompatible handle has to be incorporated into the biomacromolecules oriented on specific sites prior to photo-ligation.
 | ||
| Fig. 1 Experimental studies on the mechanism of the photo-DAFEx. (a) General scheme for the photo-click reaction of 1a in either (i) aqueous buffer with 2a or (ii) in absolute methanol. (b) 19F-NMR spectra for identifying in situ generated 5a from 1a (0.12 mmol) in ACN/H2O = 1/1 after 30 min irradiation. (c) HPLC traces for the quenching tests of the excited triplet state under various conditions. Screening for the photo-DAFEx conditions of 1a (100 μM): (d) the ratio of water in the ACN mixture; (e) temporal control by the irradiation time (311 nm, 5.9 mW cm−2). | ||
Light-triggered bioconjugation approaches also show exciting promise in both affinity-based probing17 and protein–protein interactions,18 which target canonical residues on the active site of proteins. Conventionally, diazirines, aryl azides and benzophenones are widely used photo-crosslinkers for biomedical discovery, displaying high insertion reactivity but diverse chemo-targets.19 Recently, diazocoumarins,20N-phenyltetrazole,21 acyl silanes22 and o-nitrobenzyl alcohols23 have appeared as novel photo-crosslinkers. In addition to their photo-controllability, these strategies play an important role in advancing the fluorogenic performance with tuneable chemo-specificity. However, the photon-energy utilization efficiency as well as the lifetime of photo-generated intermediate24 are the two key factors that have to be considered to achieve a photo-crosslinking method with high controllability. The prerequisites for a robust photo-crosslinking reaction also encompass biostability and compact size of the photoactivatable precursor. Furthermore, the covalent-bond formation promoted by the proximity of the photo-generated active moiety tagged on the client molecules3 is dictated by the microenvironment in which the precursor is incorporated. Given these demands, developing a photo-click reaction that satisfies all of the features in a single solution remains challenging, but desirable.
We recognized that m-trifluoromethylaniline derivatives could be activated by light to condensate into amide oligomers based on photo-defluorination.25 After careful optimization of this photochemical process, a photo-induced defluorination acyl fluorides exchange (photo-DAFEx, Scheme 1b) was discovered as a novel class of photo-click reactions for covalent conjugation towards primary/secondary amines and thiols. Distinct from conventional photo-defluorination, we found that water molecules were indispensable for polarizing the C(sp3)-F bond in m-trifluoromethylaniline in the excited triplet state, which resulted in consecutive hydrolytic C–F bond cleavages to yield acyl fluorides (Scheme 1b) in an aqueous environment. The in situ generated acyl fluorides with satisfactory stability and muted polarity are capable of exchanging with the primary/secondary amines and thiols to establish benzamide linkages toward small molecules, peptides and proteins, accompanied by a fluorescence turn-on feature (Scheme 1b). To demonstrate potential benefits in chemo-proteomic and pharmacological research, the m-trifluoromethylaniline motif has been embedded into sulfonamide inhibitors for photo-affinity labeling of endogenous hCA-II in human embryonic kidney cells (HEK-293T). Given the accessibility of m-trifluoromethylaniline derivatives, the photo-DAFEx reaction not only enriches the toolbox of photo-click chemistry, but also integrates the photo-labeling and the fluorescence tracking features into a single photo-chemical cascade.
Results and discussion
Design and optimization of the photo-DAFEx reaction
C(sp3)–F bond cleavage strategies have been realized widely in organic phases, while few reports discuss water-mediated and catalyst-free photo-defluorination strategies that are amenable to biochemical studies.26 Inspired by the photolysis of flufenamic acid,25 we found that N,N-dimethyl-3-(trifluoromethyl)aniline (1a) could be efficiently converted to 3-(dimethylamino)benzoyl fluoride (5a) after exposure to 311 nm irradiation in an ACN/H2O mixed solvent (v/v = 1/1) (Fig. 1a). However, ortho-/para-trifluoromethylaniline showed relatively low efficiency in photo-conversion (Fig. S1, ESI†). Due to the electrostatic stability of the PhCO–F bond, benzoyl fluorides show resistance to hydrolysis (half-life, t1/2 = 13.7 h, 298 K, Fig. S6, ESI†), which is akin to the sulfonyl fluorides in SuFEx.27 Thereby, its isolation after the photo-defluorination in aqueous medium was successful without strong nucleophiles (Fig. 1b). In situ photo-generated 5a could be further amidated with benzylamine (2a), furnishing N-benzyl-3-(dimethylamino)benzamide (3aa) quantitatively.Oxygen is an efficient quencher of excited triplet states; hence, nitrogen protection is often essential in many photocatalytic reactions.28 Indeed, suppression is observed in terms of the photo-conversion of 1a in the photo-DAFEx under air vs. under a nitrogen atmosphere (Fig. 1c). In particular, the photochemical conversion can be halted via oxygen saturation. However, free radical quenching by TEMPO, styrene, etc., has little influence on the photo-DAFEx (Fig. S2, ESI†). Therefore, the photo-defluorination of 1a likely occurs in an excited triplet state rather than a radical process. Unexpectedly, water molecules appear to be involved in the photo-defluorination, because the photo-conversion of 1a is negligible in an anhydrous condition (Fig. 1d). Next, we evaluated the photon utilization efficiency of the photo-DAFEx, in which the quantum yield (ΦR) was determined to be 0.17 for 1a under 311 nm irradiation (for 1a derivatives, see Table S1, ESI†). Within 60 s of irradiation, the conversion of 100 μM 1a reached completion, affording 3aa with 86% yield, indicating the high efficiency of the photo-DAFEx (Fig. 1e). To assess the biostability of m-trifluoromethylanilines, 100 μM 1a or 1e was treated with a 50-fold excess concentration of glutathione (5.0 mM GSH) in ACN/PBS solution (v/v = 1/1, pH = 7.4, 298 K). 71% of 1a and 97% of 1e remained after incubation for 72 h (Fig. S4, ESI†), suggesting excellent resistance to nucleophilic additions in a biomimicking environment in the dark. Optimization of the photo-reaction conditions, including the pH of the aqueous buffer and organic cosolvents (Table S2, ESI†), indicated that physiological conditions (PBS, pH = 7.4) are favourable in terms of the yield of 3aa (94%), while the use of an organic cosolvent brings about trivial variation in the yield, except in the case of methanol. Accordingly, absolute methanol, which acts as a protic solvent, was adopted in the photo-DAFEx, which resulted in the detection of 3-(dimethylamino)difluorobenzyl methyl ether (6a, Fig. 1a and S5, ESI†) with up to 17% HPLC yield. The successful capture of the mono-defluorination product (6a) clarified that the photo-induced dual defluorination is a stepwise process. This series of experimental phenomena aroused our interest in studying the mechanism of the photo-DAFEx reaction.
