
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable and selective Ni-catalyzed allylation of 2-oxindoles and 2-coumaranones in batch and flow chemistry†
Bouchaib
Mouhsine
ab,
Anthony
Saint Pol
a,
Abdallah
Karim
b,
Maël
Penhoat
 *d,
Clément
Dumont
ac,
Isabelle
Suisse
a and
Mathieu
Sauthier
*a
*d,
Clément
Dumont
ac,
Isabelle
Suisse
a and
Mathieu
Sauthier
*a
aUniv. Lille, CNRS, Centrale Lille, ENSCL, Univ. Artois, UMR 8181 – UCCS Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: mathieu.sauthier@univ-lille.fr
bÉquipe de Chimie de Coordination et de Catalyse, Département de Chimie, Faculté des Sciences Semlalia, Université Cadi Ayyad, BP 2390, Marrakech, Morocco
cICAM, Site de Lille, 6 rue Auber, 59016 Lille Cedex, France
dUSR 3290, MSAP (Miniaturisation pour la Synthèse, l'Analyse et la Protéomique), Univ. de Lille, F-59000 Lille, France
First published on 14th June 2023
Abstract
2-Oxindoles and 2-coumaranones could be selectively allylated in the presence of low cost catalysts based on nickel, leading selectively to different polysubstituted derivatives. Allyl alcohol was used as an allylating reagent and allowed the synthesis of compounds under neutral conditions with water produced as the sole by-product. Experiments have been performed in batch and in flow chemistry. The latter protocol in flow led to a very efficient and straightforward allylic alkylation of 2-oxindoles in only a few minutes with high chemoselectivities towards either the C,C-bisallylated product or the C,C,N-trisallylated one.
Introduction
The development of greener synthetic methods is a major challenge for chemists in order to reduce the negative impacts of chemical products and processes on human health and the environment. The use of catalysts using earth-abundant metals and the prevention of waste are important to develop economic and sustainable reactions and processes. The development of flow chemistry also presents a real opportunity for more selective and, hence, greener chemical productions.1 As a powerful tool in organic chemistry, flow chemistry has now become common in a wide range of chemical industries.2,3 This technology aims to enhance a researcher's ability to perform chemical reactions with unique control over key reaction parameters.4–82-Oxindoles and 2-coumaranones belong to an important class of molecules with a privileged aromatic heterocyclic scaffold. 2-Oxindoles are for example found in a wide range of bioactive natural products and pharmaceuticals.9–15 The introduction of substituents on the nucleophilic positions (N1 or C3) of 2-oxindoles allows the synthesis of a large panel of valuable molecules with unique biological activities such as anti-cancer, anti-microbial or anti-bacterial properties.16–19 In this context, the introduction of an allyl functionality on oxindole or coumaranone frameworks is interesting because of further possible reactions on the terminal double bond such as oxidation, hydroformylation or metathesis and the possibility of producing new valuable building blocks. The allylic alkylation reaction is one of the most relevant and efficient reactions for the construction of C–C and C–heteroatom (N/O/S) bonds.20,21 Despite the abundant studies in this field, allylation of 2-oxindoles has been poorly explored. This reaction has been performed in the presence of transition metals as palladium,22–26 molybdenum27 and iridium/copper28 as catalysts. 2-Coumaranone allylation has only been performed by stoichiometric means, typically with allyl bromide in the presence of K2CO3/Bu4NHSO4 or NaH, thus generating large amounts of salts.29,30 To the best of our knowledge, the use of nickel in the catalytic allylation of 2-oxindoles and 2-coumaranones has never been reported.31–34
Herein, we report the allylic alkylation of 2-oxindoles and 2-coumaranones with a particular focus on the sustainability of the process. The N-unprotected 2-oxindole skeleton bears two nucleophilic sites (N1 and C3) which are in competition for the allylation reaction (Scheme 1). For the first time, catalytic oxindole allylation is performed in the presence of a nickel based catalyst as a non-critical material and low cost metal. In order to prevent waste, allyl alcohol is used as an allylation reagent leading to water as the sole reaction by-product.35–39 It is also noteworthy that allyl alcohol is industrially produced40 and is also accessible from vegetal feedstock as glycerol.41–44
 | ||
| Scheme 1 The nickel-catalyzed allylation of 2-oxindole and 2-coumaranone with allyl alcohol: sustainability of the reaction/selectivity issue. | ||
Depending on the sites that are allylated and the substitution degree, several products are accessible. Diallylated products are particularly interesting as precursors of pharmaceutically active compounds.16 Herein, we wish to report that nickel-based catalytic systems are able to promote the C,C-bisallylation and C,C,N-trisallylation of 2-oxindoles with allyl alcohol under additive-free conditions. This protocol also proved to be suitable for the C,C-bisallylation of 2-coumaranones. As part of our interest in developing efficient continuous flow protocols,45,46 this reaction was implemented under flow conditions, thus allowing better activities and a highly improved selectivity control toward the products of C,C-bisallylation or C,C,N-trisallylation of 2-oxindoles.
Results and discussion
Allylation reaction of 2-oxindoles
In order to determine critical reaction parameters, the study was first focused on the allylation of 2-oxindole 1a as a model substrate with allyl alcohol 2a (2 equiv.) in MeOH at 80 °C. The catalyst was in situ generated by combining 1.5 mol% Ni(cod)2 and 3 mol% of ligand L1, L2 or L3 (Table 1, entries 1–3) which proved to be the most efficient during allylation of various substrates.35,36 The reactions with the three bidentate ligands gave a mixture of products of C,C-bisallylation and C,C,N-trisallylation, 4a and 5a, respectively. No formation of the monoallylated 2-oxindole 3a could be detected. In these cases, modest selectivities were obtained with 27–60% of the bisallylated product 4a and 33–50% of the trisallylated product 5a.|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | [Ni] (mol%) | L | T (°C) | Solvent (0.5 mL) | Equiv. 2a | Conv. 1a (%) | Yieldb (%) | ||
| 3a | 4a | 5a | |||||||
a Conditions: 1a (1.8 mmol), Ni(cod)2/ligand (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), 17 h in a sealed Schlenk tube.
b Determined by GC of the crude product using anisole as an internal standard. :2), 17 h in a sealed Schlenk tube.
b Determined by GC of the crude product using anisole as an internal standard.
|
|||||||||
| 1 | 1.5 | L1 | 80 | MeOH | 2 | 83 | — | 50 | 33 |
| 2 | 1.5 | L2 | 80 | MeOH | 2 | 85 | — | 60 | 25 |
| 3 | 1.5 | L3 | 80 | MeOH | 2 | 77 | — | 27 | 50 |
| 4 | 3 | L1 | 80 | MeOH | 3 | 100 | — | 70 | 30 |
| 5 | 3 | L1 | 100 | EtOH | 3 | 100 | — | — | >99 |
| 6 | 3 | L1 | 100 | EtOH | 2 | 81 | — | 56 | 25 |
| 7 | 3 | L1 | 100 | EtOH | 1 | 61 | 18 | 43 | — |
| 8 | 3 | L1 | 100 | Toluene | 3 | 100 | — | — | 97 |
| 9 | 3 | L1 | 100 | Dioxane | 3 | 100 | — | — | >99 |
| 10 | 3 | L1 | 100 | THF | 3 | 100 | — | — | >99 |
| 11 | 3 | L1 | 100 | Neat | 3 | 100 | — | — | 98 |
| 12 | 3 | L1 | 100 | DMSO | 3 | 100 | — | 75 | 25 |

The 1a conversions were not complete (77 to 85%) because of the total consumption of allyl alcohol to produce the polysubstituted products 4a and 5a. The ratio of allyl alcohol 2a/2-oxindole 1a and the catalyst amount were then increased (3 equiv. of 2a and 3 mol% Ni) resulting in a total 2-oxindole conversion (entry 4). This also led to the formation of 4a as the major product of reaction (yield = 70%) in the mixture with the trisallylated derivative 5a (yield = 30%). An increase of reaction temperature to 100 °C allowed the selective formation of the trisallylated product 5a with 100% 2-oxindole conversion (entry 5). In this case, EtOH was chosen instead of MeOH due to its higher boiling point. In order to evaluate the possibility of synthesizing the monosubstituted product 3a, the quantity of allyl alcohol was then decreased (entries 6 and 7). The bisallylated derivative 4a was always the major product of reaction even with only 1 equiv. of allyl alcohol. In the latter case, 3a was obtained with a low yield of 18% and the conversion of the starting material was not complete, thus showing rapid bisallylation shortly after the first allylation of 2-oxindole (entry 7). For further investigation, various solvents were also evaluated. The reaction yielded more than 97% of the trisallylated product in most solvents such as toluene, dioxane and THF as well as under solvent-less conditions (entries 8–11). However, DMSO as a polar aprotic solvent proved to be the convenient choice for the allylation of indoles38 and allowed a more selective synthesis of the C,C-bisallylated 2-oxindole 4a which was obtained with 75% yield (entry 12).
In summary, the C,C,N-trisallylated 2-oxindole could be easily produced in different solvents with 3 equiv. of allyl alcohol and 3 mol% Ni catalyst at 100 °C. The use of DMSO clearly improves the selectivity of the reaction toward the C,C-bisallylated derivative. At this point, we decided to study the scope of the reaction for the synthesis of more challenging compounds. Various 2-oxindoles have then been scrutinized as substrates under the optimized conditions for the synthesis of C,C-bisallylated derivatives (Ni(cod)2/dppf/DMSO/100 °C) (Table 2). The C,C-bisallylated 4a–g and C,C,N-trisallylated 5a–g derivatives were separated and purified by silica gel column chromatography, thus allowing access to isolated yields for both families of compounds. 2-Oxindole 1a was transformed into the 4a and 5a derivatives and the products were isolated with yields of 60% and 20% respectively (entry 1). No clear tendency corresponding to electronic effects of the oxindole 5-substitution could be highlighted. 5-Fluorooxindole 1b led to the formation of both bis and trisallylated derivatives with respectively 67% and 29% yields (entry 2). The C,C-bisallylated product 4c was isolated as the sole product with a yield of 70% from the reaction with 5-bromooxindole 1c (entry 3) and the C,C-bisallylated product 4d was also selectively obtained from 5-methoxyoxindole 1d with a yield of 95% (entry 4). The bis and trisallylated products were respectively obtained with 63% and 32% yields from the reaction with 5-aminooxindole 1e. It is very noteworthy that no allylation occurred on the amino group of the aniline moiety, very likely because of the lower acidity of this group in comparison to the amide group of the 2-oxindole (entry 5). The introduction of substituents at positions 4 and 6 of 2-oxindole led to very similar results. For example, only 36% of the C,C-bisallylated product 4f was isolated from 4-bromooxindole derivative 1f along with 30% 5f (entry 6). 4g and 5g were obtained with respectively 73% and 17% yields from 6-acyloxindole 1g (entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | 2-Oxindole | Conv.b (%) | Isolated yield (%) | |
| 1a–g | 4a–g | 5a–g | ||
| a Reaction conditions: 1a–g (1.8 mmol), 2a (5.4 mmol), Ni(cod)2 (0.054 mmol), dppf (0.108 mmol), DMSO (0.5 mL), 17 h, T = 100 °C in a sealed Schlenk tube. b Conversions determined by GC using anisole as an internal standard. | ||||
| 1 |
 1a
1a
|
100 | 60 | 20 |
| 2 |
 1b
1b
|
100 | 67 | 29 |
| 3 |
 1c
1c
|
82 | 70 | — |
| 4 |
 1d
1d
|
100 | 95 | — |
| 5 |
 1e
1e
|
100 | 63 | 32 |
| 6 |
 1f
1f
|
95 | 36 | 30 |
| 7 |
 1g
1g
|
100 | 73 | 17 |

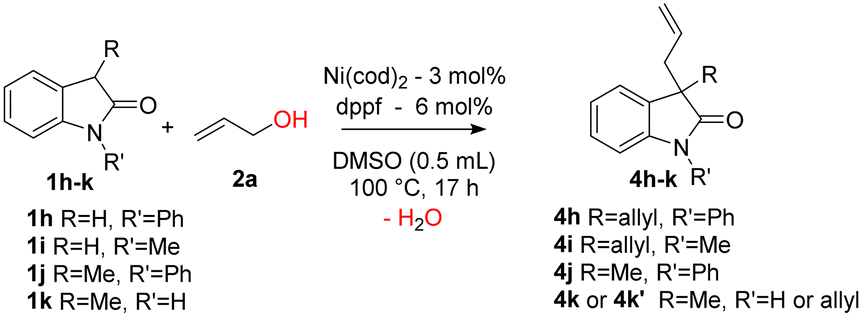
C- and/or N-substituted 2-oxindoles 1h–k were also reacted (Table 3). N-Phenyl-2-oxindole 1h was efficiently converted into the C,C-bisallylated derivative 4h with 82% isolated yield (entry 1). However, with an electron donating group on the nitrogen, the N-methyl-2-oxindole 1i was bisallylated with a moderate yield of 51% (entry 2). The 3-methyl-N-phenyl-2-oxindole 1j was readily allylated and the corresponding product 4j was isolated with a high yield of 98% (entry 3). Finally, a very good yield of 85% for derivative 4k was obtained from 3-methyl-2-oxindole 1k and only 10% of the C,C,N-trisallylated derivative 4k′ was observed (entry 4).
|
|
|||
|---|---|---|---|
| Entry | 2-Oxindole | Conv.b (%) | Isolated yield (%) |
| 1h–k | 4h–k | ||
| a Reaction conditions: 1h–k (1.8 mmol), 2a (5.4 mmol), Ni(cod)2 (0.054 mmol), dppf (0.108 mmol), DMSO (0.5 mL), 17 h, T = 100 °C in a sealed Schlenk tube. b Conversions determined by GC using anisole as an internal standard. | |||
| 1 |

|
85 | 82 |
| 2 |
 1i
1i
|
56 | 51 |
| 3 |

|
100 | 98 |
| 4 |
 1k
1k
|
100 | 4k: 85% |
| 4k′: 10% | |||
Allylation reaction of 2-coumaranones
The reactivities obtained with 2-oxindoles (cyclic amides) raised the question of the reactivity of the lactone equivalent, 2-coumaranone 1a′ (Table 4). Allylation could be performed on the C3 atom leading to C-mono and C,C-bisallylated 2-coumaranones. It should be noted that in the presence of a nickel catalyst, the allylation reaction of a linear ester such as methyl phenylacetate with allyl alcohol always led to transesterification reaction and no allylation on the alpha carbon of the ester could be observed.47 The stabilized cyclic structure of the lactone in 2-coumaranone avoids this transesterification reaction. Employing the same conditions as those used for 2-oxindole 1a allylation, 2-coumaranone 1a′ was completely and selectively transformed into the bisallylated product 3a′ which was isolated with a yield of 70%. Interestingly, no product of monoallylation was observed after 17 h. Similarly, an isolated yield of 80% for the bisallylated product 3b′ was also obtained from 5-hydroxy-2-coumaranone 1b′ with 100% conversion and a total selectivity towards 3b′. Such as in the case of the reaction with 5-aminooxindole 1e (entry 5, Table 2) that didn't show any allylation of the amino group, no allyl aryl ether is formed from 5-hydroxy-2-coumaranone 1b′. One can expect that such a type of allylation of the phenol would be reversible while the C-allylation is more likely irreversible. Dihydrocoumarin 1c′ with a six-membered cycle was also evaluated. Various approaches have already been proposed, essentially with palladium-based catalysts.48–52 We performed the nickel catalyzed allylation under the same operational conditions, but no product was observed. This lack of reactivity is attributed to the lower acidity of the proton on the carbon atom on the alpha position with respect to the carbonyl group. This shows the important stabilizing role of the aryl ring in the case of the 2-coumaranone.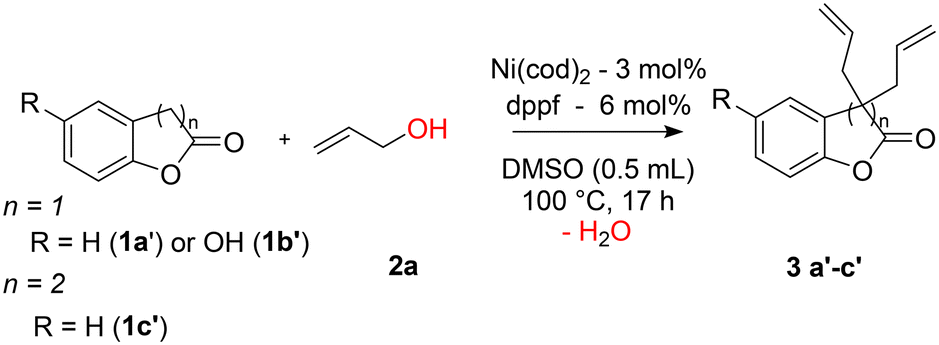

Flow/batch synthesis: kinetic profile of the allylation reaction
 | ||
| Scheme 2 Evolution curve of allylation reaction of 2-oxindole 1a and 2-coumaranone 1a′ with allyl alcohol 2a in batch (solvent THF, 0.5 mL in a sealed Schlenk tube) (top) and in flow (100 μL min−1) (bottom). Reaction conditions: 1a or 1a′ (1.8 mmol), 2a (5.4 mmol), Ni(cod)2 (0.054 mmol), dppf (0.108 mmol), 80 °C. | ||
 | ||
| Fig. 1 Evolution curves of allylation reaction of 2-oxindole 1a with allyl alcohol 2a in batch mode. | ||
 | ||
| Fig. 2 Evolution curves of allylation reaction of 2-coumaranone 1a′ with allyl alcohol 2a in batch mode. | ||
We first evaluated the 2-oxindole allylation under flow. After preparative experiments, we set up a reactor with FEP tubing (length = 1 meter; tubing i.d. = 800 μm; heated volume = 0.5 mL). The tube was immersed in a water bath at 80 °C. Substrates (2-oxindole and allyl alcohol) were introduced thanks to a unique syringe. A quenching module at the tube exit was not necessary since the reaction is immediately stopped in the presence of oxygen. A back pressure regulator (20 psi) should be installed because the reaction temperature was higher than the boiling point of the solvent (Scheme 3).
