
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-platform synthesis of ondansetron featuring process intensification in flow†
Yoshio
Hato
 ab and
Timothy F.
Jamison
*a
ab and
Timothy F.
Jamison
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: tfj@mit.edu
bShionogi Pharmaceutical Research Center, Shionogi & Co., Ltd., Toyonaka, Osaka 561-0825, Japan
First published on 1st June 2023
Abstract
Efficient and robust synthetic processes of active pharmaceutical ingredients (APIs) are highly desirable, and continuous flow chemistry is a critical component of this endeavor. The clinical importance of ondansetron, a World Health Organization essential medicine, prompted us to investigate continuous synthetic approaches to this API. Our efforts to improve the synthetic processes led to a continuous condensation step and a continuous Mannich reaction. A continuous work-up and purification process was also established for the former. A batch process was employed for an elimination and Michael addition step, as it not only accommodated the physical properties of the reaction mixtures, but also provided a high productivity of the desired product. Taken together, these findings demonstrate the complementary advantages of flow and batch chemistry in API synthesis.
Introduction
Ondansetron is a 5-HT3 receptor antagonist and used for the prevention of nausea and vomiting associated with cancer chemotherapy, radiotherapy or surgery.1 Developed by Glaxo Group Limited in 1980s2,3 and approved by the U.S. Food and Drug Administration (FDA) in 1991 as the first 5-HT3 receptor antagonist,1 ondansetron was the 20th-highest-selling brand-name pharmaceutical in the United States in 2005. The first generic version of ondansetron was approved by the U.S. FDA in 2006,4 and it has been on the World Health Organization's model list of essential medicines since 2009.5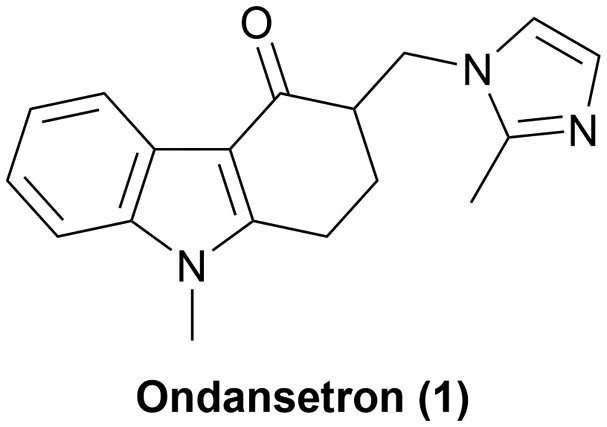
Continuous flow synthesis (flow chemistry) is an enabling platform for the synthesis of active pharmaceutical ingredients (APIs)6 for many reasons, including control of reaction parameters with greater accuracy and precision7 and efficiencies that are often realized upon scale-up of a continuous flow synthesis.8 We have recently demonstrated the continuous flow synthesis of several APIs, including tramadol, enalapril, imatinib, and linezolid.9 The ongoing interest in efficient synthetic processes for APIs and the clinical importance of ondansetron prompted us to investigate continuous synthesis approaches to ondansetron.
A recently published study demonstrated the synthesis of an ondansetron precursor (5) in a continuous flow manner, followed by a two-step, one-pot batch process to give the API in an overall 65% isolated yield (Scheme 1).10 In this report, the condensation of 1,3-cyclohexanedione (2) with 1-methyl-1-phenylhydrazine (3) and the Fisher indole synthesis of the resulted intermediate (4) were achieved in a continuous flow manner, affording the carbazolone (5) in 75% yield, which in turn was converted in batch by way of a Mannich reaction and substitution of dimethylamine with 2-methylimidazole (8), giving ondansetron in 87% yield. We envisaged that the use of alternative, readily available starting materials and continuous flow technology might lead to further improvement. Accordingly, we chose N-methylaniline (9) as a more readily available starting compound than 1-methyl-1-phenylhydrazine (3), surmising that condensation of 9 with 1,3-cyclohexanedione (2) would give enaminone 10, which could be converted into the same ondansetron precursor (5) by oxidative cyclization.11 During the course of our study, a comparable oxidative cyclization of 10 to 5 in a flow photochemical process was reported,12 prompting us to shift our focus to the other three steps in the ondansetron synthesis. Herein we describe our improvements to the synthetic process of ondansetron by the development of continuous synthesis methods for the initial condensation noted above (step 1), the Mannich reaction (step 3), and substitution with 2-methylimidazole (8) (step 4).
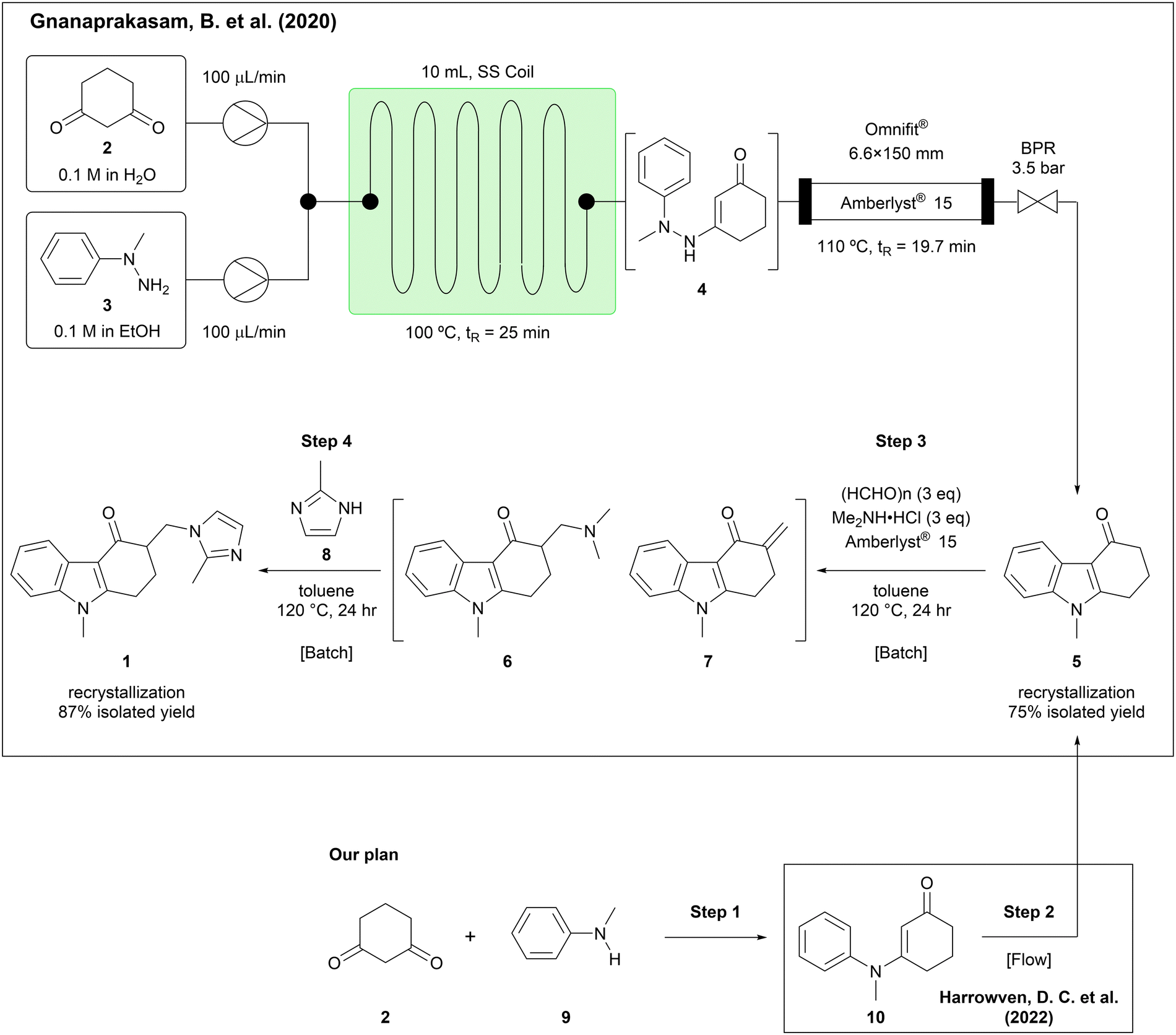 | ||
