
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic origins of accelerated hydrogenation of mixed alkylaromatics by synchronised adsorption over Rh/SiO2
Nikolay
Cherkasov
*ab,
Shusaku
Asano
 *c,
Yuta
Tsuji
e,
Kazuki
Okazawa
d,
Kazunari
Yoshizawa
d,
Hiroyuki
Miyamura
f,
Jun-ichiro
Hayashi
c,
Alexander A.
Kunitsa
g and
S. David
Jackson
h
*c,
Yuta
Tsuji
e,
Kazuki
Okazawa
d,
Kazunari
Yoshizawa
d,
Hiroyuki
Miyamura
f,
Jun-ichiro
Hayashi
c,
Alexander A.
Kunitsa
g and
S. David
Jackson
h
aSchool of Engineering, University of Warwick, Coventry, CV4 7AL, UK
bStoli Chem Ltd, Wellesbourne Campus, Wellesbourne, Coventry, CV35 9EF, UK. E-mail: n.cherkasov@stolichem.com
cInstitute for Materials Chemistry and Engineering, Kyushu University, Kasuga 816-8580, Japan. E-mail: shusaku_asano@cm.kyushu-u.ac.jp
dInstitute for Materials Chemistry and Engineering, Kyushu University, Fukuoka 819-0395, Japan
eFaculty of Engineering Sciences, Kyushu University, Kasuga 816-8580, Japan
fInterdisciplinary Research Center for Catalytic Chemistry, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Ibaraki 305-8565, Japan
gDepartment of Chemistry, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA
hDepartment of Chemistry, Joseph Black Building, University of Glasgow, Glasgow G12 8QQ, UK
First published on 14th March 2023
Abstract
Catalytic reactions of mixed substrates sometimes behave differently from those of individual substrates. For example, the hydrogenation of propylbenzene over Rh/SiO2 proceeds 120% faster in the presence of toluene. Such an acceleration effect does not agree with the well-accepted Langmuir–Hinshelwood reaction model. In this paper, we examined its mechanism experimentally and computationally. The hydrogenation experiment of vaporised aromatics confirmed that the acceleration was specific to the liquid phase with the isopropanol solvent. Direct adsorption measurements revealed that toluene adsorption synchronises with propylbenzene adsorption. Density functional theory calculations confirmed the associates of toluene and propylbenzene on the catalyst surface in the polar environment. The formation of associates increased the adsorption energy of toluene and decreased that of propylbenzene. Lowered adsorption energy reduces the activation barrier for catalytic reaction and intensifies the reaction rate beyond the Langmuir–Hinshelwood model prediction.
Introduction
Chemical processes are performed with either pure substrates or mixtures. Reactions with mixtures involve lower-value chemicals such as oil cracking or its renewable counterpart of bio-oil upgrading1 as well as higher-value chemicals such as natural products in food and cosmetics.2,3 Liquid phase reaction is common for the latter case. There are many unknowns in the interactions between the constituents in mixtures. For example, Miyamura et al.4 conducted liquid phase hydrogenation of a neat toluene–pyrrole mixture over an Rh-based catalyst with 1 atm of H2 at 50 °C. Surprisingly, the reaction rate of pyrrole hydrogenation was about three times faster than in the case where pyrrole was used as pure starting material. The reason was not discussed in their study. Sanyal et al.5 conducted the electrocatalytic hydrogenation of furfural and benzaldehyde at 25 °C in an aqueous buffer solution. The reaction was unexpectedly accelerated in the presence of phenol. They attributed the acceleration effect to the formation of H2-bonded complexes, but the evidence was limited.6 Alshehri et al.7 studied aromatic hydrogenation of toluene, ethylbenzene, and propylbenzene in isopropanol over a Rh/SiO2 catalyst. They showed that simple aromatic compounds behave unexpectedly in mixtures compared to individual compounds (Table 1). Toluene hydrogenation accelerates in a mix with ethylbenzene. Hydrogenation of propylbenzene in combination with toluene more than doubles. Similarly, a ternary mixture of aromatic compounds increases the propylbenzene hydrogenation rate, showing that this effect is widely applicable.| Toluene/ethylbenzene | Toluene/propylbenzene | Ternary mixture | |
|---|---|---|---|
| a Conditions: 4 bar H2, 25 mmol L−1 of aromatics in isopropanol, 50 °C, Rh/SiO2 catalyst. Reaction rates were based on the conversion of aromatics. | |||
| Toluene | 16% | −61% | −77% |
| Ethylbenzene | −42% | −77% | |
| Propylbenzene | +121% | +27% | |
There are extensive mechanistic studies on the hydrogenation of pure aromatic substrate.8–11 However, a reasonable mechanistic explanation for the reaction acceleration in mixtures is not found. In addition, a mixture's acceleration effect qualitatively contradicts the Langmuir–Hinshelwood model. Competitive adsorption of the substrates for the catalyst active sites is deduced from the Langmuir–Hinshelwood model.1,12–14 The competition results in the deceleration of the reaction rate for all the mixed substrates. Although many surface science studies have revealed much more complicated phenomena than the Langmuir–Hinshelwood model understanding,15 the Langmuir–Hinshelwood model has been utilized as the most accurate, mechanism-based kinetic model.16–19 Some studies have tried to expand the Langmuir–Hinshelwood model to include the mutual interaction of absorbate.20,21 However, the basic concept of competitive adsorption of the mixed substrate has not been challenged.
