
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Suitable commercial catalysts for the synthesis of oxymethylene dimethyl ethers†
Franz
Mantei
 *a,
Sebastian
Kopp
a,
Anna
Holfelder
a,
Elisa
Flad
a,
Daniela
Kloeters
a,
Matthias
Kraume
b and
Ouda
Salem
*a
*a,
Sebastian
Kopp
a,
Anna
Holfelder
a,
Elisa
Flad
a,
Daniela
Kloeters
a,
Matthias
Kraume
b and
Ouda
Salem
*a
aThermochemical Processes Department, Division Hydrogen Technologies, Fraunhofer Institute for Solar Energy Systems, Heidenhofstr. 2, 79110, Freiburg, Germany. E-mail: ouda.salem@ise.fraunhofer.de
bChair of Chemical Engineering, Technische Universität Berlin, Str. des 17. Juni 135, MAR 2-1, 10623 Berlin, Germany
First published on 13th January 2023
Abstract
OME have diesel fuel like properties with almost soot-free combustion, which can enable a reduction of nitrogen oxides. This makes them promising candidates for internal combustion engines as blends or neat fuel. Moreover, OME are addressed as environmentally benign solvents and as hydrogen dense carriers for fuel cells. OME are produced from methanol which can be produced sustainably, allowing a significant overall reduction of CO2 emissions. Various catalysts have been investigated for OME synthesis focusing on selectivity and activity. This study concentrates on commercial heterogeneous catalysts and compares not only the conversions, selectivities and the target product yield but also the activity, side product formation and thermal stability of the synthesis products. Various ion exchange resins, zeolites and Nafion catalysts were applied for the OME synthesis in a batch autoclave at 60 °C for the aqueous reaction systems methanol/paraformaldehyde and the anhydrous reaction system OME1/trioxane. Investigations of the synthesis products in a micro distillation setup showed that all applied catalysts lead to active species in the synthesis product, negatively impacting its thermal stability. This indicates that a synthesis product handling step is necessary prior to the downstream purification. Based on these investigations, ion exchange resins are identified as the most suitable for industrial OME synthesis due to their higher activity and lower side product formation.
1. Introduction
Anthropogenic climate change already causes devastating consequences all over the world with an increasing mean temperature, rising sea levels and extreme weather events. To mitigate and finally reverse the accelerating consequences our mindset, behavior and actions need to shift from a fossil-based to a carbon neutral and finally carbon negative as well as circular economy. From the six main technological avenues addressed by the IRENA world energy transitions outlook 20221 and in the net zero emissions by 2050 Scenario (NZE) of the International Energy Agency2 one major part of the solution is the comprehensive use of renewable energy resources providing sustainable heat and power that can be integrated into different sectors by storing the energy in various energy carriers.3 The power-to-X (PtX) concept enables the chemical storage of renewable electricity by the conversion of H2O to H2via water electrolysis. Combined with CO2 or N2, H2 can be converted to sustainable energy carriers and chemicals which can be used for the hard to electrify applications such as the chemical and steel industries but also for seasonal and large-scale energy storage. Furthermore, for several transportation modes such as aviation, shipping, heavy duty and other off-road machines, dense liquid fuels will be further needed. One important sustainable energy carrier is methanol which can be used as a fuel directly or upgraded to other energy carriers and chemicals, such as oxymethylene dimethyl ethers (OME). Their structure H3C–O–(CH2O)n–CH3 results in different physical and chemical properties depending on the chain length n.4 Besides their promising properties as non-toxic solvents5 and CO2 absorbents,6 OME show attractive fuel properties. Due to similar fuel properties to diesel fuel, good solubility with diesel fuel and beneficial combustion behavior, a mixture of OME with the chain lengths n = 3–5 is a promising alternative or additive to diesel fuel.7,8 The high oxygen content and absence of C–C bonds enable quasi soot-free combustion, which can be utilized to reduce NOx emissions.4 The structure of OME leads to a higher gravimetric density but reduced lower heating values (LHV) than diesel fuel leading to higher fuel consumption for the same driving distance. Due to the production of OME based on sustainably produced methanol, the carbon footprint of OME can be significantly reduced by up to 93% in comparison to diesel fuel.9–12The synthesis of OME was investigated for various catalyst systems, feed mixtures and under different reaction conditions.13–22 Besides the optimization of specific catalyst properties, some already available commercial catalysts led to promising results. However, due to the application of different feed mixture and reaction conditions a direct comparison of the results is complicated. Furthermore, besides the conversion, selectivity, activity, reaction kinetics and side product formation, the thermal stability of the synthesis product is an important parameter for the process design but was barely mentioned in the literature.
Objectives of this work
The main objective of this work is a consistent comparison of commercial heterogeneous catalysts for the OME synthesis to identify suitable catalysts available for industrial scale applications. A secondary objective is the investigation of the thermal stability of the OME synthesis products. This indicates if the synthesis product can directly be separated in a cascade of distillation columns or if a neutralization step should be considered after the synthesis to sufficiently improve the thermal stability of the mixture and prevent reverse reactions towards shorter chain OME. This is an important but little discussed aspect of the process concept development and industrial realization of the OME3–5 production.2. Theory and background
2.1. Synthesis of OME
Based on methanol (H3C–OH, MeOH), various synthesis routes for the production of OME (H3C–O–(CH2O)n–CH3) take place over acid catalysts in the liquid phase at temperatures usually between 50–100 °C.14 For the synthesis of OME, methyl capping group suppliers such as MeOH, methylal (H3C–O–(CH2O)1–CH3, OME1) or dimethyl ether (H3C–O–CH3, DME) react with a formaldehyde source (H2C–O, FA) such as formalin, paraformaldehyde (HO–(CH2O)n–H with n = 8–100, pFA), trioxane ((CH2O)3, TRI), or monomeric FA through an initiation, growth, and termination mechanism, as described by Baranowski et al.23 This leads to several simultaneous and successive reactions and the formation of intermediate and side products.In a solution of MeOH and H2O, FA is bound in poly(oxymethylene) hemiformals (HO–(CH2O)n–CH3 with n = 1–10, HFn) following eqn (1) and (2) and poly(oxymethylene) glycols (HO–(CH2O)n–H with n = 1–10, MGn) following eqn (3) and (4). These reactions are fast, even in absence of a catalyst.24–26 In solutions with MeOH and H2O the amount of monomeric FA is very small towards chemical equilibrium.26
| CH2O + CH3OH ⇌ HO(CH2O)1CH3 | (1) |
| CH2O + HO(CH2O)n−1CH3 ⇌ HO(CH2O)nCH3; n = 2–10 | (2) |
| CH2O + H2O ⇌ HO(CH2O)1H | (3) |
| CH2O + HO(CH2O)n−1H ⇌ HO(CH2O)nH; n = 2–10 | (4) |
 | (5) |
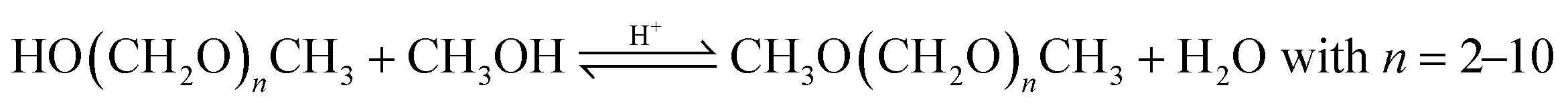 | (6) |
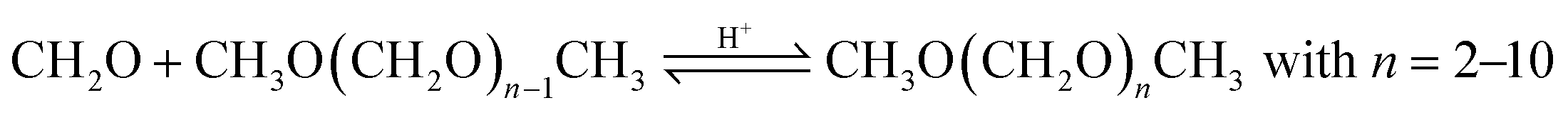 | (7) |
 | (8) |
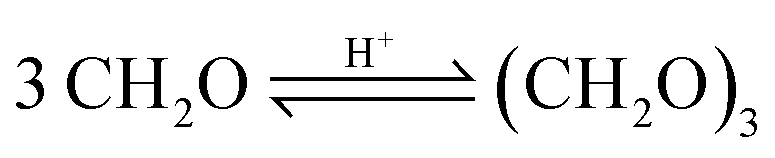 | (9) |
 | (10) |
 | (11) |
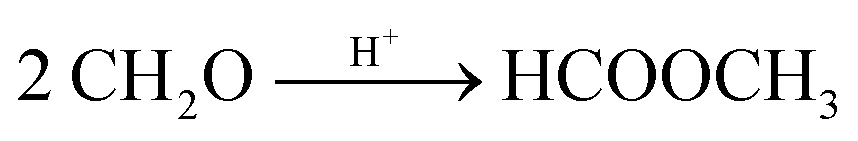 | (12) |
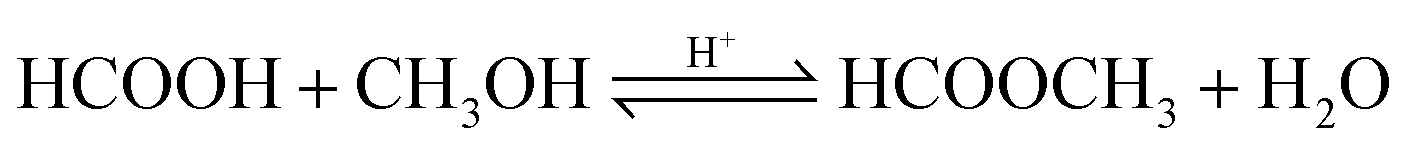 | (13) |
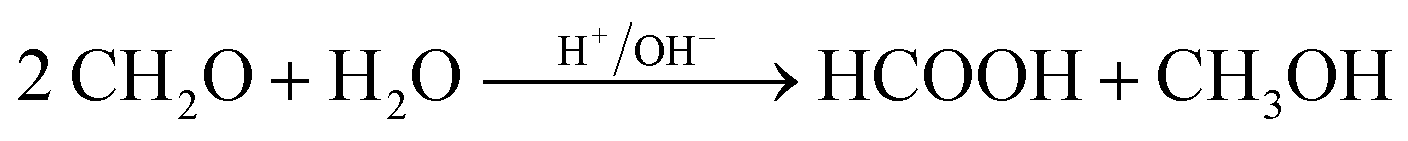 | (14) |
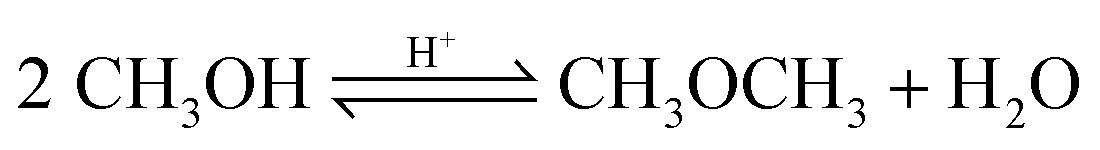 | (15) |
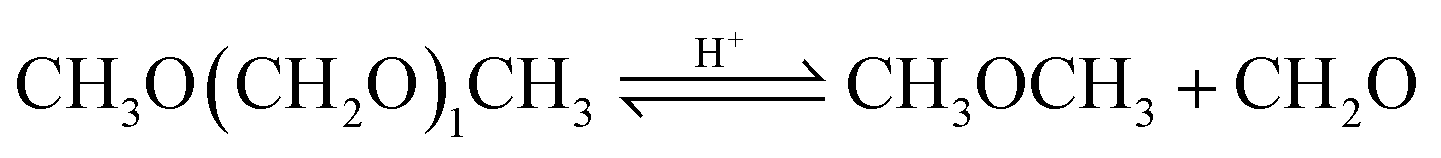 | (16) |
In the present work two reaction systems were investigated with MeOH and pFA as a typical aqueous system and OME1 and TRI as a typical anhydrous system.