DFT calculation of the photo-DAFEx mechanism in an aqueous environment
DFT calculations at the M06-2X(D3)/6-31+G** theoretical level (all Gibbs free energies below were calculated using Shermo 2.3,29 and the wavefunction analyses were performed using Multiwfn 3.8,30 for more details, see ESI computational details†) were performed to understand the mechanism of the photo-defluorination of 1a (Fig. 2a). As a comparison, a thermodynamic defluorination gives 3-(dimethylamino)difluorobenzyl alcohol (IM3) with an activation free energy barrier (ΔG‡) of as high as 49.4 kcal mol−1, which is quite harsh for a biocompatible ligation. Unlike the ground-state process, the first photo-defluorination step in the excited triplet state was found to involve a unique fluorine–oxygen swap mode, and thereby has to be subdivided: (i) the first C(sp3)–F bond breakage (TS3-T1) is evoked by hydrogen-bonding-assisted polarization in harmony with triplet-state charge delocalization to form a stabilized ionic complex (IM5-T1). (ii) Then, the C2+ atom in IM5-T1 is attacked by the adjacent oxygen atom in the water molecule with a negative charge acquired from F1− (TS4-T1) in the counter ion, affording IM6-T1. The activation barriers viaTS3-T1 (4.8 kcal mol−1) and TS4-T1 (14.7 kcal mol−1) are significantly lower than that in the ground state (ΔG‡ = 49.4 kcal mol−1). As a result, the first C(sp3)–F bond cleavage proceeds instantly in the excited triplet state via light activation. Subsequent non-radiative quenching of IM6-T1 to IM2 is computed to be thermodynamically favoured (ΔG = −69.1 kcal mol−1). Due to the instability of IM3 in an aqueous environment, the benzoyl fluoride (5a) is obtained through a fluorine atom elimination via a six-membered cyclic transition state (TS2) with a dual H-bonding activation by water molecules in the ground state (ΔG‡ = 20.5 kcal mol−1). As evidence for this pathway, 6a (Fig. 1a) with much higher stability can be detected when a mono-H-bonding donor, methanol, was used as the surrogate for water. | ||
| Fig. 2 Computational studies for deciphering the mechanism of the photo-DAFEx reaction. (a) Gibbs energy profile for the defluorination process of 1a to afford the intermediate 5a; the black-colour pathway is the thermodynamic path in the ground state (GS), while the red-colour pathway is followed in the first excited triplet state (T1); free energies were calculated at the M06-2X(D3)/ma-TZVP level (Table S8, ESI†). (b) LBO value of the key C–F bond in the denoted structures (1avs.1a-T1, IM1vs.IM1-T1), and the hydrogen bond strength (EHB) between the HO–H and the F1 atoms computed with ρBCP. (c) Spin density and ADCH charges of IM1-T1, TS3-T1 and IM5-T1 to clarify the cleavage mode of the C–F bond. | ||
To understand the synergistic effects of the photo-activation and H-bonding on the initial defluorination, we compared the Laplacian bond order (LBO)31 of the identical C–F bond in both 1a and the first excited triplet state 1a-T1, the values of which are positively correlated with the bond energy of interest (Fig. 2b; the three-dimensional structures were generated using CYLview 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32). In the excited state, one of the three C–F bonds is impaired, with LBO = 0.163 in 1a and 0.111 in 1a-T1, respectively. Meanwhile, the water molecule also accelerates the C–F bond cleavage via H-bonding interactions with a decrease in the LBO in IM1-T1 (0.097). The hydrogen bond strength between HO–H and F atom in both IM1 and IM1-T1 (triplet state) were estimated by the electron densities of the bond critical point (ρBCP) defined by AIM theory,33 which were approximately −1.45 kcal mol−1 and −2.37 kcal mol−1, respectively (Fig. 2b). These values are moderate in strength but essential in assisting the heterolysis of the C(sp3)–F bond (LBO = 0.097 in T1). Subsequently, the spin density isosurface and ADCH charge34 for the key species (IM1-T1, TS3-T1, IM5-T1) in the first defluorination process were also analysed (Fig. 2c, isosurface plots were generated using VMD 1.9.335) to evaluate the feasible electron transfer during the C–F bond cleavage. Consequently, the departing fluorine in TS3-T1 has a relatively low spin density (0.10) and a high negative charge (−0.46), indicating that the defluorination reaction is heterolysis rather than a radical process. This result is consistent with the experimental observation that radical quencher proceeds sluggishly in quenching the photo-DAFEx.
32). In the excited state, one of the three C–F bonds is impaired, with LBO = 0.163 in 1a and 0.111 in 1a-T1, respectively. Meanwhile, the water molecule also accelerates the C–F bond cleavage via H-bonding interactions with a decrease in the LBO in IM1-T1 (0.097). The hydrogen bond strength between HO–H and F atom in both IM1 and IM1-T1 (triplet state) were estimated by the electron densities of the bond critical point (ρBCP) defined by AIM theory,33 which were approximately −1.45 kcal mol−1 and −2.37 kcal mol−1, respectively (Fig. 2b). These values are moderate in strength but essential in assisting the heterolysis of the C(sp3)–F bond (LBO = 0.097 in T1). Subsequently, the spin density isosurface and ADCH charge34 for the key species (IM1-T1, TS3-T1, IM5-T1) in the first defluorination process were also analysed (Fig. 2c, isosurface plots were generated using VMD 1.9.335) to evaluate the feasible electron transfer during the C–F bond cleavage. Consequently, the departing fluorine in TS3-T1 has a relatively low spin density (0.10) and a high negative charge (−0.46), indicating that the defluorination reaction is heterolysis rather than a radical process. This result is consistent with the experimental observation that radical quencher proceeds sluggishly in quenching the photo-DAFEx.
Evaluation of the substrate scopes
To meet the requirements for widespread application in both biochemistry and material science, it is necessary to explore the tolerance of photo-DAFEx toward substrates equipped with various chemical functionalities. Therefore, we commenced to synthesize a series of m-trifluoromethylaniline derivatives containing modular units (e.g., –OH, –NHBoc, –CONH–), which were efficiently ligated with benzylamine in good yields (Table 1, 3aa–3ha, 76–87%). Oxiranemethanol and terminal alkyne were also appended on the m-trifluoromethylaniline backbone for a sequential click strategy, which afforded 60–70% yields (3ia–3ja). A variety of functional units, including an NMR tag, biotin and PEG groups were examined, and all were tolerated well under the photo-DAFEx conditions to afford the products 3ka–3ma (up to 82% yield). Afterwards, we also introduced natural products or drugs, including geraniol (3na), clofibric acid (3oa), sulfonamide (3pa), and cholic acid (3qa), in the photo-DAFEx chemistry, and obtained N-benzylbenzamide drug-conjugates in 62–90% yield. Generally, we were able to introduce diverse functionalities on the m-trifluoromethylaniline backbone without compromising the photo-defluorination in an aqueous environment.| a Substrate scopes of the photo-DAFEx reaction. Reaction conditions: m-trifluoromethylaniline derivates (0.12 mmol) and nucleophiles (0.60 mmol) in 200 mL ACN/PBS (v/v = 1/1, pH = 7.4) were irradiated with a 311 nm lamp (21.2 mW cm−2) for 1 h; isolated yields are given. b Reaction conditions: m-trifluoromethylaniline derivates (100 μM) and nucleophiles (500 μM) in 1.0 mL ACN/PBS (v/v = 1/1, pH = 7.4) were irradiated with a 311 nm lamp (5.9 mW cm−2) for 60 s, and HPLC yields were determined via the calibration with external standard method. |
|---|

|
We next evaluated the suitability of a series of nucleophiles in the photo-DAFEx. As shown in Table 1, primary/secondary amines and thiols could form a covalent linkage toward 1a smoothly and generate amides (3ab–4ai) and thioesters (5aa–5ae), respectively. However, dopamine provided a relatively poor yield of 3ak due to the photo-decomposition caused by the optical filtering effect of the pyrocatechol moiety. In comparison with p-anisidine (pKaH = 5.3, 54% yield, 3ah), pyridyl amines, such as 4-aminopyridine (pKaH = −6.30) or aminopyrazine (pKaH = −4.40), did not amidate with the benzoyl fluorides under physiological pH (= 7.4).36 Remarkably, a diverse set of pharmaceutical building blocks and natural products are well tolerated as primary amine nucleophiles (3am–3ar). Representative aliphatic and aromatic secondary amines were further employed, most of which could be conjugated with benzoyl fluoride to furnish 78–91% yields of 4aa–4ai, but pyrrole, indole and carbazole are unsuitable (Fig. S74, ESI†). Thioesters are important units in biosynthetic processes.37 Finally, we examined the substrate scope for various thiols, including 2-mercaptoethanol, protected L-cysteine and 3-azetidinethiol (the intermediate of tebipenem). Pleasingly, all of the thiols could afford the thioester conjugate in up to 84% yields (5aa–5ae). Collectively, the photo-DAFEx exhibits good tolerance and high chemo-selectivity for a specific class of nucleophiles that is appropriate for drug target fishing.
