
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel process towards the industrial realization of large-scale oxymethylene dimethyl ether production – COMET†
Franz
Mantei
 a,
Christian
Schwarz
b,
Ali
Elwalily
a,
Florian
Fuchs
a,
Andrew
Pounder
a,
Hendrik
Stein
b,
Matthias
Kraume
c and
Ouda
Salem
*a
a,
Christian
Schwarz
b,
Ali
Elwalily
a,
Florian
Fuchs
a,
Andrew
Pounder
a,
Hendrik
Stein
b,
Matthias
Kraume
c and
Ouda
Salem
*a
aSustainable Synthesis Products, Division Hydrogen Technologies, Fraunhofer Institute for Solar Energy Systems, Heidenhofstr. 2, 79110, Freiburg, Germany. E-mail: ouda.salem@ise.fraunhofer.de
bASG Analytik-Service AG, Trentiner Ring 30, 86356 Neusäss, Germany
cChair of Chemical Engineering, Technische Universität Berlin, Str. des 17. Juni 135, MAR 2-1, 10623 Berlin, Germany
First published on 31st July 2023
Abstract
Oxymethylene dimethyl ethers (OME) show promising solubility and combustion properties for applications in various chemical processes and sectors. OME enable clean and quasi soot-free combustion, which can consequently lead to considerable NOx emissions reduction. Besides reducing local emissions, OME can significantly reduce the global CO2 emissions by substituting fossil diesel fuel if their production is based on sustainable methanol. Various process concepts for the OME production were proposed and investigated, but most of them have significant bottlenecks, which prevent their demonstration and scale-up in the near future. Only the production based on OME1 and trioxane can already be demonstrated and scaled up, which, however, is complex and energy-intensive, considering a sustainable production based on H2 and CO2. Therefore, the novel COMET (clean OME technology) process concept is introduced and experimentally demonstrated utilizing only state-of-the-art process units. The COMET process relies solely on methanol and formalin as feedstock and overcomes the challenging water management aspect in the OME value chain, using a reactive distillation column. The COMET process is evaluated at a scale of 100 kilotons per annum OME3–5 product for the system boundary starting from H2O electrolysis and CO2 capture. Key performance indicators are defined and compared with alternative processes from the literature. The COMET process shows a high carbon efficiency of 88% and overall energy efficiency of 54% in comparison to the alternative OME3–5 production processes introduced in the literature. Moreover, the COMET process offers the forthwith large-scale production of OME in a relatively simple process chain and high technology readiness level.
1. Introduction
The fluctuating availability of renewable energies can be compensated by various measures, such as storage technologies like batteries and heat reservoirs. However, the need for long-term storage and long-distance transportation are met more easily and flexibly using power-to-X (PtX) technologies and products.1 Green H2 will play a key role in meeting these needs with already rising demands, as is suggested by various roadmaps, strategies, and international agreements by various governments and companies.2,3Currently, fossil energy carriers are essential to provide heat and power, and are important carbon sources for the chemical, petrochemical and plastic industries. However, at the end of the life cycle, they are usually combusted to CO2 which ends up in the atmosphere. Therefore, sustainable and circular carbon sources are required such as biogenic waste streams and CO2 capture from air. One example of a circular carbon source is direct air capture (DAC) of CO2. Due to its locally independent availability, DAC is experiencing a growing governmental interest with increasing numbers and capacities of technological demonstrations.4
A combination of captured CO2 and green H2 enables the implementation of these sustainable solutions into various sectors and hard to defossilize processes. One suitable product of CO2 and H2 are oxymethylene dimethyl ethers (OME). OME show promising fuel and physical properties for a wide range of potential applications such as solvents or diesel fuel additives or substitutes. OME3–5 have similar fuel properties to diesel fuel, a good solubility in diesel fuel and advantageous combustion behavior.5,6 This makes OME attractive as a sustainable drop-in blending component for diesel fuel. Considering the high cetane number, OME can be an interesting dual fuel or ignition promoter in marine engines. Due to the absence of C–C bonds and the high amount of molecular bound oxygen, OME combust with distinct lower levels of emissions of particle matter than diesel fuel. Therefore, the NOx and soot emission trade-off of diesel fuel can be avoided.7,8
The basis for large-scale sustainable production of OME is MeOH, which can be produced by reacting captured CO2 with H2 from renewable-powered H2O electrolysis. Following this power-to-liquid (PtL) concept, the well-to-wheel (WtW) CO2 emissions can be reduced by up to 93% compared to fossil fuels.9,10 Furthermore, Voelker et al.10 estimated an NOx reduction of 57% and an almost complete reduction of soot using OME instead of diesel fuel. Moreover, small blending rates of OME in diesel fuel already show a clearly positive impact on global CO2 emissions, as well as local NOx and soot emissions.10,11 With a worldwide demand of 26.5 million barrels diesel fuel per day,12 small blending rates of OME showcase the need for large-scale production plants. Suitable compositions of the final OME3–5 product are defined by key properties, such as density, viscosity, cetane number and flash point, which are standardized in a new fuel pre-standard for OME DIN/TS 51699.7,13
Various process concepts have been proposed to produce OME from MeOH which will be discussed and compared in detail in the following sections. One of the main challenges of these processes, in terms of technical feasibility and energy demand, is the separation of the by-product H2O from the target OME fraction. This H2O is formed in various synthesis steps from MeOH to OME. The OME production processes discussed in the literature use different techniques to separate H2O.14–25 These techniques have different advantages and disadvantages, partly coupled with methods which are still in an early phase of investigation and demonstrations. In the present work, a new process concept, clean OME technology (COMET), is proposed which solves the challenging H2O separation utilizing the state-of-the-art reactive distillation technique. This makes the COMET process a technically feasible process for large OME production capacities.
1.1. Objective of this work
The main objective of this work is the introduction of the novel COMET process concept for the production of OME from MeOH and aqueous formaldehyde (FA(aq.)) solutions, which solves the challenging H2O management problem, using a reactive distillation column. Complementary, the experimental demonstration of the main COMET process units is introduced. The work further compares the COMET process concept major technical metrics to literature discussed OME production processes.2. Theory and background
2.1. Synthesis of OME
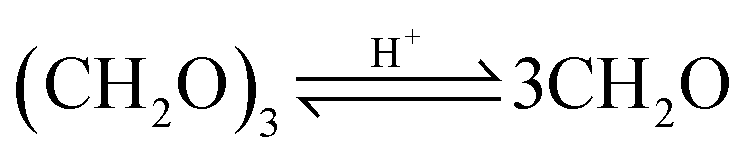
OME are synthesized in an acidic environment from two methyl capping groups and n oxymethylene groups –CH2O–. Suppliers for the methyl capping groups are methanol (H3C–OH, MeOH), methylal (H3C–O–(CH2O)1–CH3, OME1) or dimethyl ether (H3C–O–CH3, DME). For the oxymethylene groups, formalin (FA(aq.)), paraformaldehyde (HO–(CH2O)n–H with n = 8–100, pFA) or trioxane ((CH2O)3, TRI) can be used.In a solution of MeOH, FA, and H2O, poly(oxymethylene) hemiformals (HO–(CH2O)n–CH3 with n = 1–10, HFn) and poly(oxymethylene) glycols (HO–(CH2O)n–H with n = 1–10, MGn) bound most of the FA, as described by eqn (1)–(4). These are fast reactions even in absence of a catalyst. The amount of monomeric FA in solutions with MeOH and H2O is very small in chemical equilibrium.26
| CH2O + CH3OH ⇌ HO(CH2O)1CH3 | (1) |
| CH2O + HO(CH2O)n−1CH3 ⇌ HO(CH2O)nCH3; n = 2–10 | (2) |
| CH2O + H2O ⇌ HO(CH2O)1H | (3) |
| CH2O + HO(CH2O)n−1H ⇌ HO(CH2O)nH; n = 2–10 | (4) |
 | (5) |
 | (6) |
 | (7) |
 | (8) |
 | (9) |
Using DME as a feedstock for the OME synthesis, OME1 is formed by the incorporation of FA into DME, as described by eqn (10).
 | (10) |
 | (11) |
In addition to the main reaction network, side reactions lead to the formation of side products. Besides HFn, MGn, TRI and DME, the formation of tetroxane ((CH2O)4), methyl formate (HCOOCH3, MEFO) and formic acid (HCOOH, FOAC) was observed, which strongly depends on the catalyst systems and increases with increasing temperatures.32–34
2.2. OME3–5 production processes
Different feedstock combinations can be used to produce OME3–5 due to various potential suppliers of methyl groups and oxymethylene groups. Depending on the formation of H2O as a side product during the synthesis of OMEn, the reaction systems are classified as anhydrous or aqueous. Methyl group suppliers for anhydrous reaction systems are mainly DME and OME1, because their conversion to OMEn only requires a chain propagation with oxymethylene groups, as described by eqn (7) and presented in Table 1. Oxymethylene group suppliers for anhydrous reaction systems are mainly TRI and monomeric FA, which do not contain H2O or MeOH. Formalin, concentrated FA(aq.) or pFA contain H2O and are used as oxymethylene group suppliers for aqueous reaction systems.| Methyl group supplier | Oxymethylene group supplier | |
|---|---|---|
| Anhydrous | DME, OME1 | TRI, monomeric FA |
| Aqueous | MeOH, DME, OME1 | FA(aq.), pFA, TRI, monomeric FA |
Feedstocks containing MeOH generally lead to the formation of H2O as a side product in the aqueous OME synthesis, as described by eqn (5) and (6). This H2O needs to be separated and extracted from the process loop to prevent accumulation. Fig. 1 shows a simplified scheme for the production of OME3–5 from various feedstocks. It consists mainly of a reactor for the OME synthesis R, two distillation columns CO-1 and CO-2 for product purification and a H2O separation unit S for aqueous reaction systems. For the H2O separation, various methods were proposed in the literature, such as extraction, adsorption or membrane, as discussed in the following section.