| Scheme 1 Previous approaches and our experimental plan. | ||
Results and discussion
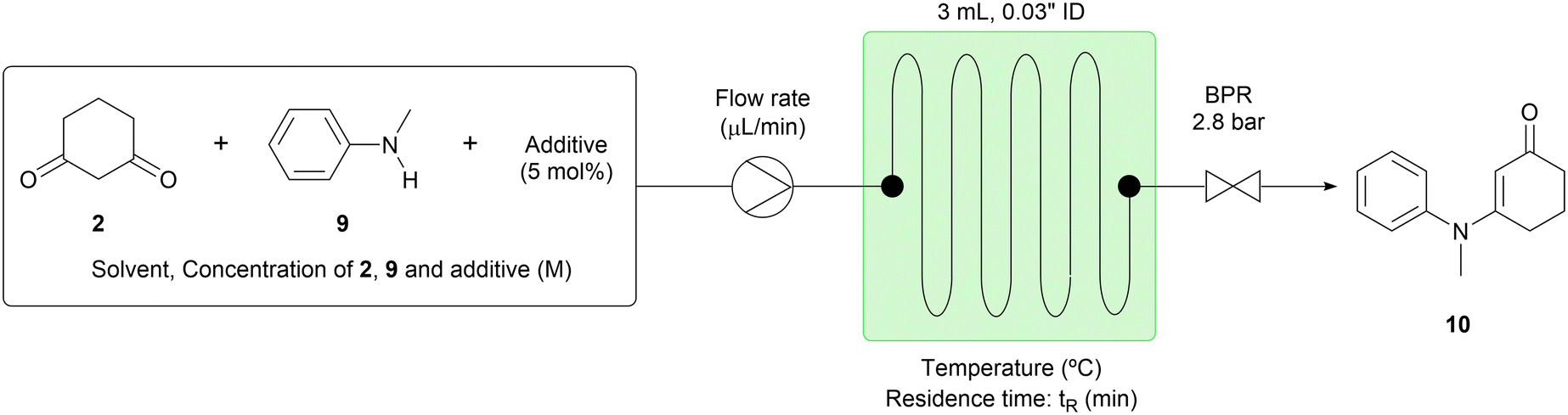
The batch conditions previously described12 were used as a starting point for our initial studies of the condensation step (step 1). We found that the reaction time of 16 hours in batch could be reduced to a 30 minute residence time in flow. Important modifications that secured the practicality of this flow approach included changing the reaction solvent from toluene to 1,2-dichloroethane (DCE), in which 1,3-cyclohexanedione (2) was much more soluble (Table 1, entry 1). The target compound (10) was afforded in 79% yield with 14% of N-methylaniline (9) remaining. We also found that increasing the reaction temperature and lengthening the residence time did not improve the yield (Table 1, entries 2–3). Next, other solvents in which 1,3-cyclohexanedione (2) has appreciable solubility, such as acetonitrile (MeCN), ethanol (EtOH) and dioxane were screened; however, conversion of 9 was low to moderate in all cases (Table 1, entries 4–6). As the use of water-miscible solvents such as MeCN and dioxane gave lower conversion than the use of DCE, we posited that conversion might be hampered by the presence of water in the reaction system and therefore was accelerated in batch by the use of the Dean–Stark apparatus.12 Nevertheless, contrary to this hypothesis, when acetic acid (AcOH) was used as a solvent, the conversion improved dramatically, affording the target compound (10) in 97% yield (Table 1, entry 7). Furthermore, similar results were observed in the absence of p-toluenesulfonic acid monohydrate (TsOH·H2O), yet 1.2 equiv. of 1,3-cyclohexanedione (2) were needed to ensure good conversion (Table 1, entries 8–9). Thus, the positive effect of AcOH as a solvent was confirmed.13 Further experimentation revealed that the concentration of the reactant solution could be increased to 2.0 M from 0.2 M and the residence time could be shortened to 5 minutes from 30 minutes (Table 1, entry 10).14 With this change, the processing rate of N-methylaniline (9) increased 60 times from 0.13 to 7.8 mL per hour. As further attempts to achieve complete conversion, such as increasing the reaction temperature and using a catalytic amount of TsOH·H2O did not improve the yield of 10,14 we turned our attention to a continuous work-up process using the reaction conditions of entry 10.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Reaction parameter | NMR yielda (%) | ||||||
| Solvent | Additive | Conc. (M) [2, 9, additive] | Temp. (°C) | t R (min) | Flow rate (μL min−1) | 9 | 10 | |
| a The NMR yield was determined using 1,3,5-trimethoxybenzene as an internal standard. b 6.9 bar of the back pressure was applied. | ||||||||
| 1 | DCE | p-TsOH·H2O | 0.2, 0.2, 0.01 | 110 | 30 | 100 | 14 | 79 |
| 2b | DCE | p-TsOH·H2O | 0.2, 0.2, 0.01 | 130 | 30 | 100 | 15 | 79 |
| 3b | DCE | p-TsOH·H2O | 0.2, 0.2, 0.01 | 130 | 60 | 50 | 11 | 81 |
| 4 | MeCN | p-TsOH·H2O | 0.2, 0.2, 0.01 | 110 | 30 | 100 | 30 | 61 |
| 5b | EtOH | p-TsOH·H2O | 0.2, 0.2, 0.01 | 110 | 30 | 100 | 64 | 25 |
| 6 | DOX | p-TsOH·H2O | 0.2, 0.2, 0.01 | 110 | 30 | 100 | 21 | 70 |
| 7 | AcOH | p-TsOH·H2O | 0.2, 0.2, 0.01 | 110 | 30 | 100 | 2.7 | 97 |
| 8 | AcOH | — | 0.2, 0.2, — | 110 | 30 | 100 | 6.7 | 90 |
| 9 | AcOH | — | 0.24, 0.2, — | 110 | 30 | 100 | 2.3 | 96 |
| 10 | AcOH | — | 2.4, 2.0, — | 110 | 5 | 600 | 3.3 | 96 |
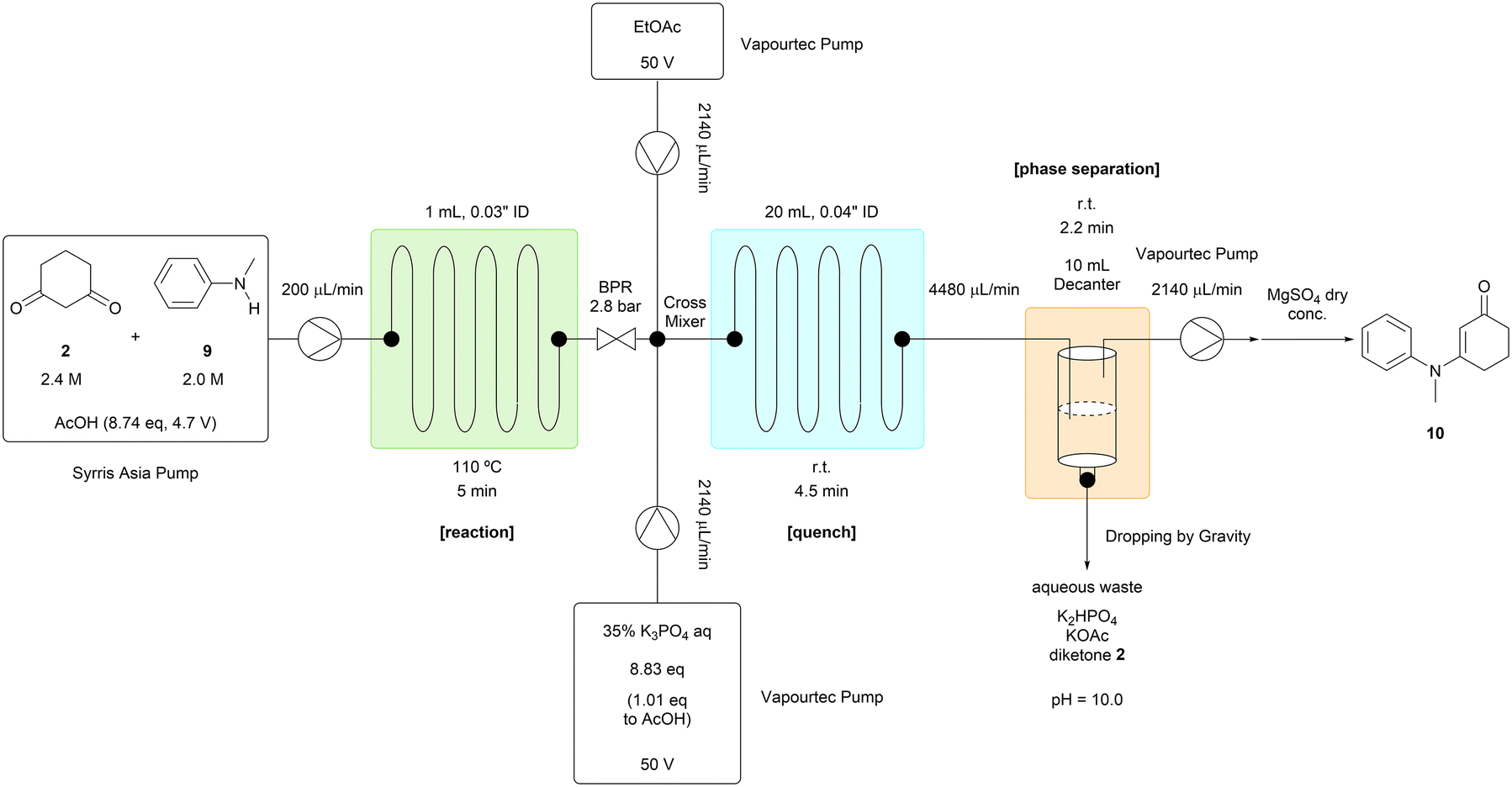
The reaction solution stream was mixed with 35% potassium phosphate aqueous solution and ethyl acetate after passing through a back pressure regulator (Table 2). This two-phase solution flowed into a decanter used to separate the phases. The organic phase containing the desired product (10) was pumped into the storage tank from the top of the decanter, and the aqueous phase was removed from the bottom of the decanter. The robustness of this process was confirmed by running the system over 7 hours with N-methylaniline (9) processed at the rate of 2.6 mL per hour. The target compound (10) was obtained at a consistent yield of around 96% during this experiment. The excess amount of 1,3-cyclohexanedione (2) was removed into the aqueous phase. AcOH was not detected in the organic phase residue by 1H NMR analysis, and we concluded that a tube reactor, rather than a continuously stirred tank reactor, was efficient enough to remove the AcOH solvent with a residence time of a few minutes. The unreacted N-methylaniline (9) was removed by filtering a slurry of the crude organic phase residue in hexane, affording the target compound (10) in an isolated yield of 94.9%.14