Spectroscopic characterization and kinetics studies
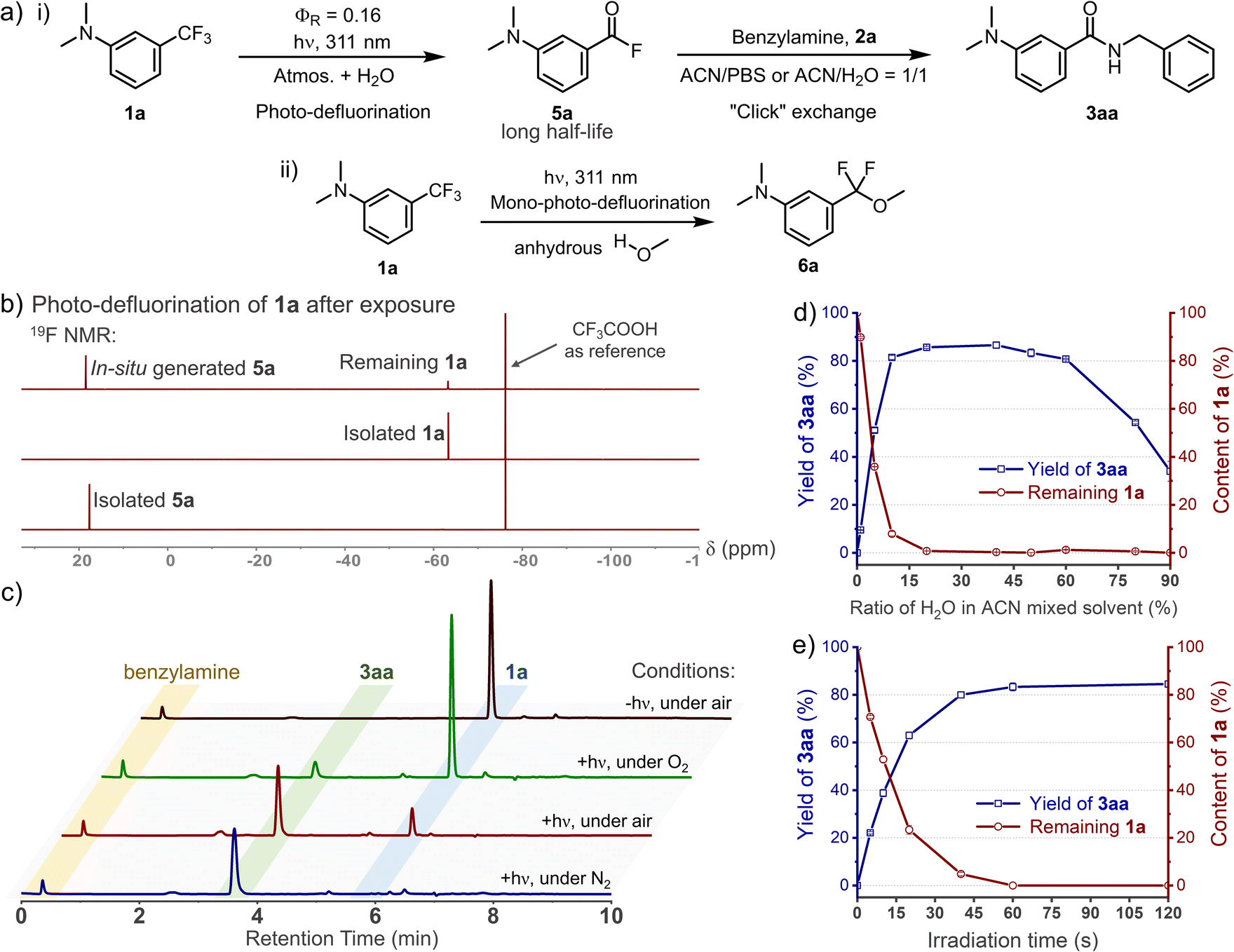
When investigating the substrate scopes, significant fluorescence turn-on was observed during each photo-conjugation (Fig. 3). In the absence of nucleophiles, the initial photo-defluorination step (1a–1h) gives the corresponding benzoyl fluorides as poor fluorophores, and this step can be monitored by real-time tracking of the absorption spectra (Fig. S7, ESI†). To clarify the fluorogenic performance of the photo-DAFEx, fluorescence emission spectra were then recorded during the course of exposure to 311 nm irradiation in the presence of benzylamine. Reagents 1a–1h were completely converted to the corresponding amides (3aa–3ha) within 60 s, accompanied by fluorogenic turn-on in a time-resolved manner (Fig. 3a, b and S8, ESI†). The turn-on ratios were determined to be 27-fold and 65-fold for 3aa and 3ca at the maximum emission wavelengths, respectively. Moreover, 3aa–3ca displayed much stronger fluorescence intensity than 3ea–3ha with two N,N-dimethylamine substitutions (Fig. S8, ESI†). Histidine, lysine, and cysteine were also subjected to the fluorogenic assessment in the photo-DAFEx, and only N-Boc-L-lysine appeared to have the turn-on character (up to 53-fold, Fig. S9 and 10, ESI†).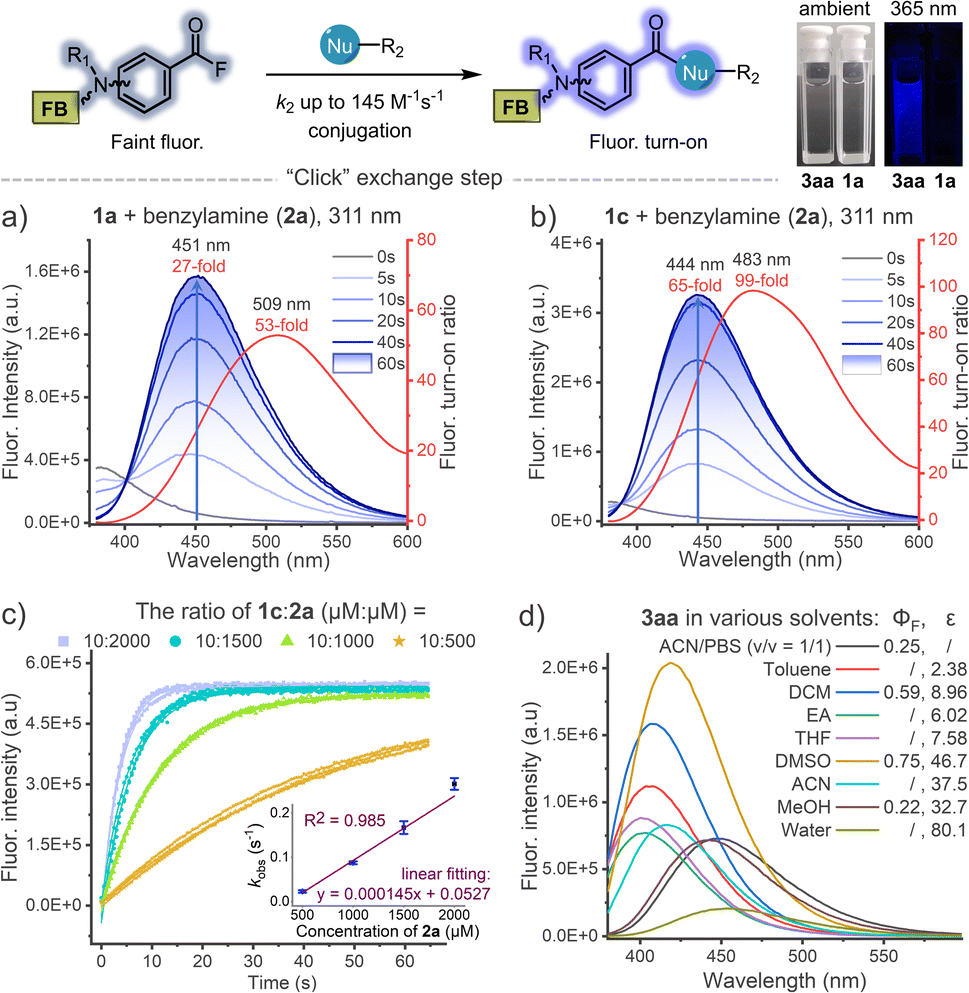 | ||
| Fig. 3 Dynamic tracking of fluorescence turn-on during the photo-DAFEx reaction between (a) 1a (100 μM) or (b) 1c (100 μM) and benzylamine (2a, 500 μM) in ACN/PBS (v/v = 1/1, pH = 7.4) under 311 nm irradiation (5.9 mW cm−2), the turn-on ratio was plotted along with the emission wavelength (red curve); for more details, see Table S3, ESI†. (c) Kinetic study of the acyl exchange step between 1c and 2a in ACN/PBS (v/v = 1/1, pH = 7.4) to obtain the second-order reaction rate; λex = 345 nm, λem = 445 nm. (d) Fluorescence emission spectra of the isolated 3aa (50 μM) in various solvents with quantum yield (ΦF) and dielectric constant (ε) of the solvent denoted; λex = 338 nm. | ||
Taking advantage of this fluorogenic feature, the second-order rate constant for the conjugation step between benzoyl fluoride and benzylamine could be facilely determined to be ∼145 M−1 s−1 (Fig. 3c), which is almost comparable to that of tetrazines-BCN IEDDA38 and faster than that of CuAAC.4 Fluorescence spectra of 3aa in various solvents were further measured, and the intensity decreased sharply along with bathochromic shift of the emission band when the dielectric constant (ε) of the solvent system was increased, except in the case of DMSO (Fig. 3d). The absolute fluorescence quantum yields (ΦF) were also then determined, and showed a consistent trend with the spectral measurements, in which 3aa in DMSO showed the highest value of up to ΦF = 0.75 (Table S3, ESI†).