2.2. Catalyst systems for the synthesis of OME
OME are synthesized in an acidic environment with Brønsted and Lewis acid sites activating the synthesis. Lewis acid sides are active for the decomposition of pFA as described by eqn (4), acetalization and chain propagation of OME as described by eqn (5)–(7). Brønsted acid sites are active for all steps of the OME synthesis, including the ring-opening of TRI.23 Various liquid and solid catalyst systems were already applied to the OME synthesis including acidic ion exchange resins (IER), zeolites and ionic liquids.Oestreich et al.13 investigated the OME synthesis from a mixture of MeOH and pFA over the IER Dowex50Wx2, Dowex50Wx4, Dowex50Wx8, Amberlyst® 36 (A36) and IR-120 and ground zeolites H-MFI 90, H-BEA 25, CBV 720, H-MFI 400 and H-MOR 30 in a batch autoclave at 80 °C. They compared the activity of the catalysts by determining the time after which 9 wt% of OME2 were obtained. Their results show that the Dowex catalysts had by far the highest activity, followed by the zeolites H-BEA 25 and H-MFI 90. A36 and IR 120 showed a lower activity and H-MFI 400 and H-MOR 30 did not reach the required OME2 concentration after 100 min. Regarding the side product formation, TRI and MEFO were detected far below 1 wt% for all IER but are pronounced for the zeolites with concentrations higher 1 wt% at 80 °C and longer retention times.
Lautenschütz14 investigated the OME synthesis from OME1 and TRI over the IER A15, A16 and A36, ground zeolites H-BEA 25, H-BEA 150, H-BEA 300, H-FER 20, H-MFI 27, H-MFI 90, H-MFI 240, H-MOR 30 and H-FAU 30 and γ-Al2O3 in a batch autoclave and fixed bed reactor at 30–100 °C. At 40 °C, A15 was more active than A36 and A16. Furthermore, Lautenschütz reported that A15, A16 and A36 were more active than A46, comparing his results to the results from Burger et al.15 Regarding the zeolites, H-BEA 25, H-BEA 150, H-BEA 300 and H-FAU 30 were active for the OME synthesis while the other zeolites only reached low conversions at the same retention time. Moreover, in a similar form and particle size the IER were still more active than the zeolites for both powder form and grain shape. However, the difference between grain shape to powder form led to a much higher activity for A36 (factor 16) and comparatively small improvements for H-BEA 25 (factor 3). γ-Al2O3 was not active for the OME synthesis from OME1 and TRI. For the reactants OME1 and pFA similar results were obtained with a higher activity for the IER followed by the BEA zeolites. However, the activity reduced significantly in comparison to the OME1 and TRI feed mixture, due to the presence of H2O which leads to the formation of several side products. For the reactants MeOH and pFA as well as MeOH and TRI the activity reduced for A36 and H-BEA 25, which was stronger pronounced for H-BEA 25. Regarding the side product formation, Lautenschütz14 reported no detection of MEFO at 40 °C for A15, A16 and A36 but detected MEFO for A36 at temperatures above 60 °C. No observations were reported regarding the TRI side product formation for OME1 and pFA or MeOH and pFA.
Burger et al.15 investigated the OME synthesis from OME1 and TRI over the IER A36 and A46 in a batch autoclave at 50–80 °C. They reported that A36 led to the formation of 1–2 wt% DME and MEFO as side products, while in the tests using A46 no DME or MEFO could be detected. Schmitz et al.16 investigated the OME synthesis from MeOH and pFA over A46 in a batch autoclave at 60–105 °C. They detected MEFO and TRI as side products. TRI concentrations of up to 2.6 wt% were obtained for feed mixtures with high FA concentrations and high reaction temperatures. The MEFO concentration did not exceed 0.06 wt% and was mainly below the detection limit. Voggenreiter et al.17 investigated the side product formation over A46 for mixtures of FA, MeOH, H2O and OME1 and published a kinetic model for the formation of MEFO, TRI and FOAC. High MEFO concentrations above 1 wt% were only detected at temperatures greater or equal to 85 °C and long residence times far after the equilibrium composition of the OME was obtained. The FOAC concentration was mainly a bit lower than the MEFO concentration but followed a similar behavior. The TRI concentration never exceeded 1 wt% and was limited by the equilibrium composition.
Zheng et al.18 investigated the OME synthesis from OME1 and pFA over the IER NKC-9, D001-CC and D72 in a batch autoclave at 20–80 °C. They reported a high activity for NKC-9. No formation of side products was reported.
Wu et al.19 investigated the OME synthesis from OME1 and TRI over the zeolite H-MFI with various Si/Al ratios in a batch autoclave at 120 °C. They reported an increasing OME1 conversion and decreasing TRI conversion for higher Si/Al ratios and a decreasing formation of MEFO, while MeOH and FA concentrations were increasing. Above the molar Si/Al ratio of 580 the MEFO concentration decreased below 1 wt%.
Wang et al.20 investigated the OME synthesis from OME1 and TRI over various homogeneous and heterogeneous catalysts, including the zeolites H-FAU, H-MFI and the IER A15, D002, D009 and CT175 in a batch autoclave at 90 °C. They reported a low activity for the zeolites and a high activity and selectivity for the IER, especially for CT175. However, they did not report the detection of side products but mentioned the formation of pFA for very low OME1 to TRI ratios.
Fink et al.21 investigated the OME synthesis from MeOH and pFA over the zeolites H-BEA 13, H-BEA 18, H-BEA 81, H-FAU 3, H-FAU 15, H-FAU 35, H-FAU 49, H-MFI 14, H-MFI 34, H-MFI 114, H-MFI 4716, H-MOR 6, H-MOR 10, H-MOR 16 in a batch autoclave at 65 °C. They did not investigate the performance of IER catalysts since in preliminary experiments, the leaching of SO3H groups was detected, leading to significant sulfur contents in the synthesis product. The catalysts with the highest activity for the OME synthesis were H-BEA 81, H-BEA 18, H-MFI 34 and H-FAU 35. The H-MOR zeolites showed far lower activities than all other zeolites except H-FAU 3 and H-MFI 4716 which showed very low and no activity, respectively. Additionally, they reported a minor formation of MEFO for all active catalysts with concentrations of about 0.1 wt% for most catalysts and 0.3–0.4 wt% for the H-MFI catalysts.
Endres et al.22 investigated the microwave-assisted OME synthesis from OME1 and TRI over the IER A15, A36, Dowex50Wx2, Dowex50Wx4 and Nafion in microwave vials at 25–100 °C. Their results show a higher activity for A15 than Nafion but a far lower formation of MEFO for Nafion than for all IER, especially at 40 °C. The observation of MeOH and FA in the samples was not reported.
This work focuses on the heterogeneous OME synthesis from commercial catalysts which could be used in industrial applications. The catalysts used for this evaluation are listed in Table 1 and were selected based on the reported performances regarding the OME synthesis from the above-mentioned investigations.
| Catalyst | Type | Form | Surface area in m2 g−1 | Acid capacity in meq g−1 | Si/Al ratio | T max in °C |
|---|---|---|---|---|---|---|
| a Assumption: diameter of 3 mm, density of 2 g cm−3, no pores. | ||||||
| Amberlyst® 15 (A15) | IER | Spherical | 5339 | 4.739 | — | 12039 |
| Amberlyst® 36 (A36) | IER | Spherical | 3340 | 5.440 | — | 15040 |
| Amberlyst® 46 (A46) | IER | Spherical | 7541 | 0.8–1.342 | — | 12014 |
| Dowex® 50WX2 (Dowex) | IER | Spherical | — | 0.8143 | — | 15043 |
| H-BEA 25 | Beta zeolite | Cylindrical | >50044 | — | 2544 | >20043 |
| H-MFI 90 | pentasil zeolite | Cylindrical | >30045 | — | 3045 | >20043 |
| Nafion™ NR40 (Nafion) | Perfluorosulfonic acid resin | Spherical | 0.001a | 1.046 | — | 20046 |
3. Materials and methods
3.1. Chemicals and catalysts
The reactants MeOH (purity ≥99.9%), granulated pFA (purity 94.5–95.5%) and OME1 (purity ≥99.9%) were purchased from Carl Roth GmbH + Co. KG. TRI (purity ≥99%) was purchased from Sigma-Aldrich Chemie GmbH. OME2 (purity ≥98.5%), OME3 (purity ≥99%), OME4 (purity ≥98.5%) and OME5 (purity ≥98.5%) were used for calibration and were supplied by ASG Analytik-Service AG. MEFO (purity 97%, 3% MeOH) was purchased from Thermo Fisher GmbH. Anhydrous sodium sulfite (purity ≥98%) and sulfuric acid (C = 0.1 mol L−1, ± 0.2%) were purchased from Carl Roth GmbH + Co. KG. The solvent EtOH (ethanol, purity ≥99.9%), the indicator thymolphthalein and the internal standard ethyl acetate (EA, purity ≥99.9%) were purchased from Carl Roth GmbH + Co. KG. HYDRANAL™-Solvent Oil and -Titrant 5 were purchased from Honeywell International Inc. Fluka. All chemicals were used without further purification.The catalysts A15, A36 and Dowex were purchased from Sigma-Aldrich Chemie GmbH, Nafion was purchased from Ion Power GmbH. A46 was provided by INAQUA Vertriebsgesellschaft mbH and the zeolites H-BEA 25 and H-MFI 90 were provided by Clariant AG. The IER and zeolites were dried overnight at 20 mbar and 30 °C before use. IER III was purchased from Merck Chemicals GmbH.
3.2. Analysis
The quantitative analysis was performed using an Agilent 7890A gas chromatograph equipped with a flame ionization detector (GC-FID) to analyze the organic components of the obtained reaction products. A sample volume of 1 μL was injected by an Agilent 7693A Autosampler onto a DB-5MS-Column (l = 30 m, di = 0.25 mm, film thickness = 0.5 μm). He (g) was used as carrier gas (flow: 202.5 mL min−1, p = 11.154 psi, split ratio = 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The GC inlet temperature was set at 290 °C, the temperature of the oven was programmed as a ramp (T5min = 30 °C, Tramp = 30 °C min−1, T7min = 270 °C, ttotal = 20 min). Calibration of the GC was achieved using EA as internal standard (Ai/AEA = Ri·wi/wEA). The components OME1–5, MeOH and TRI were calibrated using pure mixtures. MEFO was calibrated using a 97% pure mixture and subtraction of the MeOH content of 3%. The components OME6–11 were calibrated based on extrapolation and relating the peak area ratio per mass fraction ratio to the internal standard as a linear function of the number of carbon atoms of the OME molecules.