Increasing the reaction rate in mixtures raises many questions. Why the canonical Langmuir–Hinshelwood modelling of competitive adsorption fails even for the simple molecules of alkylaromatics? What is the critical factor(s) to accelerate the reaction? Could we design a catalyst or a process that efficiently utilises such effects? These questions can only be attempted after we know a phenomenon at the level of molecular chemistry on the catalyst surface. In this study, we focused on the hydrogenation of the toluene and propylbenzene mixture (Table 1) because it is the simplest case among the reports on the reaction acceleration in the mixture. Toluene and propylbenzene only differ with the side alkyl chain lengths, but their behaviour as a combination is remarkable and unpredictable. Mechanistic investigation of their interaction would have generality and applicability to many other systems. We combined hydrogenation experiments, adsorption measurements, and computational simulations to reveal the unique interactions of aromatic molecules over the catalyst.
Method
Materials
The Rh/SiO2 catalyst (M01074, Johnson Matthey) was used as in Alshehri et al.7 The catalyst characteristics supplied by Johnston Matthey show a metal loading of 2.5 wt%, a hydrogen chemisorption capacity of 4.7 ± 0.5 m2 g−1, corresponding to metal dispersion of 43% and an average Rh crystallite size of 2.6 nm. The silica support used for M01074 was supplied by Davison catalysts. It has a total surface area of 311 m2 g−1 with an average pore diameter of 13.9 nm. Toluene (>98%, Fischer Scientific), propylbenzene (>98%, Acros Organics), and isopropanol (99%, Fisher Scientific) were used as received.Gas phase hydrogenation
The gas phase hydrogenation setup (Fig. 1) contained a reactor with 41.2 mg of the catalyst placed between two layers of quartz wool. Toluene and propylbenzene were evaporated in separate vessels at 14.0 °C by passing hydrogen flows. The streams containing vapours were fed to the reactor. Hydrogen was added for further dilution. The vapours passed through the catalyst bed at 50 °C. An online gas chromatograph (GC, GC-2010, Shimadzu) analysed the outlet stream. A flame ionisation detector (FID) and a capillary column (Equity-1, 90 m × 0.53 mm × 1 μm, MilliporeSigma) were used. Concentration of toluene, propylbenzene or both were set around 0.2 vol%. The total gas flow rate was 32–40 mL min−1. In a separate experiment, isopropanol was added to the toluene vessel to study its effect on the reaction rate.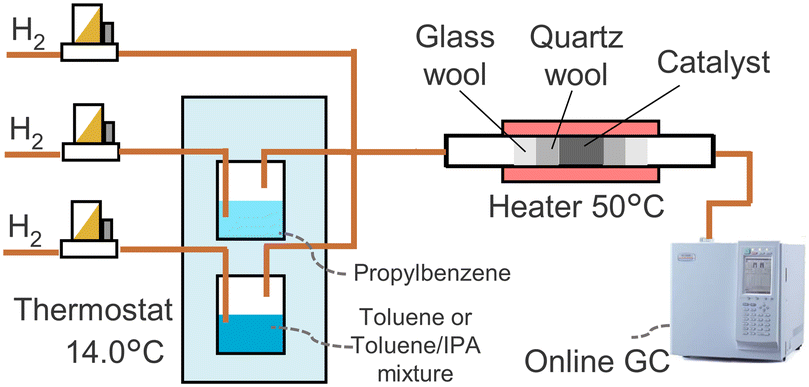 | ||
| Fig. 1 Gas phase hydrogenation setup. | ||
Liquid phase adsorption
An autonomous system controlled with an OpenFlowChem platform22 performed liquid phase adsorption experiments. The experimental system contained a syringe pump (WPI) with a 1.00 mL precision syringe (SGE) and a fraction collector (AutoSam 360, HiTec Zang) containing 13 mm vials with septa. The system executed a pre-defined list of commands, such as syringe washing and liquid injections into vials to obtain a desired concentration of compounds. 20–200 mg of the catalysts or catalyst support was placed into vials and reduced in a 5 vol% H2/N2 flow at 350 °C and passivated in 0.5 vol% O2/N2. The sample addition was performed within 2–3 hours after the catalyst reduction. Blank tests were performed using empty vials. Details of the operation procedures and blank tests were described in the ref. 23. 1 mL of isopropanol solution containing 0.6–250 μM of aromatic compounds was injected into vials. After reaching the equilibrium, the concentration of the aromatic compounds was promptly analysed by GC (GC-2010, Shimadzu) with the FID and a capillary column (CP-Sil 5, 30 m × 0.25 mm × 1 μm, Agilent).Computational analysis
The surface of Rh(111) was modelled by a periodic 18.59 Å × 18.79 Å unit cell slab consisting of three atomic layers. A 20 Å thick vacuum space was added on the surface. The coordinates of the atoms in the lowest layer in the slab were kept fixed at their optimized bulk positions. The optimized adsorption structures were visualized using VESTA.24 The initial structures for geometry optimization of adsorption structures at the DFT level were generated using the quench dynamics method with the Forcite module implemented in the Materials Studio software.25 The universal force field26 was used with the conditions of an initial temperature of 400 K, NVE ensemble, the time step of 1 fs, and the simulation time of 100 ps. The quench-step number was set to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Five most stable structures obtained in the quench dynamics were used as the initial structures for the further DFT calculations. The Vienna ab initio simulation package (VASP 5.4.4)27–29 was used to perform the DFT calculation. The Perdew–Burke–Ernzerhof (PBE)30 exchange–correlation functional was used. The Kohn–Sham equations were solved with a plane-wave basis set using the projector-augmented wave method.31,32 The cutoff energy for the plane-wave basis set was set to 400 eV. The convergence threshold for self-consistent field iteration was set to 1.0 × 10−5 eV. The atomic coordinates were relaxed until the forces on all atoms were less than 0.03 eV Å−1. The Brillouin zone was sampled at the Γ point only. Grimme's D3 dispersion correction formalism with Becke–Johnson damping was adopted.33 The effect of solvent was further investigated for the most stable adsorption structure obtained in the DFT calculations. We employed the implicit solvation method as implemented in VASP sol code.34 We used the relative permittivity value of 19.264 for the isopropanol environment. The adsorption structure was again optimized in the solvent environment.