Photo-controlled peptide cyclization and decoration
Encouraged by the high efficiency and controllability, we began to unravel the potential of photo-DAFEx in polypeptide conjugation. Based on HPLC-MS screening of free amino acids (Table S6 and Fig. S75–76, ESI†), only L-histidine, L-cysteine and L-lysine are readily targeted by reagent 5a in the photo-DAFEx, implying that about 8.83% of natural residues are available for profiling (Fig. 4a).39 To reveal the residual preference, competitive tests were further performed, which resulted in a ligation product ratio of 52:17:26 (Fig. 4b), respectively.
 | ||
| Fig. 4 Photo-DAFEx for controllable peptide cyclization and conjugation. (a) Natural abundance of each photo-clickable NAA available on proteins for the photo-DAFEx. (b) Preference of the photo-DAFEx for histidine (blue), cysteine (lime) and lysine (yellow) residues using 5a, analysed via an HPLC competition test with 1a (100 μM) and each NAA (500 μM) in 1.0 mL of ACN/PBS (v/v = 1/1, pH = 7.4); 311 nm lamp irradiation (5.9 mW cm−2) for 60 s. (c) Photo-DAFEx for peptide cyclization toward lysine and histidine residues in accessible proximity. Conditions: 100 μM peptide, 311 nm lamp irradiation (5.9 mW cm−2) for 60 s, for more details, see ESI†; (d) peptide conjugation via photo-DAFEx on L-histidine, L-lysine, free L-cysteine residues and N-terminal amines. Reaction conditions: peptide (80 μM) and 1a (1.25 equiv. for all photo-clickable NAAs residues at 100–400 μM) in ACN/PBS (v/v = 1/1, pH = 7.4) were treated with 311 nm lamp irradiation (5.9 mW cm−2) for 60–90 s; HPLC yields were determined using an external standard. AS. = atoms. | ||
The rigid conformation and topology of cyclic peptides dominate their unique functions and resistance toward enzymatic proteolysis, which has aroused considerable attention in bio-macromolecular drug design.40 To be able to control the cyclization of the unprotected peptide via the photo-DAFEx, we firstly joined the photo-DAFEx warhead onto a cysteine residue of the linear peptides via the α-halocarbonyl reagents 1r and 1s (Fig. S77–84, ESI†). Then, 311 nm irradiation could be applied to induce a temporally controlled cyclization toward available histidine/lysine residues within an acceptable proximity (Fig. 4c and S76–83, ESI†). Owing to the side-chain flexibility and stability of the resulting amide, the L-lysine residue as the cyclization juncture exhibited excellent ring-forming capability in comparison with L-histidine. However L-histidine, a reactive and reversible cyclization juncture in photo-DAFEx, is worthy of in-depth exploration for peptide medicines.
Next, the photo-DAFEx toward various functional peptides containing multiple nucleophilic residues was conducted to assess the universality for multi-decoration (Fig. 4d). Leuprorelin (9a) was first chosen to test the reactivity toward histidine residues on peptides, which afforded a conjugate in 56% yield (10aa). The resulting N-acyl imidazole as a moderate electrophile has great potential to be applied in sequential bioconjugations and enzyme activation.41,42 Disulfide bonds play a critical role in governing the conformation of proteins, and thus being inert towards them is necessary for a successful photo-bioconjugation strategy. Fortunately, no rupture of the disulfide bonds was observed after treating desmopressin (9g) with 1a under 311 nm irradiation (Fig. S91, ESI†). After the same treatments of the disulfide-bond-cyclized NoxaB protein fragments (9b/9d), no quadra-labelled products were detected in HPLC-MS, but double-decoration of both the N-terminal and the lysine residue (10ab and 10ad) were identified. In contrast, when there are two free cysteines available on FK-4 peptide analogues (9c and 9e), dual photo-thioesterification was allowed, with 74% and 60% yields, respectively. Moreover, RGD peptide analogue (9f) and Dynorphin (9h) with double lysine residues also showed good multi-ligation reactivities. For Exenatide (9i) with a long chain, a variety of multi-labelled products were detected, among which a tri-modified Exenatide (10ai) was mainly distributed in 51% yield (Fig. S93, ESI†).
