
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous-flow transfer hydrogenation of benzonitrile using formate as a safe and sustainable source of hydrogen†
Seán D.
Dempsey
 abc,
Ailbhe A.
Ryan
abc,
Megan
Smyth
b,
Thomas S.
Moody
bc,
Scott
Wharry
b,
Karen
Fahey
c,
Andrew M.
Beale
de,
Sofia
Mediavilla Madrigal
ef,
Paul
Dingwall
a,
David W.
Rooney
a,
Peter C.
Knipe
*a,
Mark J.
Muldoon
*a and
Jillian M.
Thompson
*a
abc,
Ailbhe A.
Ryan
abc,
Megan
Smyth
b,
Thomas S.
Moody
bc,
Scott
Wharry
b,
Karen
Fahey
c,
Andrew M.
Beale
de,
Sofia
Mediavilla Madrigal
ef,
Paul
Dingwall
a,
David W.
Rooney
a,
Peter C.
Knipe
*a,
Mark J.
Muldoon
*a and
Jillian M.
Thompson
*a
aSchool of Chemistry & Chemical Engineering, Queen's University Belfast, BT9 5AG, UK. E-mail: p.knipe@qub.ac.uk; m.j.muldoon@qub.ac.uk; jillian.thompson@qub.ac.uk
bAlmac Sciences Ltd., 20 Seagoe Industrial Estate, Craigavon, BT63 5QD, UK
cArran Chemical Company, Unit 1 Monksland Industrial Estate, Athlone, Co. Roscommon, Ireland
dChemistry Department, University College of London, 20 Gordon Street, London, WC1H 0AJ, UK
eUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Laboratory, Didcot, OX11 0FA, UK
fMax Planck-Cardiff Centre on the Fundamentals of Heterogeneous Catalysis FUNCAT, Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK
First published on 16th June 2023
Abstract
The continuous catalytic transfer hydrogenation of benzonitrile to benzylamine is demonstrated using a palladium on carbon catalyst with triethylammonium formate as reducing agent. Solvent choice was critical in overcoming rapid catalyst deactivation. A 15-fold increase in catalyst productivity was observed in flow compared to batch, which was achieved using an ethanol–water solvent in combination with intermittent catalyst regeneration by washing with water.
Catalytic hydrogenation is a mature technology applied across a range of industries, from large scale continuous processing in the petrochemicals industry, to smaller scale active pharmaceutical ingredient (API) production for which multi-functional batch reactors are widely used.1 The benefits of continuous processing for fine chemical production are widely recognised and increased investment in development of processes and technologies has seen rapid growth of the field.2,3
Primary amines are key synthetic intermediates and final products in the fine chemicals industry. Catalytic hydrogenation of nitriles is a useful route for the synthesis of primary amines due to the availability and relative stability of nitrile substrates and the high atom efficiency which can be achieved. Catalytic transfer hydrogenation (CTH) offers a useful alternative to processes which use H2 gas as reductant. CTH reactions make use of reducing agents, which are often stable liquids or solids at room temperature and present significantly less safety risks during storage and handling compared to H2 gas. CTH reactions can be employed as useful alternatives to high-pressure hydrogenations on an industrial scale as they do not require the use of high-pressure equipment. Formic acid and related formates have gained interest as reducing agents, due to their relative stability and high hydrogen density.4 HCOOH can also be renewably sourced as it is a by-product of biomass refineries.5
Although there are several examples of homogeneously catalysed CTH of nitriles,6–8 the work reported herein focuses on the use of heterogeneous metal catalysts, which have the advantage of easier post-reaction separation. A range of catalytic methods have previously been reported, including batch reactions utilising RANEY® nickel and highly toxic and reactive hydrazines or hydrazine formates which demonstrate good to moderate yields of primary amine.9,10 Mebane et al. reported hydrogenation of aliphatic nitriles using isopropanol as both solvent and reducing agent over RANEY® nickel in the presence of potassium hydroxide.11 Isolation of the amine salts was only achieved after acid hydrolysis and basic workup of secondary imine intermediates, which form from condensation of the amine product and acetone generated in the reaction. In 2014, Vilches-Herrera et al. reported an attractive method for aromatic nitrile reduction using a palladium on carbon (Pd/C) catalyst and HCOOH–NEt3 as the reducing agent in THF.12 This method delivered good to excellent yields of primary amines without requiring additional post-reaction modifications and employs formate as reducing agent.
Reports on CTH of nitriles in continuous-flow are more limited, with one example using a homogenous catalyst,13 but currently there are no known examples employing heterogeneous metal catalysts. Continuous-flow processes are advantageous for heterogeneously catalysed hydrogenation reactions, offering easier catalyst separation and reduced risks associated with handling potentially pyrophoric catalyst slurries. Moreover, improved control of contact time between the reaction mixture and catalyst can benefit selectivity for sequential reactions. Therefore, this work sets out to develop a continuous-flow process for the selective CTH of aromatic nitriles, using HCOOH and related formates as reducing agents.
The batch method reported by Vilches-Herrera et al. for the CTH of aromatic nitriles using HCOOH–NEt3 and Pd/C was chosen as a starting point to develop a continuous process.12 The CTH of benzonitrile was selected as a model reaction to demonstrate this chemistry. A screen of commercially available Pd catalysts in batch showed that Pd was active for the CTH of benzonitrile and that carbon was a particularly good support, giving complete conversion and 97% selectivity to benzylamine in less than 5 min (section S2, ESI†). This was in good agreement with the findings of Vilches-Herrera et al.,12 however, a small amount of toluene was also observed during our reactions (3–6% selectivity), a side product formed as a result of benzylamine hydrogenolysis (Scheme 1).14 Hydrogenolysis products have been previously reported during the reaction of aromatic nitriles with formate reducing agents over Pd catalysts.15 In addition, no formation of dibenzylamine was observed, another common side product often formed during benzonitrile hydrogenation as a result of the trans-imination reaction between the reactive imine intermediate (benzylimine) and benzylamine, and subsequent hydrogenation.16
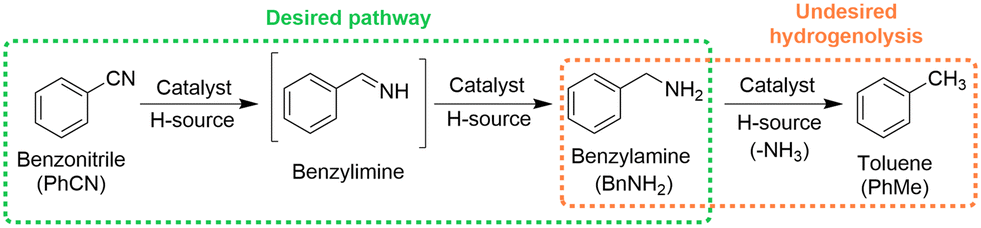 | ||
| Scheme 1 Reaction pathways associated with benzonitrile hydrogenation. | ||
To avoid build-up of pressure, reduce the risk of channelling and lessen the transfer of catalyst particles into the product stream, a commercially available granular form of 5 wt% Pd/C (particle size range of 700–900 μm) was selected as the catalyst for continuous-flow studies. Batch tests showed this granular catalyst produced benzylamine with 94% selectivity, although, a lower conversion of 56% was observed compared to the powdered form which gave 100% conversion after 30 min (Table S1†). Fig. 1 shows the experimental setup utilised for continuous-flow studies, with further experimental details provided in the ESI† (section S1). To avoid any limitations of mixing between the benzonitrile substrate in THF and HCOOH–NEt3 (section S3, ESI†), a single pump inlet configuration was employed.
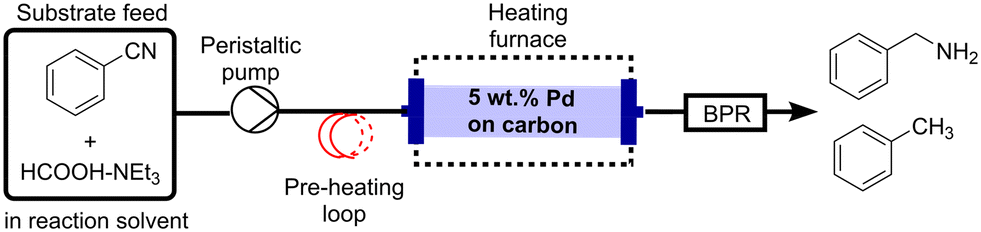 | ||
| Fig. 1 Continuous-flow setup for benzonitrile transfer hydrogenation. | ||
The conditions selected for initial application of the model reaction in continuous-flow were based on those used in preliminary batch experiments. Samples were collected periodically from the reactor outlet and offline GC analysis was used to determine conversion and product yields. Fig. 2 shows the change in reaction composition over time, with complete conversion of benzonitrile initially observed. This was accompanied by high selectivity to benzylamine of 95%, which is consistent with batch studies. However, a rapid drop in activity is observed after just 30 minutes on stream, with the conversion of benzonitrile falling to 10% within 150 min, indicating rapid deactivation of the catalyst bed. Although a back pressure of 6 bar was used to control the flow from the outlet of the reactor, it should be noted that the time on stream data for a reaction at ∼1 bar back pressure (BPR removed) resulted in a comparable profile (section 4, ESI†).
 | ||
Fig. 2 Time on stream data for the continuous-flow CTH of benzonitrile. Reaction conditions: benzonitrile (0.1 M) and HCOOH–NEt3 (HCOOH (10.0 M), NEt3 (0.55 M), 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) in THF was passed over 5 wt% Pd/C (granular, 1.0 g) at 40 °C, flow rate of 0.5 mL min−1 and 6 bar back pressure. :1 molar ratio) in THF was passed over 5 wt% Pd/C (granular, 1.0 g) at 40 °C, flow rate of 0.5 mL min−1 and 6 bar back pressure. | ||
ICP-OES analysis was carried out on both fresh and spent catalysts to determine the Pd content of the samples (5.56 wt% and 5.24 wt% respectively). This loss of Pd does not explain the near complete loss in activity observed after 150 min under continuous operation.
Deactivation during nitrile hydrogenation using H2 gas has been attributed to strong adsorption of intermediates and products.17 To investigate this, the catalyst bed was pre-treated with either benzonitrile or HCOOH–NEt3. Analysis of the reaction after pre-treatment with benzonitrile resulted in complete initial conversion and selectivity of 96% to benzylamine (Fig. S5-A, ESI†), suggesting that benzonitrile is not the poisoning species. In contrast, pre-treatment with HCOOH–NEt3 (Fig. S5-B, ESI†) resulted in little to no benzonitrile conversion, even at the beginning of the reaction, with no product formation observed. This suggests that HCOOH–NEt3 or one of the intermediates or products formed during its decomposition over the Pd catalyst is the primary poisoning species.
In the HCOOH–NEt3 mixture used, HCOOH is present in large excess of NEt3 (18:1 molar equivalents). HCOOH is known to decompose over heterogenous metal catalysts via two competing pathways:4
| HCOOH → H2 + CO2 | (1) |
| HCOOH → H2O + CO | (2) |
The dehydrogenation pathway (eqn (1)) is required for CTH reactions, however strong adsorption of CO from the undesired dehydration pathway (eqn (2)) has previously been suggested as a primary mechanism of deactivation of Pd catalysts at mild temperatures.18,19 In contrast, Caiti et al., presented evidence of formate ions being a primary contributor to deactivation in their studies of hydrogen production from formic acid over a Pd catalyst.20 This was confirmed by demonstrating that in a continuous reactor, a deactivated Pd/C catalyst bed could be regenerated by washing with water, whereas in contrast, one poisoned by CO could not be regenerated in this manner.
Similarly, it was found that catalyst regeneration could be achieved for this CTH process by simply washing the spent 5 wt% Pd/C catalyst with deionised water (Fig. 3-A). Washing the spent catalyst bed for 2 hours at 2.5 mL min−1 (totalling 300 mL of deionised water) partially restored catalyst activity back to 60% benzonitrile conversion. After completion of run 2, washing the bed for a further 16 hours (totalling 2400 mL of deionised water) resulted in nearly complete regeneration of the catalyst back to its original activity, with 91% initial conversion observed for run 3, compared to 98% for run 1. The catalyst productivity was in fact higher for run 3 compared to run 1, since some catalyst had been removed during the experiment for ex situ characterisation. These data suggest that CO poisoning is not the primary contributor to deactivation as regeneration via water washing would not overcome this type of poisoning.
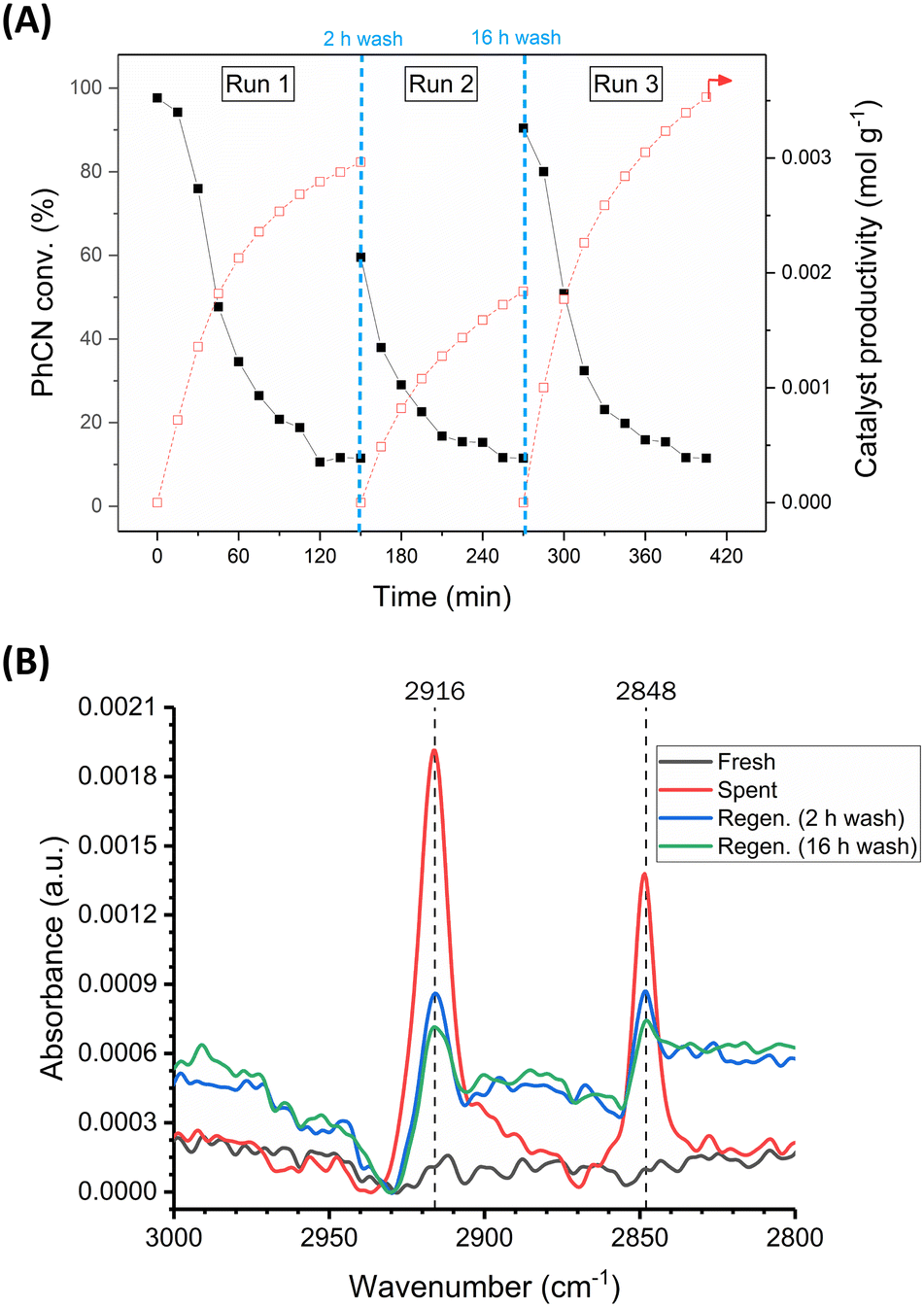 | ||
| Fig. 3 (A) Time on stream data for the continuous-flow CTH of benzonitrile demonstrating catalyst regeneration by washing with deionised water in terms of benzonitrile conversion and catalyst productivity. Reaction conditions: benzonitrile (0.1 M) and HCOOH–NEt3 (HCOOH (10.0 M), NEt3 (0.55 M), 18:1 molar ratio) in THF was passed over 5 wt% Pd/C (granular, 1.0 g for run 1, 0.8 g for run 2 and 0.6 g for run 3) at 40 °C, liquid flow rate of 0.5 mL min−1 and 6 bar back pressure. Wash procedure: deionised water at 2.5 mL min−1 for 2 or 16 h at 40 °C (denoted by blue, vertical, dashed lines). (B) Comparison of ATR-FTIR spectra of fresh, spent, and regenerated catalysts. | ||
Samples of fresh, spent, and regenerated catalyst were analysed by ATR-FTIR (Fig. 3-B). Distinct C–H stretching mode bands at 2916 and 2848 cm−1, indicative of adsorbed formate,21,22 were observed in the spectrum for the spent but not the fresh catalyst, consistent with deactivation brought about by strongly adsorbed formate species. These bands were visible in the spectra of both regenerated catalyst samples. The band intensity is significantly reduced when compared to the spent catalyst and decreases with increasing wash time, showing that the water wash helps to remove formate from the catalyst surface. This is consistent with a 16 hour wash leading to a higher increase in activity for the CTH reaction compared to a 2 hour wash.
To minimise the deactivation caused by HCOOH–NEt3, the concentration of HCOOH was decreased from 10 M to 5 M, and then to 1 M, whilst maintaining an 18-fold excess with respect to NEt3 (Fig. S6, ESI†). The time on stream data shows that although the initial benzonitrile conversion decreases from 100% to 84% with a 10-fold decrease in HCOOH concentration (10 M to 1 M), the rate of catalyst deactivation is lower, with a drop in conversion of 19% per hour using the lower concentration of HCOOH compared to 42% per hour using the higher concentration. A decrease in benzylamine selectivity, from 96% to 76%, is also observed but it was decided to keep the concentration of HCOOH as low as possible to minimise deactivation and consider other parameters to help improve selectivity.
Previous work showed that the molar ratio of HCOOH–NEt3 does not influence the activity or selectivity of the reaction in batch when using a large excess of HCOOH–NEt3 (more than 100 equivalents of HCOOH with respect to benzonitrile).12 Other studies have found the ratio of HCOOH–NEt3 can influence the activity of Pd catalysts during CTH reactions, for example in the reduction of alkenes.23 To investigate the influence of the HCOOH–NEt3 molar ratio on this continuous process, reactions were carried out with varying concentrations of NEt3, while the concentration of HCOOH was kept at 1 M (Fig. S7, ESI†). When the molar concentration of NEt3 was increased from 0.055 M (18:1 molar ratio) to 0.1 M (10:1 molar ratio) a decrease in initial benzonitrile conversion was observed from 86% to 59%, with an even further decrease to 39% observed when 0.4 M NEt3 (5:2 azeotropic molar ratio) was used. Although, increasing the molar concentration of NEt3 does have a negative effect on initial conversion, the selectivity to benzylamine increases. Interestingly, the reaction also proceeds with no NEt3 present, but similar deactivation is observed, thus confirming NEt3 is not the cause of deactivation. From these studies it was decided that a feed solution containing HCOOH (1 M) and NEt3 (0.1 M) giving a molar ratio of 10:1 was optimal to achieve high selectivity to benzylamine, while still maintaining moderate conversion of substrate.
Solvent choice is known to influence CTH reactions involving HCOOH and related formates. For example, a previous study found that for the CTH of nitrobenzene using HCOOH as reducing agent over a Pd catalyst on a graphitic carbon nitride support, the catalyst activity was much higher in protic solvents such as water and ethanol when compared to non-protic solvents.24 Hence, a solvent screen was carried out for this continuous process with benzonitrile conversion monitored over time, as shown in Fig. 4. Using ethanol as solvent led to a higher initial benzonitrile conversion of 99% when compared to aprotic solvents such as THF or ethyl acetate, for which initial conversions were 59% and 66% respectively. In addition, the rate of deactivation also decreased from 11% and 7.7% per hour for THF and ethyl acetate, to 2.3% per hour in ethanol, suggesting that the use of protic solvents enhances both the activity and lifetime of the catalyst. Further improvement was observed using mixed solvent systems consisting of water and organic solvents, with the addition of 50 vol% of water to ethanol and THF resulting in longer catalyst lifetimes compared to the corresponding pure organic solvent.
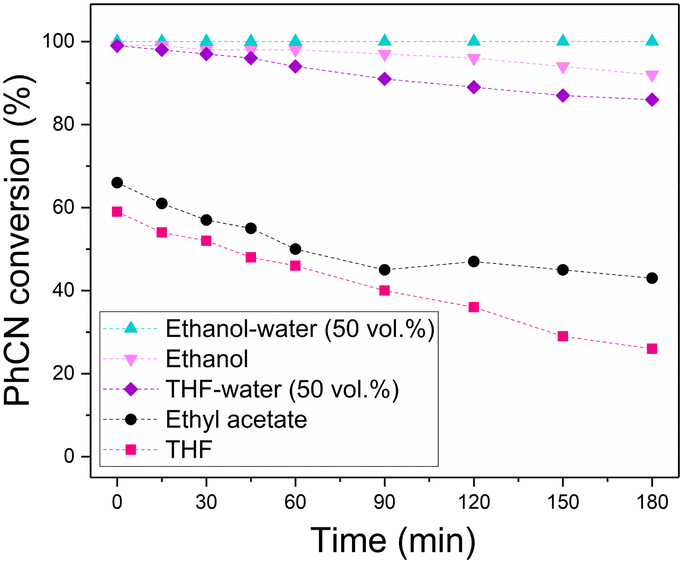 | ||
| Fig. 4 The effect of reaction solvent on benzonitrile conversion as a function of time on stream for the continuous-flow CTH of benzonitrile. Reaction conditions: benzonitrile (0.1 M) and HCOOH–NEt3 (HCOOH (1.0 M), NEt3 (0.1 M), 10:1 molar ratio) in various solvents was passed over 5 wt% Pd/C (granular, 1.0 g) at 40 °C, flow rate of 0.5 mL min−1 and 6 bar back pressure. | ||
The significant improvements observed in catalyst lifetime using protic solvents and in particular on addition of water further supports that the formate anion is the primary poisoning species in this process. The increased solubility of the formate anion in water compared to other solvents, especially aprotic, disfavours its strong adsorption to catalyst active sites. This was also observed during regeneration studies, in which water removed the formate poison from the active sites and regenerated catalyst activity.
The choice of solvent was also found to influence the selectivity, with protic solvents showing increased selectivity to benzylamine compared to aprotic solvents (Fig. S8, ESI†). Using ethanol–water (50 vol%) resulted in the highest selectivity to benzylamine, at 97%, further demonstrating the suitability of the solvent system for this CTH reaction. It is likely that the benzylamine product, once formed, is protonated in the liquid phase, and improved solubility of this species is achieved in polar, protic solvents such as water and ethanol. Therefore, adsorption of the product onto the catalyst surface is disfavoured, preventing hydrogenolysis to form unwanted toluene. Moreover, from a green chemistry perspective, ethanol–water is a more sustainable choice of solvent than THF for chemical processes.25
To determine the best solvent composition, the volume of water in the ethanol–water mixed solvent system was varied to investigate if further improvements in catalyst lifetime could be achieved (Fig. S9, ESI†). Reactions were run over fresh catalyst beds containing a reduced amount of Pd/C catalyst (0.5 g compared to 1 g used for previous studies). Ethanol–water (33 vol% H2O) was found to maintain the highest activity after 4 hours on stream.
With increased solubility of ammonium formate and sodium formate in the selected ethanol–water mixed solvent system, these were tested, along with HCOOH, as alternative reducing agents. However, no improvement in conversion or selectivity was observed over that achieved by HCOOH–NEt3 (10:1) (Fig. S10, ESI†).
With the longevity of the catalyst substantially improved using the newly developed process conditions, a continuous reaction was carried out for an extended reaction time over a catalyst bed containing half the amount of catalyst used for initial deactivation studies. This was accomplished by intermittent regeneration of the catalyst bed by simply washing with deionised water at 2.5 mL min−1. It was shown that higher flow rates during water washes led to more efficient catalyst regeneration (Fig. S11, ESI†). The results for the extended run are shown in Fig. 5, with high initial benzonitrile conversion of 99% achieved and a small decrease observed over the first 5 hours to 85%. After regeneration of the catalyst, the initial activity was completely restored, and an identical rate of deactivation was observed between 5 and 10 hours. This was repeated for three more cycles, with regeneration of the catalyst carried out every 5 hours. In this manner, material was processed for a total 25 hours, with high yields of benzylamine maintained throughout. This experiment also demonstrated that the catalyst can be regenerated successfully four times with little to no compromise in activity. In terms of catalyst productivity, or moles of benzonitrile converted per gram of catalyst, a 15-fold increase was achieved using the developed continuous-flow process (0.14 mol g−1) compared to batch reactions using 5 mol% catalyst loading (0.009 mol g−1), with even further increases in productivity possible with additional regeneration cycles.
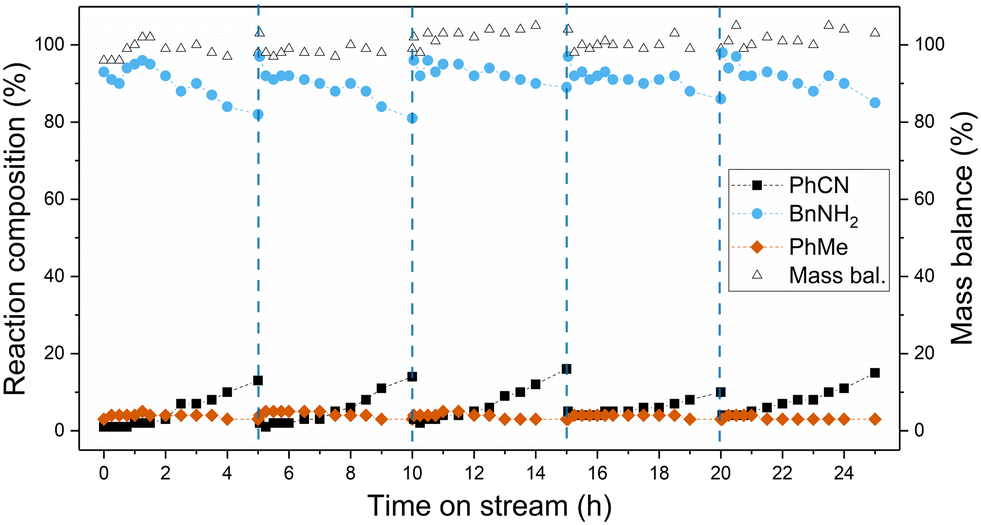 | ||
| Fig. 5 Time on stream data for the continuous-flow CTH of benzonitrile over an extended period using newly developed process conditions and intermittent washing of the catalyst bed. Reaction conditions: benzonitrile (0.1 M) and HCOOH–NEt3 (HCOOH (1.0 M), NEt3 (0.1 M), 10:1 molar ratio) in ethanol–water (33 vol% H2O) was passed over 5 wt% Pd/C (granular, 0.5 g) at 40 °C, liquid flow rate of 0.5 mL min−1 and 6 bar back pressure. Wash procedure: EtOH at 1 mL min−1 for 1 h, H2O at 2.5 mL min−1 for 16 h (denoted by blue, vertical, dashed lines). | ||
A safe and selective continuous-flow method for the transfer hydrogenation of benzonitrile to benzylamine has been developed utilising HCOOH–NEt3 as the reducing agent and a commercially available 5 wt% Pd/C catalyst. Analysis of the spent catalyst by ATR, provided strong evidence that the formate anion is the primary cause of catalyst deactivation. Through consideration of some key process parameters, the catalyst lifetime was considerably enhanced, and a much-improved catalyst productivity was achieved for this continuous CTH process. In considering scale-up, this process would benefit from running two or more fixed beds in parallel, with the CTH reaction continuously being performed on at least one fixed bed, while regeneration through washing is performed on others. This would ensure a constant stream of product and introduces the possibility of an automated process which could be incorporated into telescoped reaction sequences. The inclusion of in-line monitoring could also enable a self-optimising process to further enhance reaction productivity.
Author contributions
SD: investigation, visualization, writing – original draft, writing – review & editing, AR: writing – review & editing, MS: project administration, supervision, writing – review & editing, TM: funding acquisition, resources, supervision, writing – review & editing, SW: supervision, writing – review & editing, KF: project administration, supervision, writing – review & editing, AB: supervision, writing – review & editing, SM: investigation, writing – review & editing, PD: writing – review & editing, DR: funding acquisition, writing – review & editing, PK: funding acquisition, supervision, writing – review & editing, MM: conceptualization, funding acquisition, project administration, supervision, writing – review & editing, JT: conceptualization, funding acquisition, project administration, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Lara J. Nolan (QUB) for contributions to this article and Donal Moran (ASEP, QUB) for ICP measurements. This project received funding from the European Union's Horizon 2020 Research and Innovation Programme under Marie Skłodowska-Curie Grant Agreement 813394 & EPSRC (EP/S030468/1). This publication contains the views of the authors, and the European Commission is not responsible for any use that may be made of the information it contains.Notes and references
- E. Masson, E. M. Maciejewski, K. M. P. Wheelhouse and L. J. Edwards, Org. Process Res. Dev., 2022, 26, 2190–2223 CrossRef CAS.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS.
- J. C. McWilliams, A. D. Allian, S. M. Opalka, S. A. May, M. Journet and T. M. Braden, Org. Process Res. Dev., 2018, 22, 1143–1166 CrossRef CAS.
- R. Nie, Y. Tao, Y. Nie, T. Lu, J. Wang, Y. Zhang, X. Lu and C. C. Xu, ACS Catal., 2021, 11, 1071–1095 CrossRef CAS.
- F. Valentini, V. Kozell, C. Petrucci, A. Marrocchi, Y. Gu, D. Gelman and L. Vaccaro, Energy Environ. Sci., 2019, 12, 2646–2664 RSC.
- J. A. Garduño, M. Flores-Alamo and J. J. García, ChemCatChem, 2019, 11, 5330–5338 CrossRef.
- Z. Shao, S. Fu, M. Wei, S. Zhou and Q. Liu, Angew. Chem., Int. Ed., 2016, 55, 14653–14657 CrossRef CAS PubMed.
- K. Sarkar, K. Das, A. Kundu, D. Adhikari and B. Maji, ACS Catal., 2021, 11, 2786–2794 CrossRef CAS.
- W. W. Zajac, J. F. Sitjda, M. J. Nolan and T. M. Santostjsso, J. Org. Chem., 1971, 36, 3539–3541 CrossRef CAS.
- S. Gowda and D. C. Gowda, Tetrahedron, 2002, 58, 2211–2213 CrossRef CAS.
- R. C. Mebane, D. R. Jensen, K. R. Rickerd and B. H. Gross, Synth. Commun., 2003, 33, 3373–3379 CrossRef CAS.
- M. Vilches-Herrera, S. Werkmeister, K. Junge, A. Börner and M. Beller, Catal. Sci. Technol., 2014, 4, 629–632 RSC.
- R. Labes, D. González-Calderón, C. Battilocchio, C. Mateos, G. Cumming, O. de Frutos, J. Rincón and S. Ley, Synlett, 2017, 28, 2855–2858 CrossRef CAS.
- M. I. McAllister, C. Boulho, L. F. Gilpin, L. McMillan, C. Brennan and D. Lennon, Org. Process Res. Dev., 2019, 23, 977–989 CrossRef CAS.
- G. R. Brown and A. J. Foubister, Synthesis, 1982, 1036–1037 CrossRef CAS.
- J. J. W. Bakker, A. G. Van Der Neut, M. T. Kreutzer, J. A. Moulijn and F. Kapteijn, J. Catal., 2010, 274, 176–191 CrossRef CAS.
- C. Dai, F. Liu, W. Zhang, Y. Li, C. Ning, X. Wang and C. Zhang, Appl. Catal., A, 2017, 538, 199–206 CrossRef CAS.
- K. Jiang, K. Xu, S. Zou and W. Bin Cai, J. Am. Chem. Soc., 2014, 136, 4861–4864 CrossRef CAS PubMed.
- P. Xu, F. D. Bernal-Juan and L. Lefferts, J. Catal., 2021, 394, 342–352 CrossRef CAS.
- M. Caiti, D. Padovan and C. Hammond, ACS Catal., 2019, 9, 9188–9198 CrossRef CAS.
- A. Karelovic, G. Galdames, J. C. Medina, C. Yévenes, Y. Barra and R. Jiménez, J. Catal., 2019, 369, 415–426 CrossRef CAS.
- X. Jiang, X. Nie, X. Wang, H. Wang, N. Koizumi, Y. Chen, X. Guo and C. Song, J. Catal., 2019, 369, 21–32 CrossRef CAS.
- J. Mondal, Q. T. Trinh, A. Jana, W. K. H. Ng, P. Borah, H. Hirao and Y. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 15307–15319 CrossRef CAS PubMed.
- X. Xu, J. Luo, L. Li, D. Zhang, Y. Wang and G. Li, Green Chem., 2018, 20, 2038–2046 RSC.
- D. Prat, O. Pardigon, H. W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00195d |
| This journal is © The Royal Society of Chemistry 2023 |