000. Five most stable structures obtained in the quench dynamics were used as the initial structures for the further DFT calculations. The Vienna ab initio simulation package (VASP 5.4.4)27–29 was used to perform the DFT calculation. The Perdew–Burke–Ernzerhof (PBE)30 exchange–correlation functional was used. The Kohn–Sham equations were solved with a plane-wave basis set using the projector-augmented wave method.31,32 The cutoff energy for the plane-wave basis set was set to 400 eV. The convergence threshold for self-consistent field iteration was set to 1.0 × 10−5 eV. The atomic coordinates were relaxed until the forces on all atoms were less than 0.03 eV Å−1. The Brillouin zone was sampled at the Γ point only. Grimme's D3 dispersion correction formalism with Becke–Johnson damping was adopted.33 The effect of solvent was further investigated for the most stable adsorption structure obtained in the DFT calculations. We employed the implicit solvation method as implemented in VASP sol code.34 We used the relative permittivity value of 19.264 for the isopropanol environment. The adsorption structure was again optimized in the solvent environment.
Results
Gas phase hydrogenation
We hypothesized that interactions among substrates, catalyst surface, and solvent caused the reaction acceleration in a mixture. Alshehri et al.7 observed acceleration in the liquid phase with an isopropanol solvent. First, we investigated the gas phase reaction at the same temperature as in the previous liquid phase study to clarify whether the acceleration is specific to the liquid phase. Toluene and propylbenzene were individually hydrogenated at 100 μmol L−1 and 85 μmol L−1. Hydrogenation was also performed with a mixture of 100 μmol L−1 of toluene and 85 μmol L−1 of propylbenzene and in the presence of isopropanol at 25 μmol L−1.Table 2 summarises the reaction rate per catalyst. The reaction rate was calculated from the conversion of toluene and propylbenzene. Methylcyclohexane and propylcyclohexane were obtained, while no side product was detected in the chromatogram. The gas phase reaction showed a lower reaction rate than the liquid phase. The much lower concentration of aromatics could be the reason. In the case of gas phase reaction, hydrogenation of individual substrates showed the highest reaction rate. The hydrogenation of propylbenzene did not change in the presence of toluene, while the reaction rate of toluene decreased in the mixture with propylbenzene. Both observations are in perfect agreement with the Langmuir–Hinshelwood model understanding. Propylbenzene, a stronger-adsorbed compound, occupies the catalyst sites and displaces any toluene. Hence adsorption of propylbenzene in the gas phase and the reaction rate are not affected. Toluene, on the contrary, is displaced from the catalyst by propylbenzene. Hence, its reaction in the mixture with propylbenzene decreases. The addition of isopropanol further slowed down the reaction by blocking available sites.
| Gas phase | Liquid phase | ||||
|---|---|---|---|---|---|
| Individual | Mixture | Mixture with isopropanol | Individual | Mixture | |
| Toluene | 18.7 | 15.0 | 8.4 | 362 | 141 |
| Propylbenzene | 14.7 | 14.2 | 8.5 | 65 | 144 |
In summary, the gas phase reaction well followed the Langmuir–Hinshelwood model, and no acceleration with the mixture was observed. Thus, the interaction of intermediates on the catalyst surface, such as transfer hydrogenation,35 could not be the reason for the accelerated reaction of propylbenzene in the mixture. A deeper examination for the interactions among substrates and catalyst in the liquid phase is necessary for mechanistic study.