Fluorogenic protein decoration
To understand the conjugation efficiency of photo-DAFEx on proteins, wild-type lysozyme, super-fold green fluorescent protein (sfGFP) and bovine serum albumin (BSA) were photo-labelled by using 1a (Fig. 5a). The decoration efficiency was found to be 57%, 59%, and 66%, respectively (Fig. 5b and S17, ESI†). The photo-labeling conversion would be improved with increasing the dosage of 1a, but at the cost of site-specificity (Fig. S18, ESI†). The site precedence of the labeling was then analysed using LC-MS/MS splicing, and we found the solvent exposed lysine sites, namely, the K115 and K134 sites on lysozyme, K107 on sfGFP, and K235, K245 and K548 on BSA, were labelled (Fig. 5b and S12–16, ESI†), while other lysine residues cannot be accessed because of inappropriate proximity. Only lysine decorations could be identified by LC-MS/MS, rather than histidine or cysteine decoration, which is attributed to the reversible nature of the latter two types of conjugates and their rare occupancy, as well as the inactive disulfide form.41–43 In-gel fluorescence analysis is an intuitive way to demonstrate the fluorogenic decoration of proteins. Therefore, a concentration gradient of 1a (0–100 μM) was used for a temporally controlled modification toward the three proteins via photo-irradiation. As a result, clear blue fluorescence bands can be observed at the level of 50 μM 1a, while no signal can be detected in the absence of 311 nm irradiation (Fig. 5c(i)). Similarly, 1l with a biotin tag and 1m with a PEG tail were also successfully photo-conjugated on the three proteins with fluorescence turn-on (Fig. 5c(i–iii)). Moreover, the fluorogenic labeling of BSA occurred in a time-dependent manner, suggesting a satisfactory temporal controllability (Fig. 5c(iv)). However, no background protein decoration was observed even after 2 h incubation in the dark (Fig. S19, ESI†). Therefore, the fluorogenic photo-DEFAx of proteins could greatly facilitate visualization of the photo-conjugation process in complex biological systems without the need for washing or extra staining procedures.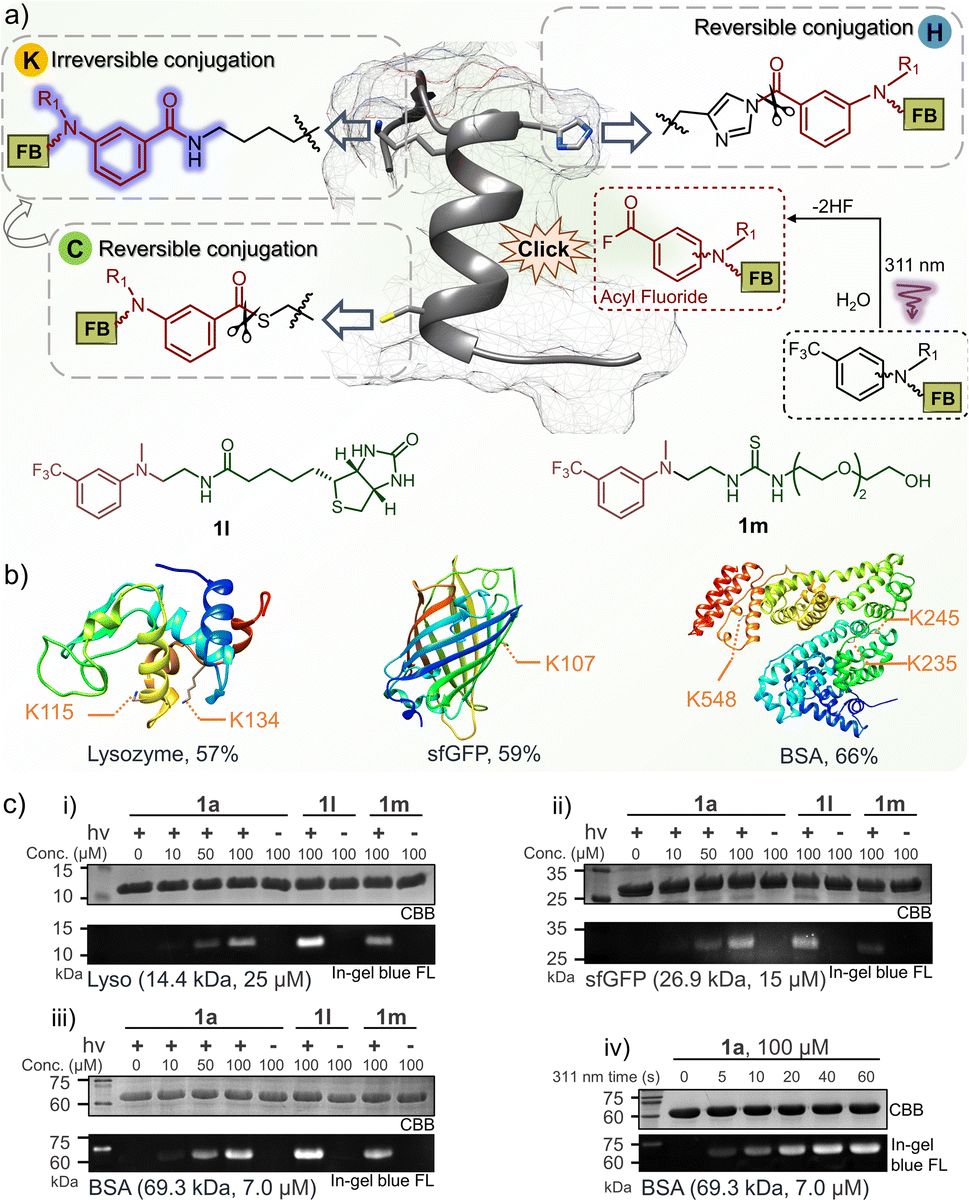 | ||
| Fig. 5 Fluorogenic and temporally controlled protein decorations via the photo-DAFEx reaction. (a) Characteristics of protein labeling toward lysine, histidine or cysteine residues, and the structure of 1l and 1m. (b) Site precedence of the photo-labeling on lysozyme, sfGFP and BSA, which was determined by LC-MS/MS analysis (Fig. S12–16, ESI†). The photo-labeling conversions were determined by deconvoluting the charge ladder of ion counting of the intact proteins in LC-MS spectra; for more details see Fig. S17, ESI†. (c) Fluorogenic protein decoration via photo-DAFEx (i–iii) visualized by in-gel fluorescence imaging and (iv) a time-course of light irradiation (5.9 mW cm−2) for 0–60 s. Reaction conditions: proteins (4.0 mg mL−1) and m-trifluoromethylaniline reagents in PBS were treated with/without 311 nm irradiation, and then resolved by SDS-PAGE for imaging. CBB = coomassie brilliant blue; FL = fluorescence channel. | ||
Photo-affinity labeling (PAL) for hCA-II
Drug-target interactions play an essential role in adjusting the functions of the targets for rescuing intrinsic cell activities. Hence, understanding of dynamic drug-target interaction networks via covalent capture offers great opportunities for therapeutics and drug discovery, especially for disease-related enzymes.19 The PAL strategy allows covalent binding to the targets with temporal control, thus enabling the identification of new drug targets and related molecular interactions.44 To demonstrate the PAL capability of the photo-DAFEx in crosslinking with lysine, we chose human carbonic anhydrase II (hCA-II) as the target owing to its availability, stability and structural clarity.45 hCA-II catalyses the reversible hydration of carbon dioxide, and it can be competitively inhibited by aryl sulfonamides, such as acetazolamide (AZA, Ki = 12.1 nM, Fig. S22, ESI†).46,47 Accordingly, photo-DAFEx-based PAL probes were designed and constructed via the fusion of m-trifluoromethylaniline into aryl sulfonamide (10a–10f, Fig. 6a and Table S7, ESI†). Before PAL, the efficacy of these probes toward hCA-II was confirmed to be IC50 = 0.95 and 1.12 μM for 10a and 10c (Fig. S22, ESI†), respectively, which