
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aromatic hydroxylation of substituted benzenes by an unspecific peroxygenase from Aspergillus brasiliensis†
Fabian
Schmitz
a,
Katja
Koschorreck
a,
Frank
Hollmann
 b and
Vlada B.
Urlacher
*a
b and
Vlada B.
Urlacher
*a
aInstitute of Biochemistry, Heinrich-Heine-University Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany. E-mail: Vlada.Urlacher@uni-duesseldorf.de
bDepartment of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629 Hz Delft, The Netherlands
First published on 1st June 2023
Abstract
Selective aromatic hydroxylation of substituted benzenes provides access to versatile phenolic synthons. Unspecific peroxygenases (UPOs) have been recognised as promising biocatalysts for synthetic chemistry. While UPOs accept diverse substrates and enable a broad range of oxygenation reactions, aromatic hydroxylation reactions catalysed by these enzymes have been rarely described. Here, we report on a UPO from Aspergillus brasiliensis (AbrUPO) heterologously expressed in Pichia pastoris at a concentration of 742 mg per litre that is able to catalyse aromatic hydroxylation of substituted benzenes. The preference of AbrUPO for aromatic or benzylic hydroxylation was found to depend on the number, chemical properties and length of existing ring substituents. While oxidation of ethylbenzene gave ring- and side-chain hydroxylation products at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, increasing the chain-length of the alkyl substituent enhanced the preference for benzylic hydroxylation. With the para-disubstituted p-cymene as a substrate, the chemoselectivity of AbrUPO strongly shifted towards aromatic hydroxylation. All tested substituted phenols resulted in exclusive aromatic hydroxylation. The observed formation of low quantities of quinones was attributed to the inherent peroxidase activity, while further oxidation of benzylic alcohols to ketones was suggested to occur due to both peroxidase and peroxygenase activity of AbrUPO. ‘Overoxidation’ due to peroxidase activity could be completely avoided by adding ascorbic acid and shortening reaction time.
:1 ratio, increasing the chain-length of the alkyl substituent enhanced the preference for benzylic hydroxylation. With the para-disubstituted p-cymene as a substrate, the chemoselectivity of AbrUPO strongly shifted towards aromatic hydroxylation. All tested substituted phenols resulted in exclusive aromatic hydroxylation. The observed formation of low quantities of quinones was attributed to the inherent peroxidase activity, while further oxidation of benzylic alcohols to ketones was suggested to occur due to both peroxidase and peroxygenase activity of AbrUPO. ‘Overoxidation’ due to peroxidase activity could be completely avoided by adding ascorbic acid and shortening reaction time.
Introduction
Over the last few years, unspecific peroxygenases (UPOs, EC 1.11.2.1) have gained increasing attention from both chemists and biotechnologists, because they enable oxidation of various organic compounds at the expense of hydrogen peroxide and do not require expensive redox cofactors.1,2 UPOs are heme-thiolate enzymes secreted by fungi and were first reported in 2004 by Hofrichter et al.2 These enzymes combine the activity of peroxidases and peroxygenases. Heme iron reacts with hydrogen peroxide to yield a ferric peroxo complex, which cleaves to give the highly reactive iron–oxo species compound I.3,4 Despite a broad range of catalysed reactions like aliphatic hydroxylation, epoxidation, sulfoxidation, N-oxidation and O-dealkylation, aromatic hydroxylations with UPOs have been rarely reported.5–7 Direct aromatic hydroxylation of substituted benzenes gives access to phenols, versatile synthons in the synthesis of dyes, pharmaceuticals and agrochemicals. Chemical aromatic oxidation remains, however, quite challenging because of low efficiency and poor selectivity.8,9 Several UPOs were reported to catalyse naphthalene oxidation yielding hydroxylated and epoxidized products.10,11 One study described benzene oxidation catalysed by UPO from Agrocybe aegerita AaeUPO. Under optimized reaction conditions phenol was formed as the main product, and benzene oxide (epoxide) was identified as the intermediate.7AaeUPO was also shown to catalyse aromatic and benzylic hydroxylation of toluene with benzyl alcohol as the main product.12 However, ethyl- and propylbenzene were exclusively oxidised with this enzyme at the alkyl side chain.13 Another group of heme-thiolate enzymes, cytochrome P450 monooxygenases have been extensively studied in this respect.14,15 Several P450s have been engineered for high activity and regioselectivity during aromatic hydroxylation of substituted benzenes and other aromatic compounds.16,17 The proposed mechanism of P450-catalysed oxidation of substituted benzenes proceeds via electrophilic and/or radical pathways, which involve an initial attack of the high-valent compound I on the π system of the aromatic species ring or arene oxide formation, accompanied by the 'NIH shift'.15,18 Interestingly, not all P450s accepting aromatic compounds as substrates enable aromatic oxidation even if the ring can access the heme group in the enzyme's active site.19 Recently, crystal structure analysis and QM/MM simulations on CYP199A2 revealed stringent geometrical requirements for efficient aromatic oxidation to occur over aliphatic hydroxylation.20 Thus, identification of further heme-thiolate enzymes such as cofactor-independent UPOs enabling aromatic hydroxylation is of great interest both, from a fundamental and a practical perspective.In this study, we identified, produced in recombinant Pichia pastoris (recently reclassified as Komagataella phaffii) and characterised an UPO from Aspergillus brasiliensis (AbrUPO). A set of substituted benzenes, phenols and other compounds were identified as substrates for AbrUPO. We showed that AbrUPO can mediate both benzylic and aromatic oxidation, and depending on ring substituents demonstrates a high chemoselectivity yielding either only ring- or only side-chain hydroxylation products.
Experimental
Strains
Escherichia coli DH5α (Clontech Laboratories Inc., Heidelberg, Germany) was used for cloning. Pichia pastoris X-33 (re-classified as Komagataella phaffii) used for expression of putative UPO genes, was purchased from Invitrogen (Carlsbad, USA).Cloning of the peroxygenases' encoding genes
The sequences encoding putative UPOs were optimized for codon usage in Saccharomyces cerevisiae using JCat (http://www.jcat.de/)21 and the synthetic gene sequences including the native signal sequences for secretion were produced by BioCat GmbH (Heidelberg, Germany) in the expression vector pPICZA (Invitrogen, Carlsbad, USA) between the BstBI and NotI restriction sites. After homologous recombination, recombinant P. pastoris X-33 transformants were selected on YPDS-agar plates containing 10 g l−1 yeast extract, 20 g l−1 peptone, 20 g l−1 glucose, 1 M sorbitol, 20 g l−1 agar, supplemented with Zeocin™ (100 μg ml−1) after 3 days incubation at 30 °C. Evolved PaDa-I from Agrocybe aegerita was produced as reported and used after solubilisation of lyophilized cell free supernatant.22Cultivation of P. pastoris
325 × g, 4 °C, 20 min), and the culture broth was concentrated by tangential flow filtration with three membrane cassettes with a cut-off value of 10 kDa. Upon this step, buffer was exchanged to 50 mM sodium phosphate buffer pH 7.0 with 2 mM MgCl2. In the next step, hydrophobic interaction chromatography (HIC) on an XK16/20 column with Butyl Sepharose HP medium (20 ml, GE Healthcare, Chicago, USA) was performed on an ÄKTApurifier FPLC-system (GE Healthcare, Chicago, USA). For characterization, AbrUPO was further purified by ion exchange chromatography (IEX) on Q Sepharose FF medium (26 ml, GE Healthcare, Chicago, USA) (ESI,† Table S2).
Estimation of enzyme concentration
Total protein concentration of samples taken in the course of fed-batch fermentation or during protein purification was measured by the Bradford method using bovine serum albumin (BSA) as standard.23 For the substrate conversion experiments concentration of purified AbrUPO was calculated from the CO-difference spectra using the extinction coefficient ε445 = 130000 M−1 cm−1 of AbrUPO.24,25 The molar extinction coefficient was calculated using Beer's law and the concentration of heme, determined by the pyridine hemochromagen assay according to Barr et al.26 The difference in concentration of purified AbrUPO determined via Bradford assay and CO-difference spectra was 6%.
Spectral properties of the purified enzyme were measured between 350–700 nm on a Lambda 35 spectrophotometer (Perkin Elmer, Waltham, USA). Peptide-N-amidase PNGase F (New England Biolabs, Frankfurt am Main, Germany) was used to deglycosylate 20 μg of purified AbrUPO under denaturing conditions according to the manufacturer's protocol.
ABTS assay
Peroxidase activity assay was performed in a total volume of 200 μl at 25 °C with 5 mM ABTS (ε420 = 36000 M−1 cm−1) as substrate in McIlvaine buffer pH 4.4 with 1.2 mM H2O2 as co-substrate. 20 μl of enzyme solution was mixed with 140 μl buffer and 20 μl substrate.27 Reaction was started by adding 20 μl H2O2. All measurements were conducted in triplicate.
pH and temperature stability
pH stability was evaluated by incubating purified AbrUPO in 100 mM Britton–Robinson buffer for 1 h at pH values ranging from pH 2.0 to 12.0. For temperature stability measurements, purified enzyme was incubated in 50 mM sodium phosphate buffer pH 7.0 with 2 mM MgCl2 between 4–80 °C for up to 240 min. In both cases, samples were taken at different time points and the residual activity towards ABTS was determined. For temperature stability determination samples were incubated on ice for 5 min prior activity measurement. The resulting data set was plotted in OriginPro 9.0 (OriginLab Corporation, Northampton, MA, USA) and the T50 value was determined by fitting the data using the Boltzmann equation.Substrate screening
Reactions were conducted in 500 μl volume in 1.5 ml reaction tubes at 25 °C and 600 rpm in triplicates. The standard reaction mixture contained 1.3 μM UPO, 4 mM H2O2, 1 mM substrate (1–22) (dissolved in acetonitrile) and 8 mM ascorbic acid in 50 mM sodium phosphate buffer pH 7.0 with 2 mM MgCl2, at a final acetonitrile concentration of 5% (v/v). If not stated otherwise, reactions were extracted with 500 μl ethyl acetate after 180 min. 500 μM 1-dodecanol was used as internal standard. For 23 500 μM substrate were used. In case of 24–27 reaction mixture contained 200 μM substrate, 500 μM H2O2 and 0.8 μM UPO. After 90 min reactions were extracted with diethyl ether.GC/MS analysis
Samples (0.5 μl) were injected to a GC/MS instrument (GC/MS-QP2010 plus, Shimadzu, Germany) equipped with an FS-Supreme-5 column (30 m × 0.25 mm × 0.25 μm, Chromatographie Service GmbH, Germany). The temperature protocols are shown in ESI,† Table S1. Conversions were calculated based on substrate depletion (control was set to 100%) and product distributions based on relative peak areas (%) in relation to the internal standard. Conversion of 23 was analysed via LC/MS (ESI,† LC/MS elution profile).Results and discussion
UPO selection and production
Sequences of seven putative UPOs from several fungi were identified by BLAST search using the amino acid sequences of rCciUPO from Coprinopsis cinerea, CglUPO from Chaetomium globosum, and AaeUPO from Agrocybe aegerita as templates (Table 1). The codon optimised sequences with their native signals for secretion were cloned in P. pastoris X-33 for heterologous expression.| UPO | Host organism | Accession number | Theoretical mol. weight [kDa] | Vol. activity [U l−1] |
|---|---|---|---|---|
| AbrUPO | Aspergillus brasiliensis | OJJ73116.1 | 29.27 | 93.0 ± 1.6 |
| CmiUPO | Coprinellus micaceus | TEB27715.1 | 41.40 | 18.2 ± 1.8 |
| GdiUPO | Gymnopilus dilepsis | PPR06026.1 | 40.99 | 2.6 ± 0.2 |
| LspUPO | Leucoagaricus sp. | KXN81291.1 | 40.68 | 4.8 ± 0.1 |
| PfiUPO | Pestalotiopsis fici | XP_007840602.1 | 27.80 | 18.0 ± 0.2 |
| PabUPO | Psathyrella aberdarensis | RXW17550.1 | 41.52 | 5.5 ± 0.2 |
| SstUPO | Sphaerobolus stellatus SS14 | KIJ32220.1 | 43.16 | 1.2 ± 0.1 |
All UPOs were expressed in P. pastoris, secreted into the culture medium and exhibited peroxidase activity with 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS) (Table 1). The UPO from Aspergillus brasiliensis (AbrUPO) that demonstrated the highest activity of 93 ± 1.6 U l−1 was chosen for further investigations. The enzyme was produced in a 7.5 l bioreactor via fed-batch cultivation of recombinant P. pastoris yielding a volumetric peroxidase activity of 23492 ± 192 U l−1 in a final volume of 4.6 l after 10 days (Fig. 1). Purified AbrUPO showed a specific activity of 31.7 ± 1.4 U mg−1 towards ABTS (ESI,† Table S2) which is comparable to other recombinant UPOs.28,29 Based on the specific activity of purified AbrUPO, a concentration of 742 mg per 1 litre of culture medium was calculated which is the highest expression level reported for a heterologously produced UPO in P. pastoris so far.30,31