Liquid phase adsorption
Then, we focused on the substrates' adsorption to the catalyst in the solvent. For reference, adsorption measurements with 50 mg of the SiO2 catalyst support were conducted with 50–250 μmol L−1 of toluene or propylbenzene solution in isopropanol. 16 measurements resulted in a 4.0 ± 3.8% “increase” in the concentration of aromatic compounds compared to the blank measurements. The increase rate was independent of the initial concentration. The preferential adsorption of isopropanol solvent onto SiO2 explain the results. SiO2 is a porous hydrophilic material that can remove water from organic solvents, including toluene.36 The water sorption capacity of SiO2 is as high as 0.3–0.8 g g−1.37 0.6 g g−1 adsorption of isopropanol onto the SiO2 can cause a 3.9% decrease in the bulk solvent volume and a 4.0% increase in the bulk concentration of the aromatic compounds. Thus, the adsorption of the aromatic compounds to the Rh surface (A) was calculated with eqn (1) considering the adsorption of the solvent by SiO2:| A = V0C0 − V1C1 = V0C0 − (V0 − αwSiO2)C1. | (1) |
Fig. 2 shows the results of the liquid phase adsorption of toluene and propylbenzene adsorbed independently over the Rh catalysts in an isopropanol solvent. The measurements were suffered from the large errors. The automated adsorption measurement system used in this study has achieved accurate adsorption measurement down to adsorption of 0.01 μmol g−1 with an error of ±0.005 μmol g−1 in our previous study.23 The catalyst support would be the reason for the significant error in this study. The previous study used Pd/CaCO3 catalyst with a BET surface area of 5 m2 g−1. No concentration change to the liquid sample was confirmed with the exposure to CaCO3 support. Disturbance in the concentration by the SiO2 support owing to its high surface area and hydrophilicity, makes it difficult to measure the adsorption on the catalyst. Regardless of the large errors, notably lower adsorption of toluene compared to propylbenzene can be confirmed. Stronger adsorption of a larger molecule is generally confirmed for benzene derivatives38 and polycyclic aromatics.39 The Langmuir isotherm is omnipresent for analysing the adsorption data. It is mandatory to measure both the high and low-saturation region to determine the two parameters of maximum adsorption capacity of Amax and adsorption constant K. However, the amount of propylbenzene adsorbed was 1 μmol gcat−1 at the maximum. The resulting aromatic-to-surface Rh site ratio was only about 0.008. It is difficult to increase concentration. Because the adsorbed amount is derived from the subtraction of two concentrations, a larger concentration requires much smaller measurement errors. As a result, the Langmuir isotherm fitting had little difference from the linear fitting as shown in Fig. 2. Thus, we focused on the adsorption behaviours at the low-saturation region with linear fittings.
 | ||
| Fig. 2 Liquid phase adsorption of toluene and propylbenzene from separate solutions in isopropanol over a Rh/SiO2 catalyst. Dashed lines are fitted using the least squares approach. A solid grey line is the Langmuir isotherm fitted for propylbenzene adsorption with the GRG nonlinear solving method minimizing the sum of absolute error, Amax = 5.24 μmol gcat−1, KL = 550 L mol−1. | ||
In the next step, we studied the adsorption of an equimolar mixture of toluene and propylbenzene to see if there are any mixture effects on the adsorption behaviour that may explain the observed reaction rates (Table 1). The adsorption behaviours in Fig. 3 show a substantial change – toluene adsorption was notably higher compared to the toluene-only solution (Fig. 2). Moreover, the adsorption values for toluene and propylbenzene are almost the same for all the concentration sets. Therefore, the adsorption of these species seems to synchronise in the 1:1 mixture. The slopes are similar to that for the adsorption of pure propylbenzene solution (Fig. 2). In other words, the adsorption of toluene increases by the co-adsorption with propylbenzene. Although a large error was inevitable for the Rh/SiO2 catalyst, the toluene and propylbenzene adsorption as mixture all resulted in similar values in Fig. 3. It implies two points. First, the primary error source in the current adsorption measurement was the adsorption of isopropanol to SiO2. Thus, a set of adsorption measurements for toluene and propylbenzene concentration in the same vial has the same relative error. Second, toluene and propylbenzene were adsorbed on the Rh surface as the equimolar associates. Otherwise, the synchronized adsorption, as in Fig. 3, is unrealistic.
 | ||
| Fig. 3 Liquid phase adsorption of toluene and propylbenzene in isopropanol from a mixture of 1:1 mol. Dashed lines are fitted using the least squares approach. | ||
When the toluene to propylbenzene ratio in the mixture decreases, the adsorption behaviour approaches that observed for the individual compounds. The adsorption of toluene was weak in a mixture of 2:25 mol toluene–propylbenzene (Fig. 4(a)). When the concentration of toluene to propylbenzene ratio increases to 10:25 mol (Fig. 4(b)), toluene adsorption becomes strong with almost the same slope as the propylbenzene. These results suggest that propylbenzene significantly influenced the adsorption of toluene, but such interaction diminished with the excess amount of propylbenzene.