 | ||
| Fig. 1 OME3–5 production process for various feedstocks, following aqueous and anhydrous reaction systems. CO, distillation column; R, reactor; S, H2O separator. | ||
Various process concepts for the OME3–5 production were proposed in patents and other publications and some of them are realized in large-scale production plants in China. However, details regarding their performance, the quality and composition of the final OME product and the long-term operation are scarce.35,36Table 2 lists the main OME3–5 production processes discussed in the literature, emphasizing the feedstock, main advantages, and hurdles. A detailed description is provided in the ESI.† A comparison with the COMET process based on their performances in terms of OME3–5 yield, energy demand and technical feasibility is discussed in the results and discussion section. Further process concepts were proposed in the literature which show significant disadvantages in comparison to the process concepts presented in Table 2, as discussed in the ESI.†
| Feedstock | Anhydrous synthesis | Aqueous synthesis | ||||
|---|---|---|---|---|---|---|
| OME1 and TRI14–16 | DME and TRI17,18 | OME1 and monomeric FA19 | MeOH and FA(aq.)20,21 | MeOH and monomeric FA19 | OME1 and FA(aq.) or pFA19 | |
| (+) Main advantages and (-) main hurdles | + High OME3–5 yield after the synthesis | + DME is cheaper than OME1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43 43 |
+ High OME3–5 yield after the synthesis | + Comparatively cheap feedstock | + Comparatively cheap feedstock | + Fairly high OME3–5 yield after the synthesis |
| + Simple product purification | - Complex and energy-intensive preparation of TRI28,39,40,42 | + Potentially simpler and cheaper production of monomeric FA | - Formation of H2O as a side product | - Similar hurdles to MeOH and FA(aq.) | ||
| - Complex and energy-intensive preparation of TRI28,39,40,42 | - High MEFO selectivity29–31,44,45 | - Very low TRL of the monomeric FA production | - Low OME3–5 yield after the synthesis | - Very low TRL of the monomeric FA production | - Similar hurdles to MeOH and FA(aq.) | |
| - Low TRL of the H2O separation methods | ||||||
2.3. H2O separation from the production of OME
One of the main challenging and energy-intensive process steps is the separation of the by-product H2O from the production chain towards OME3–5. Considering a sustainable production of OME3–5 based on MeOH produced from H2 and CO2, Fig. 2 shows how the synthesis of MeOH, intermediate products such as FA(aq.), DME and OME1, as well as the synthesis of OMEn all result in the formation of H2O. | ||
| Fig. 2 H2O separation from a sustainable production of OME3–5 based on H2 and CO2. | ||
In the MeOH synthesis from H2 and CO2, the OME1 synthesis and the DME synthesis, H2O is a by-product and separated using distillation columns.19,37,38 In the partial oxidation of MeOH towards FA, as described by eqn (15), H2O is formed as a by-product and used as a washing liquid in the absorber column. Downstream, H2O is partly separated from FA(aq.) in a concentration step using evaporation techniques.19,39 Therefore, H2O is introduced into the TRI synthesis and separated in an energy-intensive cascade of distillation columns.28,40 Only in the anhydrous FA synthesis, which is still in its very early stages, no H2O is present.41 Regarding the synthesis of OME≥2, H2O is not formed as a by-product when the oxymethylene group suppliers TRI or monomeric FA are combined with the methyl group suppliers DME or OME1, as described by eqn (7) and (8).
This simplifies the final product purification. When starting from the cheaper and established reactant FA(aq.), H2O will always be present in the OME3–5 sub-process and needs to be separated from the loop to circumvent accumulation. However, H2O cannot be separated individually simply via distillation due to a complex phase behavior of the synthesis product mixture containing mainly FA, H2O, MeOH, OME1–10, HF and MG with several azeotropes with similar boiling points. The separation of H2O from the loop is still a major challenge regarding the implementation of a potentially cheaper and scalable aqueous OME3–5 production process.
Results of Li et al.47 show the separation of the OME synthesis product using toluene. About 70% of OME are separated in the organic phase and only 14% of FA and H2O migrate in the organic phase, as indicated by the split fraction. However, the organic phase mainly consists of toluene, which needs to be separated to be recycled. Furthermore, FA and H2O still represent a large proportion of the organic phase and the aqueous phase still contains a large proportion of OME. A graphical illustration of the results of Li et al.47 is presented in the ESI† in section 2.
An extraction method for the preparation of blends of OME in diesel fuel was proposed by Oestreich et al.53 and discussed in the ESI.†
Regarding the separation of H2O from an OME3–5 production process, the adsorption has the advantage of selectively separating H2O from the loop, which enables the recycle of all other components to the OME synthesis. Due to the reaction network between H2O and FA as described by eqn (3) and (4) not only the monomeric H2O is separated, but also H2O from MGn. Therefore, a significant reduction of the overall H2O content can be achieved. However, without H2O, FA from MGn remains in the mixture and either bounds with HFn−1 to long chain HFn, or with MGn−1 to long chain MGn or it remains in monomeric form. Either way, it increases the risk of local precipitations and, therefore, deactivation of the adsorbents. Therefore, a regeneration might be necessary. To reduce the risk of precipitation the temperature can be lifted, or the remaining H2O content can be increased. The latter would, however, decrease the yield of OME3–5 in the OME synthesis and, therefore, increase the recycle streams and heat demand for separation. A suitable remaining H2O content should be experimentally investigated and confirmed by long-term stability tests with alternating sequences of adsorption and regeneration. Furthermore, the scale-up potential should be investigated to ensure its feasibility for large-scale production plants.
Regarding the heat demand for the separation of H2O via adsorption, Schemme et al.43,55 estimated that 2.1 kW h kg−1 H2O are required. Their estimations are based on the results from Schmitz et al.20,56 and assume that the adsorbents are heated up from 25 °C to 235 °C for the regeneration using high pressure steam. Furthermore, it was assumed that the heat demand is mainly based on the heat of adsorption and the heat capacity of the adsorbents.
However, Ferre et al.58 reported the application of a different membrane from DBI Gas und Umwelttechnik. The long-term stability of the membrane, selectivities in the reaction mixture and the scale-up potential should be further investigated to ensure its feasibility for large-scale production plants.
The advantages of the membrane for the separation of H2O are similar to the advantages of the adsorption with a high selectivity for H2O. However, likewise to the adsorption, this results in a higher risk for local precipitation. Therefore, a compromise might be necessary between the long-term stability and the H2O concentration of the retentate. A disadvantage of a higher H2O concentration in the retentate is an increase of the recycle streams which results in higher heat demands for the product purification and reduces the overall energy efficiency of the process.
Regarding the heat demand for the separation of H2O via membranes, Held et al.39 estimated that 0.7 kW h kg−1 H2O are required. This results from the evaporation of H2O after passing through the membrane to the reduced pressure of the permeate of 0.03 bar. Held et al.39 assumed that no external heat is required but the temperature of the process stream is reduced from 84 °C to 36 °C. In comparison to the separation of H2O via adsorption, the heat demand is significantly lower.
Table 3 summarizes the main advantages and main hurdles of the H2O separation methods extraction, adsorption, and membrane.
| Method | Extraction | Adsorption | Membrane |
|---|---|---|---|
| H2O selectivity | Low | High | High |
| Energy demand | High | High | Comparatively low |
| Long-term operation | Likely | Challenging | Challenging |
| Scale-up potential | Likely | Challenging | Challenging |
3. COMET process description
The COMET process59 is based on the commercially available MeOH and FA(aq.) feedstock and produces mainly high purity OME3–5, as illustrated in Fig. 3. For the separation of H2O from the loop, a reactive distillation column is used. | ||
| Fig. 3 COMET process concept for the production of OME3–5 from MeOH and FA(aq.) feedstocks. The light grey arrows and process units were added in this work to the FA concentration sub-process to improve the recycle of FA. CO, distillation column; E, evaporator; R, reactor. | ||
The COMET process starts at the concentration of 50–55 wt% FA(aq.) (stream 1), which can be the product stream of a state-of-the-art FA production process.19,60 The stream is mixed with the distillate of the second evaporator E-2 and the bottom of the third evaporator E-3. The FA(aq.) is then concentrated in a cascade of two evaporator stages E-1 and E-2 to provide a concentrated FA(aq.) of 85–88 wt% FA (stream 5) and an aqueous stream containing 10–25 wt% FA (stream 3). The concentrated FA(aq.) (stream 5) is used for the production of OME and mixed with the recycle streams, containing the azeotropic mixture of OME1 and MeOH (stream 10) and OME≥6 (stream 14). The mixture is converted in a fixed bed reactor R filled with an acidic heterogeneous catalyst. In contrast to the OME production process based on MeOH and FA(aq.),20 the reactor inlet stream contains mainly OME1 as a methyl capping source. This improves the selectivity towards OME3–5. The comparatively high selectivity further increases with decreasing H2O and MeOH concentrations in the concentrated FA feedstock (stream 5) and OME1 recycle (stream 10). The synthesis product mixture, mainly containing FA, H2O, MeOH and OME1–10 (stream 7), is separated in a cascade of three distillation columns. In the first distillation column CO-1, OME≥3 are separated from the more volatile components FA, H2O, MeOH and OME1–2. Importantly, OME3 cannot be completely separated to the bottom product, a small fraction remains in the distillate. In the third distillation column CO-3, OME≥6 are separated and recycled to the reactor and a final product mixture (stream 13) of OME3–5 is withdrawn at the top of CO-3. The distillate product of CO-1 is mixed with MeOH (stream 9) and sent to a reactive distillation column CO-2, to separate an azeotropic mixture of OME1 and MeOH (stream 10) from FA and H2O (stream 11). On the catalytic trays, two main conversions take place. First, OME2 and OME3 are converted to OME1 and FA over an acidic heterogeneous catalyst, as described by eqn (7). In addition, MeOH and FA are converted to OME1 and H2O, following the acetalization reaction as described by eqn (1) and (5). The mechanism on the catalytic trays is illustrated in Fig. 4. Due to the evaporation and, therefore, the separation of the volatile product OME1 from the liquid reaction mixture, the equilibrium of eqn (5) and (7) shifts, and the reactions proceed towards the production of OME1. Therefore, with sufficient retention time, OME2 is converted to a large extent to OME1, while the conversion of FA towards OME1 is limited by the amount of MeOH. The mixture is separated into the azeotropic mixture of OME1 and MeOH in the distillate (stream 10) and a mixture of FA and H2O in the bottom (stream 11). The distillate product of CO-2 is recycled back to the reactor and the bottom product is recycled to the evaporator E-2 for the FA concentration to separate H2O from the process and recycle concentrated FA back towards the OME reactor. Therefore, the reactive distillation column prevents the accumulation of H2O inside the loop and solves the challenging H2O management. In contrast to the H2O separation from the loop using adsorption or membranes, in the COMET process H2O is not separated selectively but together with the remaining FA. This significantly reduces the risk of precipitation, since enough H2O is left to convert the remaining FA to comparatively short-chain MGn which stay liquid at elevated temperature for sufficient retention time to downstream processing steps.
 | ||
| Fig. 4 H2O separation from the COMET process via reactive distillation. The left side shows the reactive distillation column with the main components of the feed and product streams. The illustration on the right side shows the interaction on a catalytic tray and was adopted from Schmitz et al.56 | ||
A similar concept for a reactive distillation column is applied in the OME1 production process by Drunsel et al.61,62 with the purpose to achieve a complete conversion of FA after the OME1 reactor.
The amount of MeOH (stream 9) added to the feed of the reactive distillation column CO-2 defines the conversion of FA and oxymethylene groups with MeOH on the catalytic trays towards OME1, following eqn (1), (5) and (7). Therefore, a variation of the amount of MeOH (stream 9) varies the amount of OME1 produced as the distillate product of the reactive distillation column CO-2. For the OME synthesis in the fixed bed reactor R a constant ratio of OME1 to concentrated FA(aq.) is required before the reactor. Therefore, the amount of MeOH (stream 9) can be defined to exactly produce the amount of OME1 required for the OME synthesis. Or the amount of MeOH (stream 9) can be increased to produce more OME1 than required by the OME synthesis and the excess OME1 can be extracted as a by-product. Another advantage of the COMET process is that the process offers a tunable product portfolio of OME. In the present work the amount of MeOH added to the reactive distillation column CO-2 was limited to only produce the required amount of distillate product (stream 10) for the OME synthesis and, therefore, achieve higher OME3–5 selectivity. Considering the production of OME1 as a target side product of the COMET process, another distillation column can be added to achieve high purities of the OME1 side product, similar to the second distillation column of the production process for OME1.61
Besides the OME3–5 product (stream 13), wastewater (stream 3) is produced with FA concentrations of about 10–25 wt%. This by-product stream is not limited to the COMET process but part of all OME3–5 production processes using FA(aq.) as an intermediate product. Instead of its disposal and to increase the carbon yield of the process, several strategies are possible to handle this stream. In the present work, the stream was partly send to the absorber column of the FA(aq.) production and concentrated in an additional distillation column CO-4. The concentrated FA stream was further concentrated in another evaporator E-3 to recycle the concentrated FA stream and to separate the stream with a low FA concentration (stream 2). This stream is also the purge stream for traces of MeOH and other volatile components to avoid accumulation in the loop. Instead of its disposal, this stream can be used to dilute an FA(aq.) product stream to prepare a stable formalin product.
3.1. Expanding the system boundary starting from H2 and CO2 feedstocks including the intermediate production of FA and MeOH
To enable a consistent basis of comparison with alternative OME3–5 production processes, the system boundary is extended to account for a sustainable OME3–5 production based on green H2 and captured CO2. The intermediate production of MeOH and FA(aq.) is described in detail by Mantei et al.19 A simplified process flow diagram of the extended COMET process concept starting from H2 and CO2 is illustrated in Fig. 5. The MeOH production starts from the compression of the feedstock H2 and CO2, the mixing with the recycle stream, and the synthesis of MeOH, as described by eqn (12)–(14).| CO2 + 3H2 ⇌ CH3OH + H2O | (12) |
| CO2 + H2 ⇌ CO + H2O | (13) |
| CO + 2H2 ⇌ CH3OH | (14) |
 | (15) |
| CH3OH ⇌ CO + 2H2 | (16) |
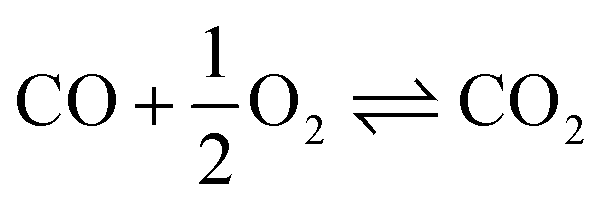 | (17) |
 | ||
| Fig. 5 COMET process concept for the production of OME3–5 from H2 and CO2 feedstock with the intermediate production of MeOH and FA. | ||
To use the heating value of the purge streams, a combustion sub-process was implemented. Following the assumptions from Mantei et al.,19 excess air was added to achieve complete combustion and keep the adiabatic temperature rise below 800 °C. The stoichiometric amount of O2 required for a complete combustion is described by eqn (18).
 | (18) |
4. Materials and methods
4.1. Experiments
4.2. Experimental setups
Before using the catalyst A46, it was stored in a mixture of FA, H2O, MeOH and OME1 to prevent further swelling inside the reactor unit.
The addition of MeOH to the OME synthesis product allowed for a stable storage and transport. The stabilization was a preventive measure to ensure a homogeneous liquid solution without precipitation even at low temperatures and long storage periods. The amount of additional MeOH was determined to meet the demand for the reactive distillation column in an integrated process, see Fig. 3 and the description in section 3. The targets of the reactive distillation column were a bottom product with a concentration of about 60 wt% FA and H2O, as well as an almost complete conversion of MeOH. The latter target assumes that OME2–3 are converted to FA and OME1 as described by eqn (7), MeOH and FA are converted to OME1 and H2O as described by eqn (1) and (5), and the distillate product is the azeotropic mixture of OME1 and MeOH at ambient pressure.
4.3. Process simulation and evaluation
The components H2, CO2, CO, N2, O2, FA, MeOH, H2O, OME1–10, HF1–10 and MG1–10 were considered. The properties for OME2–10, HF1–10 and MG1–10 were not included in the Aspen database. Their properties were introduced as new components according to a previous work of our group.19 The rest of the components properties were adopted from Aspen database. Details are described by Mantei et al.,19 who also describe the UNIFAC-based model which was used as a basis for the process simulation.
Pressure values presented in this work describe the absolute pressure.
The methodology for the process simulation including the individual simulation of all sub-processes, the material integration and interconnection, convergences, the adjustment of the production capacity and finally the heat integration followed the procedure from Mantei et al.19
For the simulation of the reactive distillation column, the kinetic model from Schmitz et al.27 for the OME synthesis over A46 for feedstocks comprising MeOH, FA, H2O and OME1 was used. The model was implemented in a Fortran subroutine and activated on the catalyzed trays inside the RadFrac column. The implementation of the kinetic model was validated with the experimental results from Drunsel,61 who investigated a reactive distillation column in the OME1 production process for a similar reactive separation task, however, using the catalyst A15 instead of A46. A good agreement was obtained between experimental and simulation results. Furthermore, the subroutine was slightly adjusted to be used in a reactor unit and was validated with the experimental results from Schmitz et al.27 with a good agreement. In contrast to the kinetics of the OME formation as described by eqn (5)–(7), the model assumes the formation of HFn and MGn as described by eqn (1)–(4) to be in chemical equilibrium at all retention times.
Regarding the side products HFn and MGn for mixtures containing FA, H2O and MeOH, the true composition was used for the process simulation, which considers the presence of HFn and MGn. The overall composition, considering the stoichiometric decomposition of HFn and MGn to their reactants MeOH, H2O and FA, was used for the evaluation and the presentation of the results.
The formation of the side products TRI, DME, MEFO, FOAC and tetroxane was not considered in the process simulation, due to very small concentrations in the synthesis product when A46 is used as a catalyst.
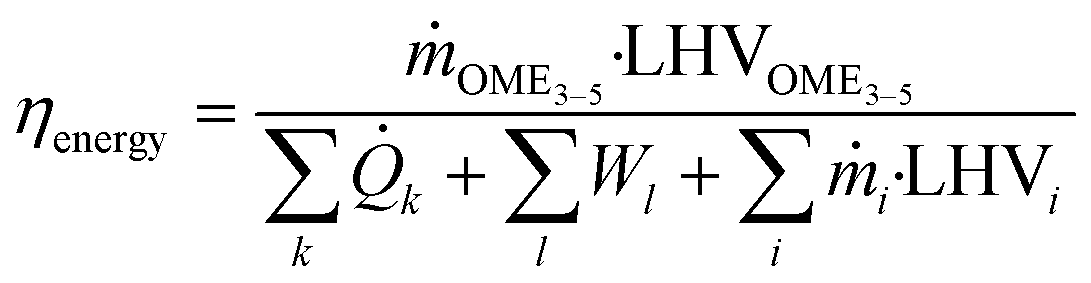 | (19) |
 | (20) |
 | (21) |
![[Q with combining dot above]](https://www.rsc.org/images/entities/i_char_0051_0307.gif) k and Wl represent the externally supplied heat flows and electric power and C denotes the number of carbon atoms.
k and Wl represent the externally supplied heat flows and electric power and C denotes the number of carbon atoms.
5. Results and discussion
5.1. Experimental demonstration
In the following sections the experimental results of the demonstration of the main COMET process units are presented and discussed. All investigations were carried using state-of-the-art experimental setups. The FA concentration units and the OME synthesis unit were interconnected. The products were collected and subsequently further processed in the distillation units. | ||
| Fig. 6 OME synthesis from OME1 and concentrated FA(aq.) over A46 (conditions: concentrated FA(aq.) with 85–89 wt% FA, (concentrated FA(aq.))/OME1 = 0.6 g g−1, A46/(OME1 + concentrated FA(aq.)) = 0.34 g g−1 h−1, approx. 3 L h−1, 90 °C, 10 bar, fixed bed reactor). F represents the feed composition. P-Sim, P1–5-Exp represent the product composition of the simulated equilibrium, two experimental preliminary products P1–2 from the starting phase and the three product barrels P3–5, respectively. | ||
The compositions of the three product barrels (P3–5-Exp) show a good agreement with the simulated equilibrium composition (P-Sim). Comparing the three product compositions among each other, a small shift towards longer chain OME≥3 and FA with increasing time on stream was observed. This is mainly a result of the slightly fluctuating FA concentration in the concentrated FA(aq.) feed stream (85–88 wt% FA) from the evaporation units and the feed stream flowrates. The OME1 flowrate was regulated to meet a constant ratio between OME1 and FA, while the FA flowrate was regulated to stabilize the level of the small storage between the second thin film evaporator and the OME synthesis sub-process. Although there was good agreement of the chemical equilibrium between simulation and experiment results, the kinetics of the OME synthesis were predicted to be much faster than those found experimentally. The simulation predicted the chemical equilibrium at a weight hourly space velocity (WHSV, feed mass flow in relation to the amount of catalyst) of about 70 h−1. However, the experiments were carried out at a WHSV of approximately 16 h−1 and 3 h−1, whereby only the lower WHSV was sufficient to obtain chemical equilibrium, as presented by P3–5-Exp in Fig. 6. The WHSV of 16 h−1 led to high amounts of unreacted FA, low concentrations of OME≥3 and, therefore, solidification of the synthesis product after cooling and without adding MeOH for stabilization, as presented by P1–2-Exp in Fig. 6. The kinetic model from Schmitz et al.27 was used for the simulation, which was initially regressed on experimental results of the OME synthesis from MeOH and FA with partly higher concentrations of H2O or OME. Therefore, the feed mixture already contained high concentrations of HF which can directly react to OME, as described by eqn (5) and (6). Furthermore, the model was based on the assumption that the reactions towards HF and MG, as described by eqn (1)–(4), are in equilibrium at all retention times since their kinetics are much faster than the kinetics of the formation of OME. In the COMET process this assumption is not met. The concentration of MeOH in the feed is very low because OME1 was used as methyl group supplier instead. Therefore, FA is bound mainly in MG which need to depolymerize to be converted to OME, as described in the ESI† in the section about the OME3–5 production process based on OME1 and FA(aq.) or pFA. This is the limiting step for the reaction kinetics of the COMET process but not significant for the OME synthesis based on MeOH and FA. Therefore, the kinetic model27 is a suitable basis but needs to be further extended to realistically describe the reaction progress of other feed mixtures, which is required to correctly design the reactor unit.
Besides the main components, small fractions of the side products MEFO, TRI and tetroxane were detected in the product barrels P3–5 of about 0.1 wt%, 0.6 wt% and 0.1 wt%, respectively. For P1–2 (during the starting phase) concentrations of about 0.1 wt%, 0.1 wt% and 0.03 wt% were obtained. Therefore, these concentrations strongly depend on the WHSV, which is a matter of investigation for high yields of OME3–5 at low concentrations of the side product.
During the investigation of the OME synthesis for about 80 h on stream, stable catalytic activity was observed. Also under reactive distillation conditions, the catalyst performance did not show an obvious deactivation for about 600 h on stream. However, further investigations are required to verify if the changing thermal stability is an initial phenomenon of the catalysts time on stream. In addition to the impact on the process design, the cause of this behavior should be further investigated. It might only be the leaching of the catalyst as emphasized by Fink et al.65,66 and Baranowski et al.,67 but it could also be influenced by the side product formation, especially formic acid, which was not analyzed in this work but reported in the literature.32
 | ||
| Fig. 7 CO-1, OME synthesis product separation (conditions: 2 L h−1, reflux/distillate = 0.5–2 s s−1, distillate/feed = 81 wt%, Montz 750 structured packing, 85–175 °C, ambient pressure). The values describe the mass fractions of the feed mixture, here P5-Exp as presented in Fig. 6, the distillate product and bottom product. | ||
The distillation setup was operated at a feed temperature of 87 °C, a condensation temperature of 85 °C and a reboiler temperature of 175 °C. The distillate to feed ratio was about 0.81 and the time-based reflux ratio was varied as a controlled variable between 0.5–2 s s−1 (time controlled) to achieve a constant condensation temperature.
Fig. 7 shows that OME2 was completely separated to the distillate product. However, also a small fraction of OME3 went to the distillate product, which was about 14% of the feed amount of OME3. Besides OME≥3, traces of FA, H2O and MeOH were detected in the bottom product which were mainly below 0.6 wt%. MEFO was not detected in the bottom product.
Regarding the continuous operation of the distillation setup, an increasing precipitation of FA inside the condenser was challenging in the initial phase but could be prevented by increasing the temperature of the cooling fluid to above 25 °C. However, as a result the temperature difference decreased between the cooling fluid and the boiling points of the most volatile components MEFO, the azeotropic mixture of OME1 and MeOH, as well as OME1. Thus, the area of the condenser was relatively small to obtain a complete condensation and a small fraction of the most volatile components accumulated in a cool trap. As a result, the ratio of OME1 to OME2 in the feed mixture P5-Exp differs from the ratio of OME1 to OME2 in the distillate product.
A representative result of the continuous reactive distillation experiment is illustrated in Fig. 8. The distillate and bottom product compositions show that the targets of the reactive distillation column were obtained. OME≥2 were converted to OME1 and FA, the composition of the distillate product is the azeotropic mixture of OME1 and MeOH and the bottom product contains mainly FA and H2O. Regarding the bottom product composition, small concentrations of MeOH of about 0.3 wt% were detected besides the desired range of FA and H2O. Furthermore, traces of OME1–6 were detected with concentrations far below 0.1 wt%. However, as a result of the high H2O and FA content in the bottom product, the quantification of traces is complex due to the fast precipitation of the bottom product solidifies fast if not heated or diluted.
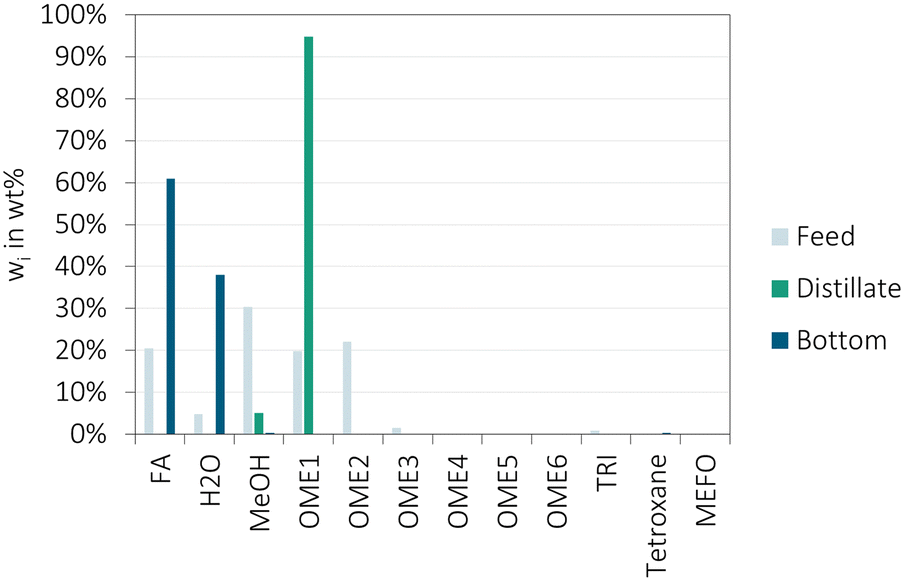 | ||
| Fig. 8 CO-2, reactive distillation of the distillate product of CO-1 over A46 (conditions: A46/(feed stream) = 0.35 g g−1 h−1, 1 L h−1, distillate/feed = 63 wt%, Montz 750 structured packing, 45–104 °C, ambient pressure). The values describe the mass fractions of the feed mixture, the distillate product and bottom product. | ||
CO-2 was operated at a condensation temperature of 45 °C and reboiler temperature of 104 °C with a distillate to feed ratio of about 0.63.
The results confirm that the reactive distillation column is a feasible instrument for the separation of H2O from the OME3–5 production loop of the OME3–5 production and that an almost complete conversion of MeOH can be achieved. Furthermore, the results indicate, that the variation of the amount of MeOH in the feed mixture to the reactive distillation column can be used to tune the amount of OME1 produced as the distillate product.
 | ||
| Fig. 9 CO-3, product separation (conditions: 5.5 L h−1, distillate/feed = 82 wt%, Montz 750 structured packing, 100–210 °C, 200 mbar). The values describe the mass fractions of the feed mixture, here the CO-1 bottom product, the distillate product and bottom product. | ||
Due to the solidification of the bottom product OME≥5 at room temperature, it was diluted in THF with a ratio of 1:10 g/g to enable the GC analysis. However, this also impairs the detection limits and accuracy of the analysis. Alternatively, to liquify the bottom product it can also be heated up. At 80 °C, the bottom product is completely liquid, which enables its recycling to the reactor, as illustrated in Fig. 3.
The distillation setup was operated at a condensation temperature of 140 °C and a reboiler temperature of 210 °C. The high distillate temperature was a result of the high feed flow rate and the limited area for condensation. As a result, a complete condensation was not obtained and small fraction of OME3 accumulated in a cool trap. In contrast to the other distillation experiments the operation pressure was reduced to 200 mbar to reduce the reboiler temperature for the separation between OME5 and OME6. The distillate to feed ratio was about 0.82.
5.2. Process simulation and evaluation
| Stream | Overall mass fractions | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | |
| T in °C | 64.9 | 30.0 | 90.0 | 30.0 | 90.4 | 90.0 | 90.0 | 81.5 | 81.0 | 41.5 | 117.4 | 200.5 | 86.6 | 194.9 |
| p in bar | 1.0 | 1.0 | 0.3 | 1.0 | 10.3 | 10.0 | 10.1 | 1.8 | 1.8 | 1.0 | 1.0 | 1.8 | 0.07 | 0.07 |
| m in kg h−1 | 18509 |
2203 | 14753 |
7870 | 22957 |
66666 |
66666 |
51796 |
5288 | 41330 |
15753 |
14871 |
12490 |
2380 |
| FA | 0.502 | 0.142 | 0.184 | 0.000 | 0.880 | 0.303 | 0.186 | 0.239 | 0.000 | 0.000 | 0.727 | 0.000 | 0.000 | 0.000 |
| H2O | 0.491 | 0.778 | 0.796 | 1.000 | 0.120 | 0.042 | 0.022 | 0.028 | 0.000 | 0.002 | 0.268 | 0.000 | 0.000 | 0.000 |
| MeOH | 0.007 | 0.080 | 0.020 | 0.000 | 0.000 | 0.028 | 0.100 | 0.129 | 1.000 | 0.045 | 0.005 | 0.000 | 0.000 | 0.000 |
| OME1 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.591 | 0.276 | 0.356 | 0.000 | 0.953 | 0.000 | 0.000 | 0.000 | 0.000 |
| OME2 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.179 | 0.230 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| OME3 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.107 | 0.017 | 0.000 | 0.000 | 0.000 | 0.419 | 0.499 | 0.000 |
| OME4 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.061 | 0.000 | 0.000 | 0.000 | 0.000 | 0.271 | 0.323 | 0.000 |
| OME5 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.033 | 0.000 | 0.000 | 0.000 | 0.000 | 0.149 | 0.177 | 0.000 |
| OME6 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.018 | 0.018 | 0.000 | 0.000 | 0.000 | 0.000 | 0.080 | 0.000 | 0.496 |
| OME7 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.009 | 0.009 | 0.000 | 0.000 | 0.000 | 0.000 | 0.042 | 0.000 | 0.262 |
| OME8 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.005 | 0.005 | 0.000 | 0.000 | 0.000 | 0.000 | 0.022 | 0.000 | 0.136 |
| OME9 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.002 | 0.002 | 0.000 | 0.000 | 0.000 | 0.000 | 0.011 | 0.000 | 0.070 |
| OME10 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.001 | 0.001 | 0.000 | 0.000 | 0.000 | 0.000 | 0.006 | 0.000 | 0.036 |
The feedstock (stream 1) containing about 50 wt% FA and 49 wt% H2O is mixed with the distillate of the second evaporator E-2 and the bottom of the third evaporator E-3. The mixture is concentrated in a cascade of two evaporators E-1 and E-2 operated at 400 and 500 mbar respectively and low retention times. The pressure levels were selected to obtain similar evaporation and condensation temperatures as experimentally verified. However, in practice, the pressure level might be lower to achieve the desired concentrations. This is a result of the simplified modelling of the evaporators which require more detailed considerations of the reaction kinetics of eqn (1)–(4) as recently introduced by Tönges and Burger.68 The FA concentration is similar to the conventional production of pFA and generates a concentrated FA solution containing about 88 wt% FA (stream 5) and a solution containing about 18 wt% FA (stream 3). Stream 3 is split to be used as a washing liquid for the FA absorber column and to be purified in the distillation column CO-4 operated at 5.5 bar to pure H2O (stream 4) (<200 ppm FA) and a concentrated FA solution with 44 wt% FA. To prevent the accumulation of MeOH and other impurities in the loop, the concentrated FA solution is sent to another evaporator E-3 operated at ambient pressure. This prepares a by-product of the COMET process with a higher MeOH concentration (stream 2) and a FA solution with a similar composition to the FA feedstock, which is recycled to the evaporator cascade. The by-product (stream 2) has a low FA concentration of about 14 wt%. Furthermore, its mass flow is about 17.6% of the mass flow of the target OME3–5 product. This high mass flow is similar to alternative OME3–5 production processes using FA(aq.) solution as an intermediate product.19
The concentrated FA product (stream 5) is pressurized to about 10 bar, then mixed with the recycle streams containing mainly OME1 and OME>5 (stream 10 and stream 14) and converted to OME in a fixed bed reactor at about 10 bar and 90 °C, over A46 catalyst, as used for the experimental demonstration. The reactor product contains about 20 wt% OME3–5, which is relatively high in comparison to the process based on MeOH and FA(aq.), which has 0 to 15 wt% OME3–5 in the reactor product,39 as presented in Table 8. The reactor product is purified in a first distillation column CO-1 operated at a slight overpressure of 1.8 bar, where OME≥3 are separated from FA, MeOH, H2O, OME1–2 and a small fraction of OME3. The slight overpressure improves the separation efficiency and reduces the losses of OME3 to the distillate product (stream 8) to about 12%. The FA concentration of the bottom product (stream 12) is reduced to about 100 ppm. In the third distillation column CO-3 operated at 70 mbar, the main product OME3–5 (stream 12) is extracted from the process with about 50 wt% OME3, 32 wt% OME4, 18 wt% OME5 and traces of FA and H2O in compliance with the pre-standard DIN/TS 51699 specifications. The distillate product (stream 8) of the first distillation column CO-1 is mixed with MeOH (stream 9) and introduced to the reactive distillation column CO-2. This column is operated at ambient pressure. The selection of the pressure level is a compromise between the condensation temperature of the distillate, the reaction kinetics on the catalytic trays and the composition of the azeotropic mixture of OME1 and MeOH in the distillate. A pressure reduction would favorably improve the azeotropic composition to higher OME1 concentrations. However, it would also lead to a reduction of the condenser temperature below 41 °C which can lead to more expensive cooling utilities and decelerate the reaction kinetics on the catalytic trays. Increased pressure levels would benefit from higher reaction kinetics due to the higher temperature level on the catalytic trays, but lower OME1 concentrations in the distillate product. This would decrease the OME3–5 selectivity in the OME synthesis reactor and necessarily increase the recycle streams and, therefore, the specific heat demand for product purification. The mixture is separated into the azeotropic mixture of 95 wt% OME1 and 4.5 wt% MeOH in the distillate (stream 10) and a mixture of 73 wt% FA and 27 wt% H2O in the bottom (stream 11). A summary of the overall mass balance of the COMET process is listed in Table 5.
| COMET | P1 | P4 | |
|---|---|---|---|
| Total input in kg kgOME3–5−1 | 6.60 | 7.54 | 8.53 |
| H2 | 0.25 | 0.27 | 0.21 |
| CO2 | 1.82 | 1.96 | 2.20 |
| Air | 4.53 | 5.32 | 5.92 |
| Total output in kg kgOME3–5−1 | 6.60 | 7.54 | 8.53 |
| OME3–5 | 1.00 | 1.00 | 1.00 |
| OME3 | 0.50 | 0.43 | 0.43 |
| OME4 | 0.32 | 0.38 | 0.36 |
| OME5 | 0.18 | 0.19 | 0.21 |
| Wastewater | 1.03 | 1.30 | 1.00 |
| Aq. FA solution | 0.18 | — | — |
| Exhaust gas | 4.39 | 5.24 | 6.54 |
For the comparison with alternative OME3–5 production processes the results from Mantei et al.19 are presented for some selected processes, namely: P1, which produced OME3–5via MeOH and a concentrated FA solution (aqueous OME synthesis) feedstock, and P4 which produces OME3–5via OME1 and monomeric FA (anhydrous OME synthesis) feedstock. The processes are described in detail in the ESI† in section 1.4 and 1.5.
The results show that the overall COMET process requires less H2 than P1 but more H2 than P4. The difference to P1 is mainly based on the FA concentration sub-process, in which the simulation of this work contains a modified separation of FA from H2O due to the addition of a distillation column and a third evaporator. This results in a smaller amount of FA, which exits the process in the form of an aqueous FA solution by-product stream (see stream 2 in Fig. 3).
The difference to P4 is mainly based on the advantages of the anhydrous FA synthesis from MeOH which produces H2 as a by- product which can be separated and recycled to the MeOH sub-process. The lower CO2 demand of the COMET process in comparison to P1 and P4 is also based on the FA concentration sub-process and the anhydrous FA synthesis. P1 requires more CO2 due to the higher amount of FA in the by-product stream (see stream 2 in Fig. 3). The lower demand of air of the COMET process is mainly a result of the consideration of smaller purge streams which are oxidized in the combustion sub-process. The oxygen demand for the partial oxidation of MeOH towards FA(aq.) is only slightly lower for the COMET process than for P1.
The composition of the final OME3–5 product mixture also shows significant differences. While Mantei et al.19 chose a composition close to the highest yield of OME3–5 after the synthesis, the composition in this work was selected to meet the requirements for the pre-standard DIN/TS 51699.
Regarding the wastewater production, the COMET process produces less wastewater than P1 but more wastewater than P4. The difference to P1 is mainly based on the composition of the wastewater. While the simulation of the COMET process produces high-purity wastewater and an aqueous FA solution by-product, Mantei et al.19 considered the aqueous FA solution to be part of the wastewater. The difference to P4 is also explained by the anhydrous FA synthesis.19
The exhaust gas flow is lower for the COMET process than for P1 and P4 which is the result of the smaller purge streams and, therefore, the lower air demand for the combustion.
The summary of the overall energy demand of the COMET process after the heat integration in comparison to P1 and P4 is listed in Table 6.
| COMET | P1 | P4 | |
|---|---|---|---|
| Total input kW h kW−1 hOME3–5,LHV−1 | |||
| H2 | 1.60 | 1.70 | 1.34 |
| Total output kW h kW−1 hOME3–5,LHV−1 | |||
| OME3–5 | 1.00 | 1.00 | 1.00 |
| Energy demand kW h kW−1 hOME3–5,LHV−1 | |||
| Electricity | 0.11 | 0.09 | 0.14 |
| LPS, 4 bar | 0.09 | −0.10 | 0.24 |
| MPS, 23 bar | 0.05 | 0.30 | −0.07 |
| Cooling water | −1.02 | −1.05 | −0.79 |
| Heat. T > 250 °C | — | — | 0.19 |
The different H2 demands between the COMET process, P1 and P4 directly reflect on the total process energy demand. Furthermore, the electricity demand of the COMET process is higher than for P1 but lower than for P4. Compared to P1, the operation conditions of the phase separators in the MeOH sub-process were adjusted resulting in higher recycling rates and, therefore, higher compression demand. Furthermore, P1 and P4 did not consider the compression demand for the combustion sub-process.19 The higher electricity demand of P4 is a result of the anhydrous FA synthesis, which requires higher recycle streams in comparison to the partial oxidation of MeOH in P1 and the COMET process.19
The demand for low pressure steam (LPS) of the COMET process is higher than the demand of P1 but lower than the demand of P4, which is mainly a result of the heat integration strategies. P1 generates more LPS than it consumes, while P4 and the COMET process show higher demands than generated. However, the demand for medium pressure steam (MPS) is lower for the COMET process. P1 shows a very high demand of MPS, while P4 generates more than it consumes. The MPS demand for the COMET process is significantly lower than for P1. The main consumers of MPS are CO-1 and CO-3 in the OME3–5 sub-process. However, MPS is also generated in the MeOH synthesis reactor and the combustion sub-process. While the combustion sub-process generates a similar amount of MPS of about −0.15 kW h kW−1 hOME3–5,LHV−1 comparing P1 and the COMET process, the amount differs for the MeOH reactor with −0.04 and −0.11 kW h kW−1 hOME3–5,LHV−1, respectively. The lower MPS generation of P1 is mainly a result of the inlet temperature to the MeOH reactor. The inlet temperature of P1 is about 185 °C and, therefore, needs to be heated up to the operation temperature of 250 °C using generated MPS and the exothermic heat of the methanol synthesis reactor. The inlet temperature of the COMET process simulation is about 240 °C, which requires a larger heat transfer area but improves the energy efficiency. Furthermore, the demand for MPS of the distillation columns in the OME3–5 sub-process differ significantly. P1 requires about 0.32 kW h kW−1 hOME3–5,LHV−1 of MPS for the purification of the OME3–5 product stream, while the COMET process requires only 0.20 kW h kW−1 hOME3–5,LHV−1. This is mainly a result of the higher OME3–5 yield of the COMET process after the OME synthesis reactor as discussed in the previous section.
The demand for cooling water is similar between P1 and the COMET process but significantly lower for P4.19 The cooling is required mainly for the temperature level between 90 °C and 30 °C and is, therefore, hardly utilizable for the heat integration of the COMET process. Mantei et al.69 proposed the utilization of heat pumps instead of cooling water and evaluated a significant enhancement potential for the overall energy efficiency.
Only P4 has a demand for heat above 250 °C, due to the endothermic anhydrous FA synthesis.
Regarding the heat demand for the separation of H2O via reactive distillation, the COMET process requires about 1.1 kWh kg−1 H2O at 117 °C. This is based on the assumption that the main target of the reactive distillation column is the separation of H2O from the loop. Therefore, the heat demand of the reboiler and the feed preheater can be allocated to the amount of H2O separated from the loop.
| COMET | P1 | P4 | |
|---|---|---|---|
| η energy in % | 54.1 | 50.3 | 54.4 |
| η C in % | 88.1 | 81.6 | 72.5 |
| η mass in % | 41.1 | 38.1 | 41.4 |
The overall energy efficiency of the COMET process is higher than P1 and similar to P4. Mantei et al.19 reported an efficiency of 49–50% for processes considering the FA(aq.) sub-process. Furthermore, the carbon efficiency is considerably higher than P1 or P4. The low carbon efficiency of P4 is mainly a result of the high CO side-product formation during the anhydrous FA synthesis.19 While the lower carbon efficiency of P1 is mainly a result of the more efficient H2O separation of the FA concentration sub-process considered for the COMET process simulation. As a result, the carbon efficiency of P1 could also be increased by adjusting the FA concentration sub-process that can be considered in a future work. The OME3–5 yield based on the feedstock H2 and CO2 is also higher for the COMET process than for P1 which is also a result of the more efficient H2O separation of the FA concentration sub-process. The OME3–5 yield is similar to P4 since H2O, formed in the FA(aq.) synthesis, is separated from the process loop, compared to the formation, separation and recycling of H2 in the anhydrous FA synthesis of P4.
| Feedstock | Anhydrous synthesis | Aqueous synthesis | |||||
|---|---|---|---|---|---|---|---|
| OME1 and TRI | DME and TRI | OME1 and monomeric FA | MeOH and FA(aq.) | OME1 and FA(aq.) or pFA | MeOH and monomeric FA | COMET | |
| a Further investigations and an adjusted process concept are required to estimate the process performance. | |||||||
| w OME3–5 in wt% | 5, 3439 | —a | 5, 2919 | 0, 1539 | 4, 1919 | 3, 1919 | 0, 20 |
| 0, 3543 | 4, 1619 | ||||||
| 0, 1443 | |||||||
| Q Reboiler/HOME3–5 in kWHeat kWOME3–5−1 | 7.6%39 | —a | 15%19 | 47%39 | 26%19 | 48%19 | 35% |
| 5.5%43 | 39%19 | ||||||
| 78%43 | |||||||
| η energy,overall in % | 29–3739 | —a | 27–3619 | 30–3639 | 26–3219 | 28–3719 | 28–34 |
| 22–2643 | 25–3119 | ||||||
| 24–2943 | |||||||
| Scale-up potential in the near future | Likely | Unlikely | Unlikely | Less likely | Less likely | Unlikely | Likely |
Regarding the electricity and heat demand for the H2O electrolysis and CO2 preparation, the assumptions from Held et al.39 were considered. For the CO2 preparation all three scenarios from Held et al.39 were considered, comprising CO2 from point sources (CPS), post combustion capture (PCC) using mono-ethanol amine scrubbing and direct air capture (DAC). The key assumptions for the expanded system boundary evaluation are summarized in ESI† in Table S28. In addition, the scale-up potential in the near future is qualitatively evaluated. The key performance parameters are based on the results from Held et al.,39 Mantei et al.19 and Schemme et al.43
Regarding the yield of OME3–5 after the reactor as illustrated in Fig. 1, the anhydrous process concepts show far higher OME3–5 concentrations than the aqueous process concepts, as indicated by wOME3–5. This also reflects on the heat demand for the OME3–5 product purification, which is compared based on QReboiler/HOME3–5. The anhydrous process concepts show significantly lower heat demands for the product purification. Exceptions are the production based on OME1 and FA(aq.) or pFA and the COMET process, which despite comparatively low yields of OME3–5 in the reactor require less heat for the separation of the target product than the other aqueous process concepts. For a consistent basis of comparison, the production of the intermediate products for different OME production processes was considered in the evaluation, indicated by ηenergy,overall. The result is a low overall energy efficiency of <40% for all processes, with minor differences between anhydrous and aqueous process concepts. Greater differences were reported between different literature sources, which is especially significant comparing the results for the OME3–5 production based on OME1 and TRI as well as MeOH and FA(aq.) from Held et al.,39 Mantei et al.19 and Schemme et al.43 Those differences are discussed in detail by Mantei et al.19 and mainly result from different heat integration strategies and the simulation procedure. Schemme et al.43 only integrated the heat between individual sub-processes, while Mantei et al.19 and Held et al.39 considered the heat integration between all sub-processes. However, Held et al.39 did use stoichiometric material balances and literature data, while Mantei et al.19 and Schemme et al.43 used the process simulation software Aspen Plus.
Regarding the low overall energy efficiency of all OME3–5 production processes, Mantei et al.69 showed the potential of including high temperature heat pumps (HTHP) to lift the temperature of the excess heat streams and, therefore, supply internal heat demands and, in addition, external heat demands. This strategy has the potential to lift the overall energy efficiency above 61% considering heat as a valuable by-product of the process. Besides only small differences in the energy efficiency, the production costs of the OME3–5 product also show no significant differences between different production processes.19,43
Regarding a sustainable large-scale production of OME3–5 in the near future the COMET process and a production based on OME1 and TRI feedstock show great potential to be scaled up today. However, the latter is comparatively complex, comprising five sub-processes for the production of MeOH, FA(aq.), TRI, OME1 and OME3–5. A sustainable OME3–5 production based on the COMET process on the other hand comprises three sub-processes for the production of MeOH, FA(aq.) and OME3–5. The OME production process based on DME and TRI requires further investigations, mainly due to the high MEFO formation during the synthesis and the low activities for the conversion of DME to OME. A fast scale-up of the processes based on monomeric FA is mainly prevented by the low TRL of the monomeric FA production. Finally, the aqueous process concepts require the separation of H2O from the loop of the OME3–5 sub-process, which is the main bottleneck for a fast scale-up. Various concepts for separating H2O from the loop were already proposed, and some show promising results, as discussed before and demonstrated in this work for the reactive distillation column. This enables a scale-up for the processes based on MeOH and FA(aq.) and OME1 and FA(aq.) or pFA. In comparison to the OME3–5 production based on OME1 and TRI, the aqueous process concepts enable a considerable simplification, which typically improves the robustness and therefore feasibility for large-scale application.
6. Conclusion
The COMET process, which solves the challenging H2O management of aqueous OME processes, was introduced in this work. The process benefits from a simple feedstock preparation, a short process chain from H2 and CO2 to OME3–5, a comparatively high OME3–5 yield after the reactor, and the possibility of extracting the by-product H2O from the loop using a state-of-the-art reactive distillation unit.Other H2O separation methods, as discussed in the literature, were presented, and their main advantages and hurdles were evaluated quantitatively. The main advantage of the H2O separation in the COMET process via reactive distillation is the scale-up potential and the feasible application in large-scale production plants.
Starting solely from MeOH and FA(aq.) commercial feedstocks, the main COMET process units, comprising all evaporation, reaction and separation process steps, were experimentally demonstrated on a pilot scale. Importantly, the technical feasibility of the reactive distillation column – the heart of the COMET process concept – was demonstrated for a long duration of around 600 h on stream. In addition, the purification of the final OME3–5 product was successfully realized with a product compliant with the pre-standard DIN/TS 51699.
The COMET process was simulated and evaluated using Aspen Plus and compared with relevant alternative OME3–5 production processes. Therefore, the system boundary was expanded, including H2 production via H2O electrolysis, CO2 capture and all intermediate production sub-processes. With an overall energy efficiency of 28–34%, depending on the CO2 source, the energy demand of the COMET process is similar to the alternative OME3–5 production processes, in which overall energy efficiencies were evaluated in the range of 25–36%. Moreover, the COMET process shows a higher carbon efficiency of 88%.
The OME market is limited by the lack of technologically feasible large-scale processes. However, compared to relevant alternative OME3–5 production processes, the novel COMET process shows the smallest technological hurdles and can already be demonstrated and scaled up.
Symbols
| C [—] | Number of carbon atoms |
| H [kW] | Energy content based on the LHV |
| m, ṁi [kg h−1] | Mass flow rate |
| p [bar] | Pressure |
|
k
[kW] | Heat flow |
| T [K] | Temperature |
| w i [wt%] | Mass fraction |
| W l [kW] | Electric power |
| η [%] | Efficiency |
Sub- and superscripts
| C | Carbon |
| i | Reactant, component |
Abbreviations
| A15 | Amberlyst® 15 |
| A46 | Amberlyst® 46 |
| CO | Distillation column |
| COMET | Clean OME technology |
| DAC | Direct air capture |
| DME | Dimethyl ether |
| EGR | Exhaust gas recirculation |
| EPDM | Ethylene propylene diene monomer |
| FA | Formaldehyde |
| FA(aq.) | Aqueous FA solution, formalin |
| FFKM | Perfluoroelastomer |
| FOAC | Formic acid |
| GC-FID | Gas chromatograph equipped with a flame ionization detector |
| GC-TCD | Gas chromatograph equipped with a thermal conductivity detectors |
| HF | Poly(oxymethylene) hemiformals |
| HTHP | High temperature heat pump |
| HVO | Hydrogenated vegetable oil |
| IER | Ion exchange resin |
| LHV | Lower heating value |
| LPS | Low pressure steam |
| MEFO | Methyl formate |
| MeOH | Methanol |
| MG | Poly(oxymethylene) glycols |
| MPS | Medium pressure steam |
| OME | Oxymethylene dimethyl ethers |
| OME1 | Methylal |
| pFA | Paraformaldehyde |
| PTFE | Polytetrafluoroethylene |
| PtL | Power-to-liquid |
| PtX | Power-to-X |
| R | Reactor |
| S | Separator |
| THF | Tetrahydrofuran |
| TRI | Trioxane |
| TRL | Technology readiness level |
| WtW | Well-to-wheel |
Author contributions
Conceptualization, F. M. and O. S.; methodology, F. M. and C. S.; experimental investigation, F. M., C. S. and O. S.; analytics, F. M., C. S. and H. S.; simulation and evaluation, F. M., A. E., F. F. and A. P.; writing – original draft preparation, F. M.; writing – review and editing, O. S. and M. K.; scientific supervision, O. S.; project administration, O. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Deutsche Bundesstiftung Umwelt (DBU) is gratefully acknowledged for funding the work of Franz Mantei (20018/541). Furthermore, ChemCom Industries B.V. is gratefully acknowledged for funding the investigation and providing the chemicals required for the investigations. Moreover, many thanks to INAQUA Vertriebsgesellschaft mbH for providing the catalyst A46.References
- C. Hank, A. Sternberg, N. Köppel, M. Holst, T. Smolinka, A. Schaadt, C. Hebling and H.-M. Henning, Sustainable Energy Fuels, 2020, 4, 2256 RSC.
- IEA, Hydrogen, Paris, 2022 Search PubMed.
- Hydrogen, https://www.iea.org/fuels-and-technologies/hydrogen, (last accessed December 2022) Search PubMed.
- Direct Air Capture, https://www.iea.org/reports/direct-air-capture, (last accessed June 2022) Search PubMed.
- M. Härtl, K. Gaukel, D. Pélerin and G. Wachtmeister, MTZ worldwide, 2017, vol. 78, p. 52 Search PubMed.
- A. Omari, B. Heuser, S. Pischinger and C. Rüdinger, Appl. Energy, 2019, 1242 CrossRef CAS.
- P. Dworschak, V. Berger, M. Härtl and G. Wachtmeister, SAE Int. J. Fuels Lubr., 2022, 15 DOI:10.4271/04-15-02-0008.
- A. D. Gelner, D. Rothe, C. Kykal, M. Irwin, A. Sommer, C. Pastoetter, M. Härtl, M. Jaensch and G. Wachtmeister, Environ. Sci.: Atmos., 2022, 2, 291 CAS.
- C. Hank, L. Lazar, F. Mantei, M. Ouda, R. J. White, T. Smolinka, A. Schaadt, C. Hebling and H.-M. Henning, Sustainable Energy Fuels, 2019, 3, 3219 RSC.
- S. Voelker, S. Deutz, J. Burre, D. Bongartz, A. Omari, B. Lehrheuer, A. Mitsos, S. Pischinger, A. Bardow and N. von der Assen, Sustainable Energy Fuels, 2022, 6, 1959 RSC.
- S. Deutz, D. Bongartz, B. Heuser, A. Kätelhön, L. Schulze Langenhorst, A. Omari, M. Walters, J. Klankermayer, W. Leitner, A. Mitsos, S. Pischinger and A. Bardow, Energy Environ. Sci., 2018, 11, 331 RSC.
- IEA, World Energy Outlook 2022, Paris, 2022 Search PubMed.
- Kraft- und Brennstoffe - Polyoxymethylendimethylether (OME) - Anforderungen und Prüfverfahren (DIN/TS 51699:2023-04 - Entwurf), Beuth Verlag GmbH, Berlin, 2023, https://www.beuth.de/de/vornorm-entwurf/din-ts-51699/364871868, (last accessed March 2023) Search PubMed.
- H. Schelling, E. Ströfer, R. Pinkos, A. Haunert, G.-D. Tebben, H. Hasse and S. Blagov, Method for producing Polyoxymethylene Dimethyl Ethers, US 20070260094A1, BASF Aktiengesellschaft, 2007 Search PubMed.
- J. Burger, A novel process for the production of diesel fuel additives by hierarchical design, Ph.D. Thesis, University of Kaiserslautern, Kaiserslautern, 2012 Search PubMed.
- J. Burger, M. Siegert, E. Ströfer and H. Hasse, Fuel, 2010, 89, 3315 CrossRef CAS.
- E. Ströfer, H. Schelling, H. Hasse and S. Blagov, Method for the Production of Polyoxymethylene Dialkylethers From Troxan and Dialkylethers, US007999140B2, 2011.
- C. F. Breitkreuz, N. Schmitz, E. Ströfer, J. Burger and H. Hasse, Chem. Ing. Tech., 2018, 90, 1489 CrossRef CAS.
- F. Mantei, R. E. Ali, C. Baensch, S. Voelker, P. Haltenort, J. Burger, R.-U. Dietrich, N. von der Assen, A. Schaadt, J. Sauer and O. Salem, Sustainable Energy Fuels, 2022, 6, 528 RSC.
- N. Schmitz, E. Ströfer, J. Burger and H. Hasse, Ind. Eng. Chem. Res., 2017, 56, 11519 CrossRef CAS.
- J. Burger, N. Schmitz, H. Hasse and E. Ströfer, Process for preparing Polyoxymethylene Dimethyl Ethers from Formaldehyde and Methanol in aqueous solutions, US 2018/0134642 A1, 2018.
- R. Palkovits, C. H. Gierlich and I. Delidovich, Verfahren zur Trennung von Oxymethylenethern, EP 3 514 134 A1, 2019.
- C. H. Gierlich, Herstellung von Oxymethylenethern anhand von alternativen Reaktionskonzepten, Ph.D. Thesis, Aachen, 2021 Search PubMed.
- G. P. Hagen and M. J. Spangler, Preparation of Polyoxymethylene Dimethyl Ethers by Catalytic Conversion of Formaldehyde Formed by Oxidation of Dimethyl Ether, US 6392102 B1, 2002.
- T. Qiang, W. Jinfu, W. Tiefeng and Z. Yanyan, Method for Preparing DMM3–5 from Hypercoagulable Polyoxymethylene Dimethyl Ether Component DMM6+ and Dimethoxymethane DMM, CN105152882 A, 2017.
- N. Schmitz, F. Homberg, J. Berje, J. Burger and H. Hasse, Ind. Eng. Chem. Res., 2015, 54, 6409 CrossRef CAS.
- N. Schmitz, J. Burger and H. Hasse, Ind. Eng. Chem. Res., 2015, 54, 12553 CrossRef CAS.
- T. Grützner, H. Hasse, N. Lang, M. Siegert and E. Ströfer, Chem. Eng. Sci., 2007, 62, 5613 CrossRef.
- M. Drexler, P. Haltenort, U. Arnold and J. Sauer, Chem. Ing. Tech., 2022, 94, 256 CrossRef CAS.
- C. F. Breitkreuz, N. Hevert, N. Schmitz, J. Burger and H. Hasse, Ind. Eng. Chem. Res., 2022, 61, 7810 CrossRef CAS.
- P. Haltenort, K. Hackbarth, D. Oestreich, L. Lautenschütz, U. Arnold and J. Sauer, Catal. Commun., 2018, 109, 80 CrossRef CAS.
- J. Voggenreiter and J. Burger, Ind. Eng. Chem. Res., 2021, 60, 2418 CrossRef CAS.
- F. Mantei, S. Kopp, A. Holfelder, E. Flad, D. Kloeters, M. Kraume and O. Salem, React. Chem. Eng., 2023, 8, 917 RSC.
- I. Bogatykh, T. Osterland, H. Stein and T. Wilharm, Energy Fuels, 2020, 34, 3357 CrossRef CAS.
- K. Hackbarth, P. Haltenort, U. Arnold and J. Sauer, Chem. Ing. Tech., 2018, 90, 1 CrossRef.
- H. Liu, Z. Wang, Y. Li, Y. Zheng, T. He and J. Wang, Appl. Energy, 2019, 233-234, 599 CrossRef CAS.
- D. Bongartz, L. Doré, K. Eichler, T. Grube, B. Heuser, L. E. Hombach, M. Robinius, S. Pischinger, D. Stolten, G. Walther and A. Mitsos, Appl. Energy, 2018, 757 CrossRef CAS.
- D. Bongartz, J. Burre and A. Mitsos, Ind. Eng. Chem. Res., 2019, 58, 4881 CrossRef CAS.
- M. Held, Y. Tönges, D. Pélerin, M. Härtl, G. Wachtmeister and J. Burger, Energy Environ. Sci., 2019, 12, 1019 RSC.
- C. F. Breitkreuz, M. Dyga, E. Forte, F. Jirasek, J. de Bont, J. Wery, T. Grützner, J. Burger and H. Hasse, Chem. Eng. Process., 2021, 108710 Search PubMed.
- J. Sauer and G. Emig, Chem. Eng. Technol., 1995, 18, 284 CrossRef CAS.
- J. Burre, D. Bongartz and A. Mitsos, Ind. Eng. Chem. Res., 2019, 58, 5567 CrossRef CAS.
- S. Schemme, J. L. Breuer, M. Köller, S. Meschede, F. Walman, R. C. Samsun, R. Peters and D. Stolten, Int. J. Hydrogen Energy, 2020, 45, 5395 CrossRef CAS.
- P. Haltenort, L. Lautenschütz, U. Arnold and J. Sauer, Top. Catal., 2019, 62, 551 CrossRef CAS.
- M. Drexler, P. Haltenort, U. Arnold, J. Sauer, S. A. Karakoulia and K. S. Triantafyllidis, Catal. Today, 2022, 113847 CrossRef.
- X. Li, J. Cao, M. A. Nawaz, Y. Hu and D. Liu, J. Chem. Eng. Data, 2019, 64, 5548 CrossRef CAS.
- X. Li, H. Tian, W. Zhang and D. Liu, International Journal of Chemical and Molecular Engineering, 2018, 536 CAS.
- L. Wang, S. Zhou, P. Li, P. Li, Q. Li and Y. Yu, J. Chem. Eng. Data, 2018, 63, 3074 CrossRef CAS.
- M. Shi, X. Yu, G. He and Q. Li, Can. J. Chem. Eng., 2018, 96, 968 CrossRef CAS.
- M. Shi, G. He, F. Gan, X. Yu and Q. Li, J. Chem. Eng. Data, 2017, 62, 2183 CrossRef CAS.
- Z. Zhuang, J. Zhang and D. Liu, Huagong Xuebao, 2016, 67 DOI:10.11949/j.issn.0438-1157.20160391.
- Z. Zhuang, J. Zhang, X. Liu and D. Liu, J. Chem. Thermodyn., 2016, 190 CrossRef CAS.
- D. Oestreich, L. Lautenschütz, U. Arnold and J. Sauer, Fuel, 2018, 214, 39 CrossRef CAS.
- A. Ferre and J. Burger, Ind. Eng. Chem. Res., 2021, 60, 15256 CrossRef CAS.
- S. Schemme, Techno-ökonomische Bewertung von Verfahren zur Herstellung von Kraftstoffen aus H2 und CO2, Ph.D. Thesis, Aachen, 2020 Search PubMed.
- N. Schmitz, Production of poly(oxymethylene) dimethyl ethers from formaldehyde and methanol, Ph.D. Thesis, Scientific Report Series, Kaiserslautern, 2018 Search PubMed.
- N. Schmitz, C. F. Breitkreuz, E. Ströfer, J. Burger and H. Hasse, J. Membr. Sci., 2018, 564, 806 CrossRef CAS.
- A. Ferre, J. Voggenreiter, Y. Tönges and J. Burger, Motortech. Z., 2021, 82, 28 CrossRef.
- F. Mantei, M. Ouda and A. Schaadt, Method for Producing Polyoxymethylene Dimethyl Ethers, WO22013132 A1, 2022, https://worldwide.espacenet.com/patent/search/family/077168217/publication/WO2022013132A1?q=pn%3DWO2022013132, (last accessed July 2022).
- Ullmann's Encyclopedia of Industrial Chemistry: Formaldehyde, ed. A. W. Franz, H. Kronemayer, D. Pfeiffer, R. D. Pilz, G. Reuss, W. Disteldorf, A. O. Gamer and A. Hilt, Wiley-VCH Verlag GmbH & Co. KG, Weinheim, 2016 Search PubMed.
- J.-O. Drunsel, Entwicklung von Verfahren zur Herstellung von Methylal und Ethylal, Ph.D. Thesis, Scientific Report Series, Kaiserslautern, 2012 Search PubMed.
- H. Hasse, J.-O. Drunsel, J. Burger and U. Schmidt, Process for the production of pure Methylal, WO 2012/062822 A1, 2012.
- I. Bogatykh, T. Osterland, H. Stein and T. Wilharm, Energy Fuels, 2019, 33, 11078 CrossRef CAS.
- Kontinuierliche Destillation PD250 AD, https://asg-analytik.de/technikum/z1-sp2/glaskolonnen, (last accessed February 2023).
- A. Fink, C. H. Gierlich, I. Delidovich and R. Palkovits, ChemCatChem, 2020, 12, 5710 CrossRef CAS.
- A. Fink, Struktur-Aktivitätsbeziehungen von Zeolithen als feste Säurekatalysatoren in der Synthese von Oxymethylendimethylethern, Ph.D. Thesis, Aachen, 2022 Search PubMed.
- C. J. Baranowski, A. M. Bahmanpour and O. Kröcher, Appl. Catal., B, 2017, 217, 407 CrossRef CAS.
- Y. Tönges and J. Burger, Chem. Eng. Res. Des., 2022, 189, 572 CrossRef.
- F. Mantei, M. Kraume and O. Salem, Chem. Ing. Tech., 2022, 25, 912 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00147d |
| This journal is © The Royal Society of Chemistry 2023 |