:1). The GC inlet temperature was set at 290 °C, the temperature of the oven was programmed as a ramp (T5min = 30 °C, Tramp = 30 °C min−1, T7min = 270 °C, ttotal = 20 min). Calibration of the GC was achieved using EA as internal standard (Ai/AEA = Ri·wi/wEA). The components OME1–5, MeOH and TRI were calibrated using pure mixtures. MEFO was calibrated using a 97% pure mixture and subtraction of the MeOH content of 3%. The components OME6–11 were calibrated based on extrapolation and relating the peak area ratio per mass fraction ratio to the internal standard as a linear function of the number of carbon atoms of the OME molecules.
The H2O content of the obtained samples was determined by Karl-Fischer titration and the content of FA was determined using the sodium sulfite method. Both methods quantify the overall composition, including H2O and FA bound in HF and MG. For a consistent data set, the analyzed H2O and FA content were not adjusted. Still, the content of the components quantified via GC-FID were normalized by proportional weighing to a sum of 1 g g−1. The sum of the overall mass fractions before this adjustment was predominantly between 0.85 g g−1 and 1.05 g g−1 for all samples. The main challenge was the precise analysis of MeOH due to the unstable side products HF which contain MeOH and were detected by the GC-FID. This was partly compensated by adjusting the MeOH calibration.
Due to their fast reaction kinetics of HF and MG as described by the eqn (1)–(4), these molecules are unstable at changing compositions and conditions. In this work, the true composition and, therefore, the formation of HF and MG is not considered. For the evaluation and presentation of the results, the overall composition is used considering the decomposition of HF and MG to their constituents MeOH, H2O and FA. This does not change the mass fraction of OME and other side products and therefore does not influence the conclusion from the results. Nevertheless, it strongly simplified the analysis of the samples.
For the OME1–TRI feed mixture the components FA, MeOH, H2O and MEFO were considered as side products. For the MeOH–pFA feed mixture only TRI and MEFO were considered as side products.
3.3. Apparatus
For the synthesis of OME, a high-pressure laboratory autoclave (Vmax = 500 mL, pmax = 100 bar) was used in combination with an integrated heating jacket (Tmax = 300 °C) and a magnetic stirrer from Carl Roth GmbH + Co. KG. The temperature was measured using a NiCr–Ni thermocouple (K-type; accuracy ± 1.5 K). The pressure was measured using a diaphragm pressure indicator (accuracy ± 0.24 bar).47 The lid of the autoclave was designed and adapted to the experimental requirements. The sampling line was cooled using a counter current heat exchanger operated with tap water to avoid evaporation of the samples. In addition, a sintered stainless-steel filter (pore size = 10 μm) near the reactor bottom ensured sampling without catalyst particles. A scheme of the reaction setup is illustrated in Fig. 1(a).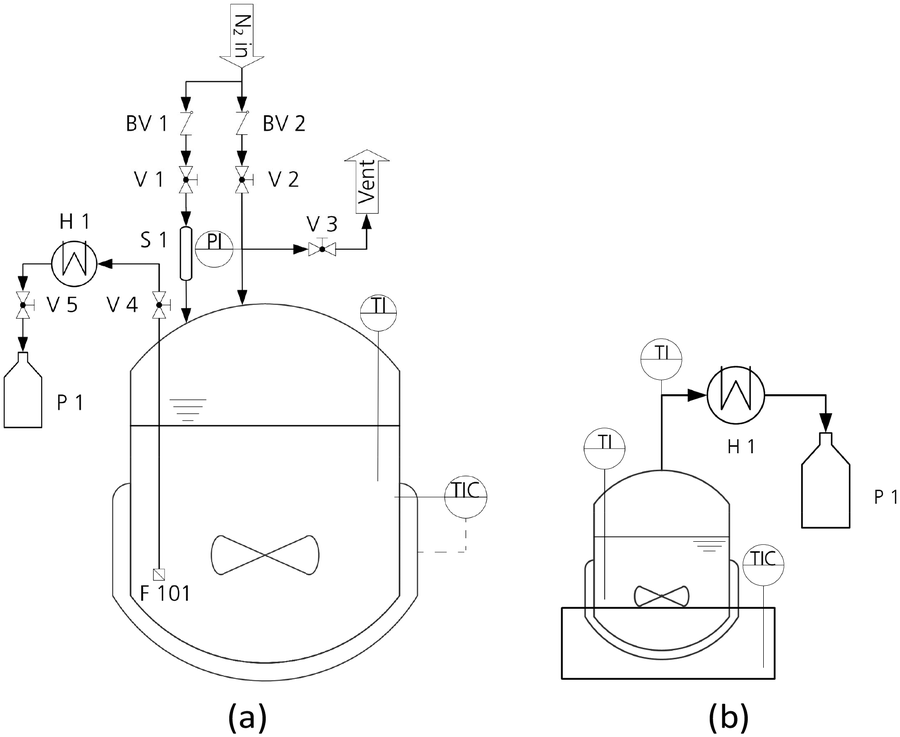 | ||
| Fig. 1 Autoclave (a) and distillation setup (b) for the investigation of the OME synthesis and thermal stability of the OME synthesis product. BV, back pressure valve; F, filter; H, heat exchanger; P, product flask; PI, pressure indicator; S, catalyst chamber; TI and TIC, temperature indicators; V, valve. | ||
For the distillation of the OME synthesis product mixtures, a micro distillation setup was used which was heated using an oil bath and a magnetic stirrer inside the 50 mL two-neck round-bottom flask was used for mixing. No column was used to enable the distillation of smaller sample amounts. A Liebig condenser with a thermometer and vacuum nozzle was used to condense the distillate and collect the product in 10 mL flasks. The temperature in the bottom product was measured using a NiCr–Ni thermocouple (K-type; accuracy ± 1.5 K). The oil bath was heated with a heating plate regulated with a thermocouple inside the oil bath and a magnetic stirrer inside the oil bath for faster heat distribution. A scheme of the distillation setup is illustrated in Fig. 1(b).
3.4. Feed preparation
The MeOH–pFA feed mixtures were prepared by dissolving pFA in MeOH at a ratio of pFA/MeOH = 1.5 g g−1 corresponding to the maximum FA solubility in a methanolic solution, similar to the procedure from Oestreich et al.13 pFA was dissolved by stirring and heating to 85 °C for up to three days until a clear solution was obtained. A condenser was placed above the round bottom flask to condense evaporating components. After cooling the mixture to room temperature, it was filtered using a pleated filter (retention range: 5 to 8 μm) to avoid solid pFA particles in the feed mixture.OME1–TRI feed mixtures were prepared by dissolving TRI in OME1 at a ratio of OME1/TRI = 2 g g−1. TRI was dissolved at room temperature by stirring for two hours until a clear solution was obtained. To avoid the evaporation of OME1, the round-bottom flask was closed with a lid and filtration of the feed mixture was omitted.
3.5. OME synthesis
A pressure test was performed before each synthesis experiment at 8 bar. For the OME synthesis, 1 wt% of catalyst (mcat = 3.5 g) was used in comparison to the mass of the feed mixture (mfeed = 350 g). The catalyst was added above the reactor head in a catalyst chamber. The prepared feed mixture was added to the reaction chamber without contact to the catalyst. After the autoclave was completely sealed again, N2 was added until a pressure of 2 bar was reached. The line above the catalyst was filled with N2 at 8 bar. The feed mixture was heated up to 60 °C inside the autoclave at constant stirring rate. At 60 °C the catalyst was added to the feed mixture by opening a ball valve above the reactor lid and N2 was added until 8 bar. With the addition of the catalyst, the synthesis experiment started. During the synthesis, samples were withdrawn through a cooled sampling line. To avoid contamination inside the sampling line, it was rinsed by withdrawing 5 mL of reaction product before withdrawing the sample. The first sample (S0) was withdrawn from the feed mixture before it was fed to the reactor. The second sample (S1) was withdrawn from the reactor when a temperature of 60 °C was reached. All further samples were withdrawn at progressively longer time intervals after the catalyst was added to the reaction chamber (t = 1, 5, 10, 15, 20, 30, 45, 60, 75, 90, 120, 150, 180, 240, 300 min). The final sample (S17) was taken together with the reaction product after 24 h. After the withdrawal of the product mixture the autoclave setup was completely cleaned. The reaction temperature of 60 °C was chosen as a compromise between the reaction kinetics of the MeOH–pFA and the OME1–TRI feed mixture, the amount of catalyst and the formation of side products. In the literature mentioned above, the OME synthesis was investigated at various temperature levels without significant changes in the final OME distribution but significant changes in the side product formation.3.6. Catalyst performance evaluation
The performance of the catalysts was evaluated based on the conversion X of the feed, the selectivity S towards OME3–5, the yield Y of OME3–5, the activity and side product formation. The conversion of the feed was evaluated with the mass fraction w of the feed before the synthesis at 0 h and after the synthesis at 24 h, as described by eqn (17).14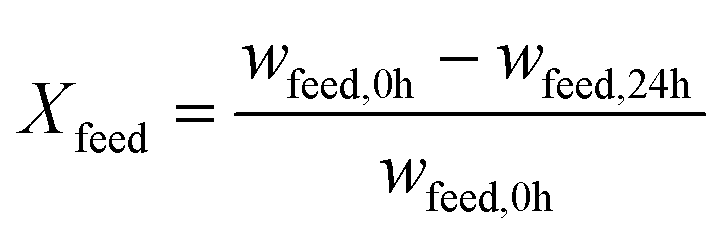 | (17) |
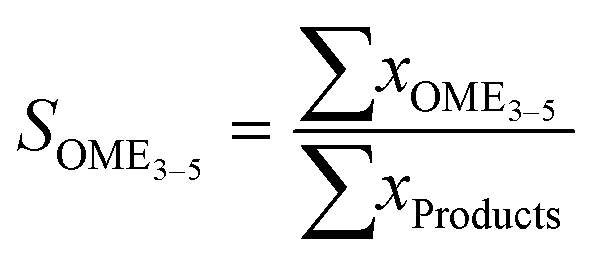 | (18) |
 | (19) |
| wOME5(tTermination) = 0.9·wOME5(24 h) | (20) |
The second indicator is the relation of the yield of OME3–5 after 30 min to the yield of OME3–5 after 24 h, as described by eqn (21).