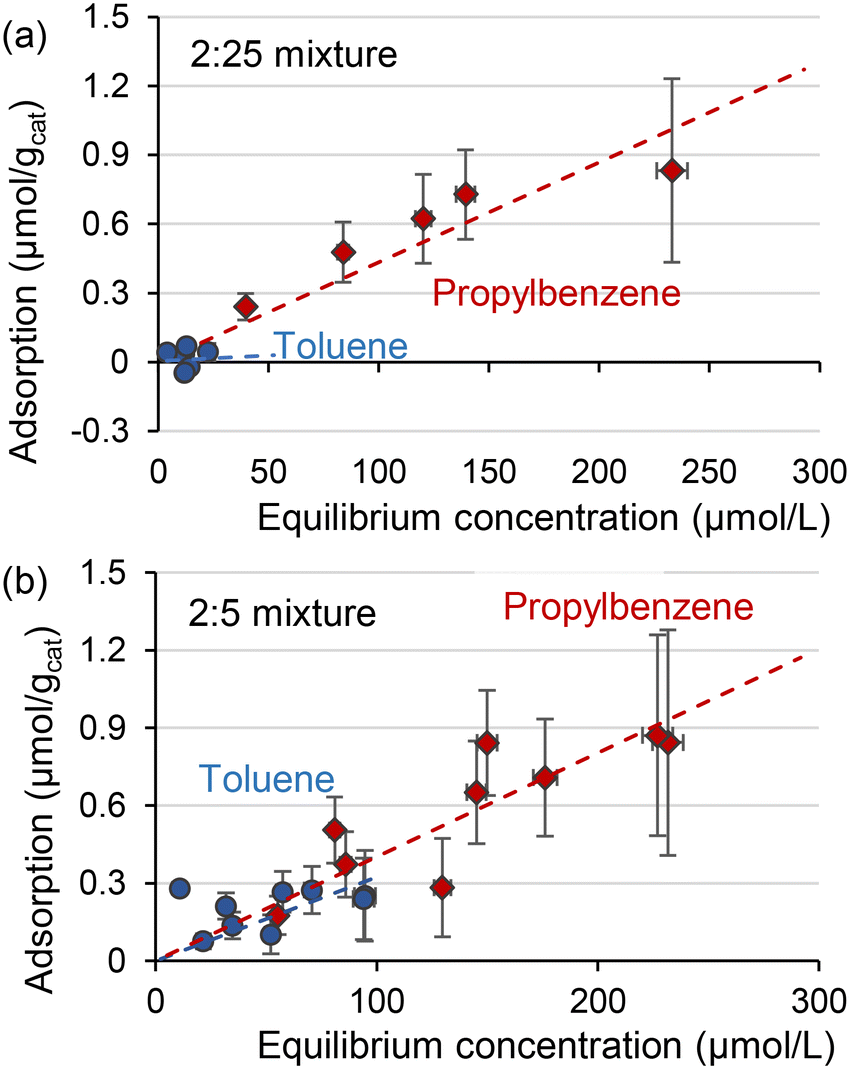 | ||
| Fig. 4 Liquid phase adsorption of toluene and propylbenzene in isopropanol from a mixture of (a) 2:25 mol or (b) 2:5 mol. | ||
Computational analysis of association and adsorption
Adsorption of one or two molecules of toluene and propylbenzene to the Rh(111) was computationally investigated. The Rh(111) surface was chosen because the (111) surface is the most abundant surface for precious metal nanoparticles,40 and the catalytic activity of Rh(111) for aromatic hydrogenation was confirmed in the previous study.41Fig. 5 shows the adsorption structures of two aromatic molecules on the Rh surface. In all the cases, two aromatic molecules spontaneously neighboured. It suggests that aromatic molecules interact as associates on the surface. The most stable arrangement of the two molecules differed with the combination. Two propylbenzene molecules were the most stable when they aligned in the same direction (Fig. 5(b)). Toluene–toluene and toluene–propylbenzene systems showed the most stable conformation with 150° rotation. The distance between the two molecules was more significant for the toluene–propylbenzene system than the others. The association between three or more molecules may have other structures. However, such investigation is problematic due to the complexity and the computational cost.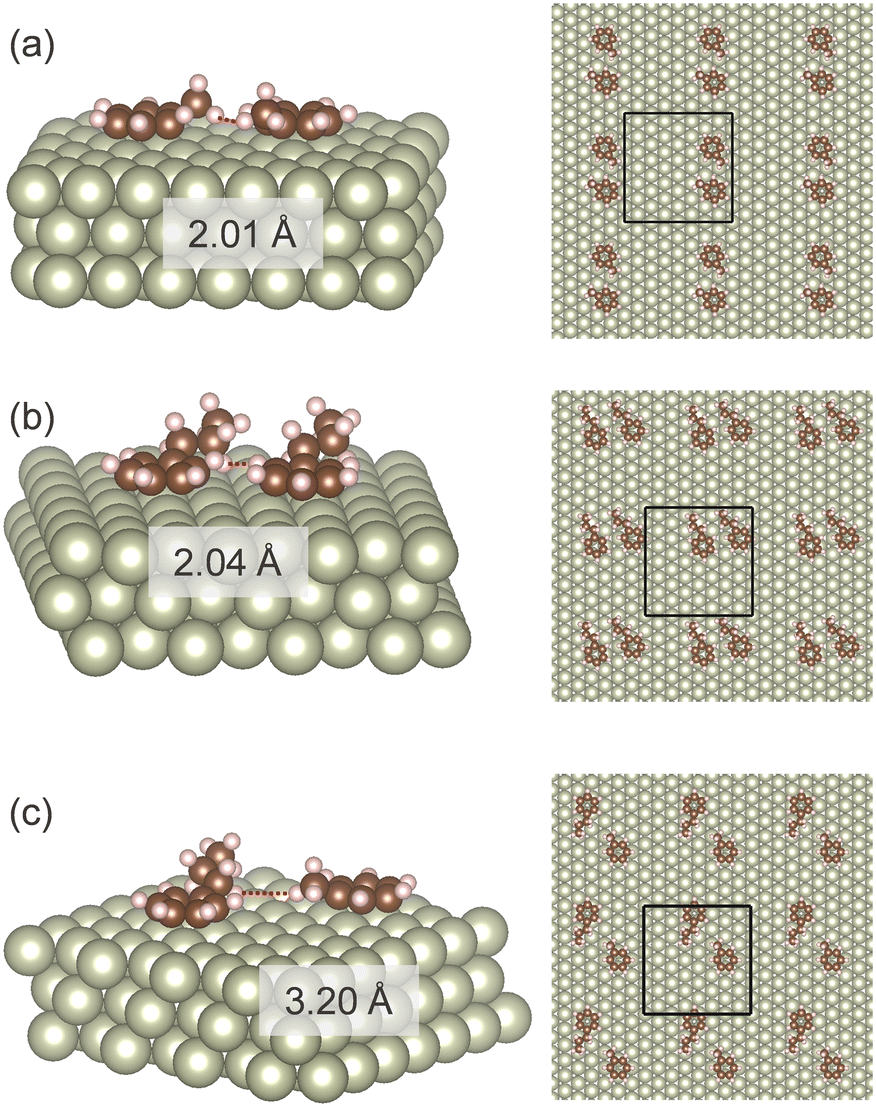 | ||
| Fig. 5 Adsorption structures of toluene and propylbenzene on Rh(111) surface under solvent effects; (a) toluene–toluene, (b) propylbenzene–propylbenzene, and (c) toluene–propylbenzene. Red dotted lines indicate distances of two molecules. Black rectangles indicate periodic boundaries. | ||
Table 3 summarises the adsorption energy obtained in the DFT calculations. The calculated adsorption energies are normalised by the number of aromatic rings to maintain a valid comparison between mono- and bi-molecular associates. A single molecule of propylbenzene showed higher adsorption energy than a single molecule of toluene owing to the larger number of interacting atoms in propylbenzene. Adsorption energy decreased for symmetrical bi-molecular associates by −0.10 to −0.02 eV. These data confirm the effect of decreasing adsorption energy with increasing surface coverage.42 Consideration of the isopropanol environment increased the adsorption energy for all the cases. Previous computational studies have also confirmed the increased adsorption energy to the metal surface in a polar protic solvent for furfural/methanol43 and phenol/water systems.44 Entry (e) in Table 3 shows that the nonsymmetric