 | ||
| Scheme 3 Fluidic reactor setup. | ||
The experimental catalytic conditions were identical to those of the batch procedures (Ni(cod)2/dppf/THF/80 °C). Thanks to the faster heat transfer, the catalytic transformation was greatly accelerated in the flow reactor with a very high chemoselectivity. Indeed, within 3 minutes, the bisallylated 2-oxindole product 4a was obtained with 100% conversion (Fig. 3). Increasing the residence time to 25 minutes in the reactor tube by using a lower flow rate allowed converting 4a to trisallylated product 5a (Fig. 3). As in the batch (Fig. 1), the monoallylated product was not observed, which shows that the second allylation step is considerably much faster than the first one. This can be explained by an increased inductive effect between the two steps. In that way, by tuning the residence time of the substrates in the reactor, either C,C-bisallylated product 4a or C,C,N-trisallylated 2-oxindole 5a could be obtained selectively in a very short time.
 | ||
| Fig. 3 Evolution curves of allylation reaction of 2-oxindole 1a with allyl alcohol 2a in flow. | ||
Using the same conditions, a similar experiment was carried out with 2-coumaranone. The solution needed to be first passed into an ultrasound bath to dissolve reagents before introduction into the flow reactor and to avoid any clogging of the system. Surprisingly, at a flow rate of 100 μL min−1, we observed that more than 80% of the C,C-bisallylated derivative 4a′ was produced after 1 minute and 95% of 2-coumaranone had been transformed to the C,C-bisallylated 2-coumaranone 4a′ after 5 minutes. In view of this noticeable result, we thought to perform the allylation reactions in batch at ambient temperature to compare. Indeed, only 2-coumaranone could be completely transformed into the diallylated product in 17 h while the other 2-oxindole substrates are unreactive under these conditions. This shows that these catalytic transformations are very sensitive to heat transfer conditions and the high level of control offered by flow chemistry enables greater selectivity to a chosen synthetic product just by varying the flow rate (Fig. 4).
 | ||
| Fig. 4 Evolution curves of allylation reaction of 2-coumaranone 1a′ with allyl alcohol 2a in flow. | ||
Experimental
General procedure for the allylation reaction under batch conditions
Ni(cod)2 (15 mg, 0.054 mmol, 3 mol%) and dppf (60 mg, 0.108 mmol, 6 mol%) were weighed in a sealed Schlenk tube under nitrogen. 2-Oxindole or 2-coumaranone (1.8 mmol) and allyl alcohol (5.4 mmol, 0.313 g, 3 equiv.) in DMSO (0.5 mL) were added. The mixture was heated for 17 h at 100 °C. After reaction, the solvent was distilled under pressure and the products were purified by silica gel column chromatography.All experimental details and product characterization data can be found in the ESI.†
Conclusion
In summary, we have developed an efficient and clean catalytic system for the allylation of 2-oxindoles and 2-coumaranones. The method is in great accordance with the principles of green chemistry allowing the synthesis of allylated 2-oxindoles and 2-coumaranones in the presence of nickel as a low cost catalyst and without any additive. The products are obtained with good to excellent yields in most cases under mild conditions. Chemoselectivities and reactions rates have been highly improved using flow chemistry conditions. Thus, this technology enables the continuous production of either C,C-bisallylated or C,C,N-trisallylated 2-oxindoles in only a few minutes. Beyond the selectivity control that can be reached thanks to the use of flow chemistry, the rising cost of energy should favor further the use of these flow processes and one can expect them to become widespread in the future. Catalytic salt free reactions with fast kinetics are particularly well adapted to this technology. In our further studies, this strategy will be developed with other substrates and catalytic reactions.Author contributions
Synthesis and catalysis, B. M. and A. S. P.; investigation, A. K., M. P., C. D. and M. S.; writing – original draft preparation, B. M. and I. S.; writing – review and editing, I. S. and M. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Ministère de l'Enseignement Supérieur de la Recherche et de l'Innovation (France) and the Ministère de la Recherche (Morocco) for financial support. We would like to thank Céline Delabre for the HR-MS analyses.References
- L. Vaccaro, D. Lanari, A. Marrocchi and G. Strappaveccia, Green Chem., 2014, 16, 3680–3704 RSC.
- D. L. Hughes, Org. Process Res. Dev., 2018, 22, 13–20 CrossRef CAS.
- S. A. May, J. Flow Chem., 2017, 7, 137–145 CrossRef CAS.
- C. A. Hone and C. O. Kappe, Chem.: Methods, 2021, 1, 454–467 CAS.
- L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- P. Plouffe, A. Macchi and D. M. Roberge, Org. Process Res. Dev., 2014, 18, 1286–1294 CrossRef CAS.
- Z. Y. Cao, Y. H. Wang, Y. P. Zeng and J. Zhou, Tetrahedron Lett., 2014, 55, 2571–2584 CrossRef CAS.
- K. Shen, X. Liu and X. Feng, Chem. Sci., 2012, 3, 327–334 RSC.
- G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed.
- R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247–7290 RSC.
- F. Zhou, Y. L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381–1407 CrossRef CAS.
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed.
- A. D. Marchese, E. M. Larin, B. Mirabi and M. Lautens, Acc. Chem. Res., 2020, 53, 1605–1619 CrossRef CAS PubMed.
- Z.-Y. Cao, F. Zhou and J. Zhou, Acc. Chem. Res., 2018, 51, 1443–1454 CrossRef CAS PubMed.
- Y.-L. Liu, X.-P. Wang, J. Wei and Y. Li, Org. Biomol. Chem., 2022, 20, 538–552 RSC.
- B. Volk, J. Barkóczy, E. Hegedus, S. Udvari, I. Gacsályi, T. Mezei, K. Pallagi, H. Kompagne, G. Lévay, A. Egyed, L. G. Hársing, M. Spedding and G. Simig, J. Med. Chem., 2008, 51, 2522–2532 CrossRef CAS PubMed.
- A. Fensome, R. Bender, J. Cohen, M. A. Collins, V. A. Mackner, L. L. Miller, J. W. Ullrich, R. Winneker, J. Wrobel, P. Zhang, Z. Zhang and Y. Zhu, Bioorg. Med. Chem. Lett., 2002, 12, 3487–3490 CrossRef CAS PubMed.
- O. Pàmies, J. Margalef, S. Cañellas, J. James, E. Judge, P. J. Guiry, C. Moberg, J.-E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Chem. Rev., 2021, 121, 4373–4505 CrossRef PubMed.
- B. M. Trost, Tetrahedron, 2015, 71, 5708–5733 CrossRef CAS PubMed.
- D. Li, S. Zhang, B. Wang, W. Sun, J. Zhao, J. Qu and Y. Zhou, Org. Chem. Front., 2022, 9, 810–815 RSC.
- C. Zhang, Y.-C. Wu, B.-D. Cui, H. Li, W.-Y. Han, N.-W. Wan and Y.-Z. Chen, Org. Biomol. Chem., 2021, 19, 4720–4725 RSC.
- K. Balaraman and C. Wolf, Angew. Chem., Int. Ed., 2017, 56, 1390–1395 CrossRef CAS PubMed.
- H. Yang, H. Zhou, H. Yin, C. Xia and G. Jiang, Synlett, 2014, 25, 2149–2254 CrossRef CAS.
- B. M. Trost and M. U. Frederiksen, Angew. Chem., Int. Ed., 2005, 44, 308–310 CrossRef CAS PubMed.
- B. M. Trost and Y. Zhang, Chem. – Eur. J., 2010, 16, 296–303 CrossRef CAS PubMed.
- T. Wang, Y. Peng, G. Li, Y. Luo, Y. Ye, X. Huo and W. Zhang, Chem. – Eur. J., 2021, 27, 10255–10260 CrossRef CAS PubMed.
- D. Nečas, M. Turský, I. Tislerová and M. Kotora, New J. Chem., 2006, 30, 671–674 RSC.
- T.-Y. Zhao, K. Li, L.-L. Yang, S.-F. Zhu and Q.-L. Zhou, Org. Lett., 2021, 23(10), 3814–3817 CrossRef CAS PubMed.
- For a review on nickel in allylation reaction: D. Ghorai, À. Cristòfol and A. W. Kleij, Eur. J. Inorg. Chem., 2022, e202100820 CAS.
- For nickel catalyzed allylation reactions with allyl alcohol: M. S. Azizi, Y. Edder, A. Karim and M. Sauthier, Eur. J. Org. Chem., 2016, 22, 3796–3803 CrossRef.
- R. Blieck, M. S. Azizi, A. Mifleur, M. Roger, C. Persyn, M. Sauthier and H. Bonin, Eur. J. Org. Chem., 2016, 6, 1194–1198 CrossRef.
- H. Bricout, J. F. Carpentier and A. Mortreux, J. Mol. Catal. A: Chem., 1998, 136, 243–251 CrossRef CAS.
- Y. Bernhard, B. Thomson, V. Ferey and M. Sauthier, Angew. Chem., Int. Ed., 2017, 56, 7460–7464 CrossRef CAS PubMed.
- B. Mouhsine, A. Karim, C. Dumont and M. Sauthier, Green Chem., 2020, 22, 950–955 RSC.
- B. Mouhsine, A. Karim, C. Dumont, I. Suisse and M. Sauthier, Adv. Synth. Catal., 2021, 363, 1457–1462 CrossRef CAS.
- B. Mouhsine, A. Karim, C. Dumont, A. Saint Pol, I. Suisse and M. Sauthier, Eur. J. Org. Chem., 2022, e202200042 CAS.
- Y.-X. Li, Q.-Q. Xuan, L. Liu, D. Wang, Y.-J. Chen and C.-J. Li, J. Am. Chem. Soc., 2013, 135, 12536–12539 CrossRef CAS PubMed.
- G. I. Panov, E. V. Starokon, M. V. Parfenov, B. Wei, V. I. Sobolev and L. V. Pirutko, ACS Catal., 2018, 8, 1173–1177 CrossRef CAS.
- E. Arceo, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2010, 132, 11408–11409 CrossRef CAS PubMed.
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef CAS PubMed.
- I. Ahmad, G. Chapman and K. M. Nicholas, Organometallics, 2011, 30, 2810–2818 CrossRef CAS.
- S. Raju, M. E. Moret and R. J. M. Klein Gebbink, ACS Catal., 2015, 5, 281–300 CrossRef CAS.
- M. Roseau, N. Dhaouadi, C. Rolando, L. Chausset-Boissarie and M. Penhoat, J. Flow Chem., 2020, 10, 347–352 CrossRef CAS.
- C. Penverne, B. Hazard, C. Rolando and M. Penhoat, Org. Process Res. Dev., 2017, 21, 1864–1868 CrossRef CAS.
- Unpublished results.
- M. Murakata, T. Jono and O. Hoshino, Tetrahedron: Asymmetry, 1998, 9, 2087–2092 CrossRef CAS.
- M. Murakata, T. Jono, Y. Mizuno and O. Hoshino, J. Am. Chem. Soc., 1997, 119, 11713–11714 CrossRef CAS.
- M. Murakata, T. Jono, T. Shoji, A. Moriya and Y. Shirai, Tetrahedron: Asymmetry, 2008, 19, 2479–2483 CrossRef CAS.
- R. Akula and P. J. Guiry, J. Org. Chem., 2016, 18, 5472–5475 CAS.
- K. Chattopadhyay, R. Jana, V. W. Day, J. T. Douglas and J. A. Tunge, Org. Lett., 2010, 12, 3042–3045 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00192j |
| This journal is © The Royal Society of Chemistry 2023 |