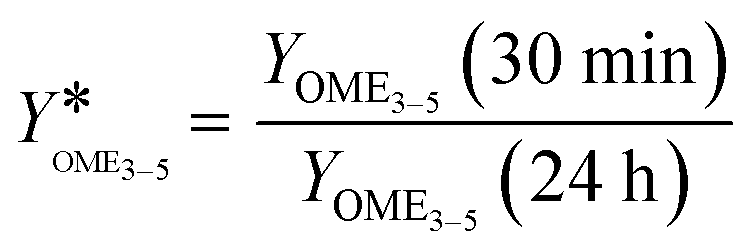 | (21) |
3.7. Distillation of the OME synthesis products
The synthesis products were directly distilled in a micro distillation setup to investigate the necessity of a product neutralization step before the thermal separation for the target product purification. The thermal stability of the product mixtures thereby determines whether the synthesis product can be directly purified in a distillation column or if the process concept needs to be extended by a neutralization step.After the micro distillation setup was mounted, the weight of the round-bottom flask was measured, and 30–50 g of the synthesis product was added to the round-bottom flask. Then, the oil bath was heated stepwise from 60 °C to 100 °C. Thereby, the temperature was increased after the distillate flow stopped. At 100 °C, the experiment continued for up to 5 h to simulate a longer retention time inside a continuous distillation process. Afterward, the setup cooled down, the round-bottom flasks were weighted, and the distillate and bottom products were withdrawn.
4. Results and discussion
The OME synthesis was investigated for various commercially available catalysts to compare their activity, selectivity and thermal stability of the synthesis product. Furthermore, the synthesis was carried out for the feed mixtures OME1–TRI as well as MeOH–pFA to consider an anhydrous and an aqueous reaction system towards OME. In addition, the synthesis products were distilled in a micro distillation setup to investigate their stability regarding downstream purification using distillation columns.4.1. Reaction progress and equilibrium composition
Fig. 2 and 3 illustrate the reaction progress and the equilibrium composition for the OME synthesis from MeOH–pFA as well as OME1–TRI, respectively, at 60 °C and 8 bar over A36. The analytic results for all investigated catalysts are presented in the ESI.†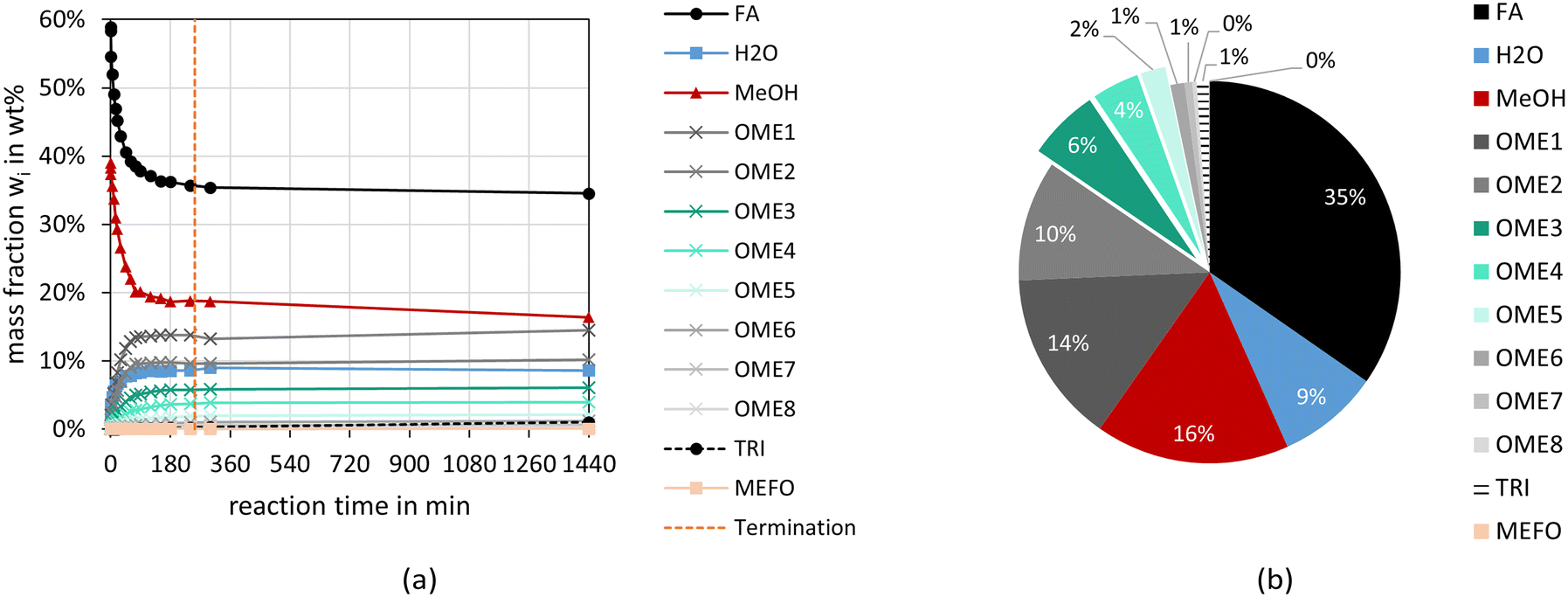 | ||
| Fig. 2 OME synthesis from MeOH–pFA over A36 (conditions: pFA/MeOH = 1.53 g g−1, A36/(MeOH + pFA) = 1.0 wt%, 60 °C, 8 bar, batch). (a) Illustrates the reaction progress and (b) the equilibrium composition after 24 h. | ||
 | ||
| Fig. 3 OME synthesis from OME1–TRI over A36 (conditions: OME1/TRI = 2.00 g g−1, A36/(OME1 + TRI) = 1.0 wt%, 60 °C, 8 bar, batch). (a) Illustrates the reaction progress and (b) the equilibrium composition after 24 h. | ||
Besides the progress of the mass fractions of FA, H2O, MeOH, OME1–8, TRI and MEFO, the termination time is indicated as vertical line in Fig. 2(a) and 3(a), which is used as an indicator for the activity of the catalysts and will be discussed in the subsequent section. The reaction progresses show that the quasi-equilibrium composition is obtained after 3–4 h for the MeOH–pFA feed mixture and after about 1 h for the OME1–TRI feed mixture. However, the side product formation shifts the equilibrium composition leading to a slightly different composition after 24 h. The presence of H2O inside the reaction mixture from MeOH–pFA leads to a reduction of catalyst activity and strongly influences the selectivity towards OME3–5. Due to the side and intermediate product formation of HF and MG, which are formed in presence of H2O and MeOH as described by eqn (1)–(4), the yield of OME3–5 reduces from 31 wt% for the OME1–TRI feed mixture to 12 wt% for the MeOH–pFA feed mixture, as illustrated by the green coloured componentes in Fig. 2(b) and 3(b). Considering the overall composition, the MeOH–pFA feed mixture leads to a large amount of unreacted feedstock in the equilibrium composition, while TRI is almost completely converted in the equilibrium composition of the OME1–TRI feed mixture.
To compare the different catalysts, Fig. 4 illustrates the yield of OME3–5 over the synthesis progress for both feed mixtures and all investigated catalysts.
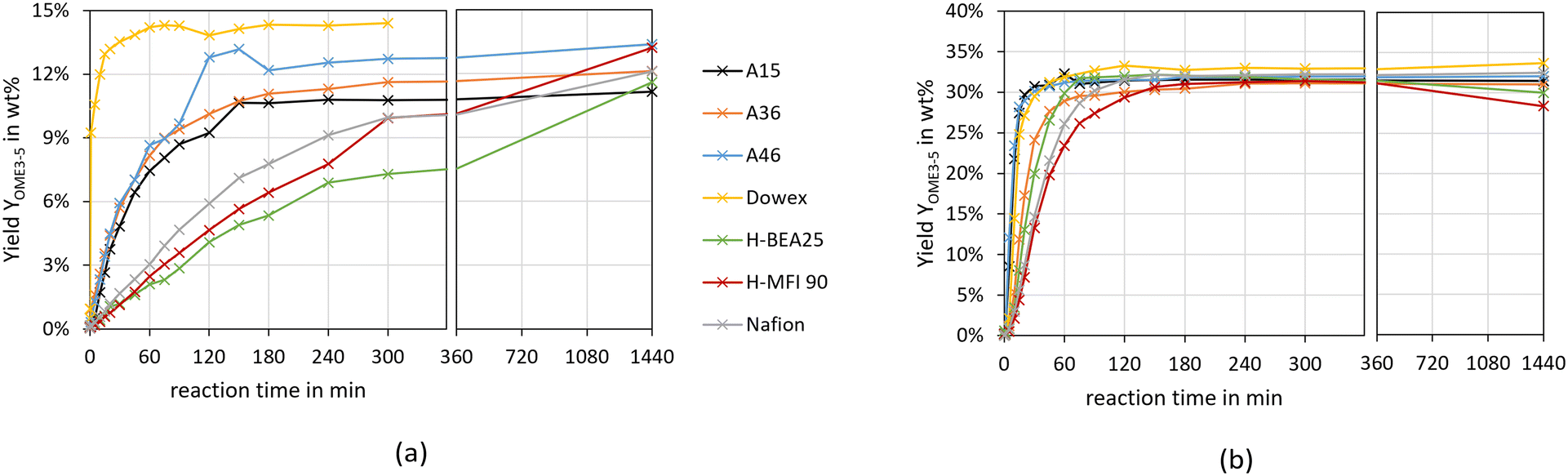 | ||
| Fig. 4 OME3–5 yield over the synthesis progress over various catalysts (conditions: pFA/MeOH = 1.5 g g−1, OME1/TRI = 2.0 g g−1, catalyst/reactants = 1.0 wt%, 60 °C, 8 bar, batch). (a) Illustrates the results for the MeOH–pFA feed mixture and (b) illustrates the results for the OME1–TRI feed mixture. | ||
The yield of OME3–5 after 24 h varies between 11–14 wt% for the MeOH–pFA feed mixture and 28–34 wt% for the OME1–TRI feed mixture. The progress of the OME3–5 yield from MeOH–pFA is much faster for the IER than for the zeolites and Nafion, with Dowex showing a significantly faster reaction than all other IER.
Furthermore, the final yield of OME3–5 using Dowex is higher than for the other catalysts, A15 and H-BEA 25 obtained the lowest yield. These differences cannot only be explained by the side product formation, which was especially prominent for the zeolites, however, those show rather good yields after 24 h. The results for the MeOH–pFA system are in contrast to those obtained by Oestreich et al.,13 who reported similar yields between the catalyst systems and also pronounced side product formations for the zeolites. However, they investigated the synthesis at higher temperatures of 80 °C and ground the zeolites before their application.
Comparing the OME3–5 yield from MeOH–pFA with OME1–TRI, a much faster progress is reached with the absence of H2O, leading to significantly higher OME3–5 yields. For the OME1–TRI feed mixture, the yield also varies between the catalysts and is led by Dowex. For the zeolites, the OME3–5 yield decreases after 5 h due to the strong MEFO formation. This phenomenon will be discussed in the following section.