toluene + propylbenzene associates are noticeably more stable than the symmetric toluene associates. Moreover, the nonsymmetric associates of entry (e) is energetically favourable than the pair of symmetric associates of entry (f). The isopropanol environment enhanced the stabilization with co-adsorption. If we assume the adsorption energy of a toluene molecule in a binary mixture as half of the total adsorption energy, as high as 0.19 eV is accounted for the toluene adsorption enhancement by the co-adsorption in isopropanol. For a propylbenzene molecule, the presence of a toluene molecule has a negative stabilization effect. The association among propylbenzene molecules is the most stable.
Adsorption energies calculated in DFT agree well with the liquid phase adsorption measurement. First, the higher adsorption energy of propylbenzene than toluene is in line with the adsorption measurements of pure substrates (Fig. 2). Second, enhanced toluene adsorption in the mixture (Fig. 3) matches the enhanced toluene adsorption in the binary combination (Table 3). Third, the highest adsorption energy of propylbenzene–propylbenzene association on the Rh surface (Fig. 5(b)) can explain the negligible adsorption of toluene with a large amount of propylbenzene (Fig. 4(a)). The Rh surface is occupied with the propylbenzene molecules if its concentration is much higher than toluene. In that case, it is hard for a toluene molecule to adsorb on the surface to associate with a propylbenzene.
Discussion
The rate equations for the hydrogenation of toluene and propylbenzene with the conventional Langmuir–Hinshelwood model are expressed by eqn (2) and (3), respectively: | (2) |
 | (3) |
1) Reaction of an adsorbed aromatic molecule and a hydrogen atom is the rate-determining step.
2) Adsorption of toluene, propylbenzene, and hydrogen follows the Langmuir isotherm and competes for an empty site.
The denominator in the equation represents competitive adsorption. The decrease in the reaction rate in the gas phase mixture (Table 2) can be explained by the increase in the denominator value. With the larger Kpbz and smaller Ktol, rtol is significantly reduced by the presence of KpbzCpbz term in the denominator. rpbz is less affected by the KtolCtol term. The addition of isopropanol vapour adds another term to the denominator to further decrease the reaction rate, as in Table 2. Note that we do not need to consider the adsorption of isopropanol with the liquid phase reaction. The high condensation enthalpy of isopropanol45 makes adsorption to the metal surface less favourable than in the gas phase.