is slightly weaker than that of commercial AZA (IC50 = 0.70 μM).46 Then, probe concentration gradients (0–120 μM) and an irradiation time-course (0–60 s) were evaluated in the photo-DAFEx toward hCA-II (Fig. 6c). In-gel fluorescence analysis clarified that an approximately 10 μM dosage of probes 10a and 10c is the minimum for in vitro studies (Fig. 6c(i)), and obvious fluorescence bands appeared after 20 s of irradiation in the time-course tracking (Fig. 6c(ii)). Thus, in vitro PAL in protein mixtures containing lysozyme, BSA and recombinant hCA-II or in cell lysates were further tested to understand the ligand-directed target specificity, and were also visualized using in-gel fluorescence (Fig. S20, ESI†). In bacterial lysates, only the lanes treated with either 10a or 10c displayed strong fluorescence bands with the correct size of hCA-II after photo-DAFEx reaction, confirming their strong affinity toward hCA-II (Fig. S21, ESI†).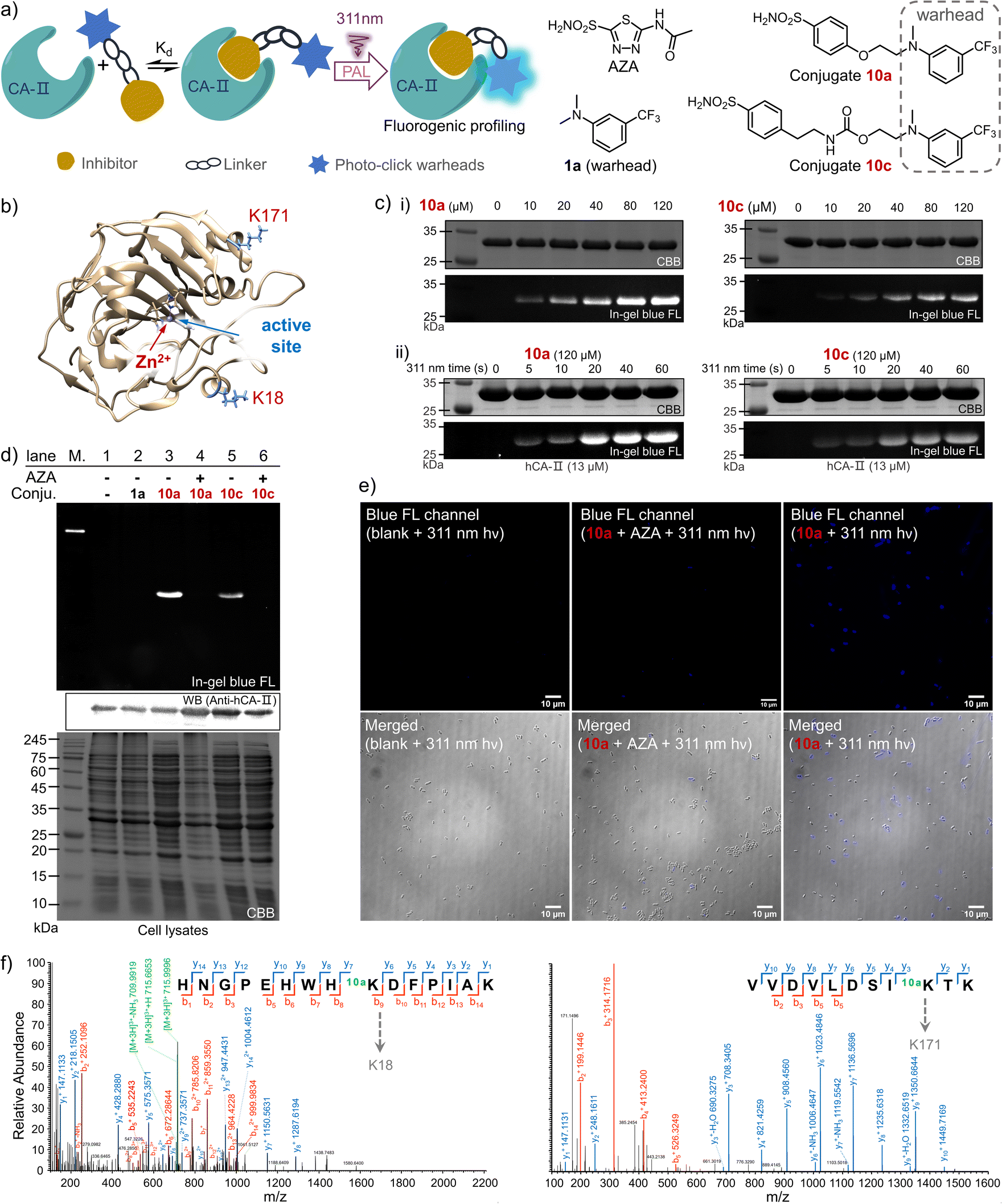 | ||
| Fig. 6 Photo-affinity labeling of hCA-II by m-trifluoromethylaniline-drug probes through the fluorogenic photo-DAFEx. (a) Schematic illustration of the PAL and the structure of the probes, commercial inhibitor and control reagent (1a). (b) Crystal structure of hCA-II showing the Zn2+ in the active site and the photo-labelled lysine residues. (c) In-gel fluorescence imaging of the SDS-PAGE for resolving the hCA-II (13 μM) after photo-labeling by probes 10a/10c with irradiation via a 311 nm lamp (5.9 mW cm−2): (i) concentration gradient after 60 s irradiation and (ii) irradiation time-course. (d) Fluorogenic photo-labeling of hCA-II in E. coli lysate via the photo-DAFEx by probe 10a/10c (20 μM) in the presence/absence of the competitive inhibitor AZA (1.0 mM) with irradiation via a 311 nm lamp (5.9 mW cm−2) for 30 s, with 1a as a negative control. (e) Fluorescence microscopic imaging of the E. coli cells (BL21(DE3)) overexpressing the hCA-II after PAL with probe 10a (5 μM) under 311 nm irradiation (5.9 mW cm−2) for 60 s. Scale bar = 10 μm; λex/λem = 365/420–470 nm, 100× oil-immersion objective. (f) LC-MS/MS analysis of the peptide fragment from trypsin digested hCA-II after photo-labeling by probe 10a. | ||
To verify the specific binding of 10a and 10c to the active pocket of hCA-II, a set of control experiments was also examined (Fig. 6d). First, no fluorescent band in lane 2 was observed when 1a without the sulfonamide drug moiety was used as a negative control, while strong fluorescence signals correlated to hCA-II were visualized in lanes 3 (10a) and 5 (10c), suggesting that their aryl sulfonamide moieties dominate the binding toward hCA-II in bacterial lysates. When the PAL probes competed against a 50-fold excess of AZA, the fluorescence bands were both quenched (lanes 4 and 6). Therefore, it is clear that the PAL probes bind competitively at the same active site of hCA-II as AZA, where the zinc(II)-histidine complex is located (Fig. 6b).47 In addition, a fluorescent western blot analysis confirmed that the hCA-II was evenly loaded among all lanes (Fig. 6d). Furthermore, the deconvoluted mass spectra also indicated that each molar equivalent of hCA-II can be crosslinked with only one molar equivalent of the probe (Fig. S23, ESI†). To determine the possible photo-labeling residues for 10a, the crosslinked hCA-II was further subjected to LC-MS/MS analysis (Fig. 6f), and the spliced peptide fragment revealed that K18 and K171 are two preferred sites for photo-crosslinking. Both are located near the entrance to the binding pocket of hCA-II (Fig. 6b and Tables S4 and 5, ESI†). Benefiting from the fluorogenic nature of photo-DAFEx, bright blue fluorescence signals could be visualized in living E. coli cells after the PAL toward hCA-II surrounded by endogenous proteins (Fig. 6e). However, the fluorescence was still diminished in the presence of the competitive AZA. Noticeably, variable expression levels of hCA-II lead to uneven turn-on signals among E. coli cells, which stems from the randomness of transcription and translation in each cell.48 Taken altogether, the PAL probe 10a is capable of fishing for the ligand-directed targets in both in vitro and in vivo studies with the added benefit of fluorogenic imaging, which could inspire utilization of the photo-DAFEx chemistry in pharmacological research.