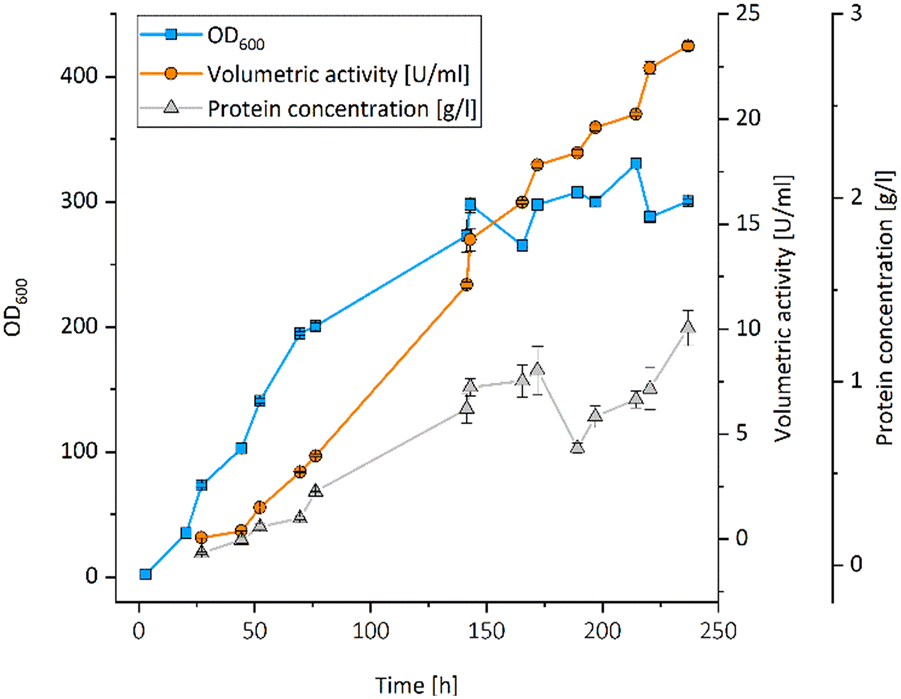 | ||
| Fig. 1 Fed-batch cultivation of recombinant P. pastoris X-33 starting from 3 l basal salt medium in a 7.5 l bioreactor to produce AbrUPO. Blue squares: OD600, orange circles: volumetric activity [U ml−1], grey triangles: protein concentration [g l−1]. | ||
Oxidised purified AbrUPO showed an absorption spectrum typical for heme-thiolate proteins with a Soret band at 421 nm, α-band at 571 nm, β-band at 540 nm and σ-band at 361 nm (ESI,† Fig. S1), similar to other UPOs.2,32–35AbrUPO belongs to the group of the so-called short UPOs.36 Accordingly, the theoretical MW of this protein is 29 kDa, however, the sequence contains 9 putative N-glycosylation sites. SDS-PAGE analysis revealed a strong band at around 70 kDa (ESI,† Fig. S1). The glycosylation degree of 55% is much higher than in other recombinant UPOs expressed in P. pastoris (ESI,† Table S3). The enzyme exhibited a T50 value of 52 °C and remained stable with over 90% of its initial activity after 60 min incubation at pH ranging from 3 to 8 (ESI,† Fig. S2), which is comparable to other UPOs.37,38
Substrate scope of AbrUPO and first conversions
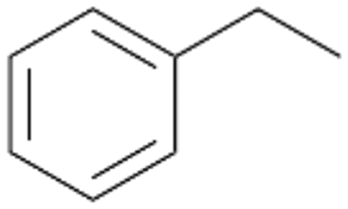
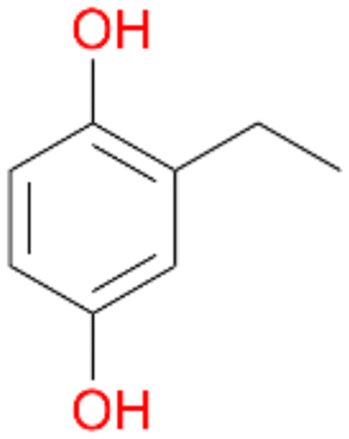
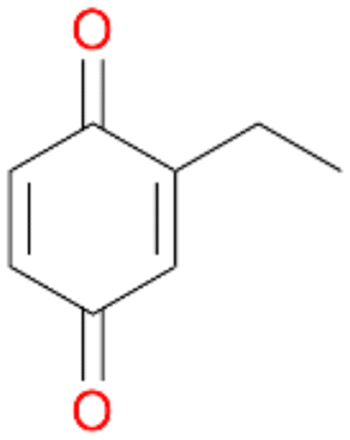
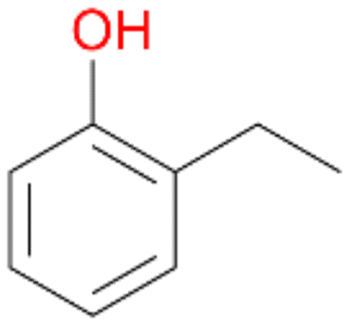
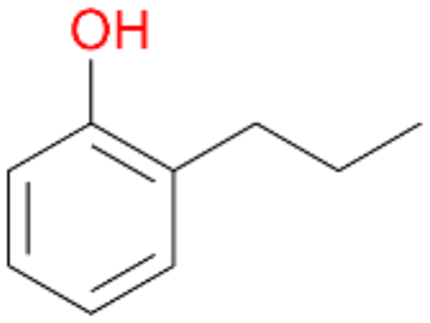
In order to assess the substrate and product spectrum of this enzyme and to investigate if AbrUPO enables aromatic hydroxylation reactions a set of 27 substances were tested. This set covered a number of substituted benzenes and phenols, sulphides, terpenes, terpenoids and fatty acids (Table 2, ESI,† Table S4 and Table S5). AbrUPO was found to possess a broad substrate spectrum and accepted almost all tested substances as substrates. After 180 min, AbrUPO-catalysed conversion of ethylbenzene 1 yielded approximately equal amounts of ring- and side-chain hydroxylation products. This is remarkable insofar as the UPOs reported to date exhibit almost strict side-chain selectivity with this substrate. Under standard reaction conditions 2-ethyl-benzene-1,4-diol 1c was the main product (44%) with low amounts of the mono-hydroxylated product 2-ethylphenol 1e (4%) and the ‘overoxidation’ product, quinone 1d (6%). Apparently, the first hydroxylation step is overall rate-limiting and the second hydroxylation proceeds faster (vide infra).| Substrate | Substrate depletion [%] | Product distribution [%] | |||||
|---|---|---|---|---|---|---|---|
| Benzylic oxidation | Aromatic oxidation | ||||||
| a Verified by MS and reference substance. b Verified by MS and NIST20 database. c Traces are detectable after 15 min. | |||||||
| 1 |
|
37 |
 1a
, 39 (ee = 18%)
1a
, 39 (ee = 18%) |
 1b
, 7
1b
, 7 |
 1c
, 44
1c
, 44 |
 1d
, 6
1d
, 6 |
 1e
, 4
1e
, 4 |
| 2 |
|
78 |
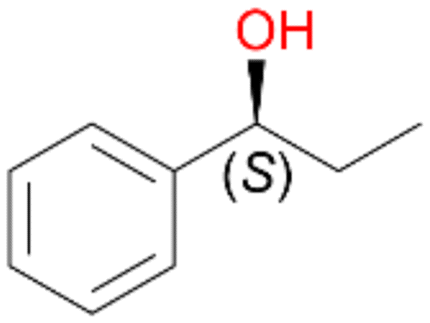 2a
, 60 (ee = 63%)
2a
, 60 (ee = 63%) |
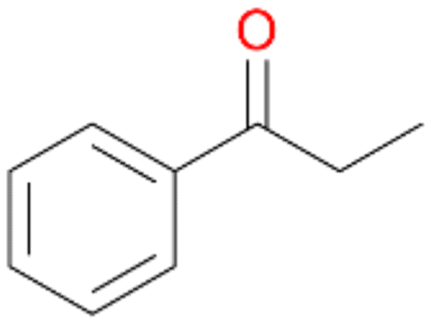 2b
, 13
2b
, 13 |
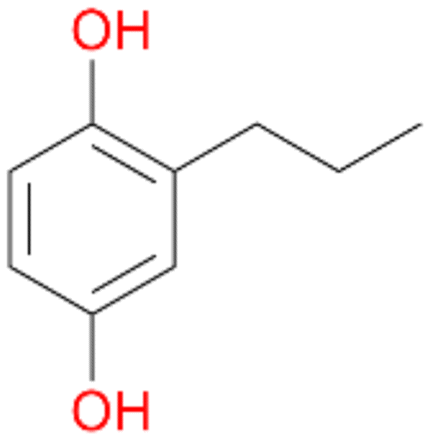 2c
, 17
2c
, 17 |
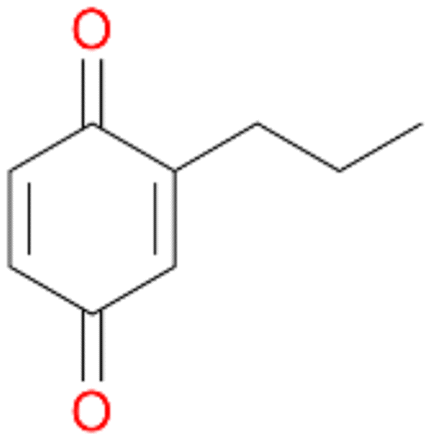 2d
, 4
2d
, 4 |
 2e
, <1c
2e
, <1c |
| 3 |
|
68 |
 3a
, 20 (ee = 99%)
3a
, 20 (ee = 99%) |
 3b
, 64
3b
, 64 |
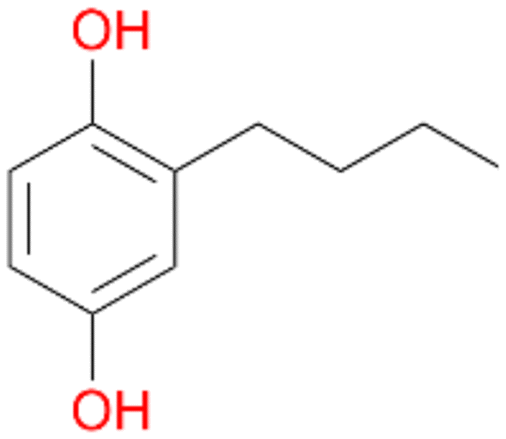 3c
, 5
3c
, 5 |
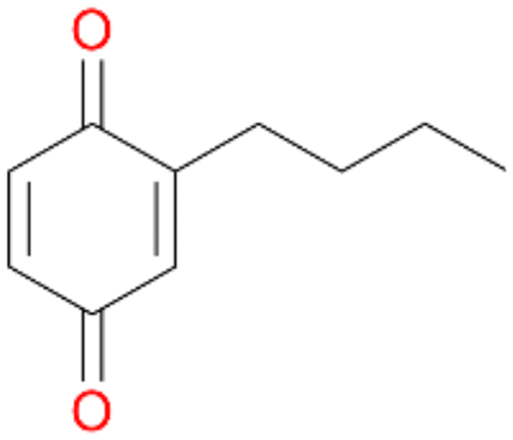 3d
, 6
3d
, 6 |
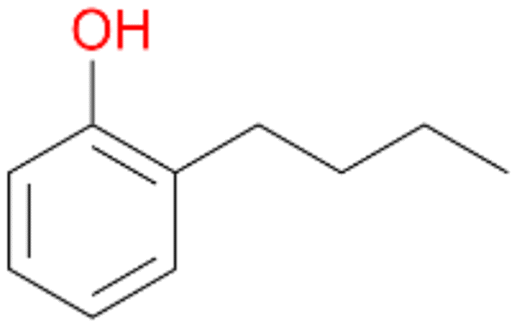 3e
, <1c
3e
, <1c |
| 4 |
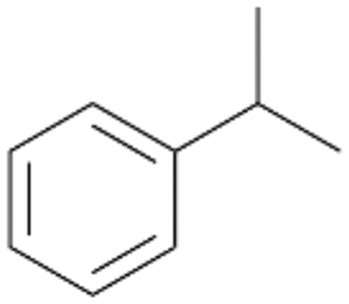
|
62 |
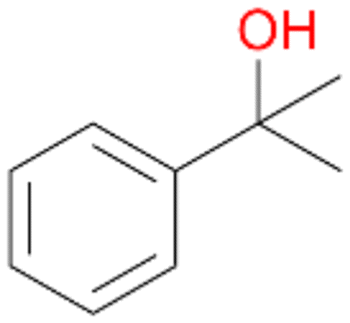 4a
, 82
4a
, 82 |
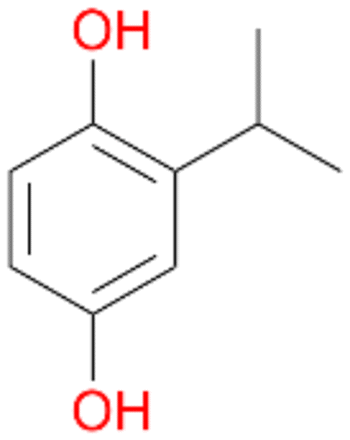 4b
, 14
4b
, 14 |
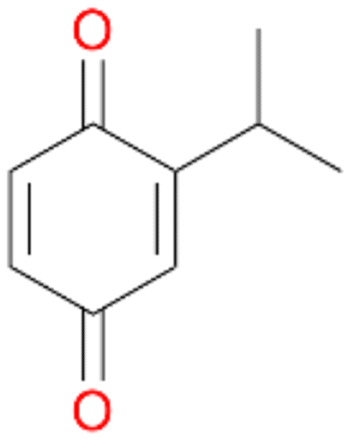 4c
, 3
4c
, 3 |
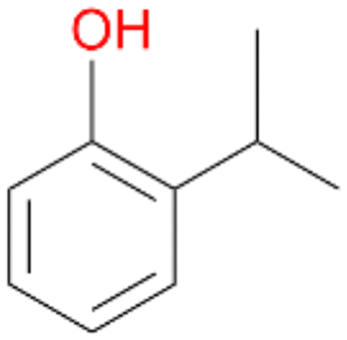 4d
, <1c
4d
, <1c |
|
| 5 |
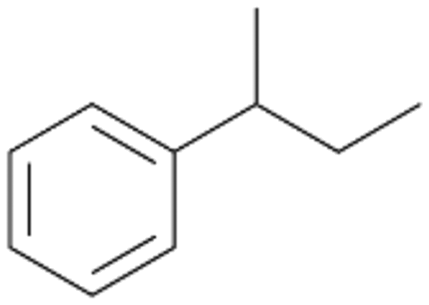
|
64 |
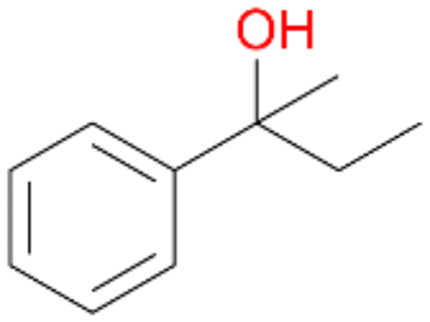 5a
, 76
5a
, 76 |
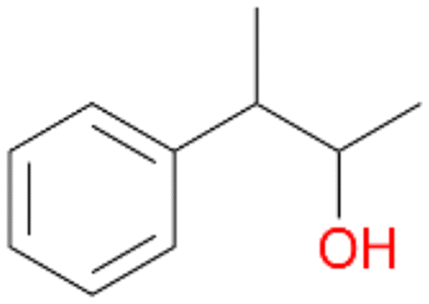 5b
, 5
5b
, 5 |
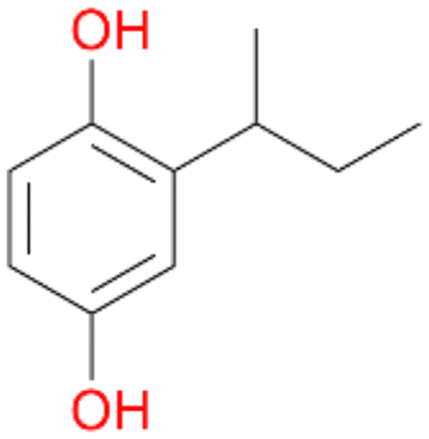 5c
, 13
5c
, 13 |
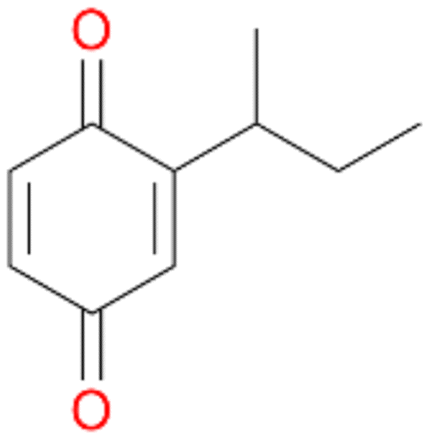 5d
, 2
5d
, 2 |
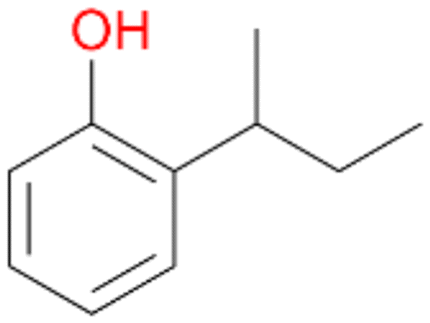 5e
, 2
5e
, 2 |
| 6 |
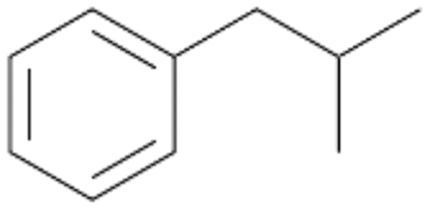
|
74 |
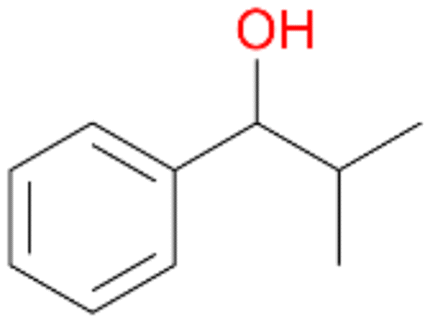 6a
, 63
6a
, 63 |
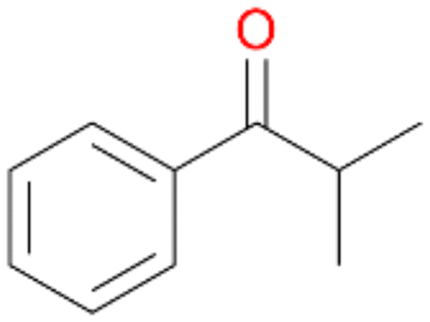 6b
, 19
6b
, 19 |
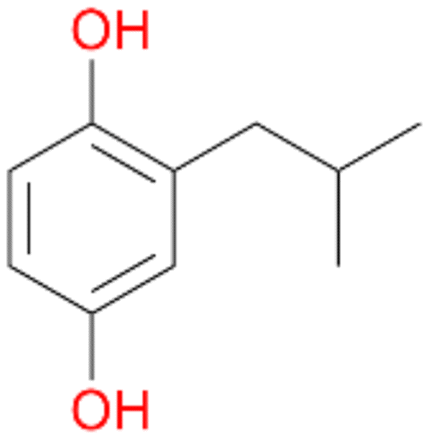 6d
, 6
6d
, 6 |
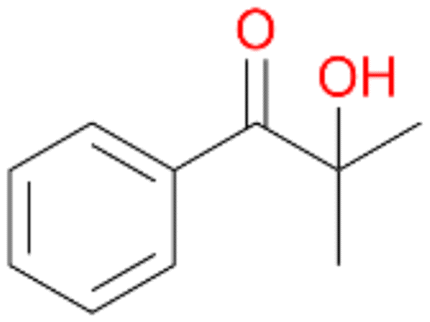 6c
, 6
6c
, 6 |
|
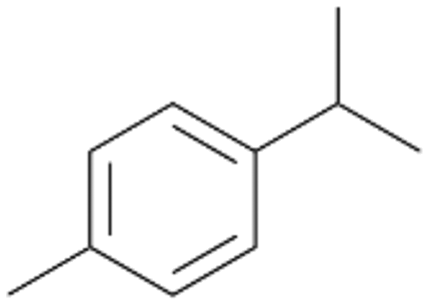
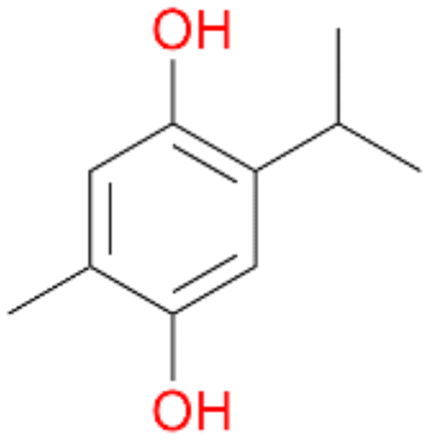
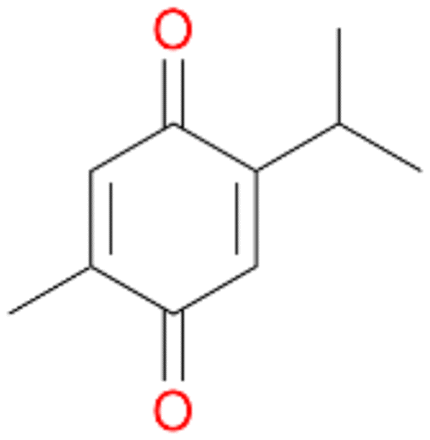
| 7 |
|
83 |
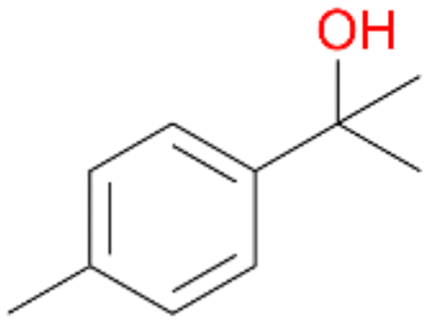 7a
, 23
7a
, 23 |
 7b
, 57
7b
, 57 |
 7c
, 7
7c
, 7 |
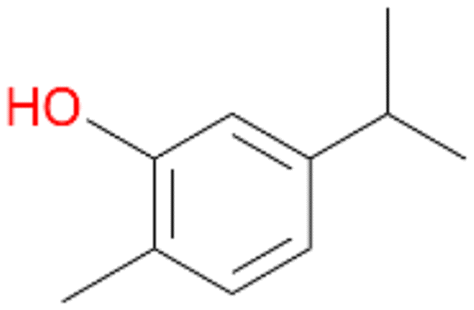 7d
, 2
7d
, 2 |
|
| 8 |
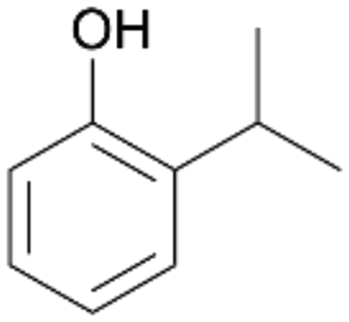
|
94 |
 4b
, 91
4b
, 91 |
 4c
, 9
4c
, 9 |
|||
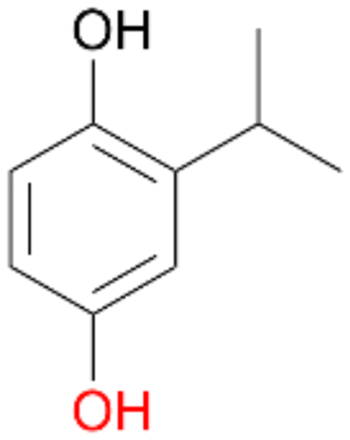
| 9 |
|
97 |
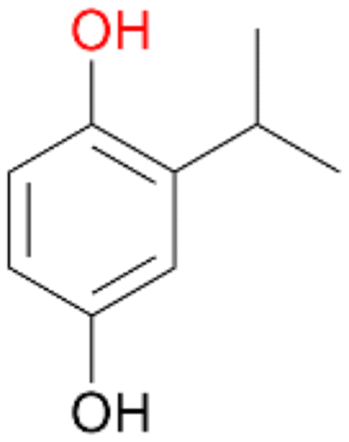 4b
, 93
4b
, 93 |
 4c
, 7
4c
, 7 |
|||
| 10 |
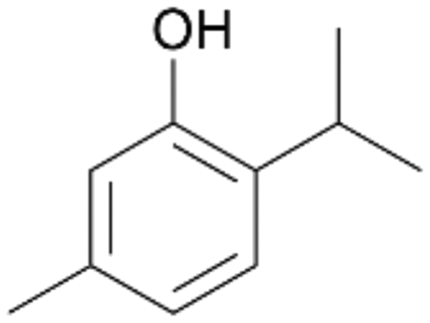
|
>99 |
 7b
, 67
7b
, 67 |
 7c
, 33
7c
, 33 |
|||
| 11 |
|
89 |
 11a
, 70 (ee = 15%)
11a
, 70 (ee = 15%) |
 11b
, 30
11b
, 30 |
|||
| 12 |
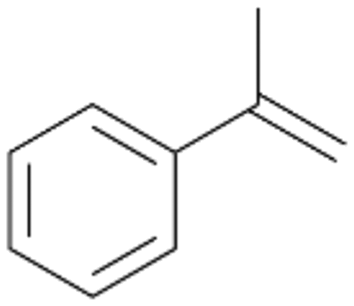
|
98 |
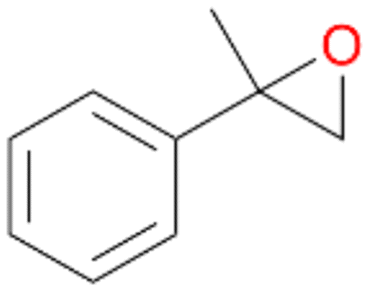 12a
, 82
12a
, 82 |
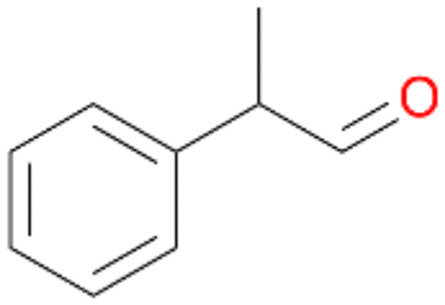 12b
, 14
12b
, 14 |
|||
| 13 |
|
94 |
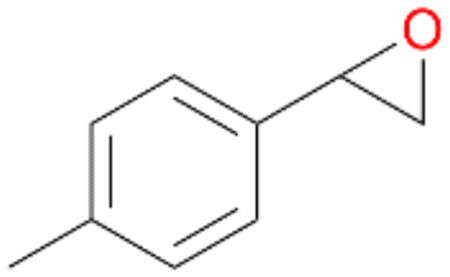 13a
, 50
13a
, 50 |
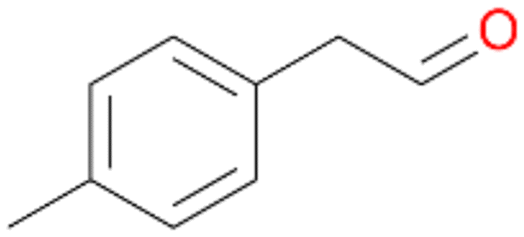 13b
, 45
13b
, 45 |
|||
| 14 |
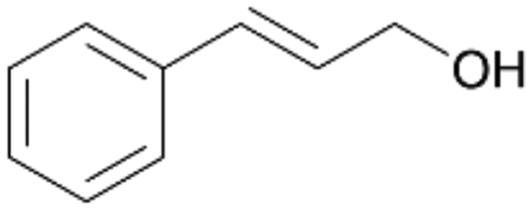
|
>99 |
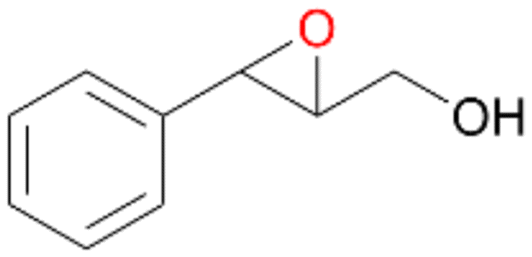 14a
, 5
14a
, 5 |
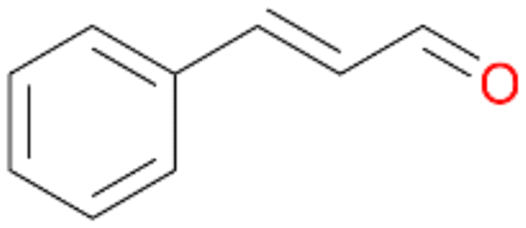 14b
, 74
14b
, 74 |
m/z = 91147 |
||
| 14c , 15 | |||||||
| 15 |
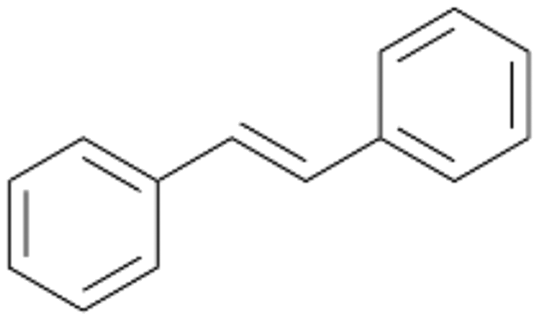
|
18 |
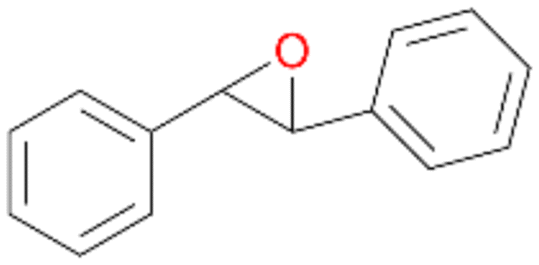 15a
, 84
15a
, 84 |
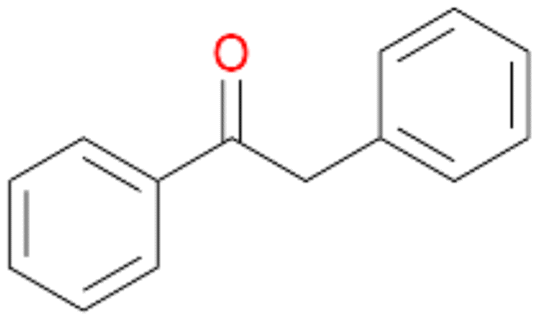 15b
, 4
15b
, 4 |
|||
| 16 |
|
93 |
 16a
, 40
16a
, 40 |
 16b
, 32
16b
, 32 |
 16c
, 28
16c
, 28 |
||
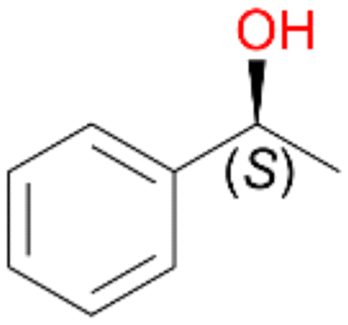
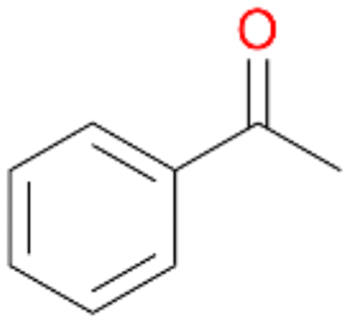
Benzylic hydroxylation of ethylbenzene 1 led to (S)-1-phenylethanol 1a (60%, ee = 18%, ESI,† Fig. S3) which was also prone to overoxidation yielding 7% acetophenone 1b. Substituted benzenes were also tested with PaDa-I, a variant of AaeUPO and one of the most studied UPOs so far, used in many UPO studies as a benchmark.39 PaDa-I led to high conversion of 1 (96%) (ESI,† Table S5), but no aromatic ring oxidation was observed. Interestingly, under the same reaction conditions PaDa-I induced a stronger further oxidation yielding 56% (R)-1-phenylethanol 1a (ee = >99%) and 44% acetophenone 1b (ESI,† Table S6).
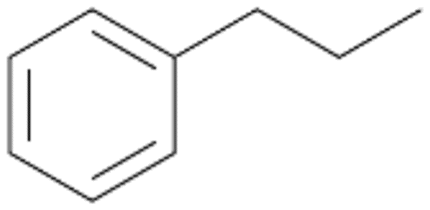
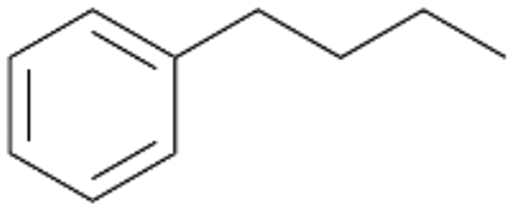
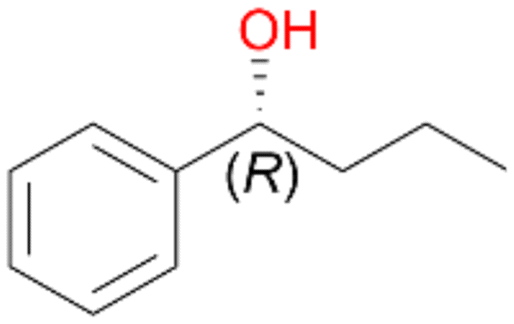
Increasing the chain-length of the alkyl substituent to propylbenzene 2 and butylbenzene 3 increased both, the preference for side chain hydroxylation over ring hydroxylation as well as the stereoselectivity of benzylic hydroxylation (Table 1). After 180 min reaction with 3 only 5% of total product accounted to 2-butylbenzene-1,4-diol 3c. Notably, the stereoselectivity switched from (S) for 1-phenyl-1-propanol 2a (ee = 63%) to (R) for 1-phenyl-1-butanol 3a (ee = 99%, ESI,† Fig. S4 and S5) for which we are currently lacking a plausible explanation. Remarkably, oxidation of 1 by chloroperoxidase CfuCPO from Caldaromyces fumago gave (R)-1-phenylethanol (ee = 97%), while oxidation of 2 led to formation of (S)-1-phenyl-1-propanol (ee = 88%).40
PaDa-I demonstrated a high stereoselectivity during hydroxylation of 2 to benzylic alcohol 2a in (R)-configuration (ee = 99%, ESI,† Table S6). In reaction with 3 PaDa-I did not furnish benzylic alcohol 3a but catalysed aliphatic hydroxylation reactions at both adjacent positions of the alkyl chain (ESI,† Table S6). Conversion of 1 and 2 by PaDa-I (ESI,† Table S6) and by several other UPOs (AaeUPO, CglUPO, MroUPO, TteUPO and MthUPO) did not give any ring oxidation products and yielded only products of benzylic oxidation.41 This indicates a strong influence of the shape and amino acid composition of the substrate binding site in UPOs on substrate positioning and enzyme chemoselectivity.
Further activation of the benzylic C–H bond by methyl or ethyl substitution favoured side-chain hydroxylation in case of 4 and 5 (compared to 1) and shifted the chemoselectivity from aromatic ring hydroxylation towards benzylic hydroxylation as observed also with 2. Percentages of ring hydroxylation products with both substrates were very similar. Conversion of isobutylbenzene 6 was similar to butylbenzene 3 and led to very low ring hydroxylation (6%). However, a high ratio of benzylic alcohol 6a to ketone 6b was observed as in the reaction with propylbenzene 2. Aromatic hydroxylation was also observed to some extent in reactions of 3, 5 and 6 with PaDa-I (ESI,† Table S6).
Among mono-substituted benzenes only for 1 the mono-hydroxylated phenol product was detected after 180 min reaction with AbrUPO and identified as 2-ethylphenol 1e. The first OH-group was introduced at ortho-position, but not at para-position as could be expected. The second OH-group was introduced at para-position related to the firstly introduced OH-group. For 2–4 only diols were detectable after 180 min reaction at higher or lower concentrations which were in some cases further oxidised to the corresponding quinones (3–6%). The structures of the diols suggest that also in these cases the first OH-group was introduced at ortho- but not at para-position of the aromatic ring. The absence of hydroxylation at para-position, also favourable for aromatic substitution, can be explained by some steric restrictions present in the active site of AbrUPO.
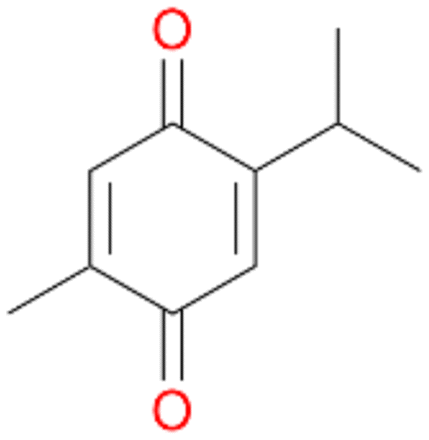
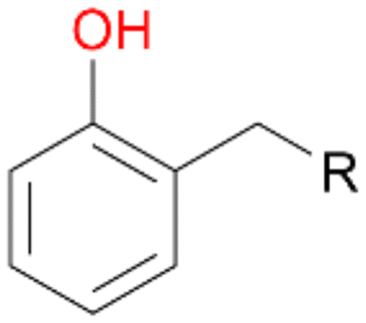
With the para-disubstituted p-cymene 7AbrUPO demonstrated higher activity than with mono-substituted benzenes, and its chemoselectivity strongly shifted towards aromatic hydroxylation, which may partially be attributed to the electron-donating (+I) effect of the additional substituent (4vs.7). The mono-hydroxylated product was identified as carvacrol (5-isopropyl-2-methylphenol, 7d), which means that p-cymene was first hydroxylated at ortho-position related to the methyl group and not to the isopropyl group. The time course analysis of the AbrUPO-catalysed conversion of 7 (ESI,† Fig. S6) revealed that carvacrol 7d was consumed over time at the benefit of the diol product (7b). The ratio of di- to mono-hydroxylated products also increased with the amount of oxidant (H2O2) applied (ESI,† Fig. S7). PaDa-I-catalysed oxidation of 7 only yielded side-chain hydroxylation products (ESI,† Table S6). Finally, using phenolic starting materials 8–10 (Table 2) resulted in exclusive ring hydroxylation in para-position to the existing OH-group. Substrates 8–9 were converted with high activities (94–97% conversion) to furnish exclusively hydroquinone 4b (91–93%) which was partially oxidised to quinone 4c (7–9%). With thymol 10 as substrate conversion of >99% was achieved. Quinone ratio was higher (33%) than in reactions with 8 and 9. After 15 min reaction 10 was nearly completely converted to the diol 7b (data not shown).
Formation of ketones and quinones
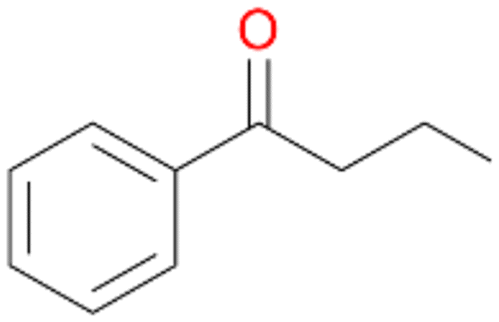
Further oxidation of the formed alcohols by the same enzyme is often considered as an issue of UPO catalysed hydroxylation reactions.42 As reported, the extent of this overoxidation varies between different UPOs and different substrates.12,43 Even though AbrUPO did not tend to strong overoxidation with most substrates, the underlying factors should be elucidated. Further oxidation of hydroquinones to quinones most likely can be attributed to the inherent peroxidase (single electron abstraction) activity of AbrUPO. To test this hypothesis, we used the hydroquinone 1c as starting material and performed the biocatalytic oxidation in the presence and absence of ascorbic acid (added to reactions to reduce transiently formed phenoxy radicals). In fact, oxidation of 1c to 1d was only observed in the absence of ascorbate (ESI,† Fig. S8). Also, aerobic and H2O2-mediated oxidation of 1c was excluded in control experiments in the absence of AbrUPO. We assume that peroxidase-mediated formation of the quinone product 1d in conversion of 1 (with addition of ascorbate) is due to instability of ascorbate in aqueous solution.44Further oxidation of benzylic alcohols to the corresponding ketones may have the same origin, but may also occur due to the peroxygenase activity of this UPO. For instance, AaeUPO from A. aegerita has been shown to catalyse oxidation of 1-phenylethanol to acetophenone via peroxygenase activity.45 Alcohol dehydrogenation activity has been reported for cytochrome P450s and proposed to occur either via two subsequent H-abstractions from the carbon atom of the alcohol or via the gem-diol formation, which then undergoes dehydration.46 We observed that increasing the chain-length of the alkyl substituent from 1 to 3 increased the ratio of the benzylic alcohol to the corresponding ketone. If in case of 1 and 2 only minor amounts of the ketones 1b and 2b were found, with 3 the ratio has turned in favour of the ketone 3b, the main product (64%) of this reaction (Table 2). In order to shed more light on this aspect, the benzylic alcohols 1-phenylethanol 1a, 1-phenyl-1-propanol 2a, and 1-phenyl-1-butanol 3a were used as substrates of AbrUPO with and without ascorbic acid (Table 3). In presence of ascorbic acid, formation of ketones 1b, 2b, and 3b was observed, which indicates that oxidation of these alcohols occurs, at least to some extent, due to the peroxygenase activity of AbrUPO. The peroxygenase activity towards benzylic alcohols increased in the row from 1a to 3a and reached its maximum with 3a, which was converted to 80% to 3b in the presence of ascorbic acid. Without ascorbic acid, formation of ketones 1b and 2b was much higher, which allows us to assume that the contribution of the peroxidase activity towards 1a and 2a is higher compared to the peroxygenase activity. With 3a only a slight increase in formation of 3b was observed without ascorbic acid, indicating that oxidation of 3a can be mainly attributed to the peroxygenase activity of AbrUPO. These results further suggest that both the peroxidase and the peroxygenase activity are substrate dependent.
| Product | Product formation [%] | |||||
|---|---|---|---|---|---|---|
| 1 R = –CH3 | 2 R = –C2H5 | 3 R = –C3H7 | ||||
| With ascorbic acid | Without ascorbic acid | With ascorbic acid | Without ascorbic acid | With ascorbic acid | Without ascorbic acid | |
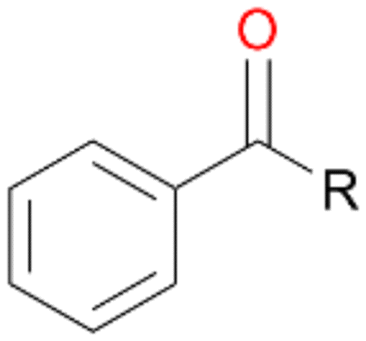
|
36 | 80 | 43 | 88 | 80 | 89 |
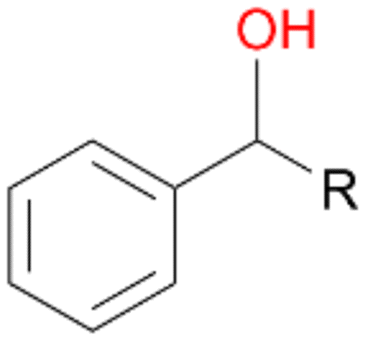
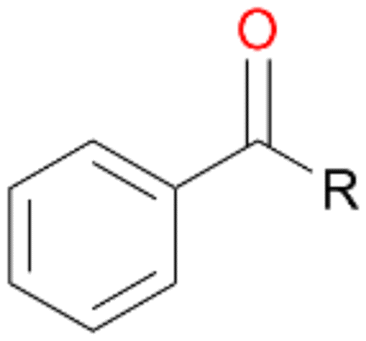
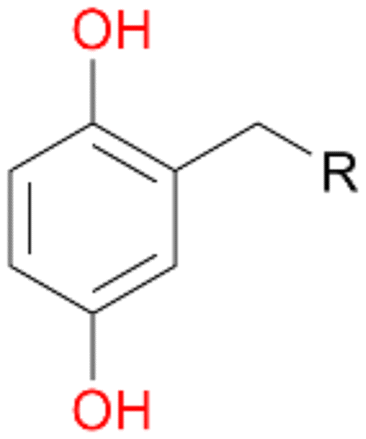
When we performed the reactions with ethylbenzene 1, propylbenzene 2 and butylbenzene 3 with and without ascorbic acid for only 15 min, the above described tendencies retained (Table 4). Quinones 1d, 2d and 3d were detected only in reactions without ascorbic acid, which confirms our previous suggestion regarding the formation of quinones due to the peroxidase activity of AbrUPO. More interesting is the ratio of benzylic alcohols to the corresponding ketones in these reactions. Lower ratios of the ketones 1b and 2b in the presence of ascorbic acid than in the absence are in line with our observation that the peroxygenase activity towards 1a and 2b is lower compared to the peroxidase activity of AbrUPO. The high ratio of 3a:3b of 1:1 after addition of ascorbic acid is in good agreement with the observed high peroxygenase activity of AbrUPO towards the alcohol 3a.
| Substrate | Ascorbic acid | Substrate depletion [%] | Product distribution [%] | ||||
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|||
| 1 R = –CH3 | + | 33 | 42 | 5 | 51 | 0 | 2 |
| − | 77 | 53 | 27 | 0 | 19 | 1 | |
| 2 R = –C2H5 | + | 60 | 53 | 10 | 27 | 0 | 2 |
| − | 95 | 30 | 47 | 0 | 10 | 2 | |
| 3 R = –C3H7 | + | 48 | 38 | 39 | 20 | 0 | <1 |
| − | 95 | 20 | 64 | 1 | 8 | 2 | |
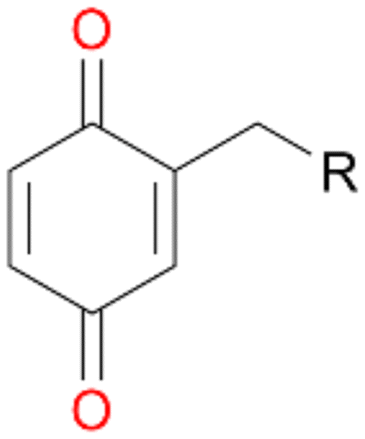
When the reactions were performed for 180 min only a minor increase in conversion of 1–3 was observed (Table 2). Destabilisation or inactivation of AbrUPO seems to contribute only marginally to this stagnation since AbrUPO retained around 60% of its initial activity after 4 h at 30 °C (ESI,† Fig. S2). Another reason for that can be the generally low enzyme activity particularly towards 1. Finally, hydrogen peroxide might become limiting in course of the reaction when the peroxidase uses up hydrogen peroxide without increasing the product concentration due to reduction of generated radicals by ascorbic acid.
Reactions with other substrates
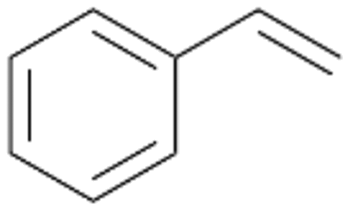
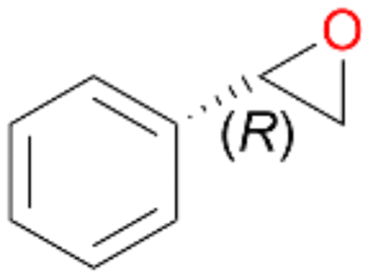
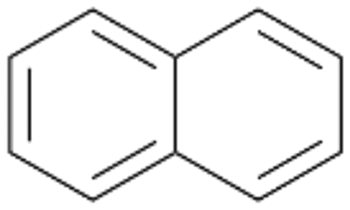
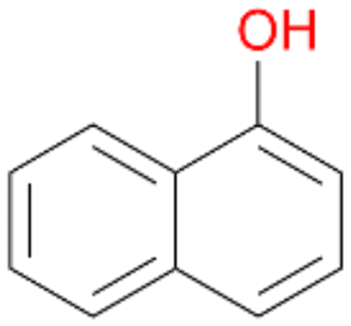
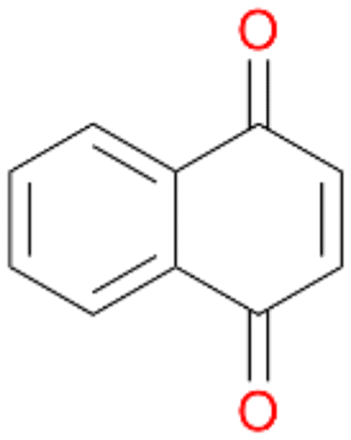
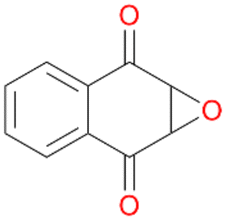
Interestingly, with styrene 11 and its derivatives 12–13, no aromatic hydroxylation was observed and conversions yielded the expected epoxides and the corresponding Meinwald rearrangement aldehyde products (Table 2). In case of styrene 11 the corresponding (R)-epoxide 11a was formed in slightly higher enantioselectivity (13% ee, ESI,† Fig. S9) as compared to PaDa-I (7% ee).47 Dual functional styrenes such as cinnamyl alcohol 14 underwent both, epoxidation as well as allylic hydroxylation/oxidation, the latter being favoured. trans-Stilbene 15 was oxidised by AbrUPO predominantly to the epoxide 15a, which was only described for CglUPO so far.48 Other UPOs like AaeUPO and MroUPO oxidised 15 at the aromatic ring yielding dihydroxy-trans-stilbene.48 Naphthalene 16 was oxidised to 1-naphthol 16a and 1,4-naphthoquinone 16b at a similar ratio as reported for e.g. CglUPO while PaDa-I mainly produced 16a.41 Additionally, 1,4-naphthoquinone-2,3-epoxide (16c) was found as a product of further oxidation.AbrUPO showed high activity for oxidation of sulphur-containing compounds 17–18 (ESI,† Table S4) as it was described for other UPOs.49 Sulfones were the main products under the investigated reaction conditions. Different terpenes and terpenoids were tested as substrates for AbrUPO as well (ESI,† Table S4). Among those, α-pinene 19 was the best substrate (conversion of 84%), but a product mixture was formed (ESI,† Fig. S28). Under the same conditions, conversion of verbenone 20 achieved 14%, while camphor (21) and valencene (22) were not oxidised at all. The bulky testosterone 23 was oxidised with 10% (ESI,† Fig. S32). The hydroxylated product could not be identified yet. Oxidation of testosterone by UPOs has only been described for CglUPO so far.50
C10–C13 fatty acids 24–27 were tested as substrates as well. Fatty acids were mainly hydroxylated at ω-1 but also at other positions (ESI,† Table S5). Interestingly, in the reactions with lauric acid 26 and tridecanoic acid 27 small amounts of lactones were formed (6–8%) (ESI,† Table S5). Other UPOs like AaeUPO and CciUPO from C. cinerea catalyzed the hydroxylation of lauric acid 26 predominantly at positions ω-1 and ω-2.35,51
In attempt to rationalise our observations we compared the active site of AbrUPO with the active sites of the long UPO PaDa-I and the two short UPOs, CglUPO and HspUPO from Hypoxylon sp. (Fig. 2).37,52 PaDa-I harbours a triad of phenylalanine residues (F69, F121 and F199) in close proximity to the heme group and two phenylalanine residues (F76 and F191) within the substrate access channel. In contrast, AbrUPO as well as HspUPO and CglUPO lack the triad of phenylalanines and possess only one or two phenylalanines close to the heme. Further, AbrUPO contains an additional glutamic acid residue at position 87, while the homologous positions in both other short UPOs are occupied by phenylalanine (F84 and F79, respectively) and in PaDa-I by alanine (A80). Less phenylalanine residues and their different location in the substrate binding site of AbrUPO compared to other UPOs might lead to an altered positioning of the aromatic substrates above the heme and facilitate aromatic oxidation. The influence of the active site amino acids in AbrUPO on chemoselectivity is currently under further investigation.
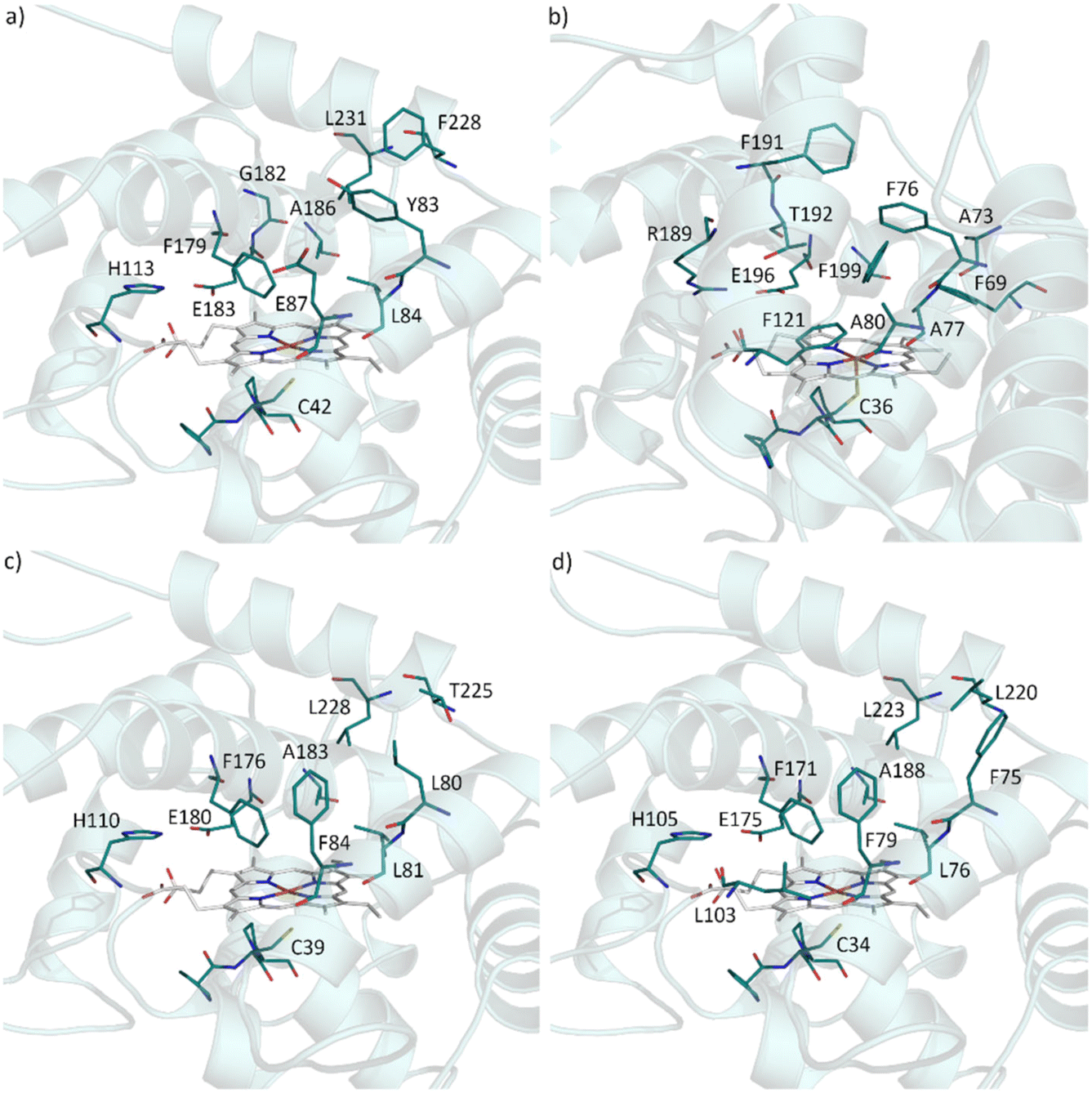 | ||
| Fig. 2 Residues present in the heme access channel and active site of AbrUPO (a), PaDa-I (b), HspUPO (c) and CglUPO (d). The 3D-models were designed from 5OXU (b) and 7O1X (c). Homology models (a) and (d) were obtained using SWISS MODEL protein structure homology-modelling server (https://swissmodel.expasy.org) with 7O1X as template. | ||
Conclusions
In summary, a UPO from Aspergillus brasiliensis was heterologously expressed in P. pastoris and secreted into the culture medium at high yields and was found to catalyse aromatic oxidation of a number of substituted benzenes. Generally, its catalytic activity was in the same range as reported for other UPOs and strongly depended on the substrate used with for example 37% conversion of ethylbenzene 1 and 78% conversion of propylbenzene 2. For comparison, conversion of propylbenzene 2 catalysed by PaDa-I was similar and achieved 72%, while activity of PaDa-I with ethylbenzene 1 was higher leading to 96% conversion. The highest activity of AbrUPO was observed towards α-methylstyrene 12 with 98% conversion and towards thymol 10 and cinnamyl alcohol 14 with over 99% conversion. The observed chemoselectivity of AbrUPO was dependent on the structure and chemical properties of the substituting group(s) at the aromatic ring. Increasing the chain length in alkyl benzenes shifted the selectivity from aromatic to benzylic hydroxylation. During aromatic hydroxylation, the first OH-group was introduced at ortho-position related to existing alkyl substituent, but not at para-position as could be expected. The second OH-group was introduced at para-position related to the firstly introduced OH-group yielding hydroquinones. The absence of hydroxylation at para-position, also favourable for aromatic substitution, can be explained by some steric restrictions present in the active site of AbrUPO.Compared to other reported UPOs, AbrUPO did not lead to a strong ‘overoxidation’. Our results indicate that further oxidation of benzylic alcohols to ketones occurs due to both peroxidase and peroxygenase activity of AbrUPO, both of which were substrate dependent. Further oxidation of hydroquinones to quinones was attributed to the inherent peroxidase activity of AbrUPO and could be completely avoided by adding ascorbic acid and shortening the reaction time. This makes AbrUPO not only an interesting biocatalyst for synthetic chemistry but also an attractive model for understanding the molecular factors governing the chemoselectivity of heme-thiolate enzymes and the starting enzyme for protein engineering studies.
Author contributions
F. S. planned and performed the experiments and drafted the manuscript. K. K., F. H. and V. B. U. designed and supervised the research work and wrote the manuscript. All authors read and approved the final manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
We thank Thomas Hilberath and Nina Jankowski for the useful discussions and the support during this work. The financial support by the Bioeconomy Science Center (BioSC, Germany) through the Ministry of Innovation, Science and Research within the framework of the NRW-Strategieprojekt BioSC (No. 313/323-400-00213) is gratefully acknowledged.Notes and references
- M. Hofrichter, H. Kellner, R. Herzog, A. Karich, C. Liers, K. Scheibner, V. W. Kimani and R. Ullrich, in Grand Challenges in Fungal Biotechnology, ed. H. Nevalainen, 2020, ch. 14, pp. 369–397 Search PubMed.
- R. Ullrich, J. Nuske, K. Scheibner, J. Spantzel and M. Hofrichter, Appl. Environ. Microbiol., 2004, 70, 4575–4581 CrossRef CAS PubMed.
- X. Wang, R. Ullrich, M. Hofrichter and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3686–3691 CrossRef CAS.
- M. Hofrichter and R. Ullrich, Curr. Opin. Chem. Biol., 2014, 19, 116–125 CrossRef CAS PubMed.
- R. Ullrich and M. Hofrichter, Cell. Mol. Life Sci., 2007, 64, 271–293 CrossRef CAS PubMed.
- P. Gomez de Santos, I. Mateljak, M. D. Hoang, S. J. Fleishman, F. Hollmann and M. Alcalde, J. Am. Chem. Soc., 2023, 145(6), 3443–3453 CrossRef CAS PubMed.
- A. Karich, M. Kluge, R. Ullrich and M. Hofrichter, AMB Express, 2013, 3, 5 CrossRef PubMed.
- L. Cheng, H. Wang, H. Cai, J. Zhang, X. Gong and W. Han, Science, 2021, 374, 77–81 CrossRef CAS PubMed.
- H. Long, T. S. Chen, J. Song, S. Zhu and H. C. Xu, Nat. Commun., 2022, 13, 3945 CrossRef CAS PubMed.
- M. Kluge, R. Ullrich, C. Dolge, K. Scheibner and M. Hofrichter, Appl. Microbiol. Biotechnol., 2009, 81, 1071–1076 CrossRef CAS PubMed.
- P. Molina-Espeja, M. Canellas, F. J. Plou, M. Hofrichter, F. Lucas, V. Guallar and M. Alcalde, ChemBioChem, 2016, 17, 341–349 CrossRef CAS PubMed.
- R. Ullrich and M. Hofrichter, FEBS Lett., 2005, 579, 6247–6250 CrossRef CAS PubMed.
- M. Hofrichter, R. Ullrich, M. Pecyna, M. Kinne, M. Kluge, E. Aranda, C. Liers, M. Poraj-Kobielska, G. Grobe, K. Scheibner, B. Bittner, K. Piontek, R. Schubert and K. Hammel, presented in part at the 16th International Conference on Cytochrome P450, Nago, Okinawa, Japan, 2009.
- T. Furuya, Y. Arai and K. Kino, Appl. Environ. Microbiol., 2012, 78, 6087–6094 CrossRef CAS PubMed.
- J. E. Stok, S. Chow, E. H. Krenske, C. Farfan Soto, C. Matyas, R. A. Poirier, C. M. Williams and J. J. De Voss, Chem. – Eur. J., 2016, 22, 4408–4412 CrossRef CAS.
- A. Dennig, N. Lulsdorf, H. Liu and U. Schwaneberg, Angew. Chem., Int. Ed., 2013, 52, 8459–8462 CrossRef CAS PubMed.
- C. J. Whitehouse, N. H. Rees, S. G. Bell and L. L. Wong, Chem. – Eur. J., 2011, 17, 6862–6868 CrossRef CAS PubMed.
- S. P. de Visser and S. Shaik, J. Am. Chem. Soc., 2003, 125, 7413–7424 CrossRef CAS PubMed.
- T. Coleman, J. Z. H. Lee, A. M. Kirk, D. Z. Doherty, M. N. Podgorski, D. K. Pinidiya, J. B. Bruning, J. J. De Voss, E. H. Krenske and S. G. Bell, Chem. – Eur. J., 2022, 28, e202201895 CrossRef CAS.
- T. Coleman, A. M. Kirk, J. H. Z. Lee, D. Z. Doherty, J. B. Bruning, E. H. Krenske, J. J. De Voss and S. G. Bell, ACS Catal., 2022, 12, 1258–1267 CrossRef CAS.
- A. Grote, K. Hiller, M. Scheer, R. Munch, B. Nortemann, D. C. Hempel and D. Jahn, Nucleic Acids Res., 2005, 33, W526–W531 CrossRef CAS PubMed.
- F. Tonin, F. Tieves, S. Willot, A. van Troost, R. van Oosten, S. Breestraat, S. van Pelt, M. Alcalde and F. Hollmann, Org. Process Res. Dev., 2021, 25, 1414–1418 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- T. A. R. S. Omura, J. Biol. Chem., 1964, 239, 2370–2378 CrossRef CAS.
- T. A. R. S. Omura, J. Biol. Chem., 1964, 239, 2379–2385 CrossRef CAS PubMed.
- I. Barr and F. Guo, Bio-Protoc., 2015, 5, e1594 Search PubMed.
- B. R. E. Childs and W. G. Bardsley, Biochem. J., 1975, 145, 93–103 CrossRef PubMed.
- A. González-Benjumea, J. Carro, C. Renau-Mínguez, D. Linde, E. Fernández-Fueyo, A. Gutiérrez and A. T. Martínez, Catal. Sci. Technol., 2020, 10, 717–725 RSC.
- D. Linde, A. Olmedo, A. González-Benjumea, M. Estévez, C. Renau-Mínguez, J. Carro, E. Fernández-Fueyo, A. Gutiérrez and A. T. Martínez, Appl. Environ. Microbiol., 2020, 86, 1–16 CrossRef.
- M. Hofrichter, H. Kellner, R. Herzog, A. Karich, J. Kiebist, K. Scheibner and R. Ullrich, Antioxidants, 2022, 11, 163 CrossRef CAS.
- A. Kinner, K. Rosenthal and S. Lutz, Front. Bioeng. Biotechnol., 2021, 9, 705630 CrossRef.
- D. H. Anh, R. Ullrich, D. Benndorf, A. Svatos, A. Muck and M. Hofrichter, Appl. Environ. Microbiol., 2007, 73, 5477–5485 CrossRef CAS.
- G. Grobe, R. Ullrich, M. J. Pecyna, D. Kapturska, S. Friedrich, M. Hofrichter and K. Scheibner, AMB Express, 2011, 1, 31 CrossRef PubMed.
- X. Wang, S. Peter, M. Kinne, M. Hofrichter and J. T. Groves, J. Am. Chem. Soc., 2012, 134, 12897–12900 CrossRef CAS.
- E. D. Babot, J. C. del Rio, L. Kalum, A. T. Martinez and A. Gutierrez, Biotechnol. Bioeng., 2013, 110, 2323–2332 CrossRef CAS PubMed.
- K. Ebner, L. J. Pfeifenberger, C. Rinnofner, V. Schusterbauer, A. Glieder and M. Winkler, Catalysts, 2023, 13, 206 CrossRef CAS.
- L. Rotilio, A. Swoboda, K. Ebner, C. Rinnofner, A. Glieder, W. Kroutil and A. Mattevi, ACS Catal., 2021, 11, 11511–11525 CrossRef CAS.
- S. Bormann, H. Kellner, J. Hermes, R. Herzog, R. Ullrich, C. Liers, R. Ulber, M. Hofrichter and D. Holtmann, Antioxidants, 2022, 11, 223 CrossRef CAS PubMed.
- P. Molina-Espeja, E. Garcia-Ruiz, D. Gonzalez-Perez, R. Ullrich, M. Hofrichter and M. Alcalde, Appl. Environ. Microbiol., 2014, 80, 3496–3507 CrossRef PubMed.
- A. Zaks and D. R. Dodds, J. Am. Chem. Soc., 1995, 117, 10419–10424 CrossRef CAS.
- P. Pullmann, A. Knorrscheidt, J. Munch, P. R. Palme, W. Hoehenwarter, S. Marillonnet, M. Alcalde, B. Westermann and M. J. Weissenborn, Commun. Biol., 2021, 4, 562 CrossRef PubMed.
- Y. Wang, D. Lan, R. Durrani and F. Hollmann, Curr. Opin. Chem. Biol., 2017, 37, 1–9 CrossRef PubMed.
- S. Peter, A. Karich, R. Ullrich, G. Gröbe, K. Scheibner and M. Hofrichter, J. Mol. Catal. B: Enzym., 2014, 103, 47–51 CrossRef CAS.
- B. Dolinska, A. Ostrozka-Cieslik, A. Caban, K. Rimantas, L. Leszczynska and F. Ryszka, Biol. Trace Elem. Res., 2012, 150, 509–512 CrossRef PubMed.
- M. Kluge, R. Ullrich, K. Scheibner and M. Hofrichter, Green Chem., 2012, 14, 440–446 RSC.
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS PubMed.
- M. G. Kluge, R. Ullrich, K. Scheibner and M. Hofrichter, Appl. Microbiol. Biotechnol., 2007, 75, 1473–1478 CrossRef CAS PubMed.
- C. Aranda, R. Ullrich, J. Kiebist, K. Scheibner, J. C. del Río, M. Hofrichter, A. T. Martínez and A. Gutiérrez, Catal. Sci. Technol., 2018, 8, 2394–2401 RSC.
- Y. Li, Y. Ma, P. Li, X. Zhang, D. Ribitsch, M. Alcalde, F. Hollmann and Y. Wang, ChemPlusChem, 2020, 85, 254–257 CrossRef CAS PubMed.
- J. Kiebist, K. U. Schmidtke, J. Zimmermann, H. Kellner, N. Jehmlich, R. Ullrich, D. Zander, M. Hofrichter and K. Scheibner, ChemBioChem, 2017, 18, 563–569 CrossRef CAS PubMed.
- A. Gutierrez, E. D. Babot, R. Ullrich, M. Hofrichter, A. T. Martinez and J. C. del Rio, Arch. Biochem. Biophys., 2011, 514, 33–43 CrossRef CAS PubMed.
- M. Ramirez-Escudero, P. Molina-Espeja, P. Gomez de Santos, M. Hofrichter, J. Sanz-Aparicio and M. Alcalde, ACS Chem. Biol., 2018, 13, 3259–3268 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00209h |
| This journal is © The Royal Society of Chemistry 2023 |