The conventional discussion with the Langmuir–Hinshelwood model assumes k, and K are independent of the solution composition. However, our experimental results and DFT calculations suggest that pure and mixed substances' adsorption energies can differ. Thus, Ktol and Kpbz are dependent on the mixture composition. In addition, KH2 would also change with the formation of aromatic associates on the surface. The configuration difference of aromatic associates on the metal surface (Fig. 5) would significantly affect the number of sites available for hydrogen. More importantly, the activation energy for surface reaction should be affected by the adsorption energy. The lowered adsorption energy of aromatic species would reduce the activation barrier for the hydrogenation reaction.
The formation of aromatic association on the Rh surface can explain the increased reaction rate of propylbenzene in the mixture (Table 1). The association with toluene changes the parameters in eqn (3). Kpbz slightly decreases while Ktol increases. The decrease in the adsorption energy pulls down the activation barrier and increases kpbz. The surface-adsorbed hydrogen concentration may also increase due to the change in the adsorption configuration of aromatics. These changes can enhance the reaction rate. For the reaction rate of toluene, ktol decreases significantly due to the increased activation barrier for hydrogenation of the stabilized adsorbed species. Judging from the gas phase reaction, the liquid phase environment in isopropanol induces the acceleration effect. As suggested by the DFT calculations, isopropanol facilitates the association of aromatic molecules by changing the environmental polarity.
The canonical kinetic modelling approach for reactions with a mixture is rate analysis with pure individual components. A kinetic and adsorption constant for a pure substrate is believed to apply to an overall rate expression for mixed substrates.46 However, an association of the mixed substrate can change these constants. Consequently, the reaction rate for mixed substrates can be unpredictable from the integration of separate experiments with pure substrates. Investigation for the mixture is necessary when the interaction of substrates matters. Adsorption analysis, as in this study, would also be essential to understand the catalytic transformation process of mixtures quantitatively. After that, we could open new possibilities for the rational design of catalysts and catalytic processes.
Conclusions
Aromatic ring hydrogenation in alkylaromatic mixtures differs substantially from the individual components. We have studied the origin of the unique reaction acceleration phenomenon in the toluene/propylbenzene mixture.Hydrogenation in a gas phase clarified that no reaction acceleration occurs in the gas phase. Thus, reactions of intermediates such as hydrogen transfer were not responsible for the acceleration. Direct adsorption studies confirmed the adsorption of toluene was coupled with that of propylbenzene. The density functional theory calculation demonstrated that toluene and propylbenzene could form associates on Rh(111) surface. The decreased adsorption energy of propylbenzene was the origin of the reaction acceleration.
We have revisited the well-accepted Langmuir–Hinshelwood model for a solid-catalysed reaction and discussed its limitations applying to a mixture. Establishing a versatile and systematic procedure to investigate the reaction of a mixture is crucial for future studies.
Author contributions
Cherkasov: conceptualization, investigation, project administration, software, writing – original draft. Asano: data curation, writing – original draft, formal analysis, validation. Tsuji: methodology, formal analysis, visualization. Okazawa: investigation. Yoshizawa: resources, supervision. Miyamura: writing – review & editing, validation. Hayashi: writing – review & editing. Kunitsa: investigation. Jackson: conceptualization, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NC is grateful to IChemE for Andrew Fellowship that greatly helped in carrying out and completing the work. NC is also grateful for the Royal society for the International Exchanges 2019 Cost Share grant, 193016. SA is grateful to a Grant-in-Aid for Transformative Research Areas (B) “Low-entropy” (grant number JP21H05083) and Grant-in-Aid for Early-Career Scientists (grant number JP22K14536). YT is grateful to a Grant-in-Aid for Transformative Research Areas (A) “Supra-ceramics” (grant number JP22H05146). KY is grateful to JST-CREST “Innovative Catalysts” (grant number JPMJCR15P5). The computations in this work were performed using the computer facilities at the Research Institute for Information Technology, Kyushu University and at the Supercomputer Center, the Institute for Solid State Physics, the University of Tokyo.References
- N. Cherkasov, V. Jadvani, J. Mann, Y. B. Losovyj, Z. B. Shifrina, L. M. Bronstein and E. V. Rebrov, Fuel Process. Technol., 2017, 167, 738–746 CrossRef CAS.
- A. P. B. Ribeiro, R. Grimaldi, L. A. Gioielli and L. A. G. Gonçalves, Food Res. Int., 2009, 42, 401–410 CrossRef CAS.
- M. Zhou, X. Chen, C. Gao, L. Ni, X. Wang, W. Zhang and S. Ren, RSC Adv., 2021, 11, 30078–30087 RSC.
- H. Miyamura, A. Suzuki, T. Yasukawa and S. Kobayashi, J. Am. Chem. Soc., 2018, 140, 11325–11334 CrossRef CAS PubMed.