For the Mannich reaction involving 5 and 2-methylimidazole (8) (steps 3 and 4), we observed similar results with a batch, one-pot procedure reported by Hesoun and Hykl, giving ondansetron (1) in a good yield after recrystallization from cold methanol (Scheme 2, top).15 Because the reaction mixture of steps 3 and 4 was a thick slurry due to limited solubility of the reactants and the products in DMF,16 we determined that these conditions would not be suitable for a plug-flow reactor system due to the likelihood of clogging by particles of different size and density.17
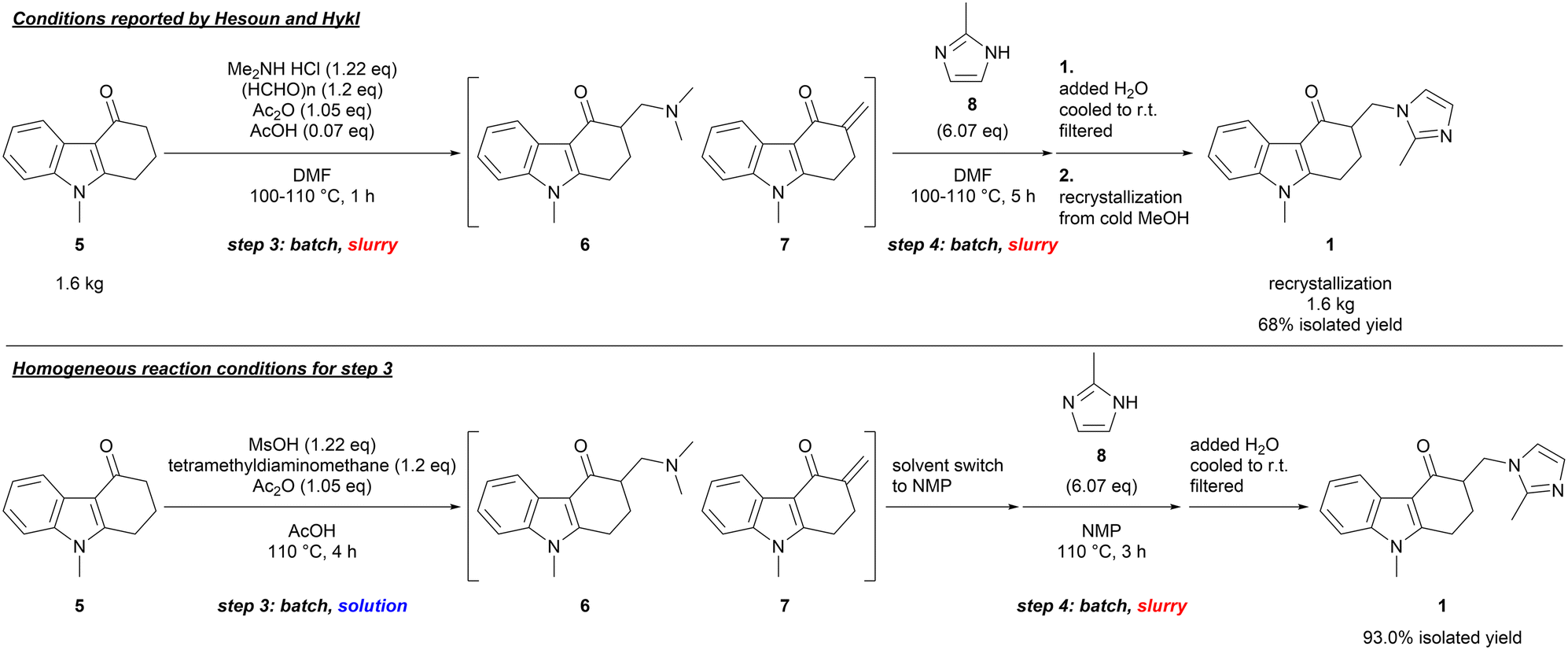 | ||
| Scheme 2 Conditions reported by Hesoun and Hykl, and homogeneous reaction conditions for step 3. | ||
To prepare a homogenous reactant solution for step 3 and to maintain the homogeneity during the last two steps, the solubility of the substrate (5) and ondansetron (1) were examined, and AcOH was chosen as a promising solvent.16 The further studies revealed that the Mannich reaction of 5 proceeded with the liquid reagents such as N,N,N,N-tetramethyldiaminomethane and methanesulfonic acid in a solvent amount of AcOH, successfully making the reaction mixture of step 3 a homogeneous solution (Scheme 2, bottom). However, the Michael addition of 2-methylimidazole (8) (step 4) became sluggish in AcOH, and a solvent switch was necessary between steps 3 and 4.14 Since AcOH could not be used for step 4 and ondansetron (1) could be crystallized from the reaction mixture because of its low solubility in other solvents, we envisaged that a flow setup for step 3 and a batch setup for step 4 could lead to a practical and robust process.
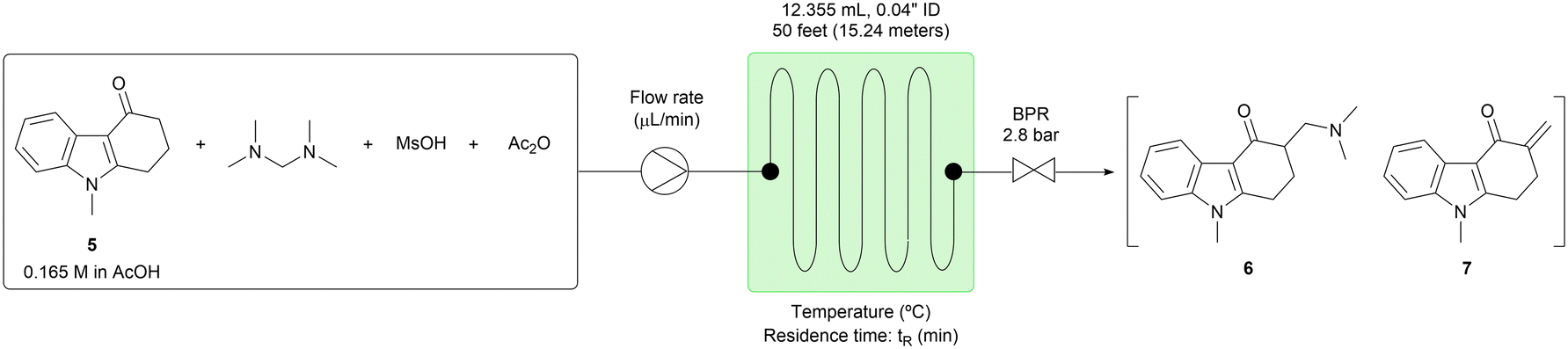
First, the homogeneous conditions shown in Scheme 2 were adapted to the flow configuration for step 3. For example, the batch reaction time was reduced from 4 hours to a 60 minute residence time in the flow system (Table 3, entry 1). However, conversion was incomplete with 8.10 area % of the starting material (5) unreacted. When the temperature was increased to 120 °C, the conversion was increased to 98% (Table 3, entry 2). Further increase of the reaction temperature to 130 °C allowed the residence time to be shortened to 30 minutes, giving 97% conversion (Table 3, entry 3). However, the impurities tended to increase at a higher temperature, and therefore reaction temperatures greater than 130 °C were not examined. Next, the stoichiometry of acetic anhydride (Ac2O) was investigated. When 3.15 equiv. of Ac2O were used, the conversion was reduced to 94% (Table 3, entry 6). In contrast, when 0 to 2.10 equiv. of Ac2O were used, the conversions were almost equal, while the product distribution between 6 and 7 was different (Table 3, entries 3–5). Product 6 was generated more and product 7 was generated less when the larger amount of Ac2O was used. It was assumed that product 6 had higher solubility than product 7 in AcOH because of the dimethylamine moiety; therefore, the conditions of entry 5 were selected for the extended-time experiments directed toward minimizing the risk of system failure.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Reaction parameter | HPLC (area%) | ||||||
| Equivalent [diamine, MsOH, Ac2O] | Temp. (°C) | t R (min) | Flow rate (μL min−1) | 5 | 6 | 7 | 5 + 6 + 7 | |
| 1 | 1.20, 1.22, 1.05 | 110 | 60 | 206 | 8.10 | 84.31 | 6.09 | 98.50 |
| 2 | 1.20, 1.22, 1.05 | 120 | 60 | 206 | 2.08 | 73.93 | 19.45 | 95.46 |
| 3 | 1.20, 1.22, 1.05 | 130 | 30 | 412 | 3.23 | 69.99 | 21.84 | 95.06 |
| 4 | 1.20, 1.22, 0 | 130 | 30 | 412 | 2.85 | 62.02 | 29.40 | 94.27 |
| 5 | 1.20, 1.22, 2.10 | 130 | 30 | 412 | 2.18 | 77.88 | 15.16 | 95.22 |
| 6 | 1.20, 1.22, 3.15 | 130 | 30 | 412 | 5.46 | 74.47 | 15.07 | 95.00 |
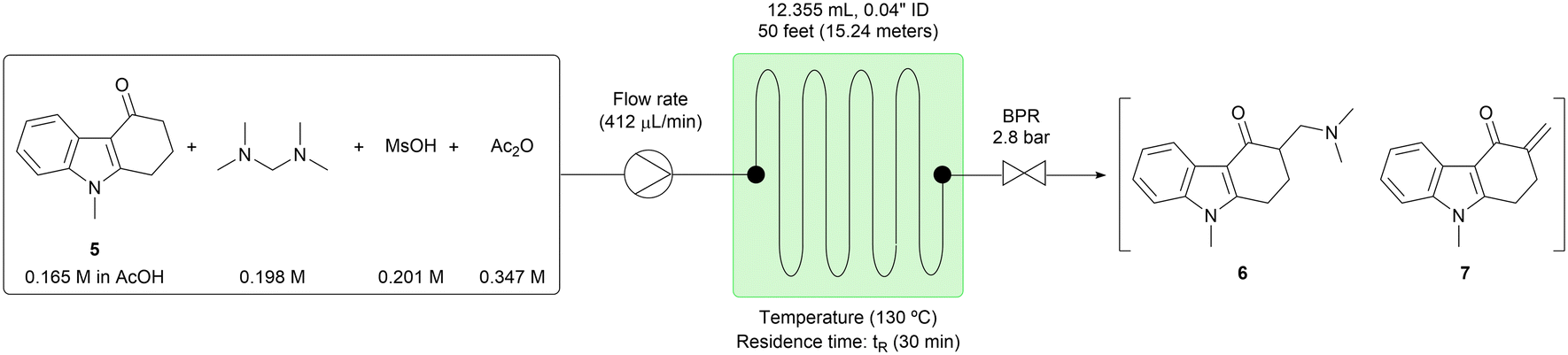
The process integrity was demonstrated over 7 hours without any clogging observed. The starting material (5) was processed at the rate of 0.81 g per hour, and the target compounds (6) and (7) were generated in consistent yields (Table 4). We also studied the stability of the reactant solution by monitoring the concentration of 5 by HPLC over time. Over 8 hours, 0.6 area% of the initial 5 were converted to the reaction products, consistent with the simple notion that the reaction of interest proceeds very slowly at room temperature. We reasoned that this slow background reaction in the reactant solution did not create any concerns that would require mitigation.
|
|
|||||
|---|---|---|---|---|---|
| Running time (h) | HPLC (area%) reactant solution | HPLC (area%) reacted solution | |||
| 5 | 5 | 6 | 7 | 5 + 6 + 7 | |
| −1.5 | 99.59 | — | — | — | — |
| 0 | 99.45 | 3.61 | 76.87 | 15.47 | 95.95 |
| 1 | 99.18 | 3.47 | 77.18 | 15.29 | 95.94 |
| 2 | 99.11 | 3.50 | 76.80 | 15.55 | 95.85 |
| 3 | 99.12 | 3.46 | 76.84 | 15.56 | 95.86 |
| 4 | 99.09 | 3.37 | 77.17 | 15.31 | 95.85 |
| 5 | 98.99 | 3.38 | 76.93 | 15.52 | 95.83 |
| 6 | 99.00 | 3.37 | 77.16 | 15.50 | 96.03 |
| 7 | 99.02 | 3.38 | 77.22 | 15.20 | 95.80 |
| All collected solution | — | 3.45 | 76.95 | 15.46 | 95.86 |
The substitution of the dimethylamine moiety by elimination and the Michael addition of 2-methylimidazole (8) (step 4) was conducted in batch after a solvent switch, affording ondansetron (1) in 91.8% yield over the last 2 steps (Scheme 3). The recrystallization of this product from ethanol gave ondansetron (1) in 93.4% yield, >99.5 area % purity.
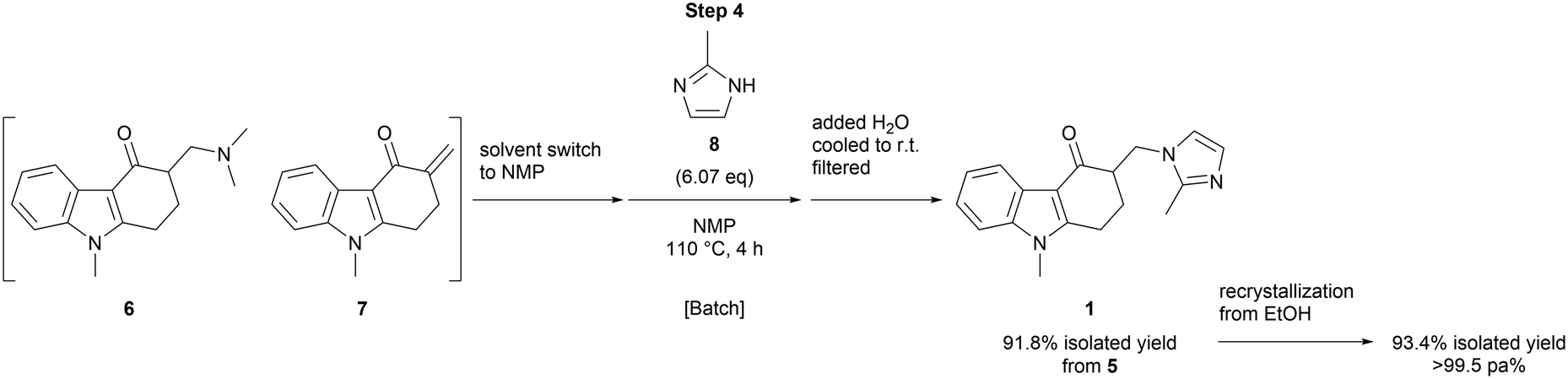 | ||
| Scheme 3 Substitution with 8 and recrystallization. | ||
Conclusions
In summary, we have developed a practical and scalable synthesis of ondansetron (1) by a multi-platform, built-for-purpose approach. Continuous flow synthesis provided significant improvements in steps 1 and 3, as did a continuous work-up process that was developed for step 1. In contrast, a batch process for step 4 was developed due to its high throughput and physical properties of the reaction mixtures. Taken together, this overall process highlights the complementary strengths of batch and flow approaches to API synthesis.Experimental section
General information
All reactions were performed with commercially available reagents and solvents that were used as received unless otherwise specified. The reagents and solvents were purchased from Sigma Aldrich, Alfa Aesar, TCI, Combi-Blocks, Thermo Scientific, Oakwood Chemical or STREM Chemicals. Batch reactions were performed in round bottom flasks under an argon or nitrogen atmosphere unless otherwise noted. Flow reactions were performed using the commercially available components supplied from IDEX Health & Science, Upchurch Scientific, Swagelok Company, Harvard Apparatus, Syrris, Vapourtec, Zaiput Flow Technologies and Luzchem Research. Reactions were monitored by thin-layer chromatography (TLC), high-performance liquid chromatography (HPLC) or proton nuclear magnetic resonance (1H NMR). Concentration and removal of solvents was performed using a Buchi Rotavapor R-210. Column chromatography was carried out using a prepackaged Teledyne ISCO RediSep High-Performance silica gel column on a Biotage Isolera chromatography system.Proton nuclear magnetic resonance (1H NMR) spectra and proton-decoupled carbon nuclear magnetic resonance (13C{1H} NMR) spectra were recorded on either a two-channel Bruker avance-III HD Nanobay spectrometer, a three-channel Bruker Avance Neo spectrometer, or a two-channel JEOL ECZ spectrometer at ambient temperature at operating frequencies of 500/400 MHz (1H) or 125/100 MHz (13C). Chemical shifts (δ) are reported in parts per million (ppm) relative to the solvent residual peak (CDCl3; 7.26 ppm for 1H NMR and 77.16 ppm for 13C NMR). Data are represented as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, o = overlap), coupling constants (J) in Hertz (Hz), integration. 1,3,5-Trimethoxybenzene and dimethyl terephthalate were used as an internal standard for quantitative 1H NMR spectroscopy. High-performance liquid chromatography (HPLC) analysis was performed using an Agilent 1200 series quaternary HPLC system with an Agilent Poroshell 120 EC-C18 2.7 μm, 4.6 × 50 mm column. The HPLC measurement conditions were as follows: flow rate: 1.000 mL min−1, UV detection wavelength: 254 and 210 nm, mobile phase: [A] is 0.1% trifluoroacetic acid containing aqueous solution and [B] is 0.1% trifluoroacetic acid containing acetonitrile solution, gradient: linear gradient of 5% to 100% solvent [B] for 10 minutes was performed and 100% solvent [B] was maintained for 2 minutes and 5% solvent [B] was maintained for 2 minutes, column temperature: 35 °C, injection volume: 2.00 μL, sample preparation: an appropriate amount of samples was dissolved in 1 mL of 80% MeCN aq. High-resolution mass spectrometry data were acquired on a JEOL AccuTOF 4F LC-plus equipped with an ionSense DART (Direct Analysis in Real Time) source. IR spectra were recorded on a Bruker Alpha II FTIR spectrometer with a Diamond Crystal ATR (attenuated total reflectance) accessory and only significant absorptions were listed.
Procedure for reaction condition screening of step 1 (Table 1, entry 10)
N-Methylaniline 9 (5.42 mL, 50 mmol, 1 eq.), 1,3-cyclohexanedione 2 (6.17 g, 55 mmol, 1.1 eq.) and p-TsOH·H2O (476 mg, 2.5 mmol, 0.05 eq.) were added to a 25 mL volumetric flask. AcOH was added to the 25 mL line of the volumetric flask. The reactor was constructed from the fluorinated ethylene propylene (FEP) tubing (1/16′′ outside diameter, 0.03′′ inside diameter, 658 cm, 3 mL), the complementary polyether ether ketone (PEEK) fittings, and the Upchurch Scientific's back pressure regulator (40 psi, 2.8 bar). The prepared reactant solutions were pumped into the reactor by the Syrris Asia pump at the designated flow rate (600 μL min−1). The reaction tube was immersed in an oil bath and heated at the designated temperature (110 °C). The reaction was equilibrated for 15 minutes, which was three times longer than the residence time. After the equilibration, 6 mL of the solution was collected for 10 minutes. To the collected solution, was added 1,3,5-trimethoxybenzene (673 mg, 4 mmol, 1/3 eq. of the collected reaction scale) as a standard compound. 0.1 mL of this solution was washed with 5% NaHCO3 aq (6 mL), extracted with EtOAc three times (6 + 6 + 6 mL), dried over MgSO4, filtered, concentrated and analyzed by 1H NMR. The methoxy peak (3.77 ppm, 9H) or the aromatic peak (6.08 ppm, 3H) of 1,3,5-trimethoxybenzene was used as a reference. The methyl peak (3.24 ppm, 3H) of the target compound 1013 and the methyl peak (2.84 ppm, 3H) of N-methylaniline 9 were used to determine the NMR yields.Procedure for continuous work-up process of step 1 (Table 2)
N-Methylaniline 9 (21.67 mL, 200 mmol, 1 eq.), 1,3-cyclohexanedione 2 (26.91 g, 240 mmol, 1.2 eq.) were added to a 100 mL volumetric flask. AcOH was added to the 100 mL line of the volumetric flask. The reactor was constructed from the fluorinated ethylene propylene (FEP) tubing (1/16′′ outside diameter, 0.03′′ inside diameter, 219 cm, 1 mL), the complementary polyether ether ketone (PEEK) fittings, and the Upchurch Scientific's back pressure regulator (40 psi, 2.8 bar). The quenching and extraction tube was constructed from the perfluoroalkoxy (PFA) tubing (1/16′′ outside diameter, 0.04′′ inside diameter, 2467 cm, 20 mL), and the complementary polyether ether ketone (PEEK) fittings. The decanter was constructed from the 10 mL glass syringe, the complementary polyether ether ketone (PEEK) fittings, and the rubber septa. The prepared reactant solutions were pumped into the reactor by the Syrris Asia pump at the designated flow rate (200 μL min−1). The reaction tube was immersed in an oil bath and heated at the designated temperature (110 °C). 35% K3PO4 aq and EtOAc were pumped into the quenching and extraction tube by the Vapourtec pumps at the designated flow rate (2.14 mL min−1). The organic phase was pumped out of the decanter into the storage tank at the designated flow rate (2.14–2.19 mL min−1). The system was equilibrated for 36 minutes, which was three times longer than the residence time. After the equilibration, 32.1 mL of the organic phase was collected for 15 minutes every hour. To each of the collected solution, was added 1,3,5-trimethoxybenzene (336.4 mg, 2 mmol, 1/3 eq. of the collected reaction scale) as a standard compound. This solution was dried over MgSO4, filtered, concentrated and analyzed by 1H NMR. The methoxy peak (3.77 ppm, 9 H) or the aromatic peak (6.08 ppm, 3 H) of 1,3,5-trimethoxybenzene was used as a reference. The methyl peak (3.24 ppm, 3H) of the target compound 1013 and the methyl peak (2.84 ppm, 3H) of N-methylaniline 9 were used to determine the NMR yields. To the 674 mL of the organic phase which was collected for 315 minutes, was added MgSO4 (6.7 g, 0.01 W). The mixture, which could contain 25.41 g of the target compound 10 theoretically, was filtered, washed with EtOAc and concentrated to 31.10 g. To the residue, was added hexane (127 mL, 5 V). The solution was concentrated to 28.63 g. To the residue, was added hexane (127 mL, 5 V). The solution was concentrated to 30.51 g. To the residue, the seed crystal was added if necessary. To the solid, was added hexane (127 mL, 5 V). The slurry was stirred at 5 °C for 1 h. The slurry was filtered, washed with cold hexane (127 mL, 5 V), and air-dried overnight to afford the title compound 1013 (24.115 g, 94.9% yield, 97.62 pa%, yellow solid, MWL loss: 1.4%). 1H NMR (500 MHz, CDCl3) δ: 7.41 (t, J = 7.7 Hz, 2H), 7.31 (t, J = 7.4 Hz, 1H), 7.15–7.11 (m, 2H), 5.32 (s, 1H), 3.24 (s, 3H), 2.31 (t, J = 6.5 Hz, 2H), 2.21 (t, J = 6.2 Hz, 2H), 1.89 (p, J = 6.3 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ: 197.7, 165.1, 145.5, 129.8 (2C), 127.6, 127.3 (2C), 100.7, 40.9, 36.3, 28.7, 22.7. IR (neat) ν: 3042, 2938, 2891, 1610, 1556 cm−1. HRMS (DART) m/z: calcd for C13H16NO+ [(M + H)+] 202.1226, found 202.1220.Procedure for homogeneous reaction conditions of step 3 (Scheme 2, bottom)
To a 250 mL round bottomed flask, were added the substrate 5 (2.00 g, 10.04 mmol, 1 eq.), and AcOH (60 mL, 30 V). The mixture was stirred at 24 °C for 10 minutes to make a homogeneous solution. To the solution, were added N,N,N,N-tetramethyldiaminomethane (1.64 mL, 12.04 mmol, 1.20 eq.), MsOH (0.80 mL, 12.25 mmol, 1.22 eq.), Ac2O (1.00 mL, 10.54 mmol, 1.05 eq., 0.5 V) in this order. The reaction mixture was warmed to 110 °C and stirred at 110 °C for 4 hours. The reaction mixture was concentrated to 13.06 g, 6.53 W. To the residue, was added toluene (60 mL, 30 V). The reaction mixture was concentrated to 8.51 g, 4.26 W. To the residue, was added toluene (60 mL, 30 V). The reaction mixture was concentrated to 6.82 g, 3.41 W. To the residue, were added NMP (8 mL, 4 V) and 2-methylimidazole 8 (5.00 g, 60.93 mmol, 6.07 eq.). The reaction mixture was warmed to 110 °C and stirred at 110 °C for 3 hours. The reaction mixture was cooled to 100 °C. To the mixture, was added H2O (62.5 mL, 31.25 V) over 10 minutes. The reaction mixture was cooled to r.t., filtered and washed with H2O (60 mL, 30 V). The solid was air-dried overnight to give ondansetron 110 (2.735 g, 93.0% yield, 97.58 pa%, off-white solid, MWL loss: 1.3%).Procedure for reaction condition screening of step 3 (Table 3)
The substrate 5 (1.64 g, 8.25 mmol, 1 eq.), N,N,N,N-tetramethyldiaminomethane (1.35 mL, 9.90 mmol, 1.2 eq.), MsOH (0.65 mL, 10.07 mmol, 1.22 eq.) and Ac2O (0 mL, 0 mmol, 0 eq., or 0.82 mL, 8.66 mmol, 1.05 eq., or 1.64 mL, 17.32 mmol, 2.10 eq., or 2.46 mL, 25.99 mmol, 3.15 eq.) were added to a 50 mL volumetric flask. AcOH was added to the 50 mL line of the volumetric flask. The reactor was constructed from the perfluoroalkoxy (PFA) tubing (1/16′′ outside diameter, 0.04′′ inside diameter, 15.24 m (= 50 feet), 12.355 mL), the complementary polyether ether ketone (PEEK) fittings, and the Upchurch Scientific's back pressure regulator (40 psi, 2.8 bar). The prepared reactant solutions were pumped into the reactor by the Syrris Asia pump at the designated flow rate (206, 275, 412, 494 or 618 μL min−1). The reaction tube was immersed in an oil bath and heated at the designated temperature (110, 120 or 130 °C). The reaction was equilibrated for 60, 75, 90, 135 or 180 minutes, which was three times longer than the residence time. After the equilibration, 6.18 mL of the solution was collected for 10, 12.5, 15, 22.5 or 30 minutes, depending on the flow rate. The collected solution was analyzed by HPLC to determine the conversion.Procedure for extended-time experiment of step 3 (Table 4)
The substrate 5 (8.22 g, 41.25 mmol, 1 eq.), N,N,N,N-tetramethyldiaminomethane (6.75 mL, 49.50 mmol, 1.2 eq.), MsOH (3.27 mL, 50.33 mmol, 1.22 eq.) and Ac2O (8.19 mL, 86.62 mmol, 2.10 eq.) were added to a 250 mL volumetric flask. AcOH was added to the 250 mL line of the volumetric flask. The reactor was constructed from the perfluoroalkoxy (PFA) tubing (1/16′′ outside diameter, 0.04′′ inside diameter, 15.24 m (= 50 feet), 12.355 mL), the complementary polyether ether ketone (PEEK) fittings, and the Upchurch Scientific's back pressure regulator (40 psi, 2.8 bar). The prepared reactant solutions were pumped into the reactor by the Syrris Asia pump at the designated flow rate (412 μL min−1). The reaction tube was immersed in an oil bath and heated at the designated temperature (130 °C). The reaction was equilibrated for 90 minutes, which was three times longer than the residence time. After the equilibration, 173.04 mL of the solution was collected for 420 minutes. The collected solution was analyzed by HPLC to determine the conversion, stored at 0 °C and used in the next step.Procedure for substitution with 8 and recrystallization (Scheme 3)
The collected solution was concentrated to 34.05 g, 5.98 W. To the residue, was added toluene (57 mL, 10 V). The reaction mixture was concentrated to 24.64 g, 4.33 W. To the residue, was added toluene (57 mL, 10 V). The reaction mixture was concentrated to 19.69 g, 3.46 W. To the residue, were added NMP (22.8 mL, 4 V) and 2-methylimidazole 8 (14.23 g, 173.30 mmol, 6.07 eq.). The reaction mixture was warmed to 110 °C and stirred at 110 °C for 4 hours. The reaction mixture was cooled to 100 °C. To the mixture, was added H2O (177.8 mL, 31.25 V) over 10 minutes. The reaction mixture was cooled to r.t., filtered and washed with H2O (170.7 mL, 30 V). The solid was air-dried overnight to give ondansetron 110 (7.69 g, 91.8% yield, 97.07 pa%, off-white solid, MWL loss: 1.3%). To a 1 L flask, were added ondansetron 1 (7.69 g, 26.21 mmol) prepared in the preceding step, activated charcoal (769 mg, 0.1 W) and EtOH (423 mL, 55 V). The slurry was stirred at 78 °C for 1 h. The hot slurry was filtered through celite and washed with hot EtOH (38 + 38 mL, 5 + 5 V). The solution was concentrated to 107.66 g, 14 W. The resulted slurry was cooled to 5 °C and stirred at 5 °C for 1 h. The slurry was filtered and washed with cold EtOH (11.5 + 11.5 mL, 1.5 + 1.5 V) to afford ondansetron 110 (7.18 g, 93.4% yield, 99.76 pa%, off-white solid, residual EtOH 4200 ppm, MWL loss: 2.9%). 1H NMR (500 MHz, CDCl3) δ: 8.29–8.22 (m, 1H), 7.36–7.28 (m, 3H), 6.93 (d, J = 1.3 Hz, 1H), 6.89 (d, J = 1.5 Hz, 1H), 4.67 (dd, J = 14.6, 4.3 Hz, 1H), 4.08 (dd, J = 14.6, 8.9 Hz, 1H), 3.71 (s, 3H), 3.01 (ddd, J = 17.2, 5.2, 3.4 Hz, 1H), 2.94–2.80 (m, 2H), 2.44 (s, 3H), 2.23–2.15 (m, 1H), 1.96–1.84 (m, 1H). 13C NMR (125 MHz, CDCl3) δ: 191.7, 151.4, 145.1, 137.8, 127.6, 124.8, 123.5, 123.1, 121.7, 119.9, 112.4, 109.5, 47.4, 45.8, 30.0, 26.7, 21.6, 13.5. IR (neat) ν: 3125, 3100, 2934, 2872, 1621, 1578, 1529, 1479, 1458 cm−1. HRMS (DART) m/z: calcd for C18H20N3O+ [(M + H)+] 294.1601, found 294.1607.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Kai Donsbach, Tai-Yuen Yue and B. Frank Gupton of Virginia Commonwealth University for helpful discussions and suggestions. YH thanks Shionogi & Co., Ltd. for a fellowship. This project has been funded in part by the Biomedical Advanced Research and Development Authority (BARDA), Assistant Secretary for Preparedness and Response (ASPR), Department of Health and Human Services (DHHS), under Contract No. 75A50120C00092.Notes and references
- G. J. Sanger and P. L. R. Andrews, Front. Pharmacol., 2018, 9, 913–947 CrossRef PubMed.
- I. H. Coates, J. A. Bell, D. C. Humber and G. B. Ewan, US Pat., 4695578, 1987 Search PubMed.
- M. B. Tyers, I. H. Coates, D. C. Humber, G. B. Ewan and J. A. Bell, US Pat., 4753789, 1988 Search PubMed.
- U.S. Food and Drug Administration, FDA Approves First Generic Ondansetron Tablets, Orally Disintegrating Tablets and Oral Solution, Silver Spring, MD, 2006 Search PubMed.
- World Health Organization model list of essential medicines: 16th list in 2009, 17th list in 2011, 18th list in 2013, 19th list in 2015, 20th list in 2017, 21st list in 2019, 22nd list, 2021.
- R. O. M. A. de Souza and P. Watts, J. Flow Chem., 2017, 7, 146–150 CrossRef CAS; R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef; M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef PubMed.
- F. M. Akwi and P. Watts, Chem. Commun., 2018, 54, 13894–13928 RSC; V. Hessel, Chem. Eng. Technol., 2009, 32, 1655–1681 CrossRef CAS.
- M. Berton, J. M. de Souza, I. Abdiaj, D. T. McQuade and D. R. Snead, J. Flow Chem., 2020, 10, 73–92 CrossRef CAS; P. D. Morse, R. L. Beingessner and T. F. Jamison, Isr. J. Chem., 2017, 57, 218–227 CrossRef.
- T. M. Monos, J. N. Jaworski, J. C. Stephens and T. F. Jamison, Synlett, 2020, 31, 1888–1893 CrossRef CAS; C. P. Breen and T. F. Jamison, Chem. – Eur. J., 2019, 25, 14527–14531 CrossRef PubMed; W. C. Fu and T. F. Jamison, Org. Lett., 2019, 21, 6112–6116 CrossRef PubMed; M. G. Russell and T. F. Jamison, Angew. Chem., Int. Ed., 2019, 58, 7678–7681 CrossRef PubMed; R. E. Ziegler, B. K. Desai, J.-A. Jee, B. F. Gupton, T. D. Roper and T. F. Jamison, Angew. Chem., Int. Ed., 2018, 57, 7181–7185 CrossRef PubMed; P. Zhang, N. Weeranoppanant, D. A. Thomas, K. Tahara, T. Stelzer, M. G. Russell, M. O'Mahony, A. S. Myerson, H. Lin, L. P. Kelly, K. F. Jensen, T. F. Jamison, C. Dai, Y. Cui, N. Briggs, R. L. Beingessner and A. Adamo, Chem. – Eur. J., 2018, 24, 2776–2784 CrossRef PubMed; A.-C. Bédard, A. R. Longstreet, J. Britton, Y. Wang, H. Moriguchi, R. W. Hicklin, W. H. Green and T. F. Jamison, Bioorg. Med. Chem., 2017, 25, 6233–6241 CrossRef PubMed; H. Lin, C. Dai, T. F. Jamison and K. F. Jensen, Angew. Chem., Int. Ed., 2017, 56, 8870–8873 CrossRef PubMed; A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef PubMed; C. Dai, D. R. Snead, P. Zhang and T. F. Jamison, J. Flow Chem., 2015, 5, 133–138 CrossRef; D. R. Snead and T. F. Jamison, Angew. Chem., Int. Ed., 2015, 54, 983–987 CrossRef PubMed; P. Zhang, M. G. Russell and T. F. Jamison, Org. Process Res. Dev., 2014, 18, 1567–1570 CrossRef; P. L. Heider, S. C. Born, S. Basak, B. Benyahia, R. Lakerveld, H. Zhang, R. Hogan, L. Buchbinder, A. Wolfe, S. Mascia, J. M. B. Evans, T. F. Jamison and K. F. Jensen, Org. Process Res. Dev., 2014, 18, 402–409 CrossRef; S. Mascia, P. L. Heider, H. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, Angew. Chem., Int. Ed., 2013, 52, 12359–12363 CrossRef PubMed; D. R. Snead and T. F. Jamison, Chem. Sci., 2013, 4, 2822–2827 RSC; B. Shen and T. F. Jamison, Org. Lett., 2012, 14, 3348–3351 CrossRef PubMed.
- N. Mohanta, K. Nair, D. V. Sutar and B. A. Gnanaprakasam, React. Chem. Eng., 2020, 5, 1501–1508 RSC.
- C. Tietcheu, C. Garcia, D. Gardette, D. Dugat and J.-C. Gramain, J. Heterocyclic Chem., 2002, 39, 965–973 CrossRef CAS.
- W. Sun, W. A. T. Raimbach, L. D. Elliott, K. I. Booker-Milburn and D. C. Harrowven, Chem. Commun., 2022, 58, 383–386 RSC.
- D. Szymor-Pietrzak, M. N. Khan, A. Pagès, A. Kumar, N. Depner and D. L. J. Clive, J. Org. Chem., 2021, 86, 619–631 CrossRef CAS PubMed.
- See ESI† for more details.
- D. Hesoun and J. Hykl, US Pat., 7041834, 2006 Search PubMed.
- See ESI† for the solubility data of the substrate (5) and ondansetron (1).
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Eur. J. Org. Chem., 2020, 7398–7406 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, including photographs of experimental setup, 1H and 13 C NMR spectra for isolated compounds, HPLC data for reaction profiles and isolated compounds (PDF). See DOI: https://doi.org/10.1039/d3re00249g |
| This journal is © The Royal Society of Chemistry 2023 |