
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The switch-off method: rapid investigation of flow photochemical reactions†
Dawid
Drelinkiewicz
 ,
Stephen T.
Alston
,
Thomas
Durand
and
Richard J.
Whitby
*
,
Stephen T.
Alston
,
Thomas
Durand
and
Richard J.
Whitby
*
School of Chemistry, Faculty of Engineering and Physical Sciences, The University of Southampton, Southampton, UK. E-mail: rjw1@soton.ac.uk
First published on 26th June 2023
Abstract
In a flow photochemical process switching off the light source and monitoring the reaction composition as it leaves the photoreactor allows the effect of all irradiation times up to a maximum to be analysed in a single experiment. The switch-off method was illustrated using three reactions: [2 + 2] intermolecular photocycloadditions between an alkene and alkyne, and between two alkenes, and a Mallory photocyclization of cis-stilbene.
Introduction
Photochemical transformations performed using continuous flow have been an area of increasing interest in recent years,1–6 due to the simplicity of reactor design and higher and more homogeneous photon flux in photochemical flow processes.7 One such design was described by Booker-Milburn et al.8 consisting of coils of UV transparent tubing wrapped around an immersion well with a high-pressure mercury UV lamp contained within. This design has been adapted with the use of low-power, wavelength-selective fluorescent tube bulbs9 to allow operational simplicity, albeit with limited scale. A second design involves placing UV LEDs over a microfluidic chip10 which has the benefit of being able to choose wavelength-specific LEDs to irradiate the sample, but the drawback is that UV LEDs below 300 nm are currently expensive. These designs are advantageous as they provide a small, precisely defined depth of reaction solution that the light must penetrate through in order to fully irradiate the solution.11 This combined with the control of the irradiation time, via changing the flow rate or reactor size, and the uniformity of irradiation,11 allows the user excellent control over the reaction conditions which can prevent over-irradiation to side products. Calculation of the quantum yield and thermal back reaction of photochemical reactions has also been demonstrated using flow photochemistry methods combined with modelling tools using a spiral-shaped microreactor and UV LEDs.12 Additionally, the benefits13 of performing reactions using flow chemistry are still present such as ease of using high pressure and temperatures,14,15 faster heat transfer, and improved mixing.16One of the drawbacks of flow chemistry is that reaction time is determined by flow rate so that, unlike batch chemistry where a reaction can be sampled at many time points to determine optimum reaction times, in flow separate experiments are required for each time point. An elegant solution has been developed based on introducing a step-change17,18 or continuous change19 in flow rate to allow many timepoints for a reaction to be observed in a single run, termed the ‘push out’ method. The use of multivariate analysis of continuously acquired data (for example IR19 or UV-vis17,20) is particularly useful for the extraction of reaction data when these methods are used.
Herein we present an operationally simple method to obtain information on all irradiance times in a single flow photochemistry experiment by switching off the light source during a run (Fig. 1) and utilizing continuous in-line or off-line analysis of collected reaction fractions.21,22 It has an advantage over the ‘push out’ method in that a step change in flow rates, which most commercial flow systems cannot automate, is not required and that exposure times from zero are captured.
 | ||
| Fig. 1 Diagram of flow photochemical reactor after light source has been switched off showing the gradient of irradiance times throughout the reactor. | ||
Results and discussion
Reactor design
The photoreactor consisted of a single layer of PFA tubing (1 mm internal diameter) coiled around a quartz tube long enough to fit a 36 W fluorescent lamp inside. UV-A, B, C, and visible light bulbs at this power are cheap and easy to purchase and install. K-type thermocouples were installed at the beginning, centre, and end of photoreactor tubing, then aluminium foil was layered over, followed by a layer of tubing for water flow to cool the reactor if needed.9Methodology
The study of the effect of irradiance time was achieved by pumping the reagent solution through the photoreactor until a steady state of the reaction mixture is reached. At this point, the reactor light is switched off creating a gradient of effective exposure time throughout the photoreactor from the maximum time, down to zero. This gradient can then be analysed as the reaction leaves the reactor either using in-line monitoring or by collecting fractions for analysis using off-line techniques.There is some drop-off in irradiation intensity at the ends of the reactor which can be dealt with by only using data from the central 90% of the tube. An alternative method was to correct for any inhomogeneity using the results from a standard reaction to give effective exposure times – i.e. what the exposure time would be for a homogeneous light source (see ESI†).