PAL of endogenous hCA-II in living cells
The ultimate goal of PAL is to capture endogenous proteins with high affinity in living cells under photo-control. Herein, we evaluated the potential of the photo-DAFEx chemistry in PAL profiling via side-by-side comparisons with diazirine photolysis in HEK-293T cells, which express hCA-II endogenously (Fig. S24, ESI†). The two types of PAL probes were first synthesized (Fig. 7a). 10g is a well-known alkyl-diazirine-based photo-affinity probe with a terminal alkyne for CuAAC fluorescence imaging;49 while 10k is a structurally similar probe but based on the photo-DAFEx warhead. In the enzymatic inhibition assay for hCA-II, both of the probes could target the active pocket, showing high inhibition efficiency (IC50 = 2.35 μM and 0.59 μM for 10h and 10k, respectively) in comparison with commercial AZA (IC50 = 1.02 μM). After CuAAC to the PAL-tagged terminal alkyne on hCA-II in vitro via azide-Alexa-647 (Fig. S25, ESI†), in-gel fluorescence imaging indicated significant brighter bands for hCA-II with the probe 10k even at 10 μM, clarifying that 10k possesses higher photo-labeling efficiency than 10h (Fig. 7b). In-gel fluorescence comparison for hCA-II in bacterial lysates also exhibited similar results, in which the photo-DAFEx (10k) displayed much stronger fluorescence band over the diazirine photolysis (10h) (Fig. S25, ESI†).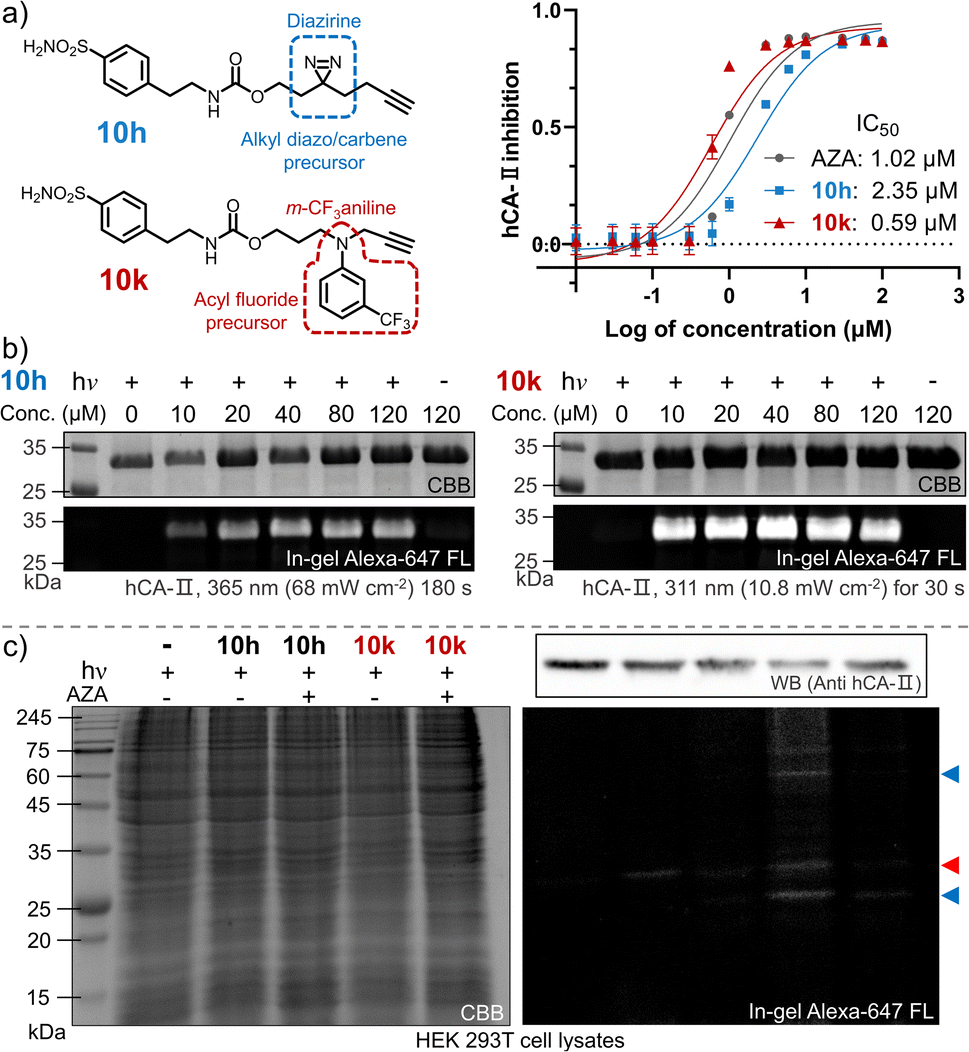 | ||
| Fig. 7 Photo-affinity labeling of hCA-II via an alkyl diazirine probe vs. m-trifluoromethylaniline-inhibitor conjugate. (a) Structural illustration of 10hvs.10k, and plot of the enzymatic activity of hCA-II to catalyse hydrolysis of p-nitrophenyl acetate vs. inhibitor concentration. Error bars indicate SD, n = 3. (b) In-gel fluorescence imaging of SDS-PAGE of the resolved hCA-II (13 μM) after photo-labeling by probes 10h (left) and 10k (right). (c) SDS-PAGE analysis of endogenous proteins after photo-labeling by 10hvs.10k (5.0 μM) in the presence/absence of AZA (25 μM) in living HEK 293T cells. Note: hCA-II is marked by a red triangle; other photo-labelled proteins are marked by blue triangles. Fluorophore attachment was realized via CuAAc with an azide-Alexa-647 conjugate. | ||
We next examined whether the in vitro efficiency differences between alkyl diazirine photolysis and photo-DAFEx corresponded to those for endogenous hCA-II in living mammalian cells. After incubating the two probes at 5.0 μM in HEK-293T cells for 2 h, the cells were exposed to irradiation, lysed, and clicked with azide-Alexa-647 for visualization using SDS-PAGE. The fluorescence bands (Fig. 7c, marked with red triangles) corresponding to hCA-II were then observed for the cells treated with the two individual probes and irradiation, and suggested good reactivity of the photo-DAFEx chemistry for drug-target profiling. Western blot (upper right panel, Fig. 7c) analysis confirmed that hCA-II was evenly expressed among the groups of cells, matching with the molecular weight of the fluorescent bands. Side-by-side in-gel comparison between the probes 10h and 10k (lower right panel, Fig. 7c) showed that the alkyl-diazirine-based PAL exhibited almost a single hCA-II band without potential off-target bands, while the photo-DAFEx displayed multiple bands. Because the acyl fluoride intermediate generated by the photo-defluorination of 10k is much more stable (t1/2 = 13.7 h) than the alkyl diazo intermediates (t1/2 ≈ 70 s)50 or the alkyl carbene intermediates (t1/2 ≈ 2 ns),24 more relevant protein targets interacting with the 10k probe with inferior affinity were able to be captured. Thus, the multiple fluorescence bands (Fig. 7c, marked with blue triangles) could possibly be attributed to secondary protein interactions with 10k in mammalian cells, which can be mostly blocked by the AZA inhibitor. The long half-life of the photo-generated intermediate might result in an increased labeling radius from the drug binding site, depending on flexibility of the linker,24 which might result in off-target ligation. In contrast, a short-lived intermediate is excellent for designing PAL probes for drug profiling with fast binding kinetics and high spatial accuracy, but also leads to high sensitivity to the microenvironment and proximity. A long-lived intermediate with a flexible linker has sufficient degrees of freedom to rotate and stretch, and subsequently to capture inferior-affinity targets, which may find use in drug/target discovery because weakly interacting targets sometimes are cofactors for drug efficacy.50 Nevertheless, these results convincingly demonstrated the photo-DAFEx chemistry could be applied in PAL for drug profiling, which expands the arsenal for target verification, especially in combination with the short-lived but reactive diazo/carbene intermediates.
Conclusions
A photo-DAFEx system was developed for convenient controllable photo-conjugation toward primary/secondary amines and thiols under mild conditions. m-Trifluoromethylaniline serving as a photo-crosslinking module produces a relatively stable and easy-to-handle intermediate, benzoyl fluoride, via water-mediated photo-defluorination. Theoretical calculations and experimental evidence revealed that the PhF2C(sp3)–F bond is weakened in the excited triplet state by water molecules. Subsequent amidation of benzoyl fluoride shows a fluorogenic feature, which allows visualization of the covalent bond formation in situ. Based on these characteristics, exploitation of the photo-DAFEx in small-molecule conjugation, peptide cyclization, direct peptide/protein modification was successful. This strategy has also been expanded in PAL for fishing for drug targets in living mammalian cells. Given the unique nucleophile preference on native residues, the advancement of photo-DAFEx holds great potential for photo-defluorination synthesis, pharmacological research and 3D photo-printing of biomaterials.Data availability
Additional figures as described in the main text, all experimental procedures, synthetic procedures, copies of spectral data for the compounds, data processing, and computational details are available in the ESI.†Author contributions
L. D., Z. S., and Z. Y. initiated the idea, designed the experiments and wrote the manuscript. L. D. synthesized the main compounds and conducted all of the experiments. C. Z., Z. S., and C. H. carried out the theoretical calculations. B. l., Z. Z., J. F., T. L., and X. Z. helped with compound synthesis and in vitro experiments. All authors contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Natural Science Foundation of China (22001181 and 22077090), the Fundamental Research Funds for the Central Universities (20826041D4117) and the Institutional Research Fund from Sichuan University (2020SCUNL105). We also express our appreciation to Dr Lunzhi Dai at State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, for help with the LC-MS/MS analysis. We also thank the Xiaoming Feng laboratory (Sichuan University) for access to equipment.Notes and references
- J. Dong, K. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430 CrossRef CAS PubMed.
- G. J. Brighty, R. C. Botham, S. Li, L. Nelson, D. E. Mortenson, G. Li, C. Morisseau, H. Wang, B. D. Hammock, K. B. Sharpless and J. W. Kelly, Nat. Chem., 2020, 12, 906 CrossRef CAS PubMed.
- Q. Li, Q. Chen, P. C. Klauser, M. Li, F. Zheng, N. Wang, X. Li, Q. Zhang, X. Fu, Q. Wang, Y. Xu and L. Wang, Cell, 2020, 182, 85 CrossRef CAS PubMed.
- (a) T. Tamura and I. Hamachi, J. Am. Chem. Soc., 2018, 141, 2782 CrossRef PubMed; (b) N. Wang, B. Yang, C. Fu, H. Zhu, F. Zheng, T. Kobayashi, J. Liu, S. Li, C. Ma, P. G. Wang, Q. Wang and L. Wang, J. Am. Chem. Soc., 2018, 140, 4995 CrossRef CAS PubMed.
- D. Medina-Cleghorn, L. A. Bateman, B. Ford, A. Heslin, K. J. Fisher, E. D. Dalvie and D. K. Nomura, Chem. Biol., 2015, 22, 1394 CrossRef CAS PubMed.
- G. S. Kumar and Q. Lin, Chem. Rev., 2020, 121, 6991 CrossRef.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915 CrossRef CAS.
- Q. Xiong, T. Zheng, X. Shen, B. Li, J. Fu, X. Zhao, C. Wang and Z. Yu, Chem. Sci., 2022, 13, 3571 RSC.
- A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G. J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769 CrossRef CAS PubMed.
- G. S. Kumar, S. Racioppi, E. Zurek and Q. Lin, J. Am. Chem. Soc., 2022, 144, 57 CrossRef CAS PubMed.
- L. Zhang, X. Zhang, Z. Yao, S. Jiang, J. Deng, B. Li and Z. Yu, J. Am. Chem. Soc., 2018, 140, 7390 CrossRef CAS PubMed.
- S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2011, 133, 5573 CrossRef CAS PubMed.
- S. V. Mayer, A. Murnauer, M. von Wrisberg, M. Jokisch and K. Lang, Angew. Chem., Int. Ed., 2019, 58, 15876 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 CrossRef CAS PubMed.
- S. S. Zalesskiy, N. S. Shlapakov and V. P. Ananikov, Chem. Sci., 2016, 7, 6740 RSC.
- J. Li, H. Kong, L. Huang, B. Cheng, K. Qin, M. Zheng, Z. Yan and Y. Zhang, J. Am. Chem. Soc., 2018, 140, 14542 CrossRef CAS PubMed.
- Z. Li, L. Qian, L. Li, J. C. Bernhammer, H. V. Huynh, J. S. Lee and S. Q. Yao, Angew. Chem., Int. Ed., 2016, 55, 2002 CrossRef CAS.
- W. Hu, Y. Yuan, C. H. Wang, H. T. Tian, A. D. Guo, H. J. Nie, H. Hu, M. Tan, Z. Tang and X. H. Chen, Chem, 2019, 5, 2955 CAS.
- H. Fang, B. Peng, S. Y. Ong, Q. Wu, L. Li and S. Q. Yao, Chem. Sci., 2021, 12, 8288 RSC.
- S. Y. Dai and D. Yang, J. Am. Chem. Soc., 2020, 142, 17156 CrossRef CAS PubMed.
- K. Cheng, J. S. Lee, P. Hao, S. Q. Yao, K. Ding and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 15044 CrossRef CAS PubMed.
- A. C. S. Page, S. O. Scholz, K. N. Keenan, J. N. Spradlin, B. P. Belcher, S. M. Brittain, J. A. Tallarico, J. M. Mckenna, M. Schirle, D. K. Nomura and F. D. Toste, Chem. Sci., 2022, 13, 3851 RSC.
- A. D. Guo, D. Wei, H. J. Nie, H. Hu, C. Peng, S. T. Li, K. N. Yan, B. S. Zhou, L. Feng, C. Fang, M. Tan, R. Huang and X. H. Chen, Nat. Commun., 2020, 11, 5472 CrossRef CAS PubMed.
- J. V. Oakley, B. F. Buksh, D. F. Fernández, D. G. Oblinsky, C. P. Seath, J. B. Geri, G. D. Scholes and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203027119 CrossRef CAS PubMed.
- S. Rafqah and M. Sarakha, J. Photochem. Photobiol., A, 2016, 316, 1 CrossRef CAS.
- T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578 CrossRef CAS.
- Y. Ogiwara and N. Sakai, Angew. Chem., Int. Ed., 2020, 59, 574 CrossRef CAS PubMed.
- V. Glembockyte, R. Lincoln and G. Cosa, J. Am. Chem. Soc., 2015, 137, 1116 CrossRef CAS PubMed.
- T. Lu and Q. Chen, Comput. Theor. Chem., 2021, 1200, 113249 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Phys. Chem. A, 2013, 117, 3100 CrossRef CAS PubMed.
- C. Y. Legault, CYLview20, Université de Sherbrooke, 2020, http://www.cylview.org Search PubMed.
- S. Emamian, T. Lu, H. Kruse and H. Emamian, J. Comput. Chem., 2019, 40, 2868 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Theor. Comput. Chem., 2012, 11, 163 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- E. K. Woodman, J. G. K. Chaffey, P. A. Hopes, D. R. J. Hose and J. P. Gilday, Org. Process Res. Dev., 2014, 13, 106 CrossRef.
- F. Seidi, R. Jenjob and D. Crespy, Chem. Rev., 2018, 118, 3965 CrossRef CAS.
- W. Mao, J. Tang, L. Dai, X. He, J. Li, L. Cai, P. Liao, R. Jiang, J. Zhou and H. Wu, Angew. Chem., Int. Ed., 2021, 60, 2393 CrossRef CAS PubMed.
- L. P. Kozlowski, Nucleic Acids Res., 2017, 45, D1112 CrossRef CAS PubMed.
- H. Y. Chow, Y. Zhang, E. Matheson and X. Li, Chem. Rev., 2019, 119, 9971 CrossRef CAS PubMed.
- A. W. Dodds, X. D. Ren, A. C. Willis and S. K. A. Law, Nature, 1996, 379, 177 CrossRef CAS PubMed.
- T. Mino, S. Sakamoto and I. Hamachi, Biosci., Biotechnol., Biochem., 2021, 85, 53 CrossRef PubMed.
- J. Kim, B. X. Li, Y. C. Huang, J. X. Qiao and D. W. C. Macmillan, J. Am. Chem. Soc., 2020, 142, 21260 CrossRef CAS PubMed.
- E. Smith and I. Collins, Future Med. Chem., 2015, 7, 159 CrossRef CAS PubMed.
- K. Teruya, K. F. Tonissen and S. A. Poulsen, MedChemComm, 2016, 7, 2045 RSC.
- A. A. Barrese, C. Genis, S. Z. Fisher, J. N. Orwenyo, M. T. Kumara, S. K. Dutta, E. Phillips, J. J. Kiddle, C. Tu, D. N. Silverman, L. Govindasamy, M. Agbandje-McKenna, R. McKenna and B. C. Tripp, Biochemistry, 2008, 47, 3174 CrossRef CAS PubMed.
- A. D. Fiore, E. Truppo, C. T. Supuran, V. Alterio, N. Dathan, F. Bootorabi, S. Parkkila, S. M. Monti and G. De Simone, Bioorg. Med. Chem. Lett., 2010, 20, 5023 CrossRef PubMed.
- M. B. Elowitz, A. J. Levine, E. D. Siggia and P. S. Swain, Science, 2002, 297, 1183 CrossRef CAS PubMed.
- Z. Li, P. Hao, L. Li, C. Y. J. Tan, X. Cheng, G. Y. J. Chen, S. K. Sze, H. M. Shen and S. Q. Yao, Angew. Chem., Int. Ed., 2013, 52, 8551 CrossRef CAS PubMed.
- B. Procacci, S. S. Roy, P. Norcott, N. Turner and S. B. Duckett, J. Am. Chem. Soc., 2018, 140, 16855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Containing details on experimental procedures, spectra property, and characterization of all new compounds. See DOI: https://doi.org/10.1039/d2sc04636a |
| This journal is © The Royal Society of Chemistry 2023 |