Fig. 5 illustrates the conversion of the reactants MeOH–pFA as well as OME1–TRI and the selectivity towards OME3–5 over various catalysts after 24 h.
 | ||
| Fig. 5 Conversion of the reactants, selectivity towards OME3–5 and yield of OME3–5 for the OME synthesis from MeOH–pFA and OME1–TRI over various catalysts (conditions: pFA/MeOH = 1.5 g g−1, OME1/TRI = 2.0 g g−1, catalyst/reactants = 1.0 wt%, 60 °C, 8 bar, 24 h, batch). (a) Illustrates the results for the MeOH–pFA feed mixture and (b) the results for the OME1–TRI feed mixture. | ||
The conversion of MeOH after 24 h in Fig. 5(a) shows substantial differences between the different catalysts, while the conversion of FA is similar. Only for the zeolite H-MFI 90 a higher conversion of FA was detected, which is mainly attributed to the higher side product formation. Besides, A15 clearly shows lower and Dowex higher conversions in comparison to the other catalysts.
Fig. 5(b) illustrates the conversions and selectivity towards OME3–5 for the OME1–TRI feed mixture. Thereby, both reactants led to very similar conversions for all catalysts with a small difference for the zeolites, which again is a result of their increased side product formation.
The selectivity towards OME3–5 was found similar between the catalyst systems for both feed mixtures. However, it was significantly lower for the MeOH–pFA mixture because of the presence of H2O and the associated side products. Furthermore, as already indicated by the conversion, the selectivity of OME3–5 was slightly lower after 24 h for the zeolites due to their comparatively high activity for the MEFO and TRI formation.
4.2. Catalyst activity
To determine the activity of the catalysts for the OME syntheses starting from MeOH–pFA as well as OME1–TRI, two indicators were evaluated. The termination time is illustrated in Fig. 6(a) and the ratio of the yield of OME3–5 after 30 min and 24 h is illustrated in Fig. 6(b).
is illustrated in Fig. 6(b).
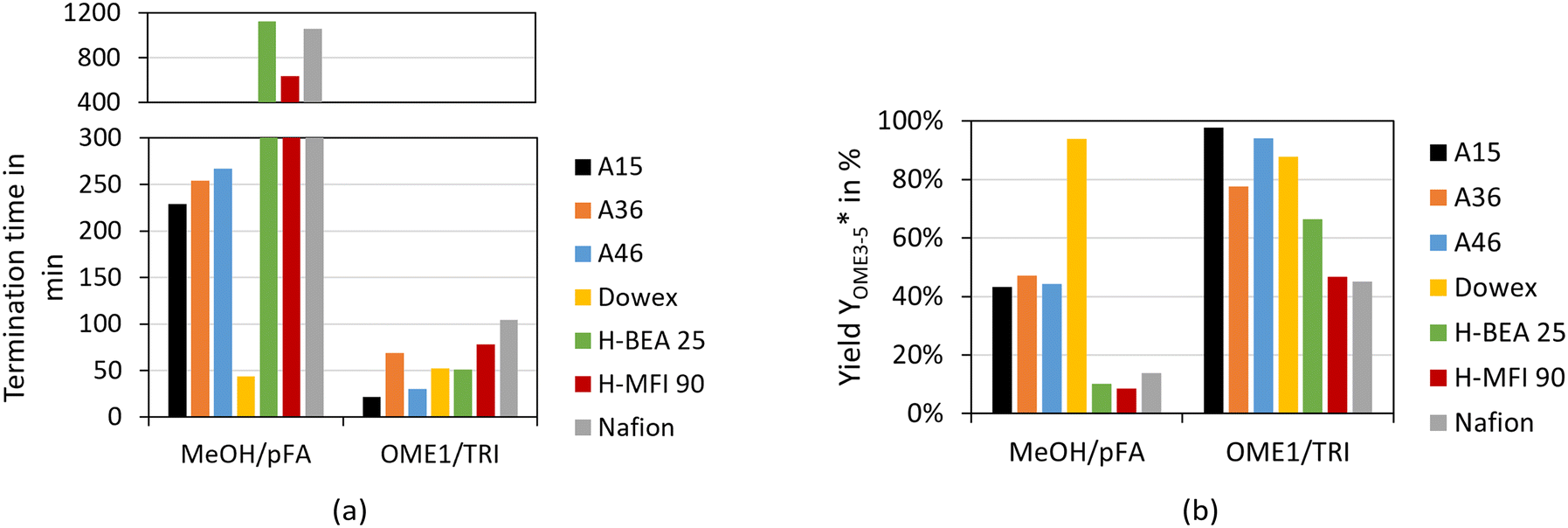 | ||
Fig. 6 Termination time (a) and yield 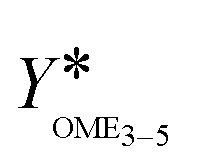 (b) of the OME synthesis from MeOH–pFA and OME1–TRI over various catalysts (conditions: pFA/MeOH = 1.5 g g−1, OME1/TRI = 2.0 g g−1, catalyst/reactants = 1.0 wt%, 60 °C, 8 bar, batch). (b) of the OME synthesis from MeOH–pFA and OME1–TRI over various catalysts (conditions: pFA/MeOH = 1.5 g g−1, OME1/TRI = 2.0 g g−1, catalyst/reactants = 1.0 wt%, 60 °C, 8 bar, batch). | ||
For the OME1–TRI feed mixture, all catalysts were very active with the shortest termination time obtained by A15 and A46 followed by H-BEA 25 and Dowex. For the MeOH–pFA feed mixture, only Dowex was very active followed by the other IER with a clear increase in the termination time. Oestreich et al.13 also reported a higher activity for the IER catalysts than for zeolites for the OME synthesis from MeOH–pFA. No clear tendency was obtained regarding the acid capacity of the IER as listed in Table 1. A36 shows the highest acid capacity but was found to be less active than A46 and Dowex, with a far lower acid capacity.
Regarding the zeolites, H-BEA 25 has a lower Si/Al ratio but is more active than H-MFI 90 for the OME1–TRI feed mixture and less active for the MeOH–pFA feed mixture. Nafion showed similar activity to the zeolites even though its surface area is significantly smaller since it contains no pores, as presented in Table 1. Therefore, smaller Nafion beads could reach the activity of the IER. The termination time was very similar for A36 and A46 for the MeOH–pFA feed mixture.
However, for the OME1–TRI feed mixture, A46 was significantly faster, even though their main difference is the degree of sulfonation which is higher for A36. A36 is sulfonated on the surface and within the micro pores of the matrix while A46 is only sulfonated on the surface.41 However, A46 has a bigger surface than A36. Dowex showed a very high activity for the MeOH–pFA feed mixture compared to the other IER but a similar activity for the OME1–TRI feed mixture. In contrast to all other catalysts, Dowex showed a higher activity for the MeOH–pFA feed mixture than for the OME1–TRI feed mixture. Therefore, the ring opening of TRI, as described by eqn (9) and the incorporation into OME as described by eqn (7) are more prominent rate determining steps than the acetalization reactions from HF to OME as described by eqn (5) and (6) and the presence and formation of the side products HF and MG as described by eqn (1)–(4). Lautenschütz14 also reported differences in the activity of different catalysts between an anhydrous and different aqueous reaction systems and explained this with the presence of H2O, which leads to additional side product formations and therefore reduces the product selectivity towards OME. In addition, H2O and MeOH inhibit the formation of OME, which was particularly apparent for H-BEA 25 in comparison to A36.
The second indicator for the activity is the yield 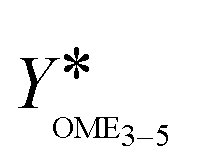 which is presented in Fig. 6(b) and shows higher values for the OME1–TRI feed mixture than for the MeOH–pFA feed mixture for all catalysts except for Dowex. These results match with those from the termination time of the catalysts. However, the results for
which is presented in Fig. 6(b) and shows higher values for the OME1–TRI feed mixture than for the MeOH–pFA feed mixture for all catalysts except for Dowex. These results match with those from the termination time of the catalysts. However, the results for 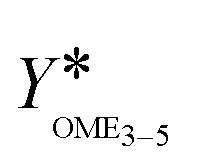 are more precise for evaluating the activity due to the evaluation of the directly measured composition instead of using linear interpolation.
are more precise for evaluating the activity due to the evaluation of the directly measured composition instead of using linear interpolation.
4.3. Side and by-product formation
For the feed mixture OME1–TRI, the side products FA, MeOH, H2O and MEFO were evaluated, whereas for the feed mixture MeOH–pFA the side products MEFO and TRI were assessed.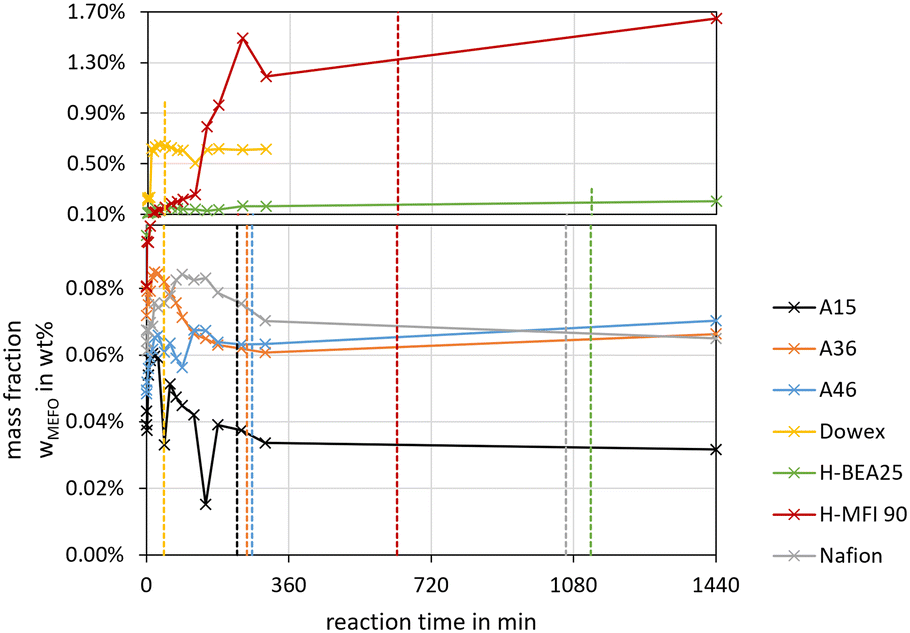 | ||
| Fig. 7 MEFO side product formation over the synthesis progress from MeOH–pFA for various catalysts (conditions: pFA/MeOH = 1.5 g g−1, catalyst/reactants = 1.0 wt%, 60 °C, 8 bar, batch). The dashed lines show the termination time of the respective catalysts. | ||
The highest concentrations of MEFO were obtained with the catalysts H-MFI 90, Dowex and H-BEA 25. All other catalysts showed very low formations of MEFO for the entire duration, with concentrations lower than 0.1 wt% MEFO at their respective termination times. Already before the catalyst was added to the reaction mixture, MEFO concentrations were detected for all syntheses. After a small increase in the initial phase of about 1–3 h, the concentration of MEFO stayed approximately constant for the rest of the synthesis without significant differences between the concentration at the termination time and after 24 h for all catalysts. This disagrees with the assumption of the irreversibility of the Tishchenko reaction as described by eqn (12) but indicates that the reversible esterification (eqn (13)) is prominent, which requires the presence of FOAC. Therefore, FOAC was likely to be part of the side products but could not be quantified with the applied analysis methods. Following eqn (13), the initial increase of the MEFO curves was influenced by the varying MeOH concentration in the reaction mixture, which also stays approximately constant after the termination time is exceeded. The negligible influence of the Tishchenko reaction for all the investigated catalysts is surprising since it leads to strong MEFO concentrations in the OME1–TRI feed mixture, as illustrated in Fig. 7. Furthermore, Voggenreiter et al.17 investigated the side product formation for the OME synthesis from MeOH, FA, OME1 and H2O for the catalyst A46. In contrast to the results obtained with all catalysts in this work, they reported a steady increase in the MEFO concentration over the synthesis progress. However, they prepared the feed mixture by dissolving pFA in a solvent using a base, sodium methoxide or sodium hydroxide to accelerate the process. The MeOH–pFA feed mixture was prepared at higher temperatures and retention times in this work. This apparently led to the formation of MEFO even without adding a catalyst. Furthermore, Voggenreiter et al.17 investigated the synthesis at lower ratios of the reactants FA and MeOH, with high concentrations of OME1 in the reactant mixture and at temperatures between 70 °C and 100 °C. For their experiment KIN3, almost no OME1 was present in the reactant mixture and a small decrease of the MEFO concentration was detected between the initial sample and the first reaction sample. However, the intervals between the samples were too big to confirm this behavior. Comparing the findings of Voggenreiter et al.17 with the results from this work, the Tishchenko reaction seems to be prominent at temperatures exceeding 70 °C. However, the reversible esterification was prominent for lower temperatures and reaction mixtures with lower OME concentrations.
For the OME1–TRI feed mixture, the MEFO concentrations were increasing over time for all catalysts without an indication of reaching an equilibrium composition. Fig. 8 shows the synthesis progress until 4 h to emphasize the initial MEFO formation until the termination time. For the OME1–TRI feed mixture, the irreversible Tishchenko reaction is prominent for the MEFO formation. The reversible esterification is not prominent due to very low MeOH and H2O concentrations in the reaction mixture. Similar to the MeOH–pFA feed mixture, the zeolites show the strongest MEFO formation. However, in contrast to the high MEFO formation in the MeOH–pFA feed mixture, MEFO could only be detected for Dowex after exceeding the termination time. Besides Dowex, the other IER and Nafion also show very low MEFO concentrations at their respective termination times.
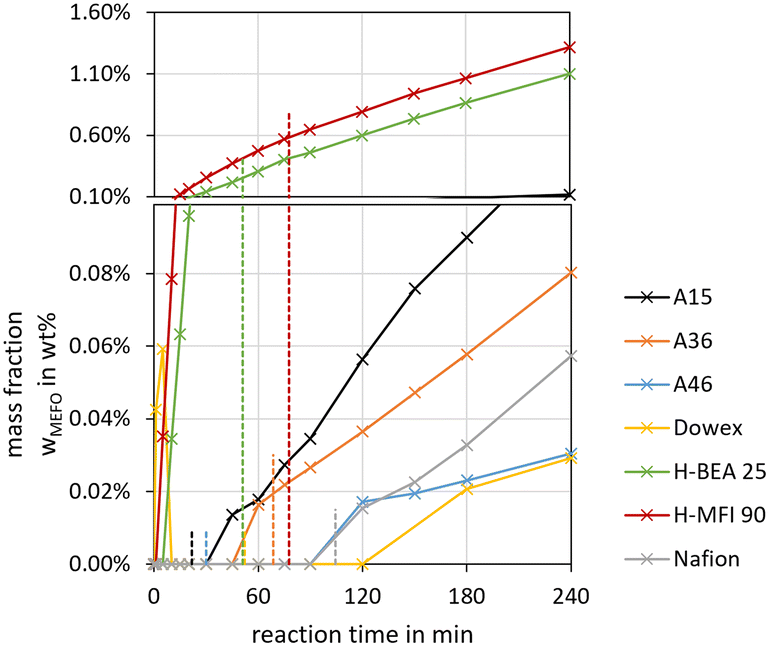 | ||
| Fig. 8 MEFO side product formation over the synthesis progress from OME1–TRI for various catalysts (conditions: OME1/TRI = 2.0 g g−1, catalyst/reactants = 1.0 wt%, 60 °C, 8 bar, batch). The dashed lines show the termination time of the respective catalysts. | ||
As a conclusion, H-MFI 90 led to very high MEFO concentrations for both feed mixtures already at the respective termination time. H-BEA 25 showed far lower MEFO concentrations for both feed mixtures at the respective termination time; however, still exceeding the concentrations obtained by the other catalysts. Only Dowex showed a higher MEFO concentration for the MeOH–pFA feed mixture but no MEFO for the OME1–TRI feed mixture. The other IER and Nafion showed similar small MEFO concentrations at their respective termination times. Unexpectedly, the irreversible Tishchenko reaction was insignificant for the MeOH–pFA feed mixture at 60 °C. Due to the irreversible Tishchenko reaction, MEFO must be extracted from the loop inside the OME3–5 production process to prevent its accumulation. However, due to a very narrow boiling point curve with OME1, the separation from the product mixture can be expensive.48,49 This is an important aspect that should be addressed by extended experimental investigations for selected catalysts and the influence of MEFO handling strategies on the complete process design should be evaluated in further investigations.
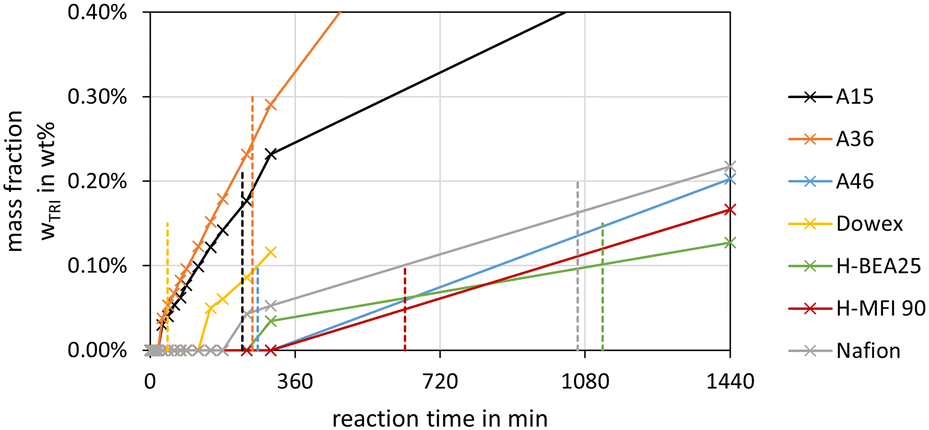 | ||
| Fig. 9 TRI side product formation over the synthesis progress from MeOH–pFA for various catalysts (conditions: pFA/MeOH = 1.5 g g−1, catalyst/reactants = 1.0 wt%, 60 °C, 8 bar, batch). The dashed lines show the termination time of the respective catalysts. | ||
The highest concentrations of TRI were obtained with the catalysts A36, A15 and Nafion at their respective termination times. The catalysts A46 and Dowex showed very low TRI concentrations, even below the detection limit. The zeolites also led to low TRI concentrations of 0.1 wt% and below at their respective termination times. In comparison to the MEFO formation, the TRI curves showed a steady increase. As described by eqn (9)–(11) the formation of TRI from FA, MG3 or OME4 is limited by an equilibrium composition which, however, was not obtained by any of the investigated catalysts at 60 °C until 24 h. Since TRI also represents a reactant for the formation of OME, it does not need to be separated from the loop inside the OME3–5 production process. Its presence influences the reaction kinetics, though, with its concentration limited by the low equilibrium concentration.16
Schmitz et al.16 reported low concentrations of TRI for the OME synthesis from MeOH–pFA over A46 at different temperatures. The concentration of TRI increased with higher concentrations of FA in the feed mixtures and at higher temperatures starting from 70 °C. Below 70 °C, no TRI was detected, which agrees with the results in this work for A46. Voggenreiter et al.17 also reported low concentrations of TRI in the OME synthesis from MeOH, FA, H2O and OME1 over A46. The amount of TRI increased with rising temperature and FA concentrations in the feed mixture but mainly did not exceed 1 wt%.
 | ||
| Fig. 10 FA (a), MeOH (b) and H2O (c) side product formation over the synthesis progress from OME1–TRI for various catalysts (conditions: OME1/TRI = 2.0 g g−1, catalyst/reactants = 1.0 wt%, 60 °C, 8 bar, batch). | ||
The highest concentration of FA and MeOH was obtained with A15, A36 and Dowex. All other catalysts lead to FA concentrations below 1 wt% at their respective termination times and even lower MeOH concentrations.
Considering the curves for the formation of MeOH, all catalysts start at very low concentrations of less than 0.2 wt%, pass through a maximum and slowly decrease towards a constant concentration. An exception is Nafion, whose MeOH concentration steadily increases towards a constant concentration. Considering the reaction network described in a previous section, the formation of MeOH requires the presence of H2O in the reaction mixture. However, Fig. 10(c) illustrates the progress of the H2O concentration and except for A36, no H2O was detected. Burger et al.15 investigated the OME synthesis from OME1–TRI over A46 and also did not detect any H2O but low concentrations of MeOH. Lautenschütz14 analyzed a blank experiment with OME1 reacting alone in the presence of A36. He reported that the product mixture contained 2 wt% MeOH and 3 wt% OME2. In a subsequent experiment, he dried OME1 before adding A36, and no MeOH or OME2 could be detected in the product mixture. Therefore, the formation of MeOH results from traces of H2O inside the reaction mixture, which is below the detection limit of about 0.02 wt% H2O. The FA formation is also influenced by the formation of MeOH from OME1 and H2O, as described by eqn (5) but additionally by the equilibrium reaction towards TRI, as described by eqn (9).
Considering the production of OME3–5 from OME1–TRI, the formation of MeOH and therefore the presence of traces of H2O inside the feed mixture is challenging for a steady state operation. Since MeOH and H2O would accumulate in the loop inside the OME3–5 production process, they need to be separated, which would strongly reduce the benefit of the anhydrous OME reaction system compared to the aqueous OME reaction system. Alternatively, the reactants need to be intensively dried before application. This would reflect on the production costs but entail the advantage of a significantly simplified OME3–5 product separation and purification.
4.4. Thermal stability of the synthesis products
Using IER to synthesize OME can lead to leaching of the active acid groups of the catalyst into the reaction product mixture.21 This was also emphasized by Baranowski et al.,50 who concluded the main drawbacks of IER to be the low thermal stability and the leaching of active species into the synthesis product when using polar solvents. Acid IER are mainly synthesized by copolymerization of styrene and divinylbenzene with a macroreticular matrix and functionalized with sulphuric acid.51 Therefore, the functional (–SO3H) groups can leach into the reaction mixture. Fink52 investigated the stability of A15 and A36 for the OME synthesis from MeOH–pFA at 65–70 °C and reported that 0.4 mol% and 0.7 mol% of the sulphur content from the sulphonic acid group was dissolved out the catalyst after 3–5 h. As a result, she concluded that IER are generally not suitable for the OME3–5 production. Furthermore, before the thermal separation of the OME synthesis product mixture using IER, Lautenschütz14 neutralized the mixture with IER III but did not mention its necessity.Active species inside the OME synthesis product enable the reactions towards and between different OME as described by eqn (5)–(8), but also side product formations as described by eqn (9)–(16), outside the reactor unit. Due to the comparatively slow kinetics of these reactions, traces of active species will not show a significant influence on the product composition at moderate temperatures. However, considering a thermal separation of the OME synthesis product to purify the desired OME3–5 fraction, high temperatures of about 200 °C (ref. 11) are required, which strongly accelerate the reactions. Furthermore, due to the separation of the more volatile components from the OME synthesis product the reaction equilibrium of eqn (5)–(8) is disturbed and the direction of the reaction will reverse. This results in the formation MeOH and HF as well as shorter chain OME and FA following the reactions describes by eqn (5)–(7). Furthermore, the transacetalization reactions as described by eqn (8) will form shorter chain OME and even longer chain OME. Depending on the reaction kinetics, temperature level and duration, the composition of the bottom product of the distillation column varies and the amount of bottom product will reduce, if active species are present. This reduces the originally produced amount of the final OME3–5 from the synthesis and needs to be prevented. A neutralization step contacting the free active acid groups with alkaline groups (OH−) after the reactor can neutralize active species and enable a stable thermal separation of the OME synthesis product.
To investigate the necessity of a neutralization step, the OME synthesis products were distilled and the composition after the distillation was compared to the composition before the distillation. Fig. 11 illustrates the results of the distillation with the filled bars showing the composition before the distillation, and the striped bars showing the composition after the distillation, which is the sum of the distillate and the bottom product.
 | ||
| Fig. 11 Synthesis product composition and added up distillation product composition of the OME synthesis from MeOH–pFA (a) and OME1–TRI (b) for various catalysts (conditions synthesis: pFA/MeOH = 1.5 g g−1, OME1/TRI = 2.0 g g−1, catalyst/reactants = 1.0 wt%, 60 °C, 8 bar, batch; conditions distillation: 30–50 g synthesis product, TOil = 60–100 °C stepwise, 5 h, batch). | ||
The synthesis product from MeOH–pFA illustrated in Fig. 11(a) shows similar compositions to the product after the distillation for all catalysts. Only the distillation product of the zeolites shows more significant differences for FA, MeOH and OME1. Unexpectedly, the concentration of OME1 reduced during the distillation of the OME synthesis product from H-MFI 90 and led to higher concentrations of MeOH and FA. Due to the lower volatility of OME1, active species inside the synthesis product should increase the total amount of OME1, which is used inside the reactive distillation of the OME1 production process.53 However, the reverse acetalization reactions described by eqn (5) and (6) might be accelerated faster than the reverse chain propagation reaction described by eqn (7). Therefore, due to kinetic limitations, this can lead to higher concentrations of MeOH and FA. H-BEA 25, on the other hand, leads to a reduction of FA, an increase of the MeOH concentration and a slight increase of the longer chain OME and by-products TRI and MEFO. A36 and A46 also obtained minor differences for FA, MeOH, H2O and OME1. In contrast to the other catalysts, the bottom product of A46 solidified at room temperature and was dissolved in MeOH for analysis. Considering the bottom composition, the FA, H2O and OME≥4 concentration is slightly higher for A46 compared to the other catalyst products, and the MeOH concentration is somewhat lower, which might exceed the solubility limits of FA and longer chain OME. The bottom product compositions after the distillation are presented in the ESI.†
Considering the result of the distillations of the synthesis product from OME1–TRI as illustrated in Fig. 11(b), more significant differences were obtained. Only A36, H-BEA 25 and Nafion show very similar results after the distillation. The distillation of the OME synthesis product of A46 and H-MFI 90 led to a reduction of the short chain OME1–2 and an increase of the longer chain OME≥4, which indicates the chain propagation coupled with the transacetalization reactions, as described by eqn (7) and (8). However, the bottom product of A46 also solidified at room temperature for the OME1–TRI based synthesis product and was dissolved in MeOH for analysis. Similar to the bottom product of the MeOH–pFA synthesis product, the concentration of longer chain OME≥4 increased for A46, which is illustrated in the ESI.† Dowex, on the other hand, leads to a substantial increase of the OME1 concentration and decreased concentrations of OME≥2 due to the reverse chain propagation described by eqn (7). The FA concentration did not change as expected according to eqn (7), however, a complete condensation of the gaseous FA without solidification is challenging, especially without the presence of H2O and MeOH. Therefore, the measured concentration of FA inside the bottom and distillate product can differ significantly from the actual amount of FA inside the setup. The bottom product of the distillation of the OME synthesis product over Dowex also solidified and was dissolved in MeOH for the analysis.
A neutralization of the synthesis products using IER III led to the expected composition after the distillation and prevented the bottom product solidification.
Overall, the results of this section indicate that some catalysts lead to OME synthesis products that are thermally unstable and consequently further react in thermal separation process steps. Other catalysts show stable product behavior, such as Dowex and Nafion for the MeOH–pFA feed mixture and A36, H-BEA 25 and Nafion for the OME1–TRI feed mixture. However, the tests were only conducted at an oil bath temperature of up to 100 °C. For the distillation separation of the OME synthesis product for the purification of OME3–5, temperatures of about 200 °C are required.11 This temperature increase coupled with the residence time inside the distillation column would lead to a substantial acceleration of the reaction kinetics and consequently reduce the OME3–5 product amount. Therefore, the thermal stability of the OME synthesis product should be tested at conditions close to the operational conditions inside the distillation column to decide the consideration of a neutralization step after the OME synthesis. Furthermore, the OME synthesis products were prepared with fresh catalysts, the thermal instability of the synthesis product might also be an initial phenomenon that might reduce with increasing time on stream after washing out instable acid groups.
Besides IER III other heterogeneous and homogeneous alkaline beds and solutions might be feasible for the neutralization of the OME synthesis product, such as alkaline IER, CaO, MgO or alkaline loaded active carbon. For further investigations not only process unit specific performance indicators such as activity, side product formation, long-term stability and regeneration should be considered, but also the influence on the overall process design and performance. Due to limited temperature stability and insertion of new components, the heat and cooling demand of the overall process might increase, and further separation might be necessary to remove side products and salts.
5. Conclusion
For the industrial production of OME, solid acid commercially available catalysts were investigated. To compare the feasibility of different IER, zeolites and Nafion catalysts for the OME synthesis in an aqueous reaction system and an anhydrous reaction system, the criteria conversion, selectivity, yield, activity, side product formation and thermal stability of the synthesis product were investigated. The synthesis product composition at the respective termination times was similar between various catalysts considered in this study. However, it showed significantly higher selectivities towards OME3–5 of about 40% for the anhydrous reaction system than for the aqueous reaction system with about 10%. The conversion of MeOH varied significantly between the catalysts, while the conversion of FA was very similar, with about 40%. Likely, the conversion of the OME1–TRI feed mixture was very similar between the catalysts and led to similar selectivities towards OME3–5. As a result, the yield of OME3–5 varied between 11–14 wt% for the MeOH–pFA feed mixture and 28–34 wt% for the OME1–TRI feed mixture.The activity varied significantly between the catalysts, with a high activity for the IER, especially Dowex, and a lower activity for the zeolites and Nafion. This was indicated by the termination time with about 50 min for Dowex for both feed mixtures and more than 1000 min and about 100 min for Nafion for the aqueous and the anhydrous reaction system, respectively. Besides Dowex, all catalysts showed significantly higher activities for the OME1–TRI feed mixture than for the MeOH–pFA feed mixture.
Regarding the formation of the side product MEFO, the reaction path varies between the aqueous and the anhydrous reaction system. For the MeOH–pFA feed mixture, high MEFO concentrations above 0.2 wt% were obtained with H-BEA 25, Dowex and H-MFI 90 after their respective termination times. All other catalysts led to MEFO concentrations below 0.1 wt% after the termination time. For the OME1–TRI feed mixture, MEFO is mainly formed following the irreversible Tishchenko reaction and, besides the zeolites, the MEFO concentration is far below 0.1 wt% for all catalysts after their respective termination times. The side product TRI was formed in comparably high concentrations above 0.1 wt% by A15, A36 and Nafion, while all other catalysts led to smaller concentrations for the MeOH–pFA feed mixture. Regarding the side products FA and MeOH for the OME1–TRI feed mixture, especially A15, A36 and Dowex show high concentrations of about 1 wt%. Even though H2O could not be detected, traces of H2O were present inside the reaction mixture below the detection limit of about 0.02 wt% H2O in all syntheses and led to the formation of FA and MeOH, even in the anhydrous reaction system.
The OME synthesis products of A36, H-BEA 25 and Nafion showed a thermal stable behavior for the OME1–TRI feed mixture. All other synthesis products indicated thermal instability during the distillation, which caused solidification of the bottom product and changes in the product composition. Therefore, a neutralization step was identified as necessary for the downstream separation using distillation columns. This neutralization step should be investigated in detail closer to the operation conditions of the distillation column and considered for the process concept for the production of OME3–5. Therefore, not only the IER lead to active species inside the synthesis product mixture but also the zeolites and Nafion. The reaction kinetics of the reverse reactions will be pronounced at increasing temperatures which will be applied in distillation columns for the synthesis product purification. IER III is suitable to neutralize the synthesis product, however, its limited thermal stability needs to be considered for the overall process design and can lead to an increasing heating and cooling demand.
The results show that all investigated catalysts are suitable for the OME synthesis for both the anhydrous and the aqueous reaction systems. However, the IER showed significantly higher activities and lower MEFO side product formations than the zeolites. A15 and A36 show higher TRI side product formations, but TRI also reacts to OME and therefore does not need to be separated from the loop inside the OME3–5 production process. Regarding the formation of FA and MeOH for the OME1–TRI feed mixture, all catalysts led to comparatively high concentrations, which can only be prevented using very dry feedstock. Without the separation of traces of H2O and MeOH inside the feed mixture of the anhydrous reaction system, H2O needs to be separated from the loop to prevent its accumulation and negative effects on the product selectivity and reaction kinetic. Regarding thermal stability, all catalysts indicated at least minor changes in the product composition after the distillation and therefore require a neutralization step before entering the separation cascade. In conclusion, the IER catalysts are identified as most suitable for the OME synthesis for both anhydrous and aqueous reaction systems, with a particular recommendation for Dowex, A15 and A46 for anhydrous reaction systems and A15, A36 and A46 for aqueous reaction systems.
Symbols used
| A [m2] | Area |
| d [m] | Diameter |
| l [m] | Length |
| m [g] | Mass |
| p [bar] | Pressure |
| S [mol mol−1] | Selectivity |
| t [min] | Time |
| T [°C] | Temperature |
| V [ml] | Volume |
| w [wt%] | Mass fraction |
| x [mol mol−1] | Mole fraction |
| X [g g−1] | Conversion |
| Y [g g−1] | Yield |
Sub- and superscripts
| cat | Catalyst |
| i | Inner |
Abbreviations
| A15 | Amberlyst® 15 |
| A36 | Amberlyst® 36 |
| A46 | Amberlyst® 46 |
| BV | Back pressure valve |
| DME | Dimethyl ether |
| Dowex | Dowex® 50WX2 |
| EA | Ethyl acetate |
| EtOH | Ethanol |
| F | Filter |
| FA | Formaldehyde |
| FOAC | Formic acid |
| GC-FID | Gas chromatograph equipped with a flame ionization detector |
| H | Heat exchanger |
| HF | Poly(oxymethylene) hemiformals |
| IER | Ion exchange resins |
| LHV | Lower heating values |
| MEFO | Methyl formate |
| MeOH | Methanol |
| MG | Poly(oxymethylene) glycols |
| Nafion | Nafion™ NR40 |
| OME | Oxymethylene dimethyl ethers |
| p | Product flask |
| pFA | Paraformaldehyde |
| PI | Pressure indicator |
| PtX | Power-to-X |
| S | Catalyst chamber, sample |
| TI, TIC | Temperature indicators |
| TRI | Trioxane |
| V | Valve |
| VLLE | Vapor–liquid–liquid equilibria |
Author contributions
Conceptualization, F. M.; methodology, F. M.; experimental investigation, S. K., A. H., E. F. and D. K.; writing—original draft preparation, F. M. and A. H.; writing—review and editing, O. S. and M. K.; scientific supervision, O. S.; project administration, O. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Deutsche Bundesstiftung Umwelt (DBU) is gratefully acknowledged for funding the PhD work of Franz Mantei (20018/541). Furthermore, many thanks to INAQUA Vertriebsgesellschaft mbH and Clariant AG for providing the catalyst A46 and the zeolites H-BEA 25 and H-MFI 90.References
- International Renewable Energy Agency (IRENA), World Energy Transitions Outlook 2022: 1.5°C Pathway: Executive summary, 2022.
- IEA, World Energy Outlook 2021, Paris, 2021.
- Net Zero by 2050, https://www.iea.org/reports/net-zero-by-2050, (last accessed September 2022).
- P. Dworschak, V. Berger, M. Härtl and G. Wachtmeister, SAE Int. J. Fuels Lubr., 2022, 15, 171–197 CrossRef CAS.
- A. Zhenova, A. Pellis, R. A. Milescu, C. R. McElroy, R. J. White and J. H. Clark, ACS Sustainable Chem. Eng., 2019, 7, 14834–14840 CrossRef CAS.
- M. Schappals, T. Breug-Nissen, K. Langenbach, J. Burger and H. Hasse, J. Chem. Eng. Data, 2017, 62, 4027–4031 CrossRef CAS.
- M. Härtl, K. Gaukel, D. Pélerin and G. Wachtmeister, MTZ worldwide, 2017, 78, 52 CrossRef.
- A. Omari, B. Heuser, S. Pischinger and C. Rüdinger, Appl. Energy, 2019, 1242 CrossRef CAS.
- C. Hank, L. Lazar, F. Mantei, M. Ouda, R. J. White, T. Smolinka, A. Schaadt, C. Hebling and H.-M. Henning, Sustainable Energy Fuels, 2019, 3, 3219 RSC.
- P. Bokinge, S. Heyne and S. Harvey, Energy Sci. Eng., 2020, 8, 2587 CrossRef.
- F. Mantei, R. E. Ali, C. Baensch, S. Voelker, P. Haltenort, J. Burger, R.-U. Dietrich, N. von der Assen, A. Schaadt, J. Sauer and O. Salem, Sustainable Energy Fuels, 2022, 6, 528 RSC.
- S. Voelker, S. Deutz, J. Burre, D. Bongartz, A. Omari, B. Lehrheuer, A. Mitsos, S. Pischinger, A. Bardow and N. von der Assen, Sustainable Energy Fuels, 2022, 6, 1959 RSC.
- D. Oestreich, L. Lautenschütz, U. Arnold and J. Sauer, Chem. Eng. Sci., 2017, 163, 92 CrossRef CAS.
- L. Lautenschütz, Neue Erkenntnisse in der Syntheseoptimierung oligomerer Oxymethylendimethylether aus Dimethoxymethan und Trioxan, Ph.D. Thesis, Ruprecht-Karls-Universität Heidelberg, Heidelberg, 2015 Search PubMed.
- J. Burger, E. Ströfer and H. Hasse, Ind. Eng. Chem. Res., 2012, 51, 12751 CrossRef CAS.
- N. Schmitz, F. Homberg, J. Berje, J. Burger and H. Hasse, Ind. Eng. Chem. Res., 2015, 54, 6409 CrossRef CAS.
- J. Voggenreiter and J. Burger, Ind. Eng. Chem. Res., 2021, 60, 2418 CrossRef CAS.
- Y. Zheng, Q. Tang, T. Wang, Y. Liao and J. Wang, Chem. Eng. Technol., 2013, 36, 1951 CrossRef CAS.
- J. Wu, H. Zhu, Z. Wu, Z. Qin, L. Yan, B. Du, W. Fan and J. Wang, Green Chem., 2015, 17, 2353 RSC.
- L. Wang, W.-T. Wu, T. Chen, Q. Chen and M.-Y. He, Chem. Eng. Commun., 2014, 201, 709 CrossRef CAS.
- A. Fink, C. H. Gierlich, I. Delidovich and R. Palkovits, ChemCatChem, 2020, 12, 5710 CrossRef CAS.
- P. Endres, S. Zechel, A. Winter, M. D. Hager and U. S. Schubert, Macromol. Chem. Phys., 2022, 2200020 CrossRef CAS.
- C. J. Baranowski, M. Roger, A. M. Bahmanpour and O. Kröcher, ChemSusChem, 2019, 12, 4421 CrossRef CAS PubMed.
- I. Hahnenstein, M. Albert, H. Hasse, C. G. Kreiter and G. Maurer, Ind. Eng. Chem. Res., 1995, 34, 440 CrossRef CAS.
- J.-O. Drunsel, M. Renner and H. Hasse, Chem. Eng. Res. Des., 2012, 90, 696 CrossRef CAS.
- N. Schmitz, J. Burger and H. Hasse, Ind. Eng. Chem. Res., 2015, 54, 12553 CrossRef CAS.
- P. Haltenort, L. Lautenschütz, U. Arnold and J. Sauer, Top. Catal., 2019, 62, 551 CrossRef CAS.
- L. Lautenschütz, D. Oestreich, P. Haltenort, U. Arnold, E. Dinjus and J. Sauer, Fuel Process. Technol., 2017, 165, 27 CrossRef.
- C. Kuhnert, Dampf-Flüssigkeits-Gleichgewichte in mehrkomponentigen formaldehydhaltigen Systemen, Ph.D. Thesis, Technischen Universität Kaiserslautern, Kaiserslautern, 2004 Search PubMed.
- T. Grützner, H. Hasse, N. Lang, M. Siegert and E. Ströfer, Chem. Eng. Sci., 2007, 62, 5613 CrossRef.
- P. Haltenort, K. Hackbarth, D. Oestreich, L. Lautenschütz, U. Arnold and J. Sauer, Catal. Commun., 2018, 109, 80 CrossRef CAS.
- K. Hackbarth, P. Haltenort, U. Arnold and J. Sauer, Chem. Ing. Tech., 2018, 90, 1 CrossRef.
- C. Cao, G. Liu, F. Xin, Q. Lei, X. Qin, Y. Yin, H. Chen and A. Ullah, Chem. Eng. Sci., 2022, 248, 117136 CrossRef CAS.
- N. Schmitz, E. Ströfer, J. Burger and H. Hasse, Ind. Eng. Chem. Res., 2017, 56, 11519 CrossRef CAS.
- N. Schmitz, C. F. Breitkreuz, E. Ströfer, J. Burger and H. Hasse, Chem. Eng. Process., 2018, 116 CrossRef CAS.
- N. Schmitz, C. F. Breitkreuz, E. Ströfer, J. Burger and H. Hasse, J. Membr. Sci., 2018, 564, 806 CrossRef CAS.
- A. Ferre and J. Burger, Ind. Eng. Chem. Res., 2021, 60, 15256 CrossRef CAS.
- C. F. Breitkreuz, M. Dyga, E. Forte, F. Jirasek, J. de Bont, J. Wery, T. Grützner, J. Burger and H. Hasse, Chem. Eng. Process., 2021, 108710 Search PubMed.
- Product Data Sheet Amberlyst 15 Wet, https://www.lenntech.com/Data-sheets/Dow-Amberlyst-15-wet-L.pdf, (last accessed August 2022).
- DUPONT, AMBERLYST™ 36WET Product Data Sheet: Form No. 177-03093, 2019, Rev. 2, pp. 1–3.
- J. Burger, A novel process for the production of diesel fuel additives by hierarchical design, Ph.D. Thesis, University of Kaiserslautern, Kaiserslautern, 2012 Search PubMed.
- Dow Chemical Company, AMBERLYST™ 46 RESIN - Sales Specification, 2019.
- D. Oestreich, Prozessentwicklung zur Gewinnung von Oxymethylenethern (OME) aus Methanol und Formaldehyd, Karlsruher Institut für Technologie, 2017 Search PubMed.
- Clariant Produkte (Deutschland) GmBH, HCZB 25 Product Data Sheet, 2019.
- Clariant Produkte (Deutschland) GmBH, HCZP 90E Product Data Sheet, 2019.
- Nafion™ NR40 and NR50 Ion Exchange Materials, https://www.nafion.com/en/-/media/files/nafion/nafion-nr40-nr50-p-16-product-info.pdf?rev=f5ee99361c214f07972e4b5a88aad413, (last accessed August 2022).
- M. Ouda, G. Yarce, R. J. White, M. J. Hadrich, D. Himmel, A. Schaadt, H. Klein, E. Jacob and I. Krossing, React. Chem. Eng., 2017, 2, 50 RSC.
- A. T. Sundberg, P. Uusi-Kyyny, M. Pakkanen and V. Alopaeus, J. Chem. Eng. Data, 2011, 56, 2634 CrossRef CAS.
- S. Lee, J. Cho and Y.-C. Kim, Fluid Phase Equilib., 2015, 397, 95 CrossRef CAS.
- C. J. Baranowski, A. M. Bahmanpour and O. Kröcher, Appl. Catal., B, 2017, 217, 407 CrossRef CAS.
- R. Soto, C. Fité, E. Ramírez, M. Iborra and J. Tejero, React. Chem. Eng., 2018, 3, 195 RSC.
- A. Fink, Struktur-Aktivitätsbeziehungen von Zeolithen als feste Säurekatalysatoren in der Synthese von Oxymethylendimethylethern, Ph.D. Thesis, Aachen, 2022 Search PubMed.
- J.-O. Drunsel, Entwicklung von Verfahren zur Herstellung von Methylal und Ethylal, Ph.D. Thesis, Scientific Report Series, Kaiserslautern, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00508e |
| This journal is © The Royal Society of Chemistry 2023 |