Three model reactions have been investigated using this methodology.
[2 + 2] photocycloaddition of alkene and alkyne
[2 + 2] photocycloadditions are an important set of reactions forming cyclobutene compounds using relatively mild conditions23,24 which can then be used as synthetic intermediates to form many different compounds,25–27 natural products,28,29 and biologically active molecules30–32 containing the cyclobutene motif. Additionally, [2 + 2] photocycloadditions have been performed under continuous flow conditions by several groups33–37 demonstrating the reactions suitability towards study via the switch-off methodology. The photochemical reaction between diphenylacetylene and 3,4-dihydro-2H-pyran38 to form 7,8-diphenyl-2-oxabicyclo[4.2.0]oct-7-ene 1 was chosen as a first model reaction due to previous reports39 that have shown that 1 can, upon further irradiation, undergo either an electrocyclic ring opening into oxocine 2, or a Mallory reaction proceeding via 6π electrocyclic ring closure and formal oxidation to form phenanthrene based compound 3 with a loss of H2 (Scheme 1). The switch-off methodology was applied with the UV-C and UV-B light sources with the effective exposure time gradient being analysed using calibrated offline gas chromatography. With UV-C light it showed that in our system diphenylacetylene had all been consumed after 80 minutes and a maximum effective exposure time of 43 minutes was suitable for maximized formation of desired cycloaddition product 1 while simultaneously limiting the formation of side-products (Fig. 2a). Further irradiation causes 1 to convert into the bis-addition compound 4 formed from a second addition of 3,4-dihydro-2H-pyran to 1 which has not been reported before. Only low amounts of 2 and 3 were detected using GC analysis. Changing the irradiation wavelength to UV-B (Fig. 2b) lowered the rate of the reaction taking approximately 225 minutes to reach >99% conversion. However, it also changed the selectivity of the reaction forming 4 as the major product of the reaction at effective exposure times longer than 30 minutes. The selectivity is due to low absorption of light by diphenylacetylene at the wavelengths used by the UV-B lamp (305 to 315 nm), whereas 1 has a broad absorption across those wavelengths (λmax = 298 nm), so 1 will become the main activated species and due to the large excess of 3,4-dihydo-2H-pyran in the reaction, 4 becomes the major product. | ||
| Scheme 1 The [2 + 2] photocycloaddition of diphenylacetylene and 3,4-dihydro-2H-pyran. | ||
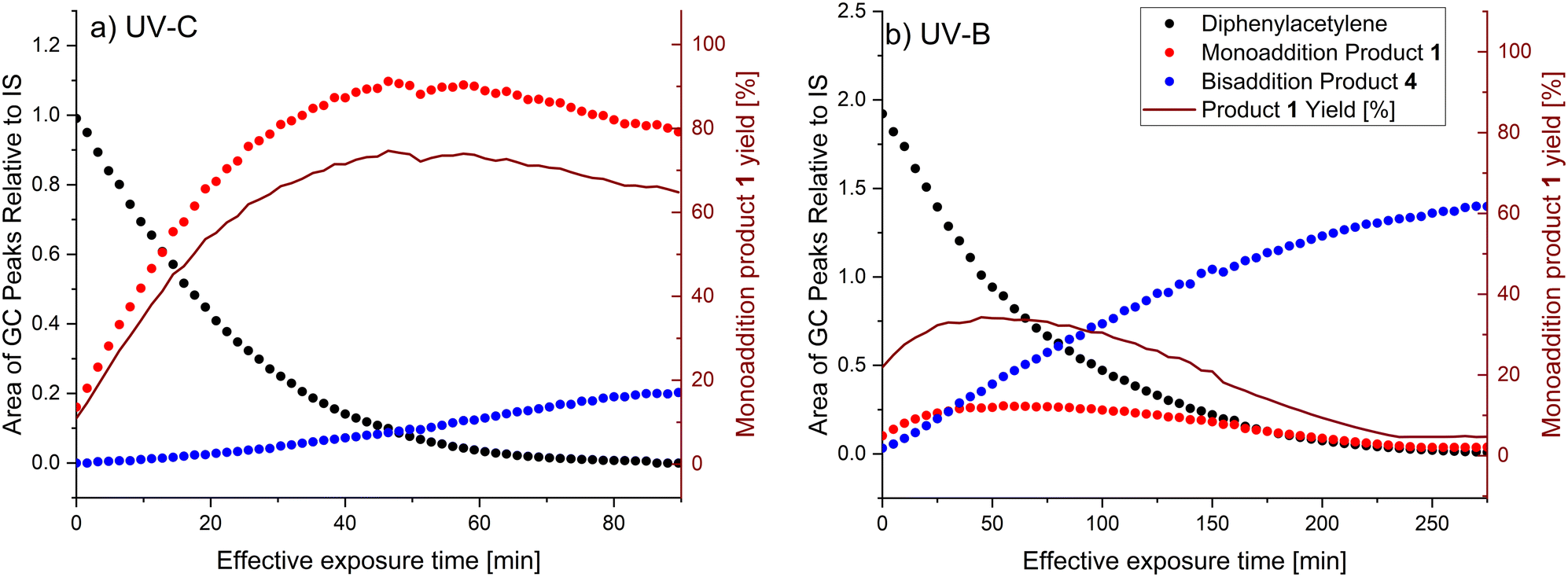 | ||
| Fig. 2 Results obtained from switch-off experiments of [2 + 2] photocycloaddition between 3,4-dihydro-2H-pyran and diphenylacetylene. Left axis corresponds to dotted lines representing area of GC peaks relative to internal standard and right axis corresponds to solid line representing percentage yield of compound 1. Relative GC areas of compounds 2 and 3 are not shown due to the low amounts formed. | ||
Two test reactions were performed with conditions obtained in switch-off experiments for both light sources to validate the results. Reaction utilizing UV-C light and effective exposure time of 43 minutes delivered product 1 in 71% isolated yield, whereas using UV-B light and 50 minutes effective exposure time delivered monocyclization product 1 in 37% isolated yield. Both results are in good agreement with result of the switch-off experiment.
[2 + 2] photocycloaddition of alkenes
A second model reaction tested was a [2 + 2] photocycloaddition between cyclohex-2-enone and tetramethylethylene (Scheme 2). In this reaction a number of compounds can be formed, including the substituted cyclobutane in a [2 + 2] photocycloaddition between alkenes (5 and 6), cyclohex-2-enone dimerization products (7 and 8), and cyclohexane (9) formed via the Norrish type II fragmentation of compounds 5 or 6.40–43 | ||
| Scheme 2 The [2 + 2] photocycloaddition between cyclohex-2-enone and tetramethylethylene. | ||
The switch-off experiments were performed using UV-A, B, and C light sources and showed that maximum yields of cyclobutane products 5 and 6 of 65%, 30%, and 22% are obtained after 49, 25, and 20 minutes of effective exposure times, respectively for UV-A, B, and C (Fig. 3). In the UV-B and C experiments, complete degradation of the photocycloaddition products was observed for long effective exposure times. The predominant formation for the less stable trans-fused cyclobutane isomer 5 is in accord with precedent40–42 and has been explained.43 The formation of 9 is precedented by some studies,41 but not others.40 Interestingly, when using UV-A light source a specific degradation of the cis-diastereoisomer 6 was observed after about 45 minutes of effective exposure time. For comparison, in the studies performed by Batten and Carless,40 probably using UV-C or broadband UV irradiation, the trans isomer 5 underwent light induced Norrish type I fragmentation much faster than the cis isomer 6, and formation of 9 was not observed.
 | ||
| Fig. 3 GC results from switch-off experiments of [2 + 2] photocycloaddition between cyclohex-2-enone and tetramethylethylene. Left axis corresponds to dotted lines representing area of GC peaks relative to internal standard and right axis corresponds to solid line representing percentage yield of compounds 5 and 6. | ||
Validation of the results was performed using UV-A light and 50 minutes effective exposure time, which resulted in formation of photocycloaddition products 5 and 6 in 62% yield (45% and 17%, respectively). Side-products 7, 8, and 9 were formed in 3%, 2%, and 8% isolated yields, respectively.
Mallory photocyclization
The third model reaction chosen for the switch-off methodology was the Mallory photocyclization of cis-stilbene to phenanthrene 10 (Scheme 3). This transformation is a highly studied area44–46 with a few examples of reactions using continuous flow methods.47–49 As stilbene absorbs across 240–320 nm, switch-off experiments were attempted using UV-A, B, and C lights and with two concentrations, 5.0 mM and 50.0 mM. In order to achieve oxidative conditions, reaction mixture was bubbled with oxygen stream prior to the reaction. | ||
| Scheme 3 Mallory photocyclization of cis-stilbene to phenanthrene 10, dimerization products 11 and 12, and phenanthrene–stilbene adduct 13. | ||
At 5.0 mM, UV-A gave 98% yield of phenanthrene 10 with effective exposure time of 24 minutes (Fig. 4), whereas UV-B and UV-C only went to 70% yield after 12 minutes (Fig. 5 and 6, respectively). The starting cis-stilbene was fully consumed after 23, 12, and 12 minutes, for UV-A, B, and C respectively, however when using UV-B light trans-stilbene was observed in approximately 16% yield after 12 minutes, which dropped down to 13% at 40 minutes. Moreover, stilbene dimers 11 and 12 were formed when using UV-B light after 5 minutes of effective exposure time in approximately 5% yield each. Experiments with UV-A afforded only traces of the 11 and 12, whereas UV-C light proved to be ineffective for formation of the dimers. The phenanthrene–stilbene adduct 13 (ref. 50) was formed in approximately 10%, 2.5%, and 1.5% yields after 10, 5, and 5 minutes of effective exposure time, respectively for UV-A, B, and C light. Interestingly, all three experiments showed that at longer exposure times the concentration of 13 drops, suggesting that it decomposes back to phenanthrene 10 and stilbene (which also transforms to 10), hence the high yield of phenanthrene when using UV-A light for long exposure times.
 | ||
| Fig. 4 GC results from the switch-off experiments of Mallory photoreaction using 5.0 mM cis-stilbene and UV-A light source. | ||
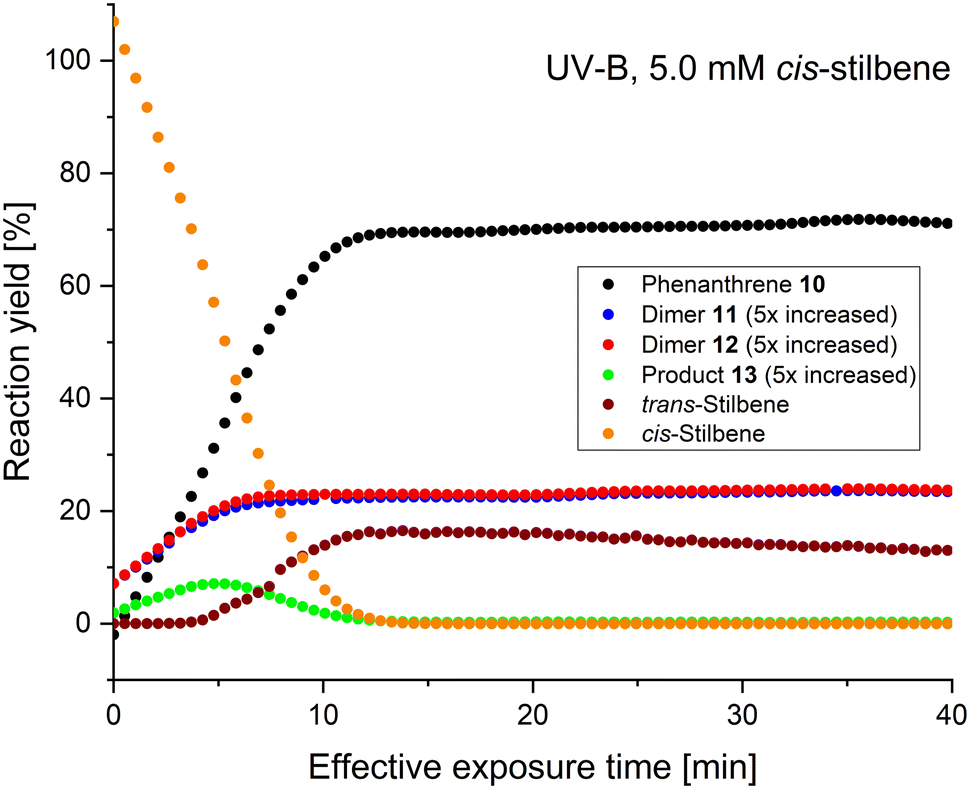 | ||
| Fig. 5 GC results from the switch-off experiments of Mallory photoreaction using 5.0 mM cis-stilbene and UV-B light source. | ||
 | ||
| Fig. 6 GC results from the switch-off experiments of Mallory photoreaction using 5.0 mM cis-stilbene and UV-C light source. | ||
At 50.0 mM cis-stilbene concentration, UV-B (Fig. 8) and C (Fig. 9) gave similar results for phenanthrene formation, affording 10 in 70% yield after 42 minutes, whereas UV-A only went to 56% yield after 150 minutes (Fig. 7). When using UV-B and UV-C light the cis-stilbene has been fully consumed after 11 and 10 minutes, respectively, and the trans-stilbene was observed only when using UV-B light, affording it in 16% yield after 42 minutes. However, when UV-A light source was used, cis-stilbene was fully consumed after 115 minutes and small amounts (<5%) of trans-stilbene were detected after 86 minutes of effective exposure time. UV-C light again showed to be ineffective for formation of dimers, however UV-A afforded the dimers 11 and 12 in 6% and 10% yield after 60 minutes, whereas UV-B light afforded the dimers in 2.1% and 4.6% yield after 17 minutes of effective exposure time. The phenanthrene–stilbene adduct 13 was formed in 2.6% yield after 34 and 13 minutes for UV-B and C respectively. Interestingly, when using UV-A light 13 was obtained in 18% yield after 86 minutes, after which the concentration of the 13 was steadily dropping. This is in agreement with the observed trans-stilbene formation after 86 minutes, and a conclusion that the adduct 13 decomposes back to phenanthrene 10 and trans-stilbene, which can then isomerise to cis-stilbene and form second molecule of 10 can be drawn, thus showing why phenanthrene was still formed even though no cis-stilbene has been observed after 115 minutes of effective exposure time.
 | ||
| Fig. 7 GC results from the switch-off experiments of Mallory photoreaction using 50.0 mM cis-stilbene and UV-A light source. | ||
 | ||
| Fig. 8 GC results from the switch-off experiments of Mallory photoreaction using 50.0 mM cis-stilbene and UV-B light source. | ||
 | ||
| Fig. 9 GC results from the switch-off experiments of Mallory photoreaction using 50.0 mM cis-stilbene and UV-C light source. | ||
In order to validate the method two test reactions were performed, the first was ran with 5.0 mM concentration, UV-A light, and 24 minutes effective exposure time. Phenanthrene 10 was isolated in 96% yield, which is consistent with the results from the switch-off experiment. Second reaction was performed using 50.0 mM concentration, UV-A light, and effective exposure time of 50 minutes, and resulted in recovery of 51.6% of cis-stilbene and formation of phenanthrene 10 in 10% yield, both cyclobutane dimers 11 and 12 in 8.5% yield, and phenanthrene–stilbene product 13 in 8.7% yield, which again, is consistent with the switch-off results.
Conclusions
A technique was designed to enable the rapid investigation of irradiance time in flow photochemical reactions using a single experiment. Reaction profiles against light exposure are obtained, as opposed to single timepoints, which greatly benefits optimisation as well as kinetic or mechanistic studies of photochemical reactions. The developed methodology was demonstrated on three different chemical systems. The [2 + 2] photocyclization of diphenylacetylene and 3,4-dihydro-2H-pyran was optimized for effective exposure time and light source giving best results when using 43 minutes and UV-C light. The selectivity of photochemical reaction was shown with the formation of a novel bis-addition product 4 being preferred when changing from UV-C to UV-B irradiation. Optimisation of the [2 + 2] photocycloaddition between cyclohex-2-enone and tetramethylethylene showed that the best results could be obtained when using UV-A light with 49 minutes of effective exposure time. The Mallory photocyclization of cis-stilbene was optimized for starting material concentration, irradiation time, and UV type giving an optimum condition of 24 minutes of effective exposure time, 5.0 mM cis-stilbene concentration, and UV-A light.Author contributions
Dawid Drelinkiewicz – conceptualization, formal analysis, investigation, validation, visualization, writing – review and editing; Stephen T. Alston – conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft; Thomas Durand – conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft; Richard J. Whitby – conceptualization, funding acquisition, methodology, resources, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank Dr Tom Corrie, Dr Matt Hughes, and Dr George Hodges (Syngenta) for useful discussions on this work. We are grateful to Syngenta (Jealott's Hill), the European Regional Development Fund (ERDF) (AI-Chem Channel within the InterReg programme) and a Southampton University Vice-Chancellors award for funding this work. Research infrastructure was supported by EPSRC (UK) core capability grant (EP/K039466).Notes and references
- K. Gilmore and P. H. Seeberger, Chem. Rec., 2014, 14, 410–418 CrossRef CAS PubMed.
- K. Booker-Milburn, Nat. Chem., 2012, 4, 433–435 CrossRef CAS PubMed.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- T. Junkers and B. Wenn, React. Chem. Eng., 2016, 1, 60–64 RSC.
- T. H. Rehm, Chem. – Eur. J., 2020, 26, 16952–16974 CrossRef CAS PubMed.
- F. N. Figueroa, A. A. Heredia, A. B. Peñéñory, D. Sampedro, J. E. Argüello and G. Oksdath-Mansilla, J. Org. Chem., 2019, 84, 3871–3880 CrossRef CAS PubMed.
- C. Sambiagio and T. Noël, Trends Chem., 2020, 2, 92–106 CrossRef CAS.
- B. D. A. Hook, W. Dohle, P. R. Hirst, M. Pickworth, M. B. Berry and K. I. Booker-Milburn, J. Org. Chem., 2005, 70, 7558–7564 CrossRef CAS PubMed.
- D. C. Harrowven, M. Mohamed, T. P. Gonçalves, R. J. Whitby, D. Bolien and H. F. Sneddon, Angew. Chem., Int. Ed., 2012, 51, 4405–4408 CrossRef CAS PubMed.
- O. Shvydkiv, A. Yavorskyy, S. B. Tan, K. Nolan, N. Hoffmann, A. Youssef and M. Oelgemöller, Photochem. Photobiol. Sci., 2011, 10, 1399–1404 CrossRef CAS PubMed.
- J. P. Knowles, L. D. Elliott and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025–2052 CrossRef CAS PubMed.
- T. Aillet, K. Loubière, O. Dechy-Cabaret and L. Prat, Chem. Eng. Technol., 2016, 39, 115–122 CrossRef CAS.
- J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC.
- E. R. Murphy, J. R. Martinelli, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 1734–1737 CrossRef CAS PubMed.
- V. Hessel, C. Hofmann, P. Löb, J. Löhndorf, H. Löwe and A. Ziogas, Org. Process Res. Dev., 2005, 9, 479–489 CrossRef CAS.
- B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300–2318 CrossRef CAS PubMed.
- T. Durand, C. Henry, D. Bolien, D. C. Harrowven, S. Bloodworth, X. Franck and R. J. Whitby, React. Chem. Eng., 2016, 1, 82–89 RSC.
- S. Mozharov, A. Nordon, D. Littlejohn, C. Wiles, P. Watts, P. Dallin and J. M. Girkin, J. Am. Chem. Soc., 2011, 133, 3601–3608 CrossRef CAS PubMed.
- J. S. Moore and K. F. Jensen, Angew. Chem., Int. Ed., 2014, 53, 470–473 CrossRef CAS PubMed.
- F. Benito-Lopez, W. Verboom, M. Kakuta, J. G. E. Gardeniers, R. J. M. Egberink, E. R. Oosterbroek, A. Van Den Berg and D. N. Reinhoudt, Chem. Commun., 2005, 2857–2859 RSC.
- T. Durand, PhD Thesis, University of Southampton, 2016 Search PubMed.
- S. T. Alston, PhD Thesis, University of Southampton, 2019 Search PubMed.
- D. I. Schuster, G. Lem and N. A. Kaprinidis, Chem. Rev., 1993, 93, 3–22 CrossRef CAS.
- S. Poplata, A. Tröster, Y. Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- D. Belluš and B. Ernst, Angew. Chem., Int. Ed. Engl., 1988, 27, 797–827 CrossRef.
- E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449–1483 CrossRef CAS PubMed.
- T. Bach, Synthesis, 1998, 1998, 683–703 CrossRef.
- Y. Y. Fan, X. H. Gao and J. M. Yue, Sci. China: Chem., 2016, 59, 1126–1141 CrossRef CAS.
- A. Sergeiko, V. V. Poroikov, L. O. Hanus and V. M. Dembitsky, Open Med. Chem. J., 2008, 2, 26–37 CrossRef CAS PubMed.
- M. Di Filippo, C. Bracken and M. Baumann, Molecules, 2020, 25, 356 CrossRef CAS PubMed.
- A. Figueras, R. Miralles-Llumà, R. Flores, A. Rustullet, F. Busqué, M. Figueredo, J. Font, R. Alibés and J. D. Maréchal, ChemMedChem, 2012, 7, 1044–1056 CrossRef CAS PubMed.
- R. Miralles-Llumà, A. Figueras, F. Busqué, A. Alvarez-Larena, J. Balzarini, M. Figueredo, J. Font, R. Alibés and J. D. Maréchal, Eur. J. Org. Chem., 2013, 2013, 7761–7775 CrossRef.
- K. G. Maskill, J. P. Knowles, L. D. Elliott, R. W. Alder and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2013, 52, 1499–1502 CrossRef CAS PubMed.
- M. Ralph, S. Ng and K. I. Booker-Milburn, Org. Lett., 2016, 18, 968–971 CrossRef CAS PubMed.
- C. Battilocchio, G. Iannucci, S. Wang, E. Godineau, A. Kolleth, A. De Mesmaeker and S. V. Ley, React. Chem. Eng., 2017, 2, 295–298 RSC.
- T. Fukuyama, Y. Hino, N. Kamata and I. Ryu, Chem. Lett., 2004, 33, 1430–1431 CrossRef CAS.
- S. Bachollet, K. Terao, S. Aida, Y. Nishiyama, K. Kakiuchi and M. Oelgemöller, Beilstein J. Org. Chem., 2013, 9, 2015–2021 CrossRef PubMed.
- H. M. Rosenberg and P. Serve, J. Org. Chem., 1968, 33, 1653–1654 CrossRef CAS.
- G. Kaupp and M. Stark, Chem. Ber., 1978, 111, 3608–3623 CrossRef CAS.
- R. J. Batten and H. A. J. Carless, J. Chem. Soc., Chem. Commun., 1985, 1146–1147 RSC.
- D. D. K. Manh, J. Ecoto, M. Fetizon, H. Colin and J.-C. Diez-Masa, J. Chem. Soc., Chem. Commun., 1981, 953–955 RSC.
- D. I. Schuster, N. Kaprinidis, D. J. Wink and J. C. Dewan, J. Org. Chem., 1991, 56, 561–567 CrossRef CAS.
- R. Shen and E. J. Corey, Org. Lett., 2007, 9, 1057–1059 CrossRef CAS PubMed.
- F. B. Mallory, C. S. Wood and J. T. Gordon, J. Am. Chem. Soc., 1964, 86, 3094–3102 CrossRef CAS.
- K. B. Jørgensen, Molecules, 2010, 15, 4334–4358 CrossRef PubMed.
- H. Meier, Angew. Chem., Int. Ed. Engl., 1992, 31, 1399–1420 CrossRef.
- H. Okamoto, T. Takane, S. Gohda, Y. Kubozono, K. Sato, M. Yamaji and K. Satake, Chem. Lett., 2014, 43, 994–996 CrossRef CAS.
- H. Okamoto, H. Takahashi, T. Takane, Y. Nishiyama, K. Kakiuchi, S. Gohda and M. Yamaji, Synthesis, 2017, 49, 2949–2957 CrossRef CAS.
- Y. Shimo, T. Mikami, S. Hamao, H. Goto, H. Okamoto, R. Eguchi, S. Gohda, Y. Hayashi and Y. Kubozono, Sci. Rep., 2016, 6, 1–13 CrossRef PubMed.
- R. A. Caldwell, K. Mizuno, P. E. Hansen, L. P. Vo, M. Frentrup and C. D. Ho, J. Am. Chem. Soc., 1981, 103, 7263–7269 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00261f |
| This journal is © The Royal Society of Chemistry 2023 |