
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA unique network of attack, defence and competence on the outer membrane of the periodontitis pathogen Tannerella forsythia†
Mirosław
Książek‡
ah,
Theodoros
Goulas‡
bc,
Danuta
Mizgalska‡
a,
Arturo
Rodríguez-Banqueri
b,
Ulrich
Eckhard
 b,
Florian
Veillard
a,
Irena
Waligórska
a,
Małgorzata
Benedyk-Machaczka
a,
Alicja M.
Sochaj-Gregorczyk
a,
Mariusz
Madej
a,
Ida B.
Thøgersen
d,
Jan J.
Enghild
d,
Anna
Cuppari
b,
Joan L.
Arolas
b,
Iñaki
de Diego
bi,
Mar
López-Pelegrín
b,
Irene
Garcia-Ferrer
b,
Tibisay
Guevara
b,
Vincent
Dive
e,
Marie-Louise
Zani
f,
Thierry
Moreau
g,
Jan
Potempa
*ah and
F. Xavier
Gomis-Rüth
*b
b,
Florian
Veillard
a,
Irena
Waligórska
a,
Małgorzata
Benedyk-Machaczka
a,
Alicja M.
Sochaj-Gregorczyk
a,
Mariusz
Madej
a,
Ida B.
Thøgersen
d,
Jan J.
Enghild
d,
Anna
Cuppari
b,
Joan L.
Arolas
b,
Iñaki
de Diego
bi,
Mar
López-Pelegrín
b,
Irene
Garcia-Ferrer
b,
Tibisay
Guevara
b,
Vincent
Dive
e,
Marie-Louise
Zani
f,
Thierry
Moreau
g,
Jan
Potempa
*ah and
F. Xavier
Gomis-Rüth
*b
aDepartment of Microbiology, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Gronostajowa 7, Kraków 30-387, Poland. E-mail: jan.potempa@icloud.com
bProteolysis Laboratory, Department of Structural Biology, Molecular Biology Institute of Barcelona (CSIC), Barcelona Science Park, c/Baldiri Reixac, 15-21, Barcelona 08028, Catalonia, Spain. E-mail: fxgr@ibmb.csic.es
cDepartment of Food Science and Nutrition, School of Agricultural Sciences, University of Thessaly, Temponera str., Karditsa 43100, Greece
dDepartment of Molecular Biology and Genetics, Aarhus University, Universitetsbyen 81, Aarhus C 8000, Denmark
eUniversité Paris-Saclay, CEA, INRAE, Département Médicaments et Technologies pour la Santé (DMTS), ERL CNRS 9004, Gif-sur-Yvette 91191, France
fDepartement de Biochimie, Université de Tours, 10 Bd. Tonellé, Tours Cedex 37032, France
gINRAE, Université de Tours, UMR BOA, Nouzilly 37380, France
hDepartment of Oral Immunology and Infectious Diseases, University of Louisville School of Dentistry, Louisville 40202, KY, USA
iSample Environment and Characterization Group, European XFEL GmbH, Holzkoppel 4, Schenefeld 22869, Germany
First published on 12th December 2022
Abstract
Periodontopathogenic Tannerella forsythia uniquely secretes six peptidases of disparate catalytic classes and families that operate as virulence factors during infection of the gums, the KLIKK-peptidases. Their coding genes are immediately downstream of novel ORFs encoding the 98–132 residue potempins (Pot) A, B1, B2, C, D and E. These are outer-membrane-anchored lipoproteins that specifically and potently inhibit the respective downstream peptidase through stable complexes that protect the outer membrane of T. forsythia, as shown in vivo. Remarkably, PotA also contributes to bacterial fitness in vivo and specifically inhibits matrix metallopeptidase (MMP) 12, a major defence component of oral macrophages, thus featuring a novel and highly-specific physiological MMP inhibitor. Information from 11 structures and high-confidence homology models showed that the potempins are distinct β-barrels with either a five-stranded OB-fold (PotA, PotC and PotD) or an eight-stranded up-and-down fold (PotE, PotB1 and PotB2), which are novel for peptidase inhibitors. Particular loops insert like wedges into the active-site cleft of the genetically-linked peptidases to specifically block them either via a new “bilobal” or the classic “standard” mechanism of inhibition. These results discover a unique, tightly-regulated proteolytic armamentarium for virulence and competence, the KLIKK-peptidase/potempin system.
1. Introduction
Excessive, deregulated, or sustained proteolytic activity correlates with histotoxic damage, apoptosis, and chronic inflammation, which contribute to neurodegenerative and vascular diseases, organ dysfunction, tumour growth and metastasis, and periodontitis. The latter is the most prevalent inflammatory infectious disease, affecting 65 million US adults (aged >30 years), with 9% progressing to severe clinical symptoms,1 and having detrimental effects on other systemic pathologies.2 Periodontitis has an immense impact on public health and quality of life, which costs tens of billions of dollars annually.3 However, the molecular basis of periodontal pathogenesis as a microbiome-shift disease is still poorly understood.4The currently accepted paradigm states that the disease is driven by dysbiotic flora, the “red complex” of oral bacteria (Porphyromonas gingivalis, Treponema denticola and Tannerella forsythia), which are assisted by a cohort of other periodontal pathogens.5 In a biofilm in the subgingival crevice, these bacteria form a tightly-knit community engaged in both competitive and cooperative interactions.6 Host neutrophils and macrophages cannot eradicate this community and fuel a chronic inflammatory response in the infected periodontium due to the massive release of pro-inflammatory factors and cytokines.7 These trigger dissolution of the periodontal ligament, alveolar bone resorption, deep periodontal pocket formation, and ultimately tooth loss in genetically susceptible individuals.8
Importantly, T. forsythia contributes directly to the pathogenicity of the dysbiotic microbial consortium by producing proteolytic enzymes as virulence factors that hinder and subvert the host-immune response.9 Most of these peptidases and other secreted virulence factors possess a ∼9 kDa C-terminal domain10 that serves as a translocation signal for a type-IX secretion system across the outer membrane to the extracellular space.11 The C-terminal domain is then removed and a lipopolysaccharide is attached to the new C-terminus, anchoring it to the outer-membrane outer leaflet.12 The first type-IX secretion system was identified in P. gingivalis11,13,14 and others have since been found in other species of Bacteroidetes, including T. forsythia.11,15 The proteomic analysis of the T. forsythia outer membrane revealed at least 26 proteins with a potential C-terminal domain.16
Recently, comparative genomics has revealed that the T. forsythia genome features two exclusive loci encoding six peptidases from five disparate families according to the MEROPS database17 (Fig. 1). One locus encodes forsilysin, a thermolysin-type M4-family metallopeptidase (MP), and miropsin-1, a trypsin-like S1D-family serine peptidase. The other encodes two metzincin-clan MPs,18 specifically mirolysin from family M43 (pappalysins;19,20) and karilysin from family M10A (matrix metalloproteinases (MMPs);21,22), as well as miropsin-2 (a trypsin-like S1D-family serine peptidase) and mirolase (a subtilisin-type S8A-family serine peptidase). Transcripts of all six active peptidases were detected in the gingival crevicular fluid of periodontitis sites.23 Their absence from non-pathogenic Tannerella species and any other bacteria, as well as their ability to degrade an array of host defence proteins, strongly supports a role in virulence. Indeed, three of them (karilysin, mirolysin, and mirolase) degraded the bactericidal LL-37 host peptide, induced the shedding of TNFα from the macrophage surface, and inactivated the host complement system.24–29 Miropsins, in turn, were strongly expressed in vivo in the subgingival dysbiotic bacterial biofilm, and the miropsin-2 transcript level correlated with the destruction of periodontal and peri-implant tissues.30 All six peptidases share the presence of a prodomain, a catalytic domain, a conserved region A, a variable region B, and the type-IX secretion system C-terminal domain, whose five C-terminal residues are identical (K–L–I–K–K). Accordingly, they were collectively named KLIKK-peptidases.23
 | ||
| Fig. 1 Arrangement of T. forsythia KLIKK-peptidase genes and upstream ORFs encoding PotA–PotE at two genomic loci. The gene distribution is based on manually curated T. forsythia ATCC 43037 sequences of the KLIKK-peptidase loci obtained by Sanger-based sequencing.23 The sequences are available at GenBank under accession numbers KP715369 and KP715368. KLIKK-peptidases are named above the genes (shown as pale grey arrows) along with the family assigned in the MEROPS database (S = serine peptidase, in blue; M = metallopeptidase, in red). The corresponding potempins (PotA–PotE) are named above the ORFs (shown as black arrows). White arrows denote other putative ORFs. The loci are drawn to scale. | ||
Here, we report the unexpected finding that each of the KLIKK-peptidase genes is preceded by a short open reading frame unrelated to any protein described thus far. We investigated their function and mechanism of action by genetic, phylogenetic, biochemical, structural, functional, and in vivo analysis, which revealed an unprecedented network of virulence and competence regulation on the T. forsythia cell surface.
2. Results and discussion
2.1 ORFs immediately upstream of the KLIKK-peptidase genes encode unique lipoproteins (potempins)
Analysis of the flanking sequences of the unique T. forsythia peptidase genes based on manually curated sequences of KLIKK-peptidase loci of T. forsythia ATCC 43037 obtained by Sanger-based sequencing23 revealed coding DNA sequences whose stop codons either overlapped with or were ≤5 base pairs upstream of the start codons of the peptidases (Fig. S1A†). The exception was the ORF preceding the gene encoding miropsin-2, which was 876 base pairs upstream. All ORFs were co-transcribed with the linked peptidase genes (Fig. S2A and B†), and the bicistronic nature of the operon was further confirmed by the comparable expression levels of the individual ORFs and the transcripts spanning both genes (Fig. S2C†).The DNA sequences preceding the KLIKK-peptidases encoded proteins of 118–152 residues (Fig. 1), including signal peptides of 20 residues that were predicted with high confidence (Fig. S1B†). To assess the distribution of these ORFs and the cognate KLIKK-peptidases, we analysed the genomes of 10 contemporary and four ancient Tannerella strains.31 The genes were absent from all non-pathogenic strains, suggesting they represent a disparate group of T. forsythia virulence factors. We therefore cloned all the novel ORFs preceding the KLIKK-peptidases and expressed the recombinant proteins for further analysis (Fig. S3 and Table S1†). We named them potempin (Pot) A, B1, B2, C, D, and E (see Fig. S4B† for the UniProt database codes (UP)). Of note, we identified several other “orphan” ORFs in T. forsythia encoding putative small lipoproteins of unknown function and structure, which shared highly similar signal peptides with the potempins. One was found in an assumed operon further encompassing a thermolysin-like protease, which may recall a potempin/KLIKK-peptidase pair (BFO_0702/BFO_0703 and FJN16_03485/FJN16_03490, in strains 92A2 and ATCC 43037, respectively). However, given the complexity of the assembly of T. forsythia genomes, which is exemplified by the misassignment of potempin and KLIKK-peptidase genes in strains ATCC 43037 and 92A2 (see Fig. S2D†), the existence of this locus needs to be experimentally verified.
Analysis of the mature sequences revealed pairwise identities among potempins of only 4–14%, with the exception of the closely-related paralogues PotB1 and PotB2, which showed 82% identity (Fig. S1B†). Sequence-based phylogenetic analysis suggested that PotA might be more closely related to PotD, followed by PotE, whereas PotC clustered weakly with PotB1 and PotB2 (Fig. S4A†). Remarkably, the signal peptides showed 65% identity, even though they are cleaved following secretion and do not affect the mature protein structure or function (Fig. S4B†). Contrary to expectations, the signal peptides of PotA and PotE, as well as those of PotC and PotD, were even identical, whereas those of the closely related PotB paralogues were the most divergent. This contrasts with the 19/20-residue signal peptides of the cognate KLIKK-peptidases, which were divergent except for a few highly-conserved residues (Fig. S4C†). The evolutionary implications of these observations are discussed later (see Section 2.12).
2.2 Potempins specifically inhibit their genetically-linked peptidases
In rare cases, the activity of bacterial peptidases is controlled by co-transcribed protein inhibitors,32 such as Staphylococcus sp. staphopains by staphostatins,33Streptococcus pyogenes streptopain by its endogenous inhibitor,34 and serralysin MPs from Erwinia, Pseudomonas, and Serratia spp. by endogenous serralysin inhibitors.35,36 We thus tested the ability of the potempins to inhibit recombinant KLIKK-peptidases (karilysin, mirolase, mirolysin, and forsilysin) in vitro. We found that each potempin inhibited its co-transcribed peptidase but not the others. The stoichiometry of inhibition was close to 1 in all cases (Fig. 2A) and the formation of stoichiometric inhibitor:peptidase complexes was confirmed by size-exclusion chromatography and gel electrophoresis (Fig. S5A and B†). Exceptionally, the PotE:forsilysin complex (stoichiometry of inhibition = 1.55 ± 0.06) contained a fraction of truncated inhibitor (cleaved at A115–I116, potempin residue numbering in superscript according to the corresponding UP, see Fig. S4B†), explaining the slightly higher stoichiometry of inhibition. In all cases, peptidase inhibition by the co-transcribed potempin followed a Michaelis–Menten kinetics (Fig. 2B) and was consistent with reversible competitive inhibition (Fig. 2C). All derived Ki values were in the nanomolar range (2–10 nM), which indicates potent inhibition (Fig. 2B). There was not enough functional miropsin-1 or miropsin-2 available to determine the inhibition kinetics, but we qualitatively observed that miropsin-1 was inhibited by PotB1—in a concentration dependent manner—but not PotB2 (Fig. S6†).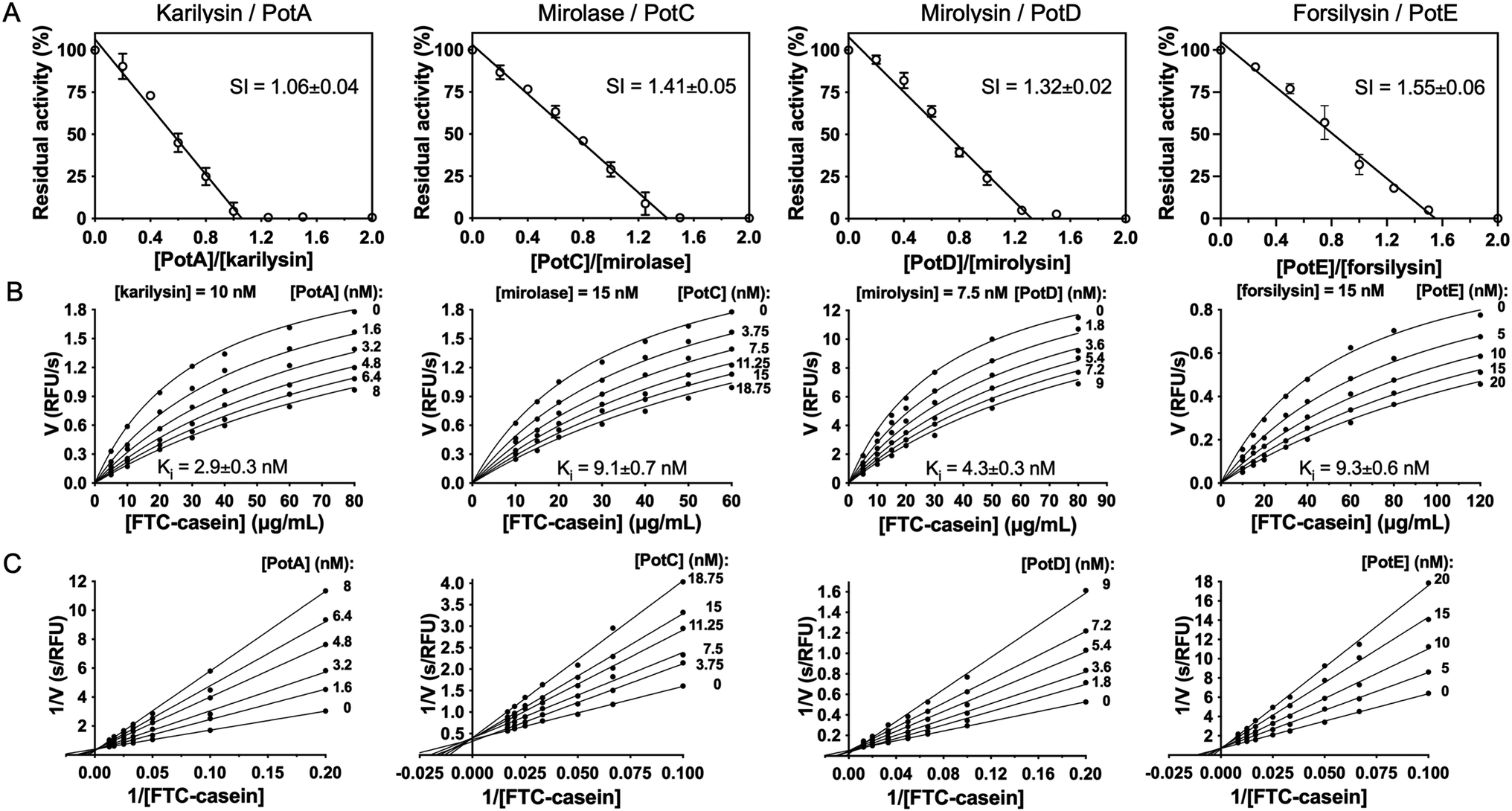 | ||
Fig. 2 Potempins efficiently inhibit their co-transcribed KLIKK-peptidases. (A) Stoichiometry of inhibition (SI) values of potempins PotA–PotD in the presence of karilysin, mirolase, mirolysin and forsilysin, respectively, indicate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes. Peptidases were incubated with increasing amounts of inhibitor for 15 min, and the residual enzymatic activity was measured and plotted against the inhibitor:peptidase molar ratio. Enzymatic activity in the absence of inhibitor was taken as 100%. Data are means ± SD (n = 3). (B) Velocities of reaction (V) for the same complexes were determined for different concentrations of fluorescence-conjugated casein (FTC-casein) and plotted against the substrate concentration at various inhibitor concentrations. The derived Ki values are means ± SD (n = 3). (C) Double reciprocal plots of V against substrate at various inhibitor concentrations showed that inhibition is reversible and competitive. :1 complexes. Peptidases were incubated with increasing amounts of inhibitor for 15 min, and the residual enzymatic activity was measured and plotted against the inhibitor:peptidase molar ratio. Enzymatic activity in the absence of inhibitor was taken as 100%. Data are means ± SD (n = 3). (B) Velocities of reaction (V) for the same complexes were determined for different concentrations of fluorescence-conjugated casein (FTC-casein) and plotted against the substrate concentration at various inhibitor concentrations. The derived Ki values are means ± SD (n = 3). (C) Double reciprocal plots of V against substrate at various inhibitor concentrations showed that inhibition is reversible and competitive. | ||
We investigated the specificity of the inhibitors in more detail by testing a cohort of other peptidases. PotA did not inhibit Staphylococcus aureus aureolysin (M4 family), Serratia sp. serralysin (M10B) or protealysin (M4), or Pseudomonas aeruginosa LasB (alias pseudolysin; M4) or aeruginolysin (M10B). Similarly, PotB1 and PotB2 did not inhibit S1-family serine peptidases (bovine trypsin, bovine chymotrypsin, porcine pancreatic elastase, human neutrophil elastase or human cathepsin G). Among S8-type serine peptidases, PotC inhibited Bacillus licheniformis subtilisin Carlsberg as a reversible competitive inhibitor with an apparent Ki of 82 nM (Fig. S7A and B†) but with a stoichiometry of inhibition of 6 (Fig. S7C†). The formation of the inhibitory complex was confirmed by size-exclusion chromatography (Fig. S7D and E†), but it was unstable and PotC was cleaved within the complex, so the inhibition efficiency was much lower than against mirolase (Fig. S7F and G†). Other serine peptidases, including the physiologically-relevant S8-family members Fusobacterium nucleatum fusolisin, Treponema denticola dentilisin, and Streptococcus gordonii challisin, as well as the aforementioned S1-family serine peptidases, were unaffected by PotC. PotD was inactive against Methanosarcina acetivorans ulilysin (M43 family) and PotE did not inhibit the M4-family MPs thermolysin from Bacillus thermoproteolyticus, aureolysin and LasB, even at a 25-fold molar excess. Taken together, these data suggest that potempins evolved as regulators to specifically and potently inhibit their genetically-linked KLIKK-peptidases.
2.3 PotA is a novel specific inhibitor of MMP-12
The MMP family is part of the metzincin clan of MPs and is responsible for many physiological and pathological functions in mammals, with 23 paralogues in humans.21 The four tissue inhibitors of metalloproteinases (TIMPs; MEROPS family I35) are the only endogenous protein inhibitors, and they broadly inhibit MMPs.37 The karilysin catalytic domain includes all the characteristic MMP features and is currently the only structurally and functionally characterized member of this family outside mammals.22,38 We therefore tested the ability of PotA to inhibit a comprehensive cohort of mammalian MMPs (Table S2†). The activity of full-length MMPs 1, 2, 3, 7, 9 and 14 was unaffected, and that of full-length MMPs 8, 10 and 13, as well as the catalytic domain of MMP-20, was only slightly impaired (Ki > 100 mM). In contrast, human and murine MMP-12 (full-length and isolated catalytic domains) were strongly inhibited by PotA (Ki = 5–10 nM). PotA is therefore a novel, potent, and highly-specific inhibitor of MMP-12. Whereas TIMPs are 22–29 kDa proteins with N-terminal and C-terminal domains responsible for broad-spectrum MMP inhibition and protein binding functions, respectively,39 PotA features a single compact 98-residue domain that fulfils the inhibitory function (Section 2.9).2.4 Potempins and their co-transcribed peptidases form stable complexes
The neutralisation of potentially damaging proteolytic activity requires the formation of stable inhibitor:peptidase complexes. Accordingly, we incubated preformed PotA:karilysin, PotC:mirolase, PotD:mirolysin and PotE:forsilysin complexes at different temperatures and assessed the integrity of the inhibitors over time by SDS-PAGE (Fig. 3A). After 24 h, intact PotA, PotC and PotD were recovered from their complexes, regardless of the incubation temperature. Furthermore, the residual activity increased only slightly – although significantly – from 1% in the control to 2–4% in samples incubated at 20 °C or 37 °C (Fig. 3B). In contrast, the PotE:forsilysin complex released the aforementioned truncated form of the inhibitor under all conditions we tested. There was generally no further degradation of the inhibitor and no significant activity was detected, although the incubation at 37 °C was an exception. This is reminiscent of the specific insect metallopeptidase inhibitor (IMPI) from Galleria mellonella, which strongly inhibits selected thermolysins despite being cleaved at its inhibitory loop.40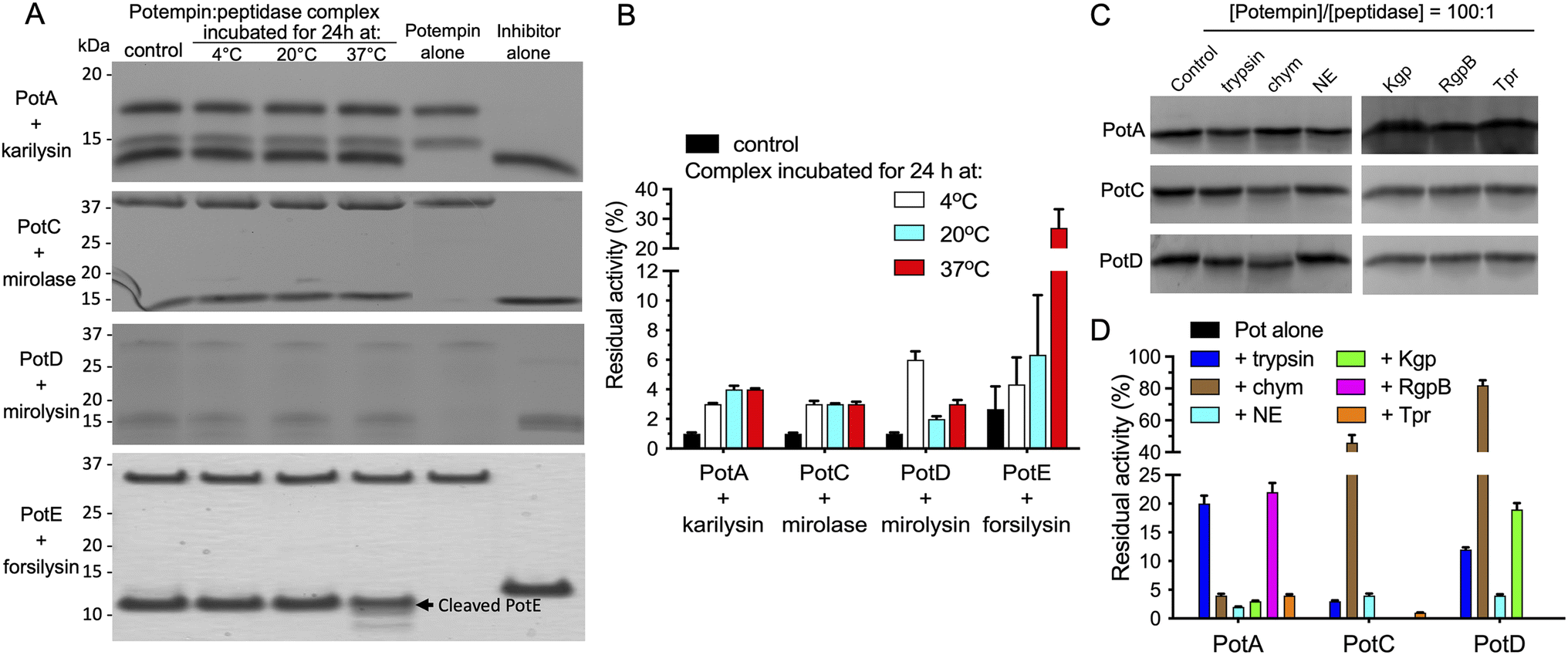 | ||
| Fig. 3 Stability of the inhibitory complexes and resistance of potempins to proteolysis. Inhibitory complexes of the potempins and their target peptidases at the stoichiometry of inhibition shown in Fig. 2 were incubated at different temperatures for 24 h. (A) The integrity of the complex components was evaluated by SDS-PAGE. (B) The residual peptidase activity released from the complex was determined with Azocoll as the substrate. As a control, we determined the residual activity of the complex incubated for 15 min at room temperature (“control”). (C) PotA, PotC or PotD (80 μM) were incubated with bovine trypsin and chymotrypsin (chym), neutrophil elastase (NE), P. gingivalis cysteine peptidase Tpr or gingipains (Kgp and RgpB) at a 100:1 molar ratio at 37 °C for 8 h to determine their resistance to proteolysis. The samples were then analysed by SDS-PAGE. (D) The residual inhibitory activity of PotA, PotB and PotC pre-incubated with non-target peptidases was determined by measuring their inhibitory potency against the targeted KLIKK-peptidases karilysin, mirolase and mirolysin, respectively, with Azocoll as the substrate. The activity of KLIKK-peptidases incubated alone was taken as 100%. Data are means ± SD (n = 3). | ||
Next, we investigated the stability of the potempins against proteolytic degradation by incubation with non-target peptidases and subsequent assessment of their capacity to inhibit their cognate KLIKK-peptidases. PotA, PotC and PotD were insensitive to cysteine peptidases secreted by P. gingivalis (Tpr and gingipains), which co-occur with T. forsythia in the subgingival biofilm, and human neutrophil elastase, which is abundant in inflammatory exudates (Fig. 3C). This resistance to proteolytic inactivation by non-targeted proteases is consistent with the competence of the potempins to control the activity of KLIKK-peptidases in the highly proteolytic environment of periodontal pockets. PotD and PotC were inactivated by the exogenous peptidase chymotrypsin, which caused ∼90% and ∼50% loss in inhibitory activity against mirolysin and mirolase, respectively, while PotA was resistant (Fig. 3D). Notably, inactivation correlated with a small decrease in the molecular mass of PotD (Fig. 3C), which suggests cleavage of the C-terminally located inhibitory segment.
2.5 Potempins are outer-membrane-anchored lipoproteins
All six potempins featured a variant of the lipobox motif [LVI][ASTVI][GAS]C at the end of the signal peptide (Fig. S4B†), which is a characteristic of bacterial lipoproteins.41 The conserved cysteine becomes the new N-terminal residue of the mature protein and undergoes lipid modification for membrane anchorage.42 Consistently, some potempins, including PotA, were identified in the proteome of the outer membrane16 and the outer-membrane vesicles of T. forsythia.43 However, the exact subcellular localisation (inner-membrane outer leaflet, outer-membrane inner leaflet or outer-membrane outer leaflet) cannot be predicted with confidence.44 Therefore, we used antibodies against PotA to detect it in T. forsythia membrane fractions enriched for the outer membrane. We used TfsA and TfsB as outer-membrane markers. These highly-glycosylated proteins (200 and 210 kDa) are secreted by the type-IX secretion system15 and are unique to the semicrystalline S-layer on the surface of T. forsythia (Fig. 4A).45 As shown in Fig. 4B and C, PotA was mainly located in the outer-membrane fraction, with the small amount in the inner-membrane fraction probably reflecting contamination with outer-membrane material during sample preparation (supported by the presence of small amounts of TfsA and TfsB). The association of PotA with the outer-membrane was confirmed by its detection in outer-membrane vesicles (Fig. 4D) in a dot-blot analysis in which intact cells and lysed cells produced equivalent signals when probed with anti-PotA antibodies. In this assay, a biotinylated inner-membrane T. forsythia protein, as well as a PotA-deficient mutant strain (potAnull), were used as controls (Fig. 4D). Moreover, using the FACS approach, we unambiguously verified the surface location of PotA (Fig. 4G). Accordingly, the proteolytic activity of karilysin, mirolase and mirolysin was inhibited by washed, intact T. forsythia cells and outer-membrane vesicles (Fig. 4E), which further confirms the cell-surface location of PotA and the other potempins. Finally, only wild-type T. forsythia but not the potAnull mutant inhibited karilysin in a concentration-depended manner (Fig. 4F). Given the small size of the potempins compared with TsfA/TfsB, the former must be intercalated between the latter close to the outer-membrane surface, either intercalated or beneath the S-layer. | ||
| Fig. 4 Potempins are located on the T. forsythia cell surface. (A) Transmission electron microscopy images of a blebbing (outer membrane vesicle forming) T. forsythia ATCC 43037 cell. CM = (inner) cell membrane, OM = outer membrane, S = S-layer composed of TfsA and TfsB glycoproteins (adapted from ref. 107 with permission). (B) Comparison of PotA distribution in T. forsythia fractions derived from whole culture (WC), washed cells (C), cell-free culture medium (M), outer-membrane vesicles (OMV), soluble proteins derived from cytoplasm and periplasm (C/P), and cell envelope (CE). (C) Detection of PotA in cell envelope (CE), outer (OM) and inner (IM) membranes by western blotting (top panel) and visualisation of TfsA/TsfB by protein staining (bottom panel). (D) Dot-blot analysis of intact wild-type T. forsythia (WT-Tf) and potAnull cells (upper row) and cells lysed by sonication (lower row) to detect PotA (left panel) and a biotinylated IM protein (right panel). Dotted circles indicate sites where dots were placed on the nitrocellulose. (E) Inhibition of karilysin, mirolase, and mirolysin proteolytic activity against Azocoll as the substrate by intact, washed T. forsythia cells (red bars) and OMV (black bars). The activity of the isolated peptidases (green bars) was taken as 100%. Data are means ± SD (n = 3). (F) Inhibition of karilysin proteolytic activity against Azocoll as the substrate by OMV produced by wild-type T. forsythia (WT-Tf) and the potAnull strain. The activity of the peptidase alone was taken as 100% and the amount of OMV was standardized based on protein concentration determined by the BCA assay. Data are means ± SD (n = 3). (G) Flow cytometry analysis showing the surface exposure of PotA in T. forsythia wild-type cells (WT-Tf) (left panel) and in the potAnull mutant strain (right panel) using anti-PotA antibodies. | ||
2.6 PotA protects surface proteins from degradation by karilysin but does not contribute to prokarilysin secretion and maturation
The surface location of the potempins may protect T. forsythia surface proteins such as TsfA/TsfB and BspA from KLIKK-peptidases. Of note, BspA is an important virulence factor of T. forsythia,46 which should not be hindered by endogenous proteins. To assess this hypothesis, we compared the SDS-PAGE protein band profile and the content of BspA in the cell envelope and outer-membrane vesicles of wild-type T. forsythia with that of the potAnull mutant strain. Although the protein pattern of the cell envelope and outer-membrane vesicles was similar in both strains (Fig. 5A), western blotting analysis revealed a significantly fainter immunoreactive band for BspA in both fractions of the mutant strain (Fig. 5B and S8†). This finding underpins that PotA protects BspA from degradation.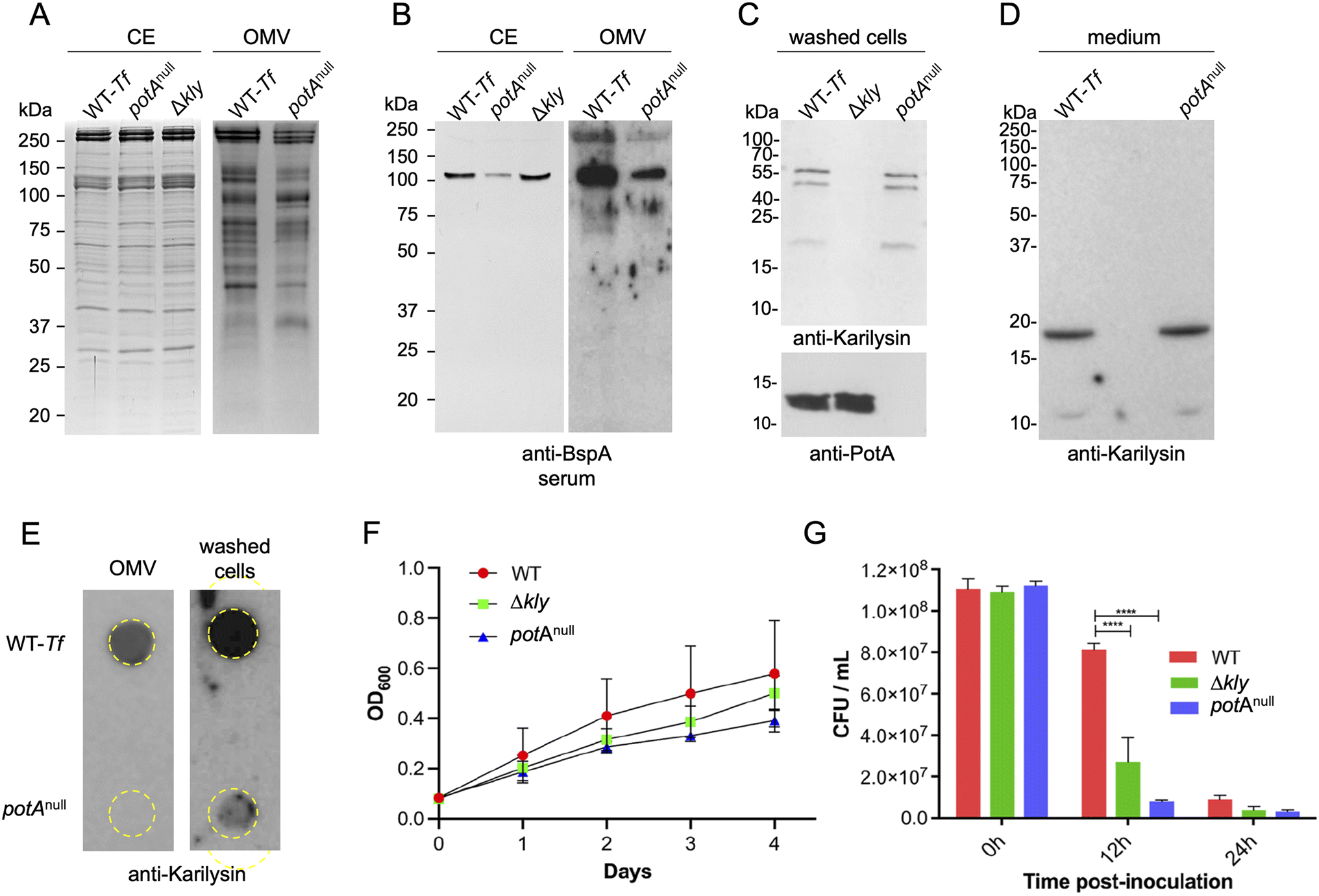 | ||
| Fig. 5 PotA protects the outer membrane against karilysin without affecting the maturation of the latter and further contributes to T. forsythia competence in vivo. (A) Analysis of proteins in the cell envelope (CE) and outer-membrane vesicles (OMV) of wild-type T. forsythia (WT-Tf) and the karilysin-null (Δkly) and potAnull strains by SDS-PAGE and (B) western blotting using rabbit antisera against BspA. Protein amounts were normalized based on total protein concentration determined by the BCA assay. Detection of karilysin in washed cells (C) and particle-free culture medium (D) of wild-type T. forsythia and potAnull strains by western blotting. The Δkly strain was used as a control for the specificity of the anti-karilysin IgGs used. (E) Dot-blot analysis of the wild-type T. forsythia and potAnull outer-membrane vesicles and intact cells. Dotted circles indicate sites where dots were placed on the nitrocellulose. The results shown are representative of 2–3 independent biological replicates (see also Fig. S8†). (F) Wild-type T. forsythia (WT) and strains lacking PotA (potAnull) or karilysin (Δkly) were inoculated into medium and the OD600 was recorded as a proxy of bacterial growth. Data are means ± SD (n = 2 technical replicates) and are representative of three independent experiments. (G) Subcutaneous chambers (six mice per group) were inoculated with 109 colony-forming units of parental (WT) and mutant (potAnull or Δkly) strains of T. forsythia. Aliquots of the chamber contents were withdrawn immediately after inoculation, after 12 h and after 24 h, and then serially diluted and plated on agar in triplicate. After 8 days, the colony-forming-unit count was determined. Data are means ± SD (n = 6; one-way ANOVA, **** = p < 0.0001). | ||
We next analysed the effect of PotA on the maturation of karilysin. Notably, KLIKK-peptidases are secreted as latent zymogens, which are autocatalytically activated.27,29,38,47–49 Western blotting of washed T. forsythia cells revealed the presence of full-length prokarilysin (55 kDa) and a partially processed form (48 kDa) as major immunoreactive bands, while only minor amounts of mature karilysin (18 kDa) were detected (Fig. 5C). Only mature karilysin was found in the culture medium (Fig. 5D). In all cases, karilysin was detected regardless of the presence of PotA. These results underpin that karilysin is processed and released to the extracellular environment in a PotA-independent manner.
We further assessed the presence of karilysin in outer-membrane vesicles and on the bacterial cell surface by dot-blot analysis using the wild type and the potAnull strain, which lacks PotA activity but expresses karilysin normally (Fig. 5C). Karilysin was detected only in intact cells and outer-membrane vesicles of the wild type but not the mutant strain (Fig. 5E). Apparently, outer-membrane vesicles are scavenging mature karilysin secreted into culture medium by forming stable inhibitory complexes, which are also present on the T. forsythia cell surface. Cumulatively, these results confirm that PotA – and most likely the other protempins – protect the surface of T. forsythia against the endogenous KLIKK-peptidases, which are secreted via the type-IX secretion system and released into the environment as mature enzymes.
2.7 PotA contributes to T. forsythia competence in vivo
Given the high selectivity of the potempins and the importance of protecting the bacterial cell surface from endogenous virulence factors (see Section 2.6), we investigated the biological function of PotA by means of the potAnull strain. Initially, this strain grew comparably to the wild type and a strain mutant for karilysin (Δkly) in rich medium in vitro (Fig. 5F). However, after 2–4 days potAnull accumulated less biomass than the wild type and Δkly, which we attribute to the unrestrained activity of karilysin on endogenous surface proteins compromising the integrity of the outer membrane. Next, we compared strain vitality using a murine model of subcutaneous chambers, in which bacteria are challenged by the host immune system (Fig. 5G). We found that the potAnull strain was severely compromised in vivo. Together with the data presented in Sections 2.2 and 2.5, this suggests that PotA (and by extrapolation the other potempins) protects the outer membrane against the endogenously secreted KLIKK-peptidases, thus providing an important element of competence. This is reminiscent of the T. forsythia serpin-type inhibitor miropin, which blocks the virulence cysteine peptidases Kgp and Tpr secreted by red-complex partner P. gingivalis and protects against the host peptidases neutrophil elastase, cathepsin G and plasmin.50–52 In the particular case of PotA, this protection probably extends to MMP-12, which is a major defence component secreted by host macrophages,53,54 thus also contributing indirectly to bacterial competence via a secondary route.2.8 Potempins feature two types of β-barrel scaffolds
To investigate the molecular determinants of potempin functionality, we obtained structural data from seven experimental crystal structures and four high-confidence comparative models (Tables S3 and S4†). This revealed that potempins are antiparallel β-barrels with hydrophobic cores traversing the structures top to bottom. Three proteins (PotA, PotC and PotD; Fig. 6B–D) adopt a five-stranded (β1–β5) Greek-key oligonucleotide/oligosaccharide-binding fold (OB-fold55), whereas the rest (PotE and the PotB proteins; Fig. 6G–I) are eight-stranded (β1–β8) up-and-down class-IV β-hairpin-repeat barrels.56 Inspection of their coulombic surfaces reveals an overall homogeneous distribution of charges, with no conspicuous charged patches (Fig. S4D†). This is consistent with these inhibitors interacting with their peptidase targets mainly through selected surface loops (see Section 2.9).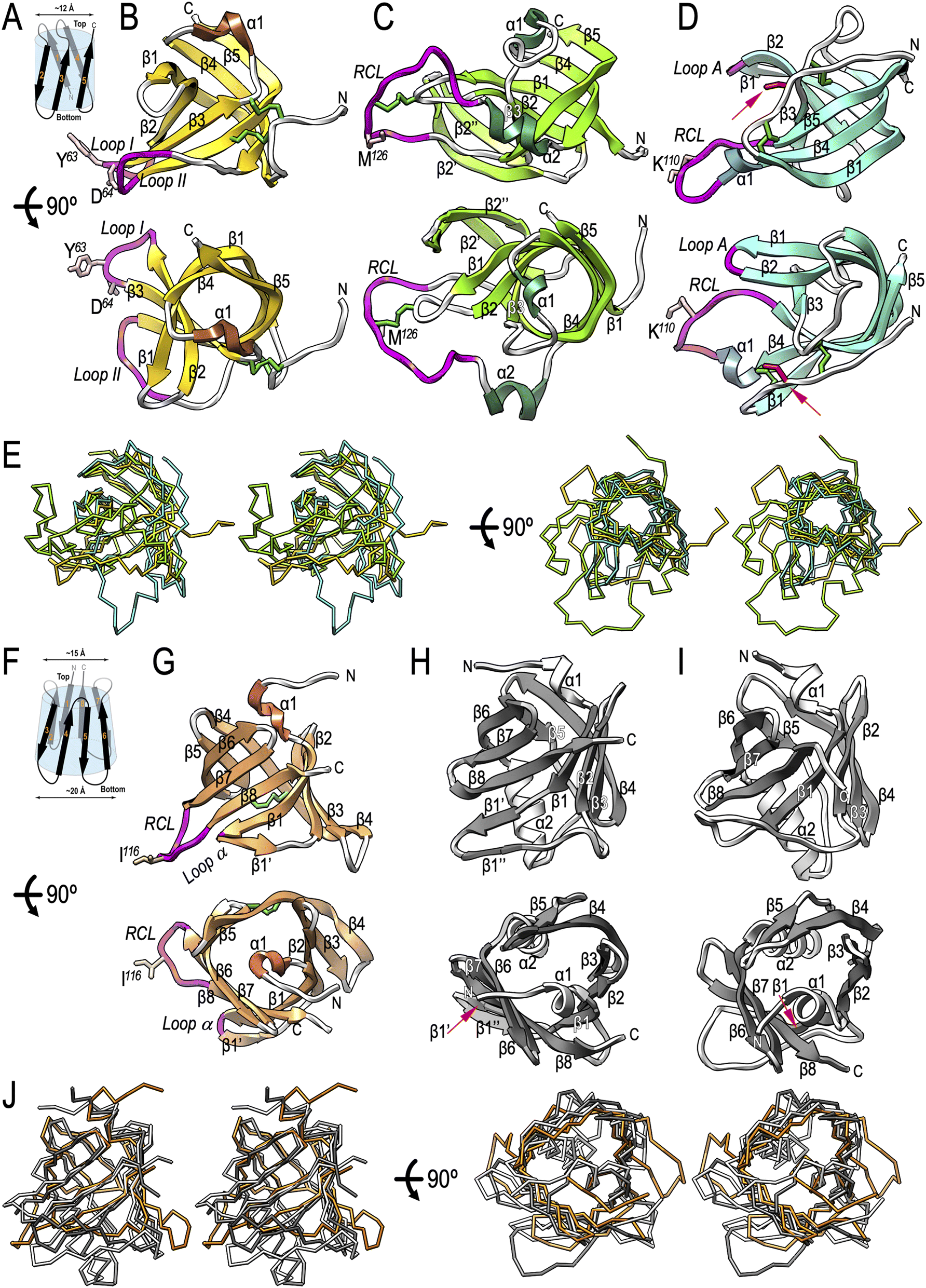 | ||
| Fig. 6 Potempin structures. (A) Topology of the five-stranded (β1–β5) Greek-key oligonucleotide/oligosaccharide binding (OB)-fold featuring the cylindrical β-barrels that scaffold the experimental structures of PotA (B), PotC (C) and PotD (D). All strands are antiparallel except edge-strands β1 and β5, and they adopt a clockwise swirl, which results in the strands being rotated ∼40° with respect to the barrel axis. Panels (B)–(D) show ribbon-type plots in two orthogonal views, parallel to (top) and down the barrel axis (bottom). (E) Superposition of the Cα traces of PotA (gold), PotC (green) and PotD (cyan) in cross-eye stereo in broadside (left) and end-on (right) views with respect to the barrel axis. (F) Topology of the antiparallel eight-stranded (β1–β8) up-and-down class-IV β-hairpin-repeat barrel shaped as a conical frustum that is found in the experimental structure of PotE (G) and the predicted structures of PotB1 (H) and PotB2 (I), which are shown in the two orthogonal orientations of (B)–(D) (top and bottom). The first four strands are rotated ∼20° with respect to the barrel axis, the second four by ∼60°. (J) Same as (E) for PotE (orange), PorB1 (white) and PotB2 (grey). In the distinct panels, regular secondary-structure elements are depicted as arrows (β-strands) or ribbons (α-helices) and are labelled, as are the N/C-termini. Regions engaged in peptidase inhibition are shown in magenta. Important functional residues are depicted for their side chain and are labelled. Disulfide bonds appear as green sticks, and an unpaired cysteine in (D) as red sticks is indicated by a red arrow. RCL = reactive-centre loop. | ||
The superposition of PotA, PotC and PotD (Fig. 6A and Table S5†) revealed that their central β-barrel scaffolds are cylinders with an inner diameter of ∼12 Å, which coincide for the five constituent β-strands, both in orientation and connectivity (Fig. 6E). The smallest potempin (PotA, 98 residue) adopts the minimal β-barrel structure (Fig. 6B). The loops (L) connecting strands β2 and β3 (Lβ2β3) and Lβ4β5 feature “loop I” (A60–D64) and “loop II” (I98–G103), respectively, which protrude ∼12 Å from the barrel surface and are engaged in target inhibition, in particular via Y63 and D64 (Section 2.9). PotC (132 residues) includes a large segment encompassing a “reactive-centre loop” (RCL; F116–I129), which is grafted between α2 and β5 and runs in a near extended conformation for E120–M126 (Fig. 6C). It projects ∼20 Å from the barrel and is linked via a disulfide bond (C42–C124) to a subjacent “scaffold loop” (T39–A46), which provides overall rigidity to the RCL around a “reactive-site bond” (ref. 57; M126–N127) that is essential for peptidase inhibition (Section 2.10). A curved β-ribbon (β2′β2′′) inserted after β2 sticks out from the bottom lateral barrel surface and is folded back to support the scaffold loop. Finally, PotD (105 residues) also contains a RCL (K106–V114) extending ∼20 Å away from the barrel surface (Fig. 6D), with K110 playing a major functional role (Section 2.11). Moreover, β-hairpin β1β2 (“loop A”) projects ∼13 Å from the surface of the barrel and may act as an ancillary inhibitory element. Finally, an unpaired cysteine (C31; Fig. 6D) gave rise to covalent dimers in the crystal structure of PotD, but these were deemed functionally irrelevant. Remarkably, all main functional loops in the OB-fold potempins – the RCL in PotC and PotD, and loop II in PotA – are inserted between strands β4 and β5 and project away from the bottom lateral barrel surface (Fig. 6B–D).
Among the β-hairpin-repeat-barrel potempins (Fig. 6F and Table S5†), PotE (107 residues) contains a RCL (D112–L119) that is inserted between β7 and β8 and is centred on I116 for inhibition (Fig. 6G and Section 2.11). Moreover, Lβ1β1′ (“loop α”) may play an ancillary role in peptidase binding. Finally, we obtained two high-confidence computational models of PotB1 and PotB2 using AlphaFold,58 which predicted they are also eight-stranded barrels (Fig. 6H and I). The superposition of PotE, PotB1 and PotB2 revealed that this subfamily has the shape of a conical frustum with top and bottom diameters of ∼15 Å and ∼20 Å, respectively (Fig. 6F and J). The β-strands of all the structures coincide in orientation and connectivity.
2.9 PotA is a novel bilobal inhibitor of karilysin and MMP-12
We solved the structures of PotA in complexes with human MMP-12 and T. forsythia karilysin (Table S3†). The inhibitor inserts like a wedge into the active-site clefts of both MMPs through small interfaces (390 Å2, ΔiG = −4.6 kcal mol−1 and 370 Å2, ΔiG = −4.7 kcal mol−1, respectively), so that loops I and II occupy primed (S1′–S3′) and non-primed (S2–S5) sub-sites of the cleft (sub-site nomenclature in bold, see ref. 59 and 60) through N61–Y63 and T99–A102, respectively (Fig. 7A–C). These residues bind the cleft is a similar manner to a true MMP substrate, except that loop-I residues bind in the reverse direction. Specifically, A102 occupies S2 and Y63 penetrates the S1′ specificity pocket, which, depending on the MMP paralogue, can accommodate medium-to-large hydrophobic side chains.61 PotA selectively and potently inhibited MMP-12 only across mammalian MMPs (Section 2.3 and Table S2†) because this orthologue has a very deep, tube-like S1′ pocket that traverses the entire molecule62 and easily allows room for the side chain of Y63. Remarkably, the S1 sub-site of the cleft is free, which explains why the inhibitor is not cleaved. In addition, the carboxylate of D64 provides a strong warhead that binds and thus blocks the catalytic zinc, and further contacts the general base/acid glutamate of MMP-12 (E219; peptidase numbering in subscript, here according to UP P39900; Fig. 7C) or karilysin (E156; for UP, see Fig. S4C†). Superposition of the MMP-12 and karilysin complexes revealed near identical binding except for a small relative rotation of the MMP moieties of ∼9° (Fig. 7B). Comparison of unbound and complexed PotA revealed that significant rearrangement occurs in loop I to allow optimal accommodation to the MMP active-site clefts by enabling zinc binding through D64 and rotation of Y63 to penetrate the S1′ pockets.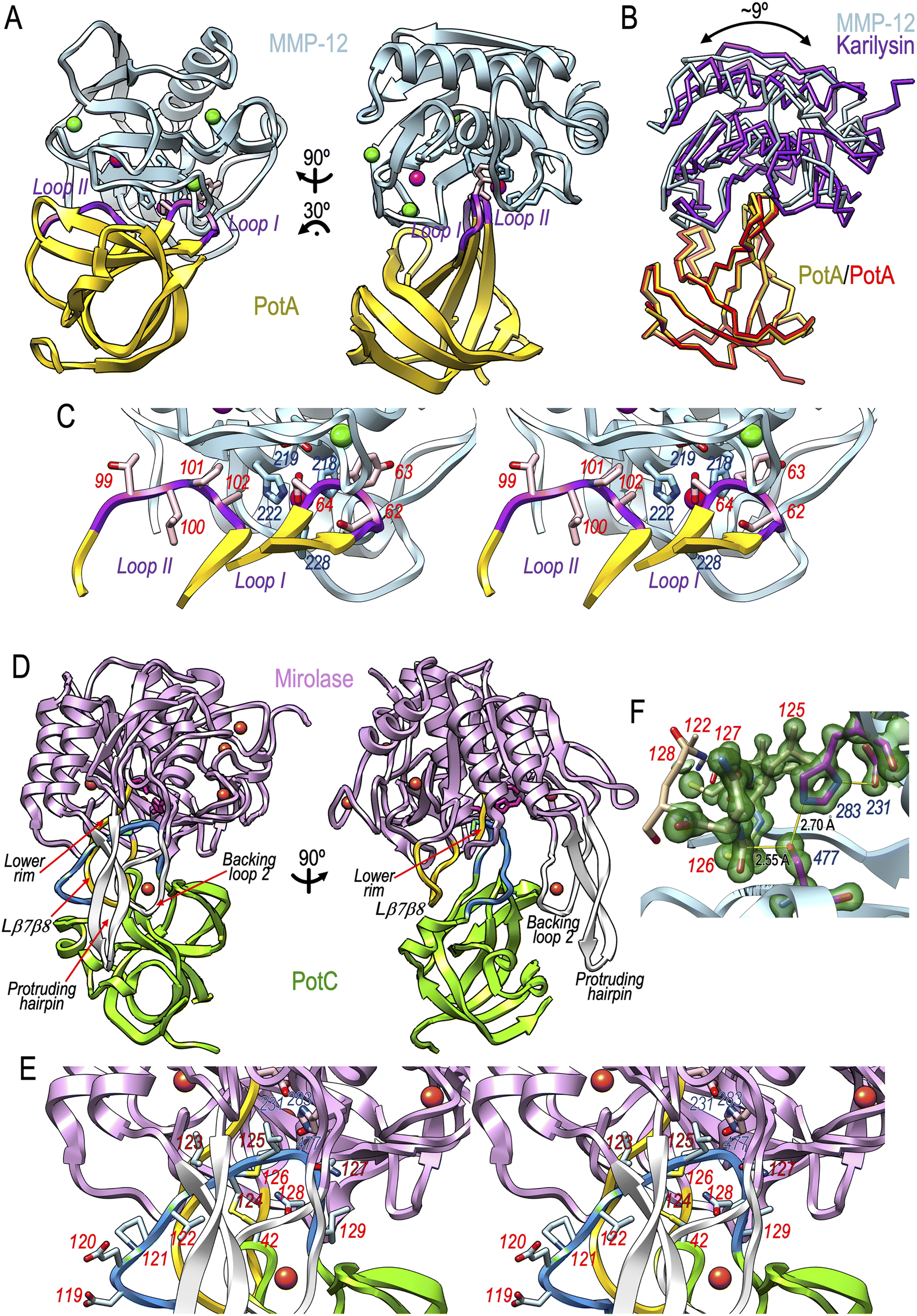 | ||
| Fig. 7 Structures of PotA and PotC complexes with peptidases. (A) Ribbon-type plot of PotA (in gold; loops I and II in purple) in a complex with the catalytic domain of human MMP-12 (in light blue) in two orientations (left and right). In the left panel, the peptidase is shown in the traditional “standard orientation”,60 in which the active site-cleft is viewed broadside running from left (non-primed side) to right (primed side), followed by a ∼60° rotation around the x-axis. The catalytic zinc of the peptidase is shown as a magenta sphere coordinated by three histidine residues (H218, H222, and H228; light blue sticks). The structural zinc (in magenta) and the three calcium cations (green spheres) are also displayed, as is the general base/acid glutamate (E219). The two most relevant residues of PotA engaged in inhibition, Y63 and D64, are highlighted as pink sticks. (B) Superposition of the Cα-traces of the PotA:MMP-12 (gold/light blue) and PotA:karilysin (red/purple) complexes in the orientation of (A, right). (C) Close up of a similar view as shown in (A, left) in stereo, focusing on the interaction between PotA and MMP-12 in the cleft. Relevant peptidase and inhibitor residues are shown as sticks and are numbered in blue and red, respectively. (D) Ribbon-plot of PotC (in green; RCL in blue) in a complex with the catalytic domain of mirolase (in plum) in two orientations (left and right). In the left panel, the peptidase is shown in a similar orientation with respect to its active-site cleft as in (A, left). The protruding hairpin and the backing loop 2 of the peptidase are shown as white ribbons and are labelled. The lower rim and loop Lβ7β8 from mirolase are shown as yellow ribbons and are labelled. The catalytic triad (D231, H283 and S477) is shown as red sticks, and the M126 side chain of PotC as cyan sticks. (E) Close up of a similar view as shown in (D, left) in stereo, displaying the side chains of the RCL residues of PotC and the three catalytic residues of the peptidase as sticks. The RCL is anchored to the subjacent scaffold loop via the disulfide C42–C124. Residue numbers are in blue and red for the peptidase and the inhibitor, respectively. (F) Detail of the mirolase active site showing the catalytic triad with magenta carbons and black labels and PotC segment V122–Q128 (selected residues are numbered in red). The final (2mFobs–DFcalc)-type Fourier map at 1.10 Å resolution is shown as a semi-transparent green surface contoured at 1.5σ above threshold for the depicted residues only. The hydrogen bonds of the catalytic triad (S477Oγ–H283Nε2 and H283Nδ1–D231Oδ2) are shown as yellow lines and are labelled with the corresponding distance in Å, as is the interaction between S477Oγ and the scissile carbonyl carbon, M126C. | ||
Overall, PotA inhibits MMPs using a “bilobal mechanism” (Fig. 7A and C), which is distantly reminiscent of the “raised elephant trunk mechanism” described for the specific inhibition of astacin MPs by fetuin-B63,64 and contains elements of the “aspartate-switch mechanism” of latency found in certain MP zymogens.65 Finally, an OB-fold β-barrel scaffold has also been found in the otherwise unrelated structures of TIMPs.37,39 However, TIMPs use their N-terminal domain to bind the catalytic zinc of MMPs via the N-terminal α-amino group and only block the primed side of the cleft.
2.10 PotC is a new standard-mechanism inhibitor of subtilases
We also solved the structure of PotC in a complex with T. forsythia mirolase, a peptidase herein reported for its structure for the first time (ESI Results and Fig. S9†). Briefly, mirolase contains a 348-residue subtilisin-type S8-family catalytic domain centred on a catalytic serine-peptidase triad comprising D231, H283 and S477. Despite its broad substrate specificity similar to other subtilisins27 (Section 2.2), the structure of mirolase differs substantially from that of the archetypal Bacillus subtilisin in the loops conforming the active-site cleft. The Tannerella enzyme contains 25% more residues and four new calcium-binding sites (Fig. S9 and Table S6†). In particular, mirolase contains a “protruding hairpin” projecting ∼30 Å from the surface (Fig. S9†) rather than the classic “upper-rim segment” framing the top of the non-primed side of the cleft as found in subtilases. This hairpin is supported by “backing loop 1” and “backing loop 2”.In the complex, PotC inserts like a shim into the active-site cleft of mirolase (Fig. 7D and E) and uses a substantially larger interface than PotA (1327 Å2, ΔiG = −17.1 kcal mol−1). Its RCL is pinched between the protruding hairpin and backing loop 2 in front, and by loop Lβ7β8 and the lower rim of the cleft behind. The RCL interacts with this lower rim (S361–Y364) in an extended, antiparallel, substrate-like conformation (see ESI Results† for details on the mirolase structure and Fig. S9†) between I123 in S4 and N127 in S1′ (Fig. 7E), and further contacts calcium site 1 within backing loop 2. The RCL flanks the reactive-site bond (M126–N127), with the side chain of M126 nestling into the S1 specificity pocket of the enzyme (Fig. 7E). The overall architecture and geometry of the complex conforms to the “standard-mechanism” widely described for serine-peptidase inhibitors66,67 and for a few MP inhibitors40 but here applying to an inhibitor with an uncharacterized fold. These inhibitors bind to target enzymes in a substrate-like manner, adopting an extended, “canonical” conformation. The reactive-site bond of the RCL is cleaved very slowly due to the high stability of the Michaelis complex, yielding very low dissociation rate constants.68 The complex can therefore dissociate to produce either the intact or cleaved forms of the inhibitor. Indeed, the high resolution of the PotC:mirolase crystal structure (1.1 Å; see Table S3†) revealed that the complex corresponds to a cleavage-reaction intermediate in which the catalytic nucleophile S477Oγ is very close (2.55 Å) to the scissile carbonyl carbon and perpendicular to the plane of the carbonyl group (Fig. 7F). This distance exceeds the length of a covalent C–O bond but is shorter than the sum of the van der Waals radii, so that it represents an intermediate of the nucleophilic addition preceding the tetrahedral intermediate. Moreover, the scissile carbonyl oxygen was stabilised by oxyanion-hole atoms N399Nδ2 and S477N (ESI Results†).
Finally, the orientation of the inhibitor β-barrel with respect to the peptidase is similar in the PotA and PotC complexes: the barrel axis is roughly perpendicular to the peptidase active-site cleft (compare left panels of Fig. 7A and D).
2.11 PotD and PotE are probably standard-mechanism inhibitors of MPs
We obtained a high confidence docking model of the PotD:mirolysin complex based on the experimental structures of PotD (this work) and a mirolysin product complex.48 We also calculated a reliable computational model of the PotE:forsilysin complex based on the experimental structure of PotE (this work), a high-confidence computational model of forsilysin, and the complex between the M4-peptidase thermolysin and IMPI.40In the PotD:mirolysin complex (Fig. 8A), the RCL would insert in a substrate-like manner into the active site of the enzyme and interact with the cleft's upper-rim segment, here formed by strand β7 (D179–Q185; see ref. 48 for structural details of mirolysin), in an antiparallel manner between W108 and D112 (Fig. 8B). This inhibition would conform to the standard mechanism, which for MPs has been described at the structural level only for IMPI.40,69 Most relevantly, the side chain of K110, which flanks the reactive-site bond, would intrude into the S1′ specificity pocket, thus matching the preference of the enzyme for basic residues,29 and would form a salt bridge with D289 at the pocket bottom. In addition, D170 would bind R111, and the side chains of I109 and W108 would occupy the S1 and S2 sub-sites by interacting with M147plus L180 and F186plus F188, respectively. Atom Y286Oη would bind the reactive-site bond carbonyl. The shape of PotD further suggests that, farther on the primed side of the cleft, the tip of loop A (R55–R56) may contact loop Lβ8α4 of the MP, which protrudes from the surface and shapes the outermost cleft region,48 possibly through a salt bridge (R55–E213). Loop A is kept in a competent conformation for interaction by an ionic network involving the RCL (D58–R56–D112; Fig. 8B).
 | ||
| Fig. 8 Models of PotD and PotE complexes with peptidases. (A) Ribbon-plot of PotD (in cyan; RCL and loop A in plum) in the modelled complex with the catalytic domain of mirolysin (in tan) in two orientations (left and right). In the left panel, the peptidase is shown in a similar orientation with respect to its active-site cleft as MMP-12 in Fig. 7A. The catalytic zinc of the peptidase is shown as a magenta sphere coordinated by three histidine residues (H224, H228 and H234; brown sticks). The two calcium cations (green spheres) are also displayed, as are the general base/acid glutamate (E225) and E213. The most relevant residue of PotD potentially engaged in inhibition (K110) , as well as ancillary R55, are further displayed as pink sticks. (B) Close up of a similar view as shown in (A, left) in stereo, focusing on the predicted interaction between PotD and mirolysin in the active-site cleft. Relevant residues of the peptidase and inhibitor are shown as sticks and are numbered in blue and red, respectively. (C) Ribbon-plot of PotE (in orange; RCL and loop α in blue) in the modelled complex with the forsilyin catalytic domain model (in light green) in two orientations (left and right). In the left panel, the peptidase is shown in a similar orientation with respect to its active-site cleft as MMP-12 in Fig. 7A. The catalytic zinc of the peptidase is shown as a magenta sphere coordinated by three residues (H348, H352 and E372; green sticks). A predicted calcium cation (red sphere) is also displayed, as is the general base/acid glutamate (E349). The most relevant residues of PotE potentially engaged in inhibition (I116 from the RCL and R49 from loop α) are displayed as cyan sticks. (D) Close up of a similar view as shown in (A, left) in stereo, focusing on the predicted interaction between PotE and forsilysin in the active-site cleft. Relevant residues of the peptidase and inhibitor are shown as sticks and are numbered in blue and red, respectively. | ||
Like PotD, PotE would insert its RCL to interact with the upper-rim strand of the forsilysin cleft (D317–N322) via segment I114–I116 (Fig. 8C). The side chain of residue I116, which flanks the reactive-site bond (A115–I116), would penetrate the S1′ pocket, thus matching the general specificity of thermolysin-type MPs for middle-sized hydrophobic side chains (Fig. 8D). As found in the IMPI:thermolysin complex40 and our in vitro inhibition studies (Section 2.2), PotE is cleaved at A115–I116 during complex formation while retaining its inhibitory capacity. Moreover, R117 in S2′ might form a salt-bridge with D336 of the MP. Finally, and likewise reminiscent of PotD, the tip of a β-ribbon adjacent to the RCL (here loop α) might assist in binding of the primed side of the cleft via interactions involving S46 and R49.
Taken together, as for the OB-fold barrels PotA and PotC, the β-hairpin-repeat-barrels PotD and PotE would also display a similar orientation relative to the peptidase in the corresponding complexes, here with the barrel axis roughly parallel to the active-site cleft (compare left panels of Fig. 8A and C).
2.12 Corollary
To thrive in the harsh ecological niche of the gingival crevice, periodontopathogenic T. forsythia has uniquely evolved six co-localized and co-transcribed peptidases and specific inhibitors, the KLIKK-peptidase/potempin system. The secreted peptidases (representing five families and two chemical classes) act as soluble virulence factors targeting the host and bacterial competitors for resources in the extracellular environment. The inhibitors, which are inserted close to the outer-membrane surface beneath the S-layer and in outer-membrane vesicles, contribute to bacterial competence and fitness in vivo by protecting surface proteins from proteolysis before the peptidases are released to the extracellular space. Notably, although potempins solely use the regular secretory pathway and remain anchored on the outer leaflet of the outer membrane, their target peptidases are soluble and use the latter pathway plus a C-terminal domain-mediated type-IX secretion system for export to the extracellular space.Given their disparate signal peptides, as often found in unrelated proteins,70 KLIKK-peptidases appear to have been acquired gradually over long evolutionary timescales, possibly by horizontal gene transfer from mammalian hosts or other bacteria in the oral or gut microbiomes.27,38 In contrast, potempins appear to have arisen from a small number of events close in time to regulate the co-transcribed KLIKK-peptidases.
Structurally, potempins adopt two β-barrel architectures that are new for peptidase inhibitors and inhibit their target peptidases either through a bilobal mechanism or the standard mechanism, using surface loops in two distinct orientations of their β-barrel axes relative to the active-site clefts. Most potempins potently and selectively inhibit only their co-transcribed KLIKK-peptidase by forming a firm complex. Exceptionally, PotA also strongly inhibits MMP-12, but not other MMPs. MMP-12 is almost exclusively expressed in macrophages, which trigger the primary line of defence and the inflammatory response to invading periodontopathogens, including T. forsythia.71,72 Moreover, MMP-12 participates in bacterial clearance when challenged with Gram-positive bacteria,53 and it is secreted to the extracellular space.54 Thus, PotA offers a strategy to escape the human host response and adds up to the cognate broad-spectrum TIMPs as a novel, specific, and physiologically relevant MMP inhibitor that may be suitable for the treatment of MMP-12-mediated diseases.
3. Experimental procedures
3.1 Reagents
Human neutrophil elastase, cathepsin G, and α2-macroglobulin were purchased from Athens Research and Technology. Succinyl-A–A–P–F-pNA, batimastat, ecotin, subtilisin Carlsberg, bovine pancreatic trypsin, porcine pancreatic elastase, B. thermoproteolyticus thermolysin, and bovine pancreatic α-chymotrypsin were from Sigma-Aldrich. Serratia sp. serralysin and protealysin, P. aeruginosa LasB and aeruginolysin, F. nucleatum fusolisin, T. denticola dentilysin, and S. gordonii challisin were commercially expressed and purified by Creative Enzymes. Azocoll was from EMD-Millipore, fluorescein-labelled casein (FTC-casein), the PageRuler pre-stained protein ladder (10–180 kDa), restriction endonucleases BamHI and XhoI, and Phusion DNA polymerase were from Thermo Fisher Scientific. The expression vector pGEX-6P-1, glutathione-Sepharose 4 fast flow resin, and 3C (PreScission) protease were from Cytiva. Full-length human MMPs 1, 2, 3, 7, 8, 9, 10, 12, 13, 14, and full-length murine MMP-12 were purchased from R&D Systems Europe. The catalytic domain of human MMP-20 was from Enzo Fischer Scientific. Primers (Table S1†) were synthesized by Genomed. All other chemical reagents, unless stated otherwise, were from BioShop Canada.3.2 Recombinant peptidase production, purification, and titration
P. gingivalis gingipains (Kgp and RgpB) and Tpr peptidase, S. aureus aureolysin, M. acetivorans ulilysin, KLIKK-peptidases (mirolase, karilysin, mirolysin, forsilysin, miropsin-1 and miropsin-2), and the catalytic domains of human and murine MMP-12 were produced and/or purified as previously described.23,27,29,47,69,73,74 Peptidases were active-site titrated with α2-macroglobulin (mirolase, mirolysin, forsilysin and subtilisin Carlsberg), ecotin (chymotrypsin, trypsin, elastase and cathepsin G) or batimastat (karilysin).3.3 Cloning, expression, and purification of recombinant potempins
With the exception of potD, the sequences encoding the potempins, without the predicted signal peptides and with the N-terminal cysteine (C21) replaced with alanine, were directly amplified by PCR from genomic DNA extracted from T. forsythia ATCC 43037 using the primers listed in Table S1.† The potD gene was amplified by nested PCR. Next, vector pGEX-6P-1 and the PCR products were digested with BamHI/NotI (potA, potB1, potB2, potC and potE) or EcoRI/NotI (potD) and ligated. Escherichia coli strain DH5α was transformed with the recombinant plasmids, and positive clones were selected. The integrity of the genetic constructs, including an N-terminal glutathione-S-transferase-tag for purification and a PreScission peptidase cleavage site for tag removal, was confirmed by DNA sequencing. Expression plasmids were introduced into E. coli strain BL21 (DE3) (EMD-Millipore), and the bacteria were grown in lysogeny broth (Lennox) containing 100 μg mL−1 ampicillin at 37 °C to an OD600 of 0.75–1 and then cooled down to 20 °C. Recombinant fusion protein expression was induced with 0.1 mM isopropyl-1-β-D-thiogalactopyranoside. After 16 h at 20 °C, the cells were collected by centrifugation (6000×g, 10 min, 4 °C), resuspended in phosphate-buffered saline supplemented with 0.02% sodium azide, and lysed by sonication (one cycle of 30 × 0.5 s pulses at 70% power output per pellet from 1 L culture) using a Branson digital 450 sonifier (Branson Ultrasonics). Cell lysates were clarified by centrifugation (50000×g, 50 min, 4 °C) and loaded onto a 5 mL column with pre-equilibrated glutathione-Sepharose 4 fast-flow matrix at 4 °C. The glutathione-S-transferase-tag was removed by in-column cleavage with PreScission protease, which left five or eight (only for PotE) vector-derived additional residues (G–P–L–G–S or G–P–L–G–S–P–E–F) at the N-terminus of the recombinant potempins. The eluted proteins were concentrated to 2 mL and size-exclusion chromatography was carried out at a flow rate of 1.5 mL min−1 on a HiLoad 16/600 Superdex 75 pg column (Cytiva) connected to an ÄKTA Pure FPLC system (Cytiva), and previously equilibrated with 5 mM Tris·HCl, 50 mM sodium chloride, 0.02% sodium azide, pH 8.0. Protein concentrations were determined by averaging the values obtained with the BCA Protein Assay Kit (Thermo Fisher Scientific) and those resulting from measuring A280 with a NanoDrop device (Thermo Fisher Scientific) applying the theoretical extinction coefficient calculated with ProtParam (http://web.expasy.org).
3.4 Inhibition assays
Karilysin, mirolysin, mirolase, subtilisin Carlsberg, chymotrypsin, trypsin or elastase (200 nM), as well as miropsin-1 or forsilysin (2 μM), were pre-incubated for 15 min at 37 °C in assay buffer (50 mM Tris·HCl, 150 mM sodium chloride, 2.5 mM calcium chloride, 0.02% sodium azide, pH 7.6), alone or in the presence of a 10-fold molar excess of the corresponding potempin. The residual peptidase activity was determined using 200 μL Azocoll (15 mg mL−1 suspension) as a substrate according to the manufacturer's instructions. All other peptidases were incubated with a 10-fold weight excess of the corresponding potempin and the residual activity was determined with Azocoll. With respect to the inhibition by intact cells and outer-membrane vesicles, karilysin, mirolysin and mirolase (at 1 nM) were incubated with fractions for 15 min at 37 °C in assay buffer (50 μL), and 200 μL of Azocoll substrate suspension (15 mg mL−1) was added.3.5 Stoichiometry of inhibition
Mirolase, mirolysin, karilysin (all at 200 nM) or subtilisin (50 nM) were mixed with increasing concentrations of Pot C, PotD, PotA or PotC, respectively, in 50 μL reactions to yield enzyme:inhibitor molar ratios of 0–9. After pre-incubation for 30 min at 37 °C, residual peptidase activity was determined using Azocoll as the substrate.3.6 Determination of inhibition modes and inhibition constants
Karilysin (10 nM), mirolase (15 nM), mirolysin (7.5 nM), subtilisin (0.5 nM) or forsilysin (15 nM) were incubated at 37 °C in microtiter plates containing 100 μL assay buffer per well, in the presence of increasing amounts of PotA (0–8 nM), PotC (0–18.75 nM), PotD (0–9 nM for mirolysin and 0–192 nM for subtilisin) or PotE (0–20 nM), respectively. After incubation for 15 min at 37 °C, we added 100 μL FTC-casein (0–120 μg mL−1) and residual proteolytic activity was recorded (λexc = 485 nm; λem = 538 nm) for 30 min using a SpectraMax Gemini XPS microplate reader (Molecular Devices). The modes of inhibition of the target peptidases were determined graphically with a Lineweaver–Burk plot constructed using eqn (1): | (1) |
 | (2) |
3.7 Assessment of complex formation by size-exclusion chromatography
Mixtures of karilysin and PotA (at molar ratios of 1:1 and 1:5), mirolase and PotC (1:0.75 and 1:1.5), subtilisin and PotC (1:4 and 1:6), mirolysin and PotD (1:0.75 and 1:1.5), and forsilysin and PotE (ratios 1:0.8 and 1:1.6) were pre-incubated for 15 min at 20 °C in buffer (5 mM Tris·HCl, 50 mM sodium chloride, 2.5 mM calcium chloride, 0.02% sodium azide, pH 7.6). Each reaction mixture contained 75 μg peptidase and inhibitor at concentrations below and above the corresponding stoichiometry of inhibition. The mixtures (200 μL) and each component separately were analysed by size-exclusion chromatography on a Superdex 75 10/300 GL column (Cytiva) attached to an ÄKTA Pure FPLC system operated at a flow rate of 0.75 mL min−1. The column was calibrated using the LMW and HMW Calibration Kits (Cytiva), and the protein elution profiles were recorded at λ = 280 nm. We collected 0.5 mL fractions and analysed 30 μL aliquots by 10% SDS-PAGE (acrylamide/bis-acrylamide ratio = 33:1) using a Tris·HCl/Tricine buffer system.75
3.8 Kinetic studies of the inhibition of MMPs by PotA
MMPs were incubated with increasing concentrations of recombinant PotA up to a 1:5 enzyme:inhibitor molar ratio for 15 min at 30 °C in activity buffer, which was 50 mM Tris·HCl, 10 mM calcium chloride, 150 mM sodium chloride, 0.05% Brij35, pH 7.5 for all MMPs except MMP-14 (50 mM Tris·HCl, 3 mM calcium chloride, 1 μM zinc chloride, pH 8.5). Residual enzymatic activity was measured as previously reported73 using the fluorogenic substrates Abz-R–P–L–A–L–W–R–S–Q–E–D–Dnp (for MMP-2, MMP-10, human and mouse MMP-12, and MMP-13), Abz-R–P–L–G–L–W–G–A–Q–E–D–Dnp (for MMPs 1, 7, 8, 9, 14 and 20) or Mca-R–P–K–P–V–E–Nva–W–R–K(Dnp)-NH2 (for MMP-3). MMPs were first titrated with the small-molecule active-site inhibitor GM6001 (R&D Systems Europe). Ki values were derived from non-linear inhibition curves as previously described.76 Data were plotted considering tight-binding inhibition,77 and the associated Ki values were determined graphically (Table S2†).
3.9 Stability of potempins, isolated and in complex with target peptidases
Mixtures of karilysin, mirolase, mirolysin or subtilisin (10 μM) with PotA (12 μM), PotC (15 μM), PotD (15 μM) or PotC (75 μM), respectively, in 25 μL assay buffer were incubated at 4, 20 or 37 °C for 24 h before SDS-PAGE analysis and the determination of the residual proteolytic activity against Azocoll. Isolated peptidases or their complexes with inhibitors incubated for 15 min (“fresh complexes”) served as controls for the activity of the free enzymes and the residual activity of the complexes, respectively. To assess susceptibility to proteolytic degradation and/or inactivation by non-target peptidases, potempins (10 μM) were incubated alone or with neutrophil elastase, trypsin, chymotrypsin, Kgp, RgpB or Tpr protease at a 1:100 enzyme:inhibitor molar ratio for 8 h at 37 °C in assay buffer. For cysteine peptidases (gingipains and Tpr), the assay buffers also contained 10 mM cysteine. After incubation, potempin integrity was determined by 10% SDS-PAGE and inhibitory activity was tested against the corresponding KLIKK-peptidases with Azocoll as the substrate.
3.10 Cultivation of T. forsythia
T. forsythia strain ATCC 43037 was grown at 37 °C in anaerobic chambers (Whitley A85 Workstation) on tryptic soy broth (30 g L−1) with 5 g L−1 yeast extract supplemented with 0.5 mg L−1 haemin, 1 mg L−1 menadione, and 10 mg L−1 N−1-acetylmuramic acid. Solid cultures were further supplemented with 5% sheep blood and solidified with 1.5% agar. For liquid cultures, we added 5% foetal bovine serum.3.11 Cell fractionation, western blotting, and dot blot analysis
T. forsythia cultures and cells were fractioned as previously described for P. gingivalis78 except that inner membranes were solubilized with a solution containing 0.1% sarcosyl instead of 1% Triton X-100. Protein concentration was determined by the BCA assay (Sigma-Aldrich). Fractions resolved by 10% SDS-PAGE were electrotransferred onto polyvinylidene difluoride membranes (Bio-Rad, 0.22 μm pores) at 15 V for 40 min using a semi-dry transfer unit (Bio-Rad) and a transfer buffer comprising 50 mM Tris·HCl, 40 mM glycine, 0.04% SDS and 10% methanol. The membranes were blocked with 5% (w/v) skimmed milk in 20 mM Tris·HCl, 0.5 M sodium chloride, 0.1% Tween-20, pH 7.5. Blots were probed with 1 μg mL−1 anti-PotA or anti-karilysin antibody, as well as through anti-BspA rabbit serum (at 1:2000 dilution), in blocking solution, followed by incubation at room temperature for 1 h with a 1:20000 dilution of a horseradish-peroxidase-conjugated goat anti-rabbit polyclonal secondary antibody in blocking solution. The signal was developed using the Pierce ECL western blotting substrate (Thermo Fisher Scientific). For dot-blot analysis, T. forsythia cells were harvested, washed and resuspended in cold phosphate-buffered saline. The OD600 in suspensions of washed cells was adjusted to 1.0. To determine the cellular localisation of PotA by immunostaining, phosphate-buffered saline was supplemented with cOmplete EDTA-free protease inhibitor cocktail (Roche). Half of the intact cell suspensions were sonicated to disrupt the cells, and 5 μL of the intact or sonicated cell suspensions were spotted onto 0.22 μm nitrocellulose membranes (Bio-Rad), air-dried, and analysed as described above.
3.12 Mutagenesis of T. forsythia
Mutations were introduced into the T. forsythia genome by natural homologous recombination. A master plasmid (pKO-PINA-ermF), which introduces the antibiotic resistance cassette (ermF) before the potA-karilysin operon, was created from PCR-amplified inserts by the ligation of restriction-digested DNA fragments into the pUC19 suicide vector. The ermF cassette was amplified from vector pURgpB-E (primers are listed in Table S1†). Flanking regions of the antibiotic resistance gene were amplified from T. forsythia sp. 43037 genomic DNA. An upstream 664-base-pair fragment covered the region preceding the regulatory sequences of the potA-karilysin operon, and a downstream 1727-base-pair fragment spanned the proposed promoter region (502 base pairs), the whole potA coding DNA sequence, and a part of the karilysin coding DNA sequence. Next, vectors for complete potA deletion (pdelKO-PINA) and for the replacement of the N-terminal cysteine for lipid anchoring with alanine (pKO-PotA_C21A) were generated from the KO-PINA master plasmid in a single PCR step by Gibson assembly (https://www.neb.com/protocols/2012/12/11/gibson-assembly-protocol-e5510) or SLIM mutagenesis. After verification by sequencing, the vectors were introduced into T. forsythia by electroporation, and recombinant clones were selected on medium supplemented with 5 μg mL−1 erythromycin.26 Only the cysteine point mutant showed a complete lack of PotA protein without altered karilysin expression levels (Fig. 5C). This mutant was designated the potAnull strain.
3.13 In vivo fitness of T. forsythia
All animal studies took place in the Animal Facility of the Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Kraków, Poland, in accordance with the protocols laid down by the Institutional Animal Care and Use II Regional Ethics Committee on Animal Experimentation, Kraków, Poland (approval no. 249/2019). Eight-week-old female C57BL/6 mice were obtained from Janvier Labs and housed in individually ventilated cages, with a 12 h photoperiod and a constant temperature of 22 ± 1 °C. Animals were fed a standard laboratory diet and water ad libitum under specific pathogen-free conditions. Mice were randomly assigned to experimental groups, and control and bacteria-infected mice were housed in separate cages. Mice were quarantined for at least 7 days prior to the experiment. Mid-dorsal subcutaneous implantation of surgical-grade titanium coil chambers (length 1.5 cm, diameter 5.0 ± 0.08 mm) was carried out under anaesthesia with ketamine and xylazine (100 and 10 mg kg−1, respectively). After 10 days, 1 × 108 colony-forming units of wild-type (WT) T. forsythia, karilysin-depleted T. forsythia (Δkly) or PotA-null T. forsythia (potAnull) in 100 μL sterile phosphate-buffered saline were injected into the chambers with a 27-gauge syringe. Chamber exudates were harvested from mice at 0, 1, 2 and 24 h post-infection with the same syringe and 10-fold serial dilutions were prepared. All samples were plated on agar, and the plates were incubated anaerobically for 10 days at 37 °C. Visible colonies were counted to determine the total viable cell number.3.14 Crystallization and diffraction data collection
Initial crystallization conditions were determined at the joint IBMB/IRB Automated Crystallography Platform (https://www.ibmb.csic.es/en/platforms/automated-crystallographic-plattform) using the sitting-drop vapour diffusion method to test up to ∼1800 reservoir solutions, which were prepared by a Tecan robot in 96-well 2 mL DeepWell plates. Crystallization drops of 100 nL each of protein and reservoir solution were dispensed on Swissci 96-well 2-drop MRC crystallization plates (Molecular Dimensions) by a Phoenix nanodrop robot (Art Robbins) or a Cartesian Microsys 4000 XL robot (Genomic Solutions). Plates were stored in Bruker steady-temperature crystal farms at 20 or 4 °C. Best hits were scaled up whenever possible to drops containing 0.5, 1 or 2 μL of protein and reservoir solutions using Swissci 48-well MRC Maxi optimization plates (Molecular Dimensions). Crystals were cryoprotected by rapid passage through drops containing up to 25% (v/v) glycerol prior to vitrification in liquid nitrogen for transport to synchrotrons for data collection.Isolated PotA (∼1.8 mg mL−1 in 50 mM sodium chloride, 5 mM Tris·HCl, pH 8.0) was crystallised using 20% [w/v] polyethylene glycol (PEG) 2000, 10 μM nickel chloride, 100 mM Tris·HCl, pH 8.5 as the reservoir solution. The PotA:karilysin complex (∼15 mg mL−1 in 50 mM sodium chloride, 5 mM calcium chloride, 0.02% sodium azide, 5 mM Tris·HCl, pH 8.0) was crystallized using 25% PEG 6000, 100 mM MES, pH 6.0. The PotA:MMP-12 complex (∼8.5 mg mL−1 in 2.5 mM calcium chloride, 150 mM sodium chloride, 20 mM Tris·HCl, pH 7.5) was crystallized using 30% PEG 3000, 200 mM sodium chloride, 100 mM Tris·HCl, pH 7.0. The PotC:mirolase complex (∼10 mg mL−1 in 2 mM calcium chloride, 50 mM sodium chloride, 5 mM Tris·HCl, pH 8.0) was crystallized using 19% PEG monomethyl ether 2000 in 100 mM mixed succinic acid, sodium dihydrogen phosphate and glycine at a molar ratio of 2:7:7 (pH 8.0). Isolated selenomethionine-derivatised PotD mutant I53M (∼14 mg L−1 in 5 mM Tris·HCl, 50 mM sodium chloride, 0.02% sodium azide, pH 8.0) was crystallized using 20% PEG 3350, 0.2 M diammonium hydrogen citrate. Finally, native and selenomethionine-derivatised PotE (∼9 mg mL−1 and ∼10 mg mL−1, respectively, in 50 mM sodium chloride, 5 mM Tris·HCl, pH 8.0) were crystallized using 20% PEG 8000, 100 mM HEPES, pH 7.5 and 20% PEG 1000, 100 mM Tris·HCl, pH 8.5, respectively.
Diffraction datasets were collected at 100 K on beam line ID23-1 of the ESRF synchrotron (Grenoble, France) or beam line XALOC of the ALBA synchrotron (Cerdanyola, Catalonia, Spain) on PILATUS 6M detectors (Dectris). Data were processed using Xds79 and Xscale, and were transformed with Xdsconv to MTZ-format for the Phenix80 and Ccp4 (ref. 81) program packages. Table S3† provides statistical details for crystal parameters and data processing.
3.15 Structure solution, model building and refinement
The structure of the PotA:karilysin complex was solved first by likelihood-scored molecular replacement with Phaser.82 The coordinates of the protein part of the karilysin catalytic domain (Protein Data Bank access code (PDB) 4IN938,83) were used as a searching model to find the only molecule present in the asymmetric unit (a.u.) of the crystal. The phases derived from this solution were used for density modification and automatic model building with the Arp/wArp suite.84 The structure of the PotA:MMP-12 complex was solved by molecular replacement with the coordinates of PotA from the PotA:karilysin complex and the protein part of the human MMP-12 catalytic domain (PDB 1JK362) to find the two complexes contained in the a.u. Finally, the structure of unbound PotA, with one protomer per a.u., was also solved by molecular replacement with the coordinates of PotA from the PotA:karilysin complex.The structure of the PotC complex with mirolase was solved by molecular replacement with Phaser. A searching model to locate the only peptidase protomer in the a.u. was constructed based on the coordinates of the alkaline protease subtilisin BL from Bacillus lentus (PDB 1ST385), which is the closest relative by sequence similarity whose structure is available (as determined with Blast86). The side chains were trimmed with Chainsaw87 in Ccp4 according to a sequence alignment with mirolase performed with Multalin.88 Phases derived from the rotated and translated model were used for density modification and automatic model building as described above.
The structure of isolated selenomethionine-derivatized PotE, with two protomers in the a.u., was solved by single-wavelength anomalous diffraction by means of the Autosol procedure of the Phenix package89 using data collected at the selenium absorption peak and processed with separate Friedel mates. These calculations found the four selenium atoms in the a.u. through the Hyss substructure search protocol90 and produced phases with an estimated mean figure-of-merit of 0.29. Subsequently, Autobuild calculations within Phenix91 produced a Fourier map with phases with a mean figure-of-merit of 0.59 after density modification and twofold averaging. The structure of native PotE in a different space group with one protomer in the a.u. was solved by molecular replacement with the coordinates of selenomethionine-derivatised PotE.
Finally, the structure of the isolated selenomethionine-derivatized PotD mutant I53M, whose inhibitory ability was indistinguishable from WT PotD, was solved by molecular replacement with Phaser. Structure solution was hindered by the presence of six protomers in the a.u., an insufficient anomalous signal (six selenium atoms with partial occupancy per 79 kDa total molecular mass), and the presence of a strong peak (72% height of origin peak) at fractional coordinates 0.5, 0.5, 0.0, which resulted from translational non-crystallographic symmetry (NCS) and distorted the mean intensity distribution. At this point, a fragment encompassing ∼50% of a model predicted by AlphaFold58 served to find all the copies in the a.u. We selected parts of this model with values of the predicted local-distance difference test (pLLDT;ref. 92), which reliably estimates how well the prediction agrees with an experimental structure, scoring >85 (main chain and side chains) or >70 (main chain only). The correctness of the solution was verified by an anomalous Fourier map, which revealed a strong peak for the side chain of residue 53 of each protomer. The partial model solution and the structure factors (with separate Friedel mates) were then fed into Autosol, which found 10 heavy-atom positions and derived phases with an estimated mean figure-of-merit of 0.88.
The Fourier maps after the structure-solution procedures were used for manual model building and completion with the Coot program,93 alternating with crystallographic refinement using Refine in Phenix94 and Buster/Tnt,95 until the final models were obtained. Both procedures included non-crystallographic symmetry restraints where required and translation/libration/screw-motion refinement. The final structures were validated with the wwPDB Validation Service. Table S3† provides statistics for the refinement and validation parameters, as well as the respective PDB access codes.
3.16 Comparative modelling and structure prediction
A high-confidence homology model of the forsilysin catalytic domain was computed with Raptor-X,96 which uses the Modeller program.97 The model was based on Thermus thermophilus thermolysin (PDB 4M65; p = 2.38 × 10−12; 29% sequence identity) and was validated using Lomets,98 with a root-mean-square deviation of 0.9 Å.For PotB1 and PotB2, models were predicted using AlphaFold.58 The average pLLDT values were 90.8 and 91.1 for all atoms, respectively. These values exceed the high-accuracy cut-off of 90,92 and are thus classed as high confidence. Moreover, the structural similarity of PotB1 and PotB2 observed with PotE (Section 2.8) is unbiased because their structures were absent from the PDB sample on which AlphaFold was trained. This further underpins the high quality of the predictions for PotB1 and PotB2.
A homology model of the PotD:mirolysin complex was obtained by fitting protomer A of the PotD crystal structure into the active-site cleft of mirolysin based on the coordinates of the latter in complex with a C-terminal 14-residue cleavage product of PotD in the primed side of the cleft (PDB 6R7W48), which had identified I109–K110 as the reactive-site bond.
The PotE:forsilysin complex was modelled based on the experimental coordinates of PotE, the comparative model of forsilysin, the complex between the standard-mechanism inhibitor IMPI and B. thermoproteolyticus thermolysin (PDB 3SSB40), and the knowledge of the forsilysin cleavage site in forsilysin (A115–I116; Section 2.2), which was identified as the reactive-site bond.
All computational models and predictions were visually inspected with Coot, manually corrected for clashes and chemical inconsistencies, regularised with Coot or the Geometry_minimization routine of Phenix, and validated with Molprobity (Table S4†). They can be downloaded as part of the ESI Materials†.
3.17 Miscellaneous
Structural similarity was assessed using Dali99 and PDBeFold at pdbe.org/fold based on program Ssm.100 Structures were superimposed using Ssm in Coot, with Fatcat101 or with Lsqkab.102 Figures and coulombic surfaces were prepared using Chimera,103 and protein interfaces were calculated using Pisa104 at https://www.ebi.ac.uk/pdbe/pisa/. The interacting surface of a complex was defined as half the sum of the buried surface areas of either molecule. Phylogeny was assessed using Multalin88 based on a sequence alignment using standard parameters. This alignment was also used to compute pairwise sequence identities, with and without signal peptides, with the Sequence Manipulation Suite.105 Lipoprotein signal peptides were predicted using LipoP v1.0.106Data availability
The coordinates of the herein determined experimental structures are available from the Protein Data Bank (access codes in Table S3†). The homology models mentioned under Section 3.16 are retrievable from the ESI.† All herein developed biochemical reagents and tools, if not commercial, are available from the corresponding authors upon reasonable request.Author contributions
J. P. and F. X. G.-R. conceived and supervised the project; M. K., I. W., M. M., A. S.-G., F. V. and T. M. produced and purified proteins, and performed functional studies in vitro; D. M. cloned the potempin genes, prepared mutants, and analysed operon structures and gene expression; M. B.-M. performed functional studies in vivo; I. B. T. and J. J. E. performed proteomics studies; A. C., T. Go., I. G.-F., M. L.-P., A. R.-B., J. L. A., I. d. D. and T. Gu. repurified and crystallized proteins; T. Go., A. R.-B. and U. E. collected diffraction data; J. P. analysed and interpreted biochemical and in vivo data; V. D. provided reagents; F. X. G.-R. solved and refined crystal structures, and obtained homology models; and J. P. and F. X. G.-R. wrote the manuscript with contributions from all authors.Conflicts of interest
The authors declare no financial or non-financial conflicts of interest.Acknowledgements
The authors thank Laura Company, Xandra Kreplin and Joan Pous from the joint IBMB/IRB Automated Crystallography Platform and the Protein Purification Service for assistance during purification, crystallization and SEC-MALLS experiments. The authors also thank the ESRF and ALBA synchrotrons for beamtime and the beamline staff for assistance during diffraction data collection and Dr Ashu Sharma (University of Buffalo, Buffalo, NY, USA) for providing rabbit antiserum against the BspA protein of T. forsythia. This study was supported in part by grants from Polish (National Science Centre and Ministry of Science and Higher Education), US (NIH/NIDR), Spanish, Danish and Catalan public and private bodies (grant/fellowship references PID2019-107725RG-I00 and PDC2022-133344-I00 by MICIN/AEI/10.13039/501100011033, 2017SGR3, 2016/21/B/NZ1/00292, 1306/MOB/IV/2015/0, 2019/35/B/NZ1/03118, DE026280, Fundació “La Marató de TV3” 201815, and Novo Nordisk Foundation grant NNF18OC0032724). The authors thank Richard M. Twyman for editing the manuscript.References
- P. I. Eke, B. A. Dye, L. Wei, G. D. Slade, G. O. Thornton-Evans, W. S. Borgnakke, G. W. Taylor, R. C. Page, J. D. Beck and R. J. Genco, J. Periodontol., 2015, 86, 611–622 CrossRef.
- G. Hajishengallis and T. Chavakis, Nat. Rev. Immunol., 2021, 21, 426–440 CrossRef CAS PubMed.
- S. Listl, J. Galloway, P. A. Mossey and W. Marcenes, J. Dent. Res., 2015, 94, 1355–1361 CrossRef CAS PubMed.
- G. Hajishengallis, Nat. Rev. Immunol., 2015, 15, 30–44 CrossRef CAS PubMed.
- R. P. Darveau, Nat. Rev. Microbiol., 2010, 8, 481–490 CrossRef CAS PubMed.
- A. Endo, T. Watanabe, N. Ogata, T. Nozawa, C. Aikawa, S. Arakawa, F. Maruyama, Y. Izumi and I. Nakagawa, ISME J., 2015, 9, 629–642 CrossRef CAS PubMed.
- R. Cortés-Vieyra, C. Rosales and E. Uribe-Querol, J. Immunol. Res., 2016, 2016, 1396106 Search PubMed.
- R. J. Lamont and G. Hajishengallis, Trends Mol. Med., 2015, 21, 172–183 CrossRef CAS PubMed.
- J. Potempa, A. Banbula and J. Travis, Periodontol, 2000, 24, 153–192 CrossRef CAS PubMed.
- I. de Diego, M. Ksiazek, D. Mizgalska, L. Koneru, P. Golik, B. Szmigielski, M. Nowak, Z. Nowakowska, B. Potempa, J. A. Houston, J. J. Enghild, I. B. Thøgersen, J. Gao, A. H. Kwan, J. Trewhella, G. Dubin, F. X. Gomis-Rüth, K. A. Nguyen and J. Potempa, Sci. Rep., 2016, 6, 23123 CrossRef CAS PubMed.
- A. M. Lasica, M. Ksiazek, M. Madej and J. Potempa, Front. Cell. Infect. Microbiol., 2017, 7, 215 CrossRef PubMed.
- D. Mizgalska, T. Goulas, A. Rodríguez-Banqueri, F. Veillard, M. Madej, E. Małecka, K. Szczesniak, M. Ksiazek, M. Widziołek, T. Guevara, U. Eckhard, M. Solà, J. Potempa and F. X. Gomis-Rüth, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2103573118 CrossRef CAS PubMed.
- K. Sato, E. Sakai, P. D. Veith, M. Shoji, Y. Kikuchi, H. Yukitake, N. Ohara, M. Naito, K. Okamoto, E. C. Reynolds and K. Nakayama, J. Biol. Chem., 2005, 280, 8668–8677 CrossRef CAS PubMed.
- P. D. Veith, N. A. Nor Muhammad, S. G. Dashper, V. A. Likic, D. G. Gorasia, D. Chen, S. J. Byrne, D. V. Catmull and E. C. Reynolds, J. Proteome Res., 2013, 12, 4449–4461 CrossRef CAS.
- Y. Narita, K. Sato, H. Yukitake, M. Shoji, D. Nakane, K. Nagano, F. Yoshimura, M. Naito and K. Nakayama, Microbiology, 2014, 160, 2295–2303 CrossRef CAS.
- P. D. Veith, N. M. O'Brien-Simpson, Y. Tan, D. C. Djatmiko, S. G. Dashper and E. C. Reynolds, J. Proteome Res., 2009, 8, 4279–4292 CrossRef CAS PubMed.
- N. D. Rawlings and A. Bateman, Protein Sci., 2021, 30, 83–92 CrossRef CAS PubMed.
- N. Cerdà-Costa and F. X. Gomis-Rüth, Protein Sci., 2014, 23, 123–144 CrossRef.
- H. B. Boldt, M. T. Overgaard, L. S. Laursen, K. Weyer, L. Sottrup-Jensen and C. Oxvig, Biochem. J., 2001, 358, 359–367 CrossRef CAS PubMed.
- F. X. Gomis-Rüth, J. Biol. Chem., 2009, 284, 15353–15357 CrossRef PubMed.
- C. Tallant, A. Marrero and F. X. Gomis-Rüth, Biochim. Biophys. Acta, Mol. Cell Res., 2010, 1803, 20–28 CrossRef CAS PubMed.
- L. Marino-Puertas, T. Goulas and F. X. Gomis-Rüth, Biochim. Biophys. Acta, Mol. Cell Res., 2017, 1864, 2026–2035 CrossRef CAS.
- M. Ksiazek, D. Mizgalska, S. Eick, I. B. Thøgersen, J. J. Enghild and J. Potempa, Front. Microbiol., 2015, 6, 312 Search PubMed.
- J. Koziel, A. Y. Karim, K. Przybyszewska, M. Ksiazek, M. Rapala-Kozik, K. A. Nguyen and J. Potempa, J. Innate Immun., 2010, 2, 288–293 CrossRef CAS.
- M. Jusko, J. Potempa, A. Y. Karim, M. Ksiazek, K. Riesbeck, P. Garred, S. Eick and A. M. Blom, J. Immunol., 2012, 188, 2338–2349 CrossRef CAS.
- D. Bryzek, M. Ksiazek, E. Bielecka, A. Y. Karim, B. Potempa, D. Staniec, J. Koziel and J. Potempa, Mol. Oral Microbiol., 2014, 29, 294–306 CrossRef CAS PubMed.
- M. Ksiazek, A. Y. Karim, D. Bryzek, J. J. Enghild, I. B. Thøgersen, J. Koziel and J. Potempa, Biol. Chem., 2015, 396, 261–275 CAS.
- M. Jusko, J. Potempa, D. Mizgalska, E. Bielecka, M. Ksiazek, K. Riesbeck, P. Garred, S. Eick and A. M. Blom, J. Immunol., 2015, 195, 2231–2240 CrossRef CAS.
- L. Koneru, M. Ksiazek, I. Waligorska, A. Straczek, M. Lukasik, M. Madej, I. B. Thøgersen, J. J. Enghild and J. Potempa, Biol. Chem., 2017, 398, 395–409 CAS.
- M. Eckert, D. Mizgalska, A. Sculean, J. Potempa, A. Stavropoulos and S. Eick, Mol. Oral Microbiol., 2018, 33, 240–248 CrossRef CAS.
- A. Philips, I. Stolarek, L. Handschuh, K. Nowis, A. Juras, D. Trzcinski, W. Nowaczewska, A. Wrzesinska, J. Potempa and M. Figlerowicz, BMC Genomics, 2020, 21, 402 CrossRef CAS PubMed.
- T. Kantyka, N. D. Rawlings and J. Potempa, Biochimie, 2010, 92, 1644–1656 CrossRef CAS PubMed.
- G. Dubin, B. Wladyka, J. Stec-Niemczyk, D. Chmiel, M. Zdzalik, A. Dubin and J. Potempa, Biol. Chem., 2007, 388, 227–235 CAS.
- T. F. Kagawa, P. W. O'Toole and J. C. Cooney, Mol. Microbiol., 2005, 57, 650–666 CrossRef CAS PubMed.
- U. Baumann, M. Bauer, S. Letoffe, P. Delepelaire and C. Wandersman, J. Mol. Biol., 1995, 248, 653–661 CrossRef CAS.
- R. E. Feltzer, R. D. Gray, W. L. Dean and W. M. Pierce Jr, J. Biol. Chem., 2000, 275, 21002–21009 CrossRef CAS.
- H. Nagase, R. Visse and G. Murphy, Cardiovasc. Res., 2006, 69, 562–573 CrossRef CAS PubMed.
- N. Cerdà-Costa, T. Guevara, A. Y. Karim, M. Ksiazek, K. A. Nguyen, J. L. Arolas, J. Potempa and F. X. Gomis-Rüth, Mol. Microbiol., 2011, 79, 119–132 CrossRef.
- F. X. Gomis-Rüth, K. Maskos, M. Betz, A. Bergner, R. Huber, K. Suzuki, N. Yoshida, H. Nagase, K. Brew, G. P. Bourenkov, H. Bartunik and W. Bode, Nature, 1997, 389, 77–81 CrossRef PubMed.
- J. L. Arolas, T. O. Botelho, A. Vilcinskas and F. X. Gomis-Rüth, Angew. Chem., Int. Ed. Engl., 2011, 50, 10357–10360 CrossRef CAS PubMed.
- G. B. Cole, T. J. Bateman and T. F. Moraes, J. Biol. Chem., 2021, 296, 100147 CrossRef CAS.
- C. L. Noland, M. D. Kattke, J. Diao, S. L. Gloor, H. Pantua, M. Reichelt, A. K. Katakam, D. Yan, J. Kang, I. Zilberleyb, M. Xu, S. B. Kapadia and J. M. Murray, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6044–E6053 CrossRef CAS.
- P. D. Veith, Y. Y. Chen, D. Chen, N. M. O'Brien-Simpson, J. D. Cecil, J. A. Holden, J. C. Lenzo and E. C. Reynolds, J. Proteome Res., 2015, 14, 5355–5366 CrossRef CAS.
- S. I. Narita and H. Tokuda, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2017, 1862, 1414–1423 CrossRef CAS PubMed.
- S.-W. Lee, M. Sabet, H.-S. Um, J. Yang, H. C. Kim and W. Zhu, Gene, 2006, 371, 102–111 CrossRef CAS PubMed.
- S. R. Myneni, R. P. Settem, T. D. Connell, A. D. Keegan, S. L. Gaffen and A. Sharma, J. Immunol., 2011, 187, 501–509 CrossRef CAS.
- A. Y. Karim, M. Kulczycka, T. Kantyka, G. Dubin, A. Jabaiah, P. S. Daugherty, I. B. Thogersen, J. J. Enghild, K. A. Nguyen and J. Potempa, Biol. Chem., 2010, 391, 105–117 CAS.
- T. Guevara, A. Rodríguez-Banqueri, M. Ksiazek, J. Potempa and F. X. Gomis-Rüth, IUCrJ, 2020, 7, 18–29 CrossRef CAS.
- K. M. Zak, M. J. Bostock, I. Waligorska, I. B. Thøgersen, J. J. Enghild, G. M. Popowicz, P. Grudnik, J. Potempa and M. Ksiazek, J. Enzyme Inhib. Med. Chem., 2021, 36, 1267–1281 CrossRef CAS PubMed.
- M. Ksiazek, D. Mizgalska, J. J. Enghild, C. Scavenius, I. B. Thøgersen and J. Potempa, J. Biol. Chem., 2015, 290, 658–670 CrossRef CAS PubMed.
- T. Goulas, M. Ksiazek, I. Garcia-Ferrer, A. M. Sochaj-Gregorczyk, I. Waligorska, M. Wasylewski, J. Potempa and F. X. Gomis-Rüth, J. Biol. Chem., 2017, 292, 10883–10898 CrossRef CAS.
- A. Sochaj-Gregorczyk, M. Ksiazek, I. Waligorska, A. Straczek, M. Benedyk, D. Mizgalska, I. B. Thøgersen, J. J. Enghild and J. Potempa, FASEB J., 2020, 34, 619–630 CrossRef CAS.
- A. M. Houghton, W. O. Hartzell, C. S. Robbins, F. X. Gomis-Rüth and S. D. Shapiro, Nature, 2009, 460, 637–641 CrossRef CAS PubMed.
- M. Aristorena, E. Gallardo-Vara, M. Vicen, M. de Las Casas-Engel, L. Ojeda-Fernández, C. Nieto, F. J. Blanco, A. C. Valbuena-Díez, L. M. Botella, P. Nachtigal, A. L. Corbi, M. Colmenares and C. Bernabeu, Int. J. Mol. Sci., 2019, 20, 3107 CrossRef CAS PubMed.
- V. Arcus, Curr. Opin. Struct. Biol., 2002, 12, 794–801 CrossRef CAS PubMed.
- D. B. Roche, P. D. Viet, A. Bakulina, L. Hirsh, S. C. E. Tosatto and A. V. Kajava, J. Struct. Biol., 2018, 201, 130–138 CrossRef CAS.
- K. Ozawa and M. Laskowski Jr, J. Biol. Chem., 1966, 241, 3955–3961 CrossRef CAS.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS.
- I. Schechter and A. Berger, Biochem. Biophys. Res. Commun., 1967, 27, 157–162 CrossRef CAS.
- F. X. Gomis-Rüth, T. O. Botelho and W. Bode, Biochim. Biophys. Acta, 2012, 1824, 157–163 CrossRef.
- A. Gimeno, R. Beltrán-Debón, M. Mulero, G. Pujadas and S. Garcia-Vallvé, Drug Discovery Today, 2020, 25, 38–57 CrossRef CAS PubMed.
- R. Lang, A. Kocourek, M. Braun, H. Tschesche, R. Huber, W. Bode and K. Maskos, J. Mol. Biol., 2001, 312, 731–742 CrossRef CAS.
- A. Cuppari, H. Körschgen, D. Fahrenkamp, C. Schmitz, T. Guevara, K. Karmilin, M. Kuske, M. Olf, E. Dietzel, I. Yiallouros, D. de Sanctis, T. Goulas, R. Weiskirchen, W. Jahnen-Dechent, J. Floehr, W. Stoecker, L. Jovine and F. X. Gomis-Rüth, IUCrJ, 2019, 6, 317–330 CrossRef CAS.
- U. Eckhard, H. Körschgen, N. von Wiegen, W. Stöcker and F. X. Gomis-Rüth, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2023839118 CrossRef CAS.
- J. L. Arolas, T. Goulas, A. Cuppari and F. X. Gomis-Rüth, Chem. Rev., 2018, 118, 5581–5597 CrossRef CAS PubMed.
- M. Laskowski Jr and I. Kato, Annu. Rev. Biochem., 1980, 49, 593–626 CrossRef CAS PubMed.
- W. Bode and R. Huber, Eur. J. Biochem., 1992, 204, 433–451 CrossRef CAS.
- M. Laskowski Jr and M. A. Qasim, Biochim. Biophys. Acta, 2000, 1477, 324–337 CrossRef PubMed.
- S. R. Mendes, U. Eckhard, A. Rodríguez-Banqueri, T. Guevara, P. Czermak, E. Marcos, A. Vilcinskas and F. X. Gomis-Rüth, Comput. Struct. Biotechnol. J., 2022, 20, 534–544 CrossRef CAS PubMed.
- Y.-D. Li, Z.-Y. Xie, Y.-L. Du, Z. Zhou, X.-M. Mao, L.-X. Lv and Y.-Q. Li, Gene, 2009, 436, 8–11 CrossRef CAS.
- C. Bodet, F. Chandad and D. Grenier, Microbes Infect., 2006, 8, 27–35 CrossRef CAS.
- H. Hasturk, A. Kantarci and T. E. van Dyke, Front. Immunol., 2012, 3, 118 Search PubMed.
- A. S. Lamort, R. Gravier, A. Laffitte, L. Juliano, M. L. Zani and T. Moreau, Biol. Chem., 2016, 397, 469–484 CAS.
- C. Tallant, R. García-Castellanos, J. Seco, U. Baumann and F. X. Gomis-Rüth, J. Biol. Chem., 2006, 281, 17920–17928 CrossRef CAS PubMed.
- H. Schägger and G. von Jagow, Anal. Biochem., 1987, 166, 368–379 CrossRef PubMed.
- T. Moreau, N. Gutman, A. el Moujahed, F. Esnard and F. Gauthier, Eur. J. Biochem., 1986, 159, 341–346 CrossRef CAS PubMed.
- L. N. Easson and E. Stedman, Proc. R. Soc. London, Ser. B, 1936, 121, 142–164 CAS.
- A. M. Lasica, T. Goulas, D. Mizgalska, X. Zhou, I. de Diego, M. Ksiazek, M. Madej, Y. Guo, T. Guevara, M. Nowak, B. Potempa, A. Goel, M. Sztukowska, A. T. Prabhakar, M. Bzowska, M. Widziolek, I. B. Thøgersen, J. J. Enghild, M. Simonian, A. W. Kulczyk, K. A. Nguyen, J. Potempa and F. X. Gomis-Rüth, Sci. Rep., 2016, 6, 37708 CrossRef CAS.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- T. Guevara, M. Ksiazek, P. D. Skottrup, N. Cerda-Costa, S. Trillo-Muyo, I. de Diego, E. Riise, J. Potempa and F. X. Gomis-Rüth, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2013, 69, 472–476 CrossRef CAS PubMed.
- G. G. Langer, S. Hazledine, T. Wiegels, C. Carolan and V. S. Lamzin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 635–641 CrossRef CAS PubMed.
- D. W. Goddette, C. Paech, S. S. Yang, J. R. Mielenz, C. Bystroff, M. E. Wilke and R. J. Fletterick, J. Mol. Biol., 1992, 228, 580–595 CrossRef CAS PubMed.
- S. F. Altschul and E. V. Koonin, Trends Biochem. Sci., 1998, 23, 444–447 CrossRef CAS PubMed.
- N. Stein, J. Appl. Crystallogr., 2008, 41, 641–643 CrossRef CAS.
- F. Corpet, Nucleic Acids Res., 1988, 16, 10881–10890 CrossRef CAS.
- T. C. Terwilliger, P. D. Adams, R. J. Read, A. J. McCoy, N. Moriarty, R. W. Grosse-Kunstleve, P. V. Afonine, P. H. Zwart and L. W. Hung, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 582–601 CrossRef CAS PubMed.
- R. W. Grosse-Kunstleve and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 1966–1973 CrossRef CAS PubMed.
- T. C. Terwilliger, R. W. Grosse-Kunstleve, P. V. Afonine, N. W. Moriarty, P. H. Zwart, L. W. Hung, R. J. Read and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 61–69 CrossRef CAS.
- K. Tunyasuvunakool, J. Adler, Z. Wu, T. Green, M. Zielinski, A. Zidek, A. Bridgland, A. Cowie, C. Meyer, A. Laydon, S. Velankar, G. J. Kleywegt, A. Bateman, R. Evans, A. Pritzel, M. Figurnov, O. Ronneberger, R. Bates, S. A. A. Kohl, A. Potapenko, A. J. Ballard, B. Romera-Paredes, S. Nikolov, R. Jain, E. Clancy, D. Reiman, S. Petersen, A. W. Senior, K. Kavukcuoglu, E. Birney, P. Kohli, J. Jumper and D. Hassabis, Nature, 2021, 596, 590–596 CrossRef CAS PubMed.
- A. Casañal, B. Lohkamp and P. Emsley, Protein Sci., 2020, 29, 1069–1078 CrossRef.
- D. Liebschner, P. V. Afonine, M. L. Baker, G. Bunkóczi, V. B. Chen, T. I. Croll, B. Hintze, L.-W. Hung, S. Jain, A. J. McCoy, N. W. Moriarty, R. D. Oeffner, B. K. Poon, M. G. Prisant, R. J. Read, J. S. Richardson, D. C. Richardson, M. D. Sammito, O. V. Sobolev, D. H. Stockwell, T. C. Terwilliger, A. G. Urzhumtsev, L. L. Videau, C. J. Williams and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2019, 75, 861–877 CrossRef CAS PubMed.
- O. S. Smart, T. O. Womack, C. Flensburg, P. Keller, W. Paciorek, A. Sharff, C. Vonrhein and G. Bricogne, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 368–380 CrossRef CAS PubMed.
- M. Källberg, H. Wang, S. Wang, J. Peng, Z. Wang, H. Lu and J. Xu, Nat. Protoc., 2012, 7, 1511–1522 CrossRef.
- B. Webb and A. Šali, Methods Mol. Biol., 2017, 1654, 39–54 CrossRef CAS PubMed.
- W. Zheng, C. Zhang, Q. Wuyun, R. Pearce, Y. Li and Y. Zhang, Nucleic Acids Res., 2019, 47, W429–W436 CrossRef CAS.
- L. Holm and P. Rosenström, Nucleic Acids Res., 2010, 38, W545–W549 CrossRef CAS PubMed.
- E. Krissinel and K. Henrick, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2256–2268 CrossRef CAS.
- Z. Li, L. Jaroszewski, M. Iyer, M. Sedova and A. Godzik, Nucleic Acids Res., 2020, 48, W60–W64 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 922–923 CrossRef.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- E. Krissinel and K. Henrick, J. Mol. Biol., 2007, 372, 774–797 CrossRef CAS.
- P. Stothard, Biotechniques, 2000, 28, 1102–1104 CrossRef CAS PubMed.
- A. S. Juncker, H. Willenbrock, G. Von Heijne, S. Brunak, H. Nielsen and A. Krogh, Protein Sci., 2003, 12, 1652–1662 CrossRef CAS PubMed.
- V. Friedrich, C. Gruber, I. Nimeth, S. Pabinger, G. Sekot, G. Posch, F. Altmann, P. Messner, O. Andrukhov and C. Schäffer, Mol. Oral Microbiol., 2015, 30, 451–473 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04166a |
| ‡ These authors shared first authorship. |
| This journal is © The Royal Society of Chemistry 2023 |