- U. Sanyal, K. Koh, L. C. Meyer, A. Karkamkar and O. Y. Gutiérrez, J. Appl. Electrochem., 2021, 51, 27–36 CrossRef CAS.
- U. Sanyal, S. F. Yuk, K. Koh, M.-S. Lee, K. Stoerzinger, D. Zhang, L. C. Meyer, J. A. Lopez-Ruiz, A. Karkamkar, J. D. Holladay, D. M. Camaioni, M.-T. Nguyen, V.-A. Glezakou, R. Rousseau, O. Y. Gutiérrez and J. A. Lercher, Angew. Chem., 2021, 133, 294–300 CrossRef.
- F. Alshehri, H. M. Weinert and S. D. Jackson, React. Kinet., Mech. Catal., 2017, 122, 699–714 CrossRef CAS.
- M. Saeys, M. F. Reyniers, J. W. Thybaut, M. Neurock and G. B. Marin, J. Catal., 2005, 236, 129–138 CrossRef CAS.
- R. A. Sánchez-Delgado and M. Rosales, Coord. Chem. Rev., 2000, 196, 249–280 CrossRef.
- T. Takahashi, K. Yamashita, T. Kai and I. Fujiyoshi, Can. J. Chem. Eng., 1986, 64, 1008–1013 CrossRef CAS.
- S.-C. Qi, X.-Y. Wei, Z.-M. Zong and Y.-K. Wang, RSC Adv., 2013, 3, 14219 RSC.
- C. Berguerand, I. Yuranov, F. Car and L. Kiwi-Minsker, J. Phys. Chem. C, 2014, 10 Search PubMed.
- N. Cherkasov, A. O. Ibhadon, A. McCue, J. A. Anderson and S. K. Johnston, Appl. Catal., A, 2015, 497, 22–30 CrossRef CAS.
- P. D. Vaidya and V. V. Mahajani, Ind. Eng. Chem. Res., 2003, 42, 3881–3885 CrossRef CAS.
- M. Bowker, ACS Nano, 2007, 1, 253–257 CrossRef CAS PubMed.
- R. Krishna, R. Baur and J. M. van Baten, React. Chem. Eng., 2017, 2, 324–336 RSC.
- S. Mondal, H. Malviya and P. Biswas, React. Chem. Eng., 2019, 4, 595–609 RSC.
- A. S. May, S. M. Watt and E. J. Biddinger, React. Chem. Eng., 2021, 6, 2075–2086 RSC.
- B. Jia, X. Sun, M. Chen, J. Jian, K. You, H. Luo, Y. Huang, X. Luo, B. Jin, N. Wang and Z. Liang, React. Chem. Eng., 2021, 6, 1854–1868 RSC.
- R. Krishna and R. Baur, Chem. Eng. Sci., 2005, 60, 1155–1166 CrossRef CAS.
- A. M. Gómez-Marín and J. P. Hernández-Ortiz, J. Phys. Chem. C, 2014, 118, 2475–2486 CrossRef.
- N. Cherkasov, Y. Bai, A. J. Expósito and E. V. Rebrov, React. Chem. Eng., 2018, 3, 769–780 RSC.
- N. Cherkasov, D. Yu. Murzin, C. R. A. Catlow and A. Chutia, Catal. Sci. Technol., 2021, 11, 6205–6216 RSC.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Dassault Systèmes, BIOVIA Materials Studio, Version 2020, 2020 Search PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- I. Shimada, C. Uno, Y. Watanabe and T. Takatsuka, Fuel Process. Technol., 2022, 232, 107267 CrossRef CAS.
- D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351–8354 CrossRef CAS PubMed.
- M. Suzuki, Adsorption engineering, Kodansha, Elsevier, Tokyo, Amsterdam, 1990 Search PubMed.
- M. A. Hernández, J. A. Velasco, M. Asomoza, S. Solís, F. Rojas and V. H. Lara, Ind. Eng. Chem. Res., 2004, 43, 1779–1787 CrossRef.
- S. J. Jenkins, Proc. R. Soc. A, 2009, 465, 2949–2976 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- C. J. Kliewer, M. Bieri and G. A. Somorjai, J. Phys. Chem. C, 2008, 112, 11373–11378 CrossRef CAS.
- S. G. Podkolzin, R. M. Watwe, Q. Yan, J. J. de Pablo and J. A. Dumesic, J. Phys. Chem. B, 2001, 105, 8550–8562 CrossRef CAS.
- J. Wang, C.-Q. Lv, J.-H. Liu, R.-R. Ren and G.-C. Wang, Int. J. Hydrogen Energy, 2021, 46, 1592–1604 CrossRef CAS.
- J. Akinola, I. Barth, B. R. Goldsmith and N. Singh, ACS Catal., 2020, 10, 4929–4941 CrossRef CAS.
- S. Zhou, C. Qian and X. Chen, Catal. Lett., 2011, 141, 726–734 CrossRef CAS.
- M. Kawase, H. Fujitsuka, H. Nakanishi, T. Yoshikawa and K. Miura, Ind. Eng. Chem. Res., 2010, 49, 10341–10347 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |