
Efficient electrosynthesis of dimethyl carbonate from urea and methanol under mild conditions†
Mingjuan
Sun
a,
Fengjiao
Li
 *ab,
Yanhui
Cui
a,
Xiaolin
Zhao
a,
Haiwei
Chen
a and
Shuting
Liang
c
*ab,
Yanhui
Cui
a,
Xiaolin
Zhao
a,
Haiwei
Chen
a and
Shuting
Liang
c
aShenzhen Automotive Research Institute, Beijing Institute of Technology, Shenzhen 518118, Guangdong, China. E-mail: lifengjiao@szari.ac.cn
bNational Engineering Laboratory for Electric Vehicles, Beijing Institute of Technology, Beijing 100081, Beijing, China
cChongqing Key Laboratory of Environmental Materials & Remediation Technologies, Chongqing University of Arts and Sciences, Chongqing 402160, China
First published on 31st December 2022
Abstract
Dimethyl carbonate (DMC) has versatile applications in chemical, pharmaceutical, and new energy industries. Synthesis of DMC from low-cost and abundant urea and methanol is an attractive and promising strategy via indirect utilization of CO2. However, high temperature and pressure are usually required for the thermo-synthesis of DMC from urea and methanol. Herein, mild and efficient synthesis of DMC has been firstly developed by electrolyzing urea and methanol over Pt (+)|Ti (−) electrodes in an undivided cell. Effects of various electrosynthesis conditions, supporting electrolytes, and electrode materials were systematically investigated. Under optimized conditions, the highest DMC yield of 50.4% with 100% selectivity was achieved. Furthermore, a possible mechanism was proposed for the electrosynthesis of DMC from urea and methanol. Other important organic carbonates such as diethyl carbonate (DEC), dipropyl carbonate (DPC), dibutyl carbonate (DBC), ethylene carbonate (EC), and propylene carbonate (PC) were also successfully synthesized by this mild electrosynthesis method.
1. Introduction
Dimethyl carbonate (DMC) is an environment-friendly and versatile chemical with wide applications mainly due to the merits of high oxygen content of 53.3%, low toxicity, non-corrosiveness, and biodegradability.1,2 For instance, DMC can be employed as a benign and important intermediate and green solvent to produce polycarbonate (PC), polyurethane (PU), and pharmaceuticals.3–5 Besides, DMC is also a potential additive to fuel oil6,7 and an excellent electrolyte solvent in lithium/sodium-ion batteries of new energy industries.8–10 In particular, the market demand for DMC has been increasing drastically as a result of the exploding growth of the PC industry and electric vehicles.11To date, many methods have been developed to synthesize DMC, including phosgenation of methanol,12 carbonylation of methanol,13,14 and direct/indirect utilization of CO2 with methanol.15–22 However, the first two routes suffer from obvious shortcomings, such as toxic and corrosive reactants. In contrast, CO2 is an abundant, nontoxic, and economical C1 resource. Therefore, the capture and utilization of CO2 to value-added DMC has attracted worldwide attention as one of the most popular and sustainable methods.1,2,23,24 Nonetheless, owing to obstacles in the activation of highly stable CO2 molecule and thermodynamic restrictions, the direct conversion of CO2 and methanol is usually operated under high pressure, such as 50 atm, to obtain DMC22,25,26 and has not been commercialized on a large-scale. On the contrary, indirect utilization of CO2via transesterification of ethylene carbonate (EC) or propylene carbonate (PC) intermediate with methanol has been well adopted as the main method for industrial production of DMC.19,27,28 However, the cost of EC/PC has been increasing very recently due to the expensive and explosive ethylene oxide (EO) or propylene oxide (PO) derived from refinery factories. Hence, developing greener and more economical technical routes is still highly desired in the future.
At present, industrial production of urea directly from CO2 and ammonia is very mature, and thus alcoholysis of CO2-derived urea is an attractive, promising, and economical alternative for the synthesis of DMC, which possesses advantages of abundant resources, low-cost reactants, avoiding the formation of methanol–water–DMC ternary azeotrope, and simplified subsequent separation processes.20,21,29 The urea alcoholysis route is composed of two steps, i.e., the formation of methyl carbamate (MC) intermediate from urea and methanol and the reaction of MC with methanol to form DMC, with the latter one being rate-limiting.21,30 Up to now, intensive research has been focused on developing efficient catalysts such as ZnO based solid base catalysts for urea alcoholysis route via a thermo-synthesis way, which requires harsh conditions of over 400 K and 10 atm (Fig. 1).20 In addition, N-methyl methyl carbamate (NMMC) is also formed as a by-product from DMC and MC due to high reaction temperature, usually leading to severe pipeline block.20,21,30
 | ||
| Fig. 1 Comparison of electrosynthesis of DMC from urea and methanol with the traditional thermo-synthesis way using CO2 as the starting material. | ||
Electrosynthesis is a green and advantageous alternative to traditional thermo-synthesis, which can take place under mild conditions and replace dangerous and toxic chemicals with an electric current or “clean” electrons.31 Electrosynthesis can also well handle the selectivity of a process by imposing a certain cell voltage and enabling sustainable transformations when electricity is harvested from renewable energies like wind or solar.32–34 In addition, electrosynthesis can be readily stopped at any time by cutting off the electrical power.35 Therefore, electrosynthesis has been gaining increasing influence, both at the laboratory and industrial scales, in wide areas of fine chemicals, environmental mitigation, pharmaceuticals, etc. Recently, the electrosynthesis of DMC from CO2 and methanol has been gaining growing attention, most of which use a carcinogenic alkylating agent such as CH3I to achieve satisfactory yields.34 Otherwise, very low yields (below 5%) are obtained in the absence of CH3I.36 Thus, more efforts should be devoted to the mild, green, and efficient electrosynthesis of DMC.
Herein, following our continuous interest in the synthesis of organic carbonates via urea alcoholysis route,37–40 we first reported and disclosed a novel, green, effective, and practical electrochemical method for DMC synthesis from urea and methanol over Pt (+)|Ti (−) electrodes under mild conditions (≤298 K, 1 atm) (Fig. 1). Effects of various electrosynthesis conditions, supporting electrolytes, and electrode materials were systematically investigated. A possible mechanism was also proposed for this electrosynthesis reaction. In addition, this mild method could also be employed to synthesize diverse organic carbonates such as diethyl carbonate (DEC), dipropyl carbonate (DPC), dibutyl carbonate (DBC), ethylene carbonate (EC), and propylene carbonate (PC). This electrosynthesis method is green, mild, oxidant-free, and easy to operate and scale up.
2. Experimental
2.1. Reagents and materials
Methanol, ethanol, n-propanol, n-butyl alcohol, ethylene glycol, 1,2-propanediol, sodium bromide (NaBr), sodium sulfate (Na2SO4), sodium hydroxide (NaOH), sodium iodide (NaI), sodium chloride (NaCl), sodium bromate (NaBrO3), sodium methoxide (CH3ONa) and urea were purchased from Shanghai Aladdin Chemical Reagent Co., Ltd., China. Standards of DMC, DEC, DPC, DBC, EC, and PC were also obtained from Shanghai Aladdin Chemical Reagent Co., Ltd., China. Alcohols were used after drying and storing in 4 Å molecular sieves. Commercial platinum plate and titanium foil were burnished with abrasive paper, treated with 1 mol L−1 HCl, thoroughly washed with distilled water and ethanol, and dried in an oven prior to the electrosynthesis experiments. The reagents and solvents were directly used as received without further purification unless otherwise noted.2.2. Electrosynthesis of DMC from urea and methanol
The electrosynthesis of DMC from urea and methanol was conducted in an undivided one-compartment cell, which was equipped with a platinum plate anode (Pt, 10 mm × 10 mm × 1 mm) and a titanium foil cathode (Ti, 10 mm × 10 mm × 0.08 mm). In a typical electrosynthesis experiment, urea (0.05 mol L−1), NaBr (0.15 mol L−1), and methanol (20 mL) were added to the cell. Ar was used to purge air out of the cell before electrosynthesis, which could minimize the effect of air on the electrosynthesis reaction (Table S1†). The Pt plate anode and Ti foil cathode were connected to a DC regulated power supply. Then, the electrosynthesis of DMC was carried out at a current density of 20 mA cm−2 and a low reaction temperature of 288 K for 12 h under 1 atm. After the mild electrosynthesis reaction was finished, the resultant liquid mixtures were analysed by gas chromatography (GC), 1H nuclear magnetic resonance spectroscopy (1HNMR), and gas chromatography-mass spectrometry (GC-MS).GC tests were conducted on GC9800 (Shanghai Kechuang Chromatographic Instrument Co., LTD., China) equipped with a flame-ionization detector (FID) and an FFAP capillary column (inner diameter: 0.32 mm, length: 30 m, film thickness: 0.50 μm). Typically, for the analysis of DMC from the electrochemical conversion of urea with methanol, n-butanol was used as the internal standard and N2 as the carrier gas. The temperature was detected from 313 to 453 K at a rate of 20 K min−1. 1HNMR spectra were obtained by NMR spectrometer (Bruker-400 M, 400 MHz). The CDCl3 was used as the solvent and the internal standard to calibrate the chemical shift. GC-MS tests were performed on GCMS-QP 2020 NX (SHIMADZU) with an SH-Stabilwax-DA capillary column (inner diameter: 0.25 mm, length: 30 m, film thickness: 0.25 μm).
2.3. Expandability of electrosynthesis of diverse organic carbonates
In order to investigate the expandability of electrosynthesis of diverse organic carbonates from urea and alcohols, electrosynthesis of DEC, DPC, DBC, EC, and PC were also carried out and analysed similar to procedures of DMC, except that DEC and DBC used n-propanol as the internal standard for GC analysis. The corresponding equations are shown in ESI.†3. Results and discussion
3.1. Product analysis
The scheme for electrosynthesis of DMC from urea and methanol is shown in Fig. 2a, using NaBr as the supporting electrolyte, Pt plate as an anode, and Ti foil as a cathode. At first, (I) electrosynthesis of DMC from urea and methanol was performed over Pt (+)|Ti (−) electrodes under the conditions of Curea = 0.15 mol L−1, CNaBr = 0.15 mol L−1, T = 298 K, current density = 20 mA cm−2, and time = 36 h (shorted as “CH3OH + NaBr + urea”). For comparison, the following two blank experiments were also performed: (II) the electrochemical reaction of methanol and NaBr without urea (shorted as “CH3OH + NaBr”), and (III) the electrochemical reaction of urea and methanol without NaBr (shorted as “CH3OH + urea”). As observed in Fig. 2b and c and S1,† after the electrosynthesis reaction was carried out at a constant current density, apart from MC intermediate and DMC product, a new compound of methyl formate (MF) was also detected by GC in the experiment of “CH3OH + NaBr + urea”. In addition, no NMMC by-product was found in this electrosynthesis system, also greatly differing from the thermo-synthesis way. As expected, no DMC was detected in the two blank experiments. However, with the help of NaBr supporting electrolyte, MF existed in the solutions containing methanol and NaBr (Fig. S1c and d†), mainly due to the oxidation of pure methanol in the Pt anode.41 The identities of MC, DMC, and MF were further confirmed by 1HNMR and GC-MS. As displayed in Fig. 2d, 1HNMR spectra of the liquid mixture showed a strong peak around 3.4 ppm, indicative of the presence of the –CH3 group.42 The enlarged characteristic peak at 3.62 ppm corresponded to the –OCH3 group in DMC and MC molecules. Besides, a peak at 3.75 ppm was assigned to the –OCH3 group in the MF molecule. The other two peaks at 5.15 and 8.07 ppm were attributed to –NH2 group of MC and H(CO)– group of MF, respectively. These results were in excellent agreement with their corresponding 1HNMR spectra of standards (Fig. S2†). Moreover, GC-MS analysis results also verified the formation of those three compounds (Fig. 2e and S1f†). | ||
| Fig. 2 (a) Scheme for the electrosynthesis of DMC from urea and methanol over Pt (+)|Ti (−) electrodes using NaBr as the supporting electrolyte; (b and c) GC results on (I) “CH3OH + NaBr + urea” experiment under the conditions of Curea = 0.15 mol L−1, CNaBr = 0.15 mol L−1, T = 298 K, current density = 20 mA cm−2, and time = 36 h, (II) “CH3OH + NaBr” experiment under the same conditions in the absence of urea, (III) “CH3OH + urea” experiment under the same conditions in the absence of NaBr, in comparison with standards of DMC and MC; (d) 1HNMR and (e) mass spectra of the liquid mixture from “CH3OH + NaBr + urea” experiment. | ||
3.2. Effect of electrosynthesis conditions
Effects of electrosynthesis conditions such as NaBr concentration, urea concentration, current density, reaction temperature, and reaction time were systematically investigated to optimize the electrosynthesis procedure.At first, the effect of NaBr concentration on the electrosynthesis of DMC from urea and methanol was performed over Pt (+)|Ti (−) electrodes under the conditions of Curea = 0.15 mol L−1, methanol (20 mL), T = 298 K, current density = 20 mA cm−2, time = 36 h. High conductivity was obtained for the mixture of CH3OH, NaBr, and urea (Table S2†). As seen in Fig. 3a, NaBr concentration had a significant impact on DMC yield. DMC yield greatly increased with the increasing NaBr concentration from 0 to 0.15 mol L−1, and reached the optimum yield at 0.15 mol L−1 of NaBr concentration, mainly due to the improved and suitable conductivity of the mixture and the possible catalytic effect. However, further increasing NaBr concentration from 0.15 to 0.30 mol L−1, DMC yield slightly decreased to 27.4% at 0.30 mol L−1, which might be due to much larger amount of MF was produced at a higher concentration of NaBr (Fig. S3†), indicating that too excess NaBr was disadvantageous to the production of DMC. Therefore, 0.15 mol L−1 of NaBr was chosen in the following experiments.
 | ||
| Fig. 3 Effects of electrosynthesis conditions on DMC yield over Pt (+)|Ti (−) electrodes. (a) Effect of NaBr concentration. Reaction conditions: Curea = 0.15 mol L−1, methanol (20 mL), T = 298 K, current density = 20 mA cm−2, time = 36 h; (b) effect of urea concentration. Reaction conditions: CNaBr = 0.15 mol L−1, methanol (20 mL), T = 298 K, current density = 20 mA cm−2, time = 36 h; (c) effect of current density. Reaction conditions: CNaBr = 0.15 mol L−1, Curea = 0.05 mol L−1, methanol (20 mL), T = 298 K, time = 36 h; (d) effect of reaction temperature. Reaction conditions: CNaBr = 0.15 mol L−1, Curea = 0.05 mol L−1, methanol (20 mL), current density = 20 mA cm−2, time = 36 h; (e) effect of reaction time. Reaction conditions: CNaBr = 0.15 mol L−1, Curea = 0.05 mol L−1, methanol (20 mL), current density = 20 mA cm−2, T = 288 K; (f) comparison of electrosynthesis of DMC directly from urea and methanol with reported thermo-synthesis results in Table S7.† | ||
The effect of urea concentration in the range of 0.01–0.40 mol L−1 on the electrosynthesis of DMC was investigated over Pt (+)|Ti (−) electrodes under the conditions of CNaBr = 0.15 mol L−1, methanol (20 mL), T = 298 K, current density = 20 mA cm−2, time = 36 h. As displayed in Fig. 3b, when urea concentration was increased from 0.01 to 0.05 mol L−1, DMC yield was greatly boosted from 15.5% to 42.4% since higher urea concentration was beneficial to shift the electrosynthesis equilibrium to the side of DMC formation. Nonetheless, DMC yield gradually decreased with the urea concentration increasing to 0.40 mol L−1. Interestingly, no MC intermediate was detected when urea concentration is ≤0.05 mol L−1, suggesting that MC intermediate was completely converted with 100% of DMC selectivity at urea concentration lower than 0.05 mol L−1 (Fig. S4†). Therefore, the optimum urea concentration was selected as 0.05 mol L−1.
The effect of current density was investigated at current densities of 5–40 mA cm−2 under the conditions of CNaBr = 0.15 mol L−1, Curea = 0.05 mol L−1, methanol (20 mL), T = 298 K, time = 36 h. As exhibited in Fig. 3c, DMC yield increased rapidly when the current density was increased from 5 to 20 mA cm−2 and reached 42.4% at 20 mA−2, mainly attributed to the faster electrosynthesis rate at higher current density. However, a slight decrease was observed at the current density from 30 to 40 mA cm−2 due to the occurrence of side reactions. Hence, the best current density was 20 mA cm−2 to ensure an excellent DMC product yield.
Electrosynthesis experiments were carried out from 278 to 308 K under the conditions of CNaBr = 0.15 mol L−1, Curea = 0.05 mol L−1, methanol (20 mL), current density = 20 mA cm−2, time = 36 h, in order to investigate the effect of reaction temperature on DMC yield. As displayed in Fig. 3d, DMC yield slightly increased when the reaction temperature was increased from 278 to 288 K, mainly ascribing to faster reaction kinetics at a higher reaction temperature. However, too high temperature is not advantageous to electrosynthesis of DMC, and DMC yield decreased to 35.2% at 308 K, which could be due to the formation of less MC intermediate and generation of more MF side-product, thus leading to the lower DMC yield at the temperature over 298 K. Generally, high DMC yields of >40% could be obtained at low and wide reaction temperatures of 278–298 K. The best reaction temperature was chosen as 288 K.
The effect of reaction time was investigated from 6 to 48 h under the conditions of CNaBr = 0.15 mol L−1, Curea = 0.05 mol L−1, methanol (20 mL), current density = 20 mA cm−2, T = 288 K. As presented in Fig. 3e, DMC yield rapidly increased when the reaction time was prolonged from 6 to 12 h, but further increasing reaction time could not improve DMC yield, implying that the electrosynthesis process had approached equilibrium. Therefore, the highest DMC yield of 50.4% with 100% selectivity was obtained under the optimized conditions of CNaBr = 0.15 mol L−1, Curea = 0.05 mol L−1, 20 mA cm−2, 288 K, and 12 h (Table 1, entry 1). More importantly, no MF was detected when the reaction time was shorter than 12 h (Fig. S5†), indicating that only DMC was produced under the optimized electrosynthesis conditions.
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | DMC yieldb |
| a Pt plate anode, Ti foil cathode, urea (0.05 mol L−1), NaBr (0.15 mol L−1), methanol (20 mL), current density = 20 mA cm−2, 288 K, 12 h, 1 atm. b Yield was analysed by GC-MS and GC with an internal standard, n. d. = not detected, n. r. = no reaction. | ||
| 1 | None | 50.4% |
| 2 | No urea | n. r. |
| 3 | No NaBr | n. r. |
| 4 | No electricity | n. r. |
| 5 | NaOH instead of NaBr | n. d. |
| 6 | NaI instead of NaBr | n. d. |
| 7 | CH3ONa instead of NaBr | n. d. |
| 8 | Na2SO4 instead of NaBr | n. d. |
| 9 | NaCl instead of NaBr | 17.8% |
| 10 | NaBrO3 instead of NaBr | 8.2% |

3.3. Effect of supporting electrolytes
The effect of supporting electrolytes on DMC yield was studied. Certainly, no DMC would be produced in the absence of urea (Table 1, entry 2). The electrosynthesis reaction of DMC could not take place without NaBr or electricity at 288 K and 1 atm (Table 1, entries 3–4), also suggesting that NaBr with high solubility in methanol is an excellent and necessary conductive supporting electrolyte, which could enable the electron transfer and efficiently facilitate the electrosynthesis of DMC from urea and methanol. Furthermore, with the contribution of electricity and NaBr, Br−/Br2 were generated as mediators to significantly influence the formation of DMC, resulting in a high DMC yield of 50.4%. In contrast, no desired DMC product was detected when NaBr was replaced by other supporting electrolytes such as NaOH, NaI, CH3ONa, and Na2SO4 (Table 1, entries 5–8), suggesting that NaOH, NaI, CH3ONa, and Na2SO4 were not suitable in this system. It should be noted that Na2SO4 is insoluble in methanol solution and then could not provide efficient conductivity in this system. I2 and Br2 were also tried for the electrosynthesis of DMC. As expected, since I2 and Br2 are not conductive, the electrosynthesis reaction could not proceed when I2 or Br2 was used instead of NaBr (Tables S3 and S4†). Therefore, it can be concluded that an excellent supporting electrolyte should possess a high electrical conductivity as well as high solubility in methanol, both of which are essential for the electrosynthesis of DMC. In addition, when using NaCl or NaBrO3 instead of NaBr, the yields of DMC greatly dropped to 17.8% and 8.2%, respectively (Table 1, entries 9 and 10), also verifying that NaBr was the best supporting electrolyte in the electrosynthesis of DMC from urea and methanol. More significantly, apart from improving electrical conductivity, NaBr may also function as an efficient catalyst to increase the electrosynthesis reaction rate and improve the product yield.In a word, DMC with an excellent yield of 50.4% and 100% selectivity has been successfully prepared by electrolyzing inexpensive and abundant urea and methanol over Pt (+)|Ti (−) electrodes with the help of NaBr under mild electrochemical conditions, which can be comparable to those via a thermo-synthesis way (Fig. 3f). Details are summarized in Table S7.† Besides, in comparison with the reported electrosynthesis of DMC from CO2 and methanol (Table S8†), the electrosynthesis of DMC from urea and methanol also achieved good product yield and selectivity, exhibiting great potential application in future commercialization.
3.4. Effect of electrode materials
To begin with, in order to investigate the effect of cathode materials on the electrosynthesis of DMC from urea and methanol, different materials such as Cu foam, Ni foam, Zn sheet, Pt plate, glassy carbon (GC), and graphite were used as the cathodes under the same reaction conditions for comparison. As shown in Fig. 4, among the investigated electrode systems, the Pt (+)|Ti (−) electrode system showed the highest performances toward DMC electrosynthesis. In contrast, Pt (+)|Cu (−), Pt (+)|Ni (−), Pt (+)|Zn (−), Pt (+)|Pt (−), and Pt (+)|graphite (−) electrode systems exhibited relatively lower DMC yields in the range of 32.6–48.4%, indicative of the relatively inferior performances of Cu, Ni, Zn, Pt, and graphite as the cathode. Especially, the Pt (+)|GC (−) electrode system exhibited the lowest electrocatalytic activity with only 2.6% of DMC yield, which was far smaller than the Pt (+)|Ti (−) electrode system. These results suggested that cathode materials had a great effect on the electrosynthesis of DMC, mainly attributing to the different adsorption abilities of the reactants on the cathodes, which thus led to different reactivities of the reactants on different cathodes.36,43,44 In addition, other materials, such as graphite and GC, were also used as the anode materials in comparison with the Pt anode. As shown in Table S5,† the graphite (+)|Ti (−) and GC (+)|Ti (−) electrode systems exhibited DMC yields of 45.0% and 25.7%, respectively, both of which are lower than that of the Pt (+)|Ti (−) electrode system, which could be ascribed to the better stability of Pt anode during the electrosynthesis process. As confirmed by ICP-MS and SEM results, the Pt element is hardly dissolved in the non-aqueous solution after the electrosynthesis (Table S6†) or deposited on the surface of Ti foil cathode (Fig. S6†). The above results indicated that Pt was an excellent and stable anode material. In summary, Pt (+)|Ti (−) was found to be the most effective electrode system for the electrosynthesis of DMC. | ||
| Fig. 4 Effects of various cathode materials on the electrosynthesis of DMC from urea and methanol. Reaction conditions: Pt plate anode, urea (0.05 mol L−1), methanol (20 mL), NaBr (0.15 mol L−1), current density = 20 mA cm−2, 288 K, 12 h, 1 atm. | ||
3.5. Possible reaction mechanism for electrosynthesis of DMC
In order to disclose the reaction mechanism for the electrosynthesis of DMC from urea and methanol, cyclic voltammetry (CV) and methoxide capture experiments were also performed.At first, CV experiments were executed to gain insight into the electrosynthesis process. As shown in Fig. S7a and b,† no oxidation peak was obviously seen in the absence of NaBr. After NaBr was dissolved in methanol, an oxidation peak was observed at ∼0.8 V vs. SCE (Fig. S7c†) due to the electro-oxidation of Br− ion.45 Similar oxidation peak was also found after adding urea into the mixture of methanol and NaBr (Fig. S7d†). Besides, similar phenomena were also observed using a different reference electrode, such as Ag/AgCl (Fig. S8†). In combination with the electrosynthesis results, it could be deduced that urea and methanol could be electrochemically activated and converted into DMC in the presence of NaBr supporting electrolyte.
Furthermore, conversion from CH3OH to methoxide anion (CH3O−) was confirmed by the methoxide capture experiment, which was employed in an H-cell reactor with a Nafion-117 membrane to separate the anode and cathode and the 1-iodobutane as the capturing agent. As displayed in Fig. S9,† there was an obvious peak corresponding to the butyl methyl ether after adding the 1-iodobutane into the cathode solution, suggesting that conversion from CH3OH to CH3O− happened in the Ti cathode during the electrosynthesis process of DMC, also in agreement with other reported works.46 Therefore, methoxide anion could be formed by deprotonation of methanol,46–50 and thus acted as a nucleophile to participate in the electrosynthesis of DMC.
Based on the above results, a possible mechanism for the electrosynthesis of DMC from urea and methanol was proposed in Fig. 5. Br−/Br2, as mediators, played a crucial role in the electrosynthesis reaction.46,51 Initially, the Br− ion was oxidized to Br2 in the Pt anode (Fig. S10 and S11†). Meantime, methanol adsorbed on the surface of the Ti cathode and was electro-activated to CH3O− (I). The formed Br2 could be consumed by direct reduction to Br− in the Ti cathode or by reaction with I to produce CH3OBr.46 It was worth noting that MF was also co-produced in this electrosynthesis system due to the oxidation and direct dehydrogenation of methanol in the Pt anode, accompanied by the formation of H+. Then, I acted as a nucleophile to react with II to form an MC intermediate with the help of H+ and simultaneously release a molecule of NH3. After that, MC was electro-activated to III and then reacted with I and H+ to obtain the targeted DMC product, accompanying the release of NH3.
 | ||
| Fig. 5 Possible reaction mechanism for electrosynthesis of DMC over Pt (+)|Ti (−) electrodes in the presence of NaBr supporting electrolyte. | ||
3.6. Expandability of electrosynthesis of diverse organic carbonates
The effectiveness and expandability of electrosynthesis of diverse organic carbonates such as diethyl carbonate (DEC), dipropyl carbonate (DPC), dibutyl carbonate (DBC), ethylene carbonate (EC), and propylene carbonate (PC) from urea and corresponding alcohols were also performed, and the results are presented in Fig. 6. DEC, DPC, DBC, EC, and PC were successfully produced with yields of >6%, indicative of the high versatility of this electrosynthesis method. However, these yields are much lower than DMC, mainly ascribing to the low solubility of NaBr in their corresponding alcohols, the inferior conductivity, as well as the occurrence of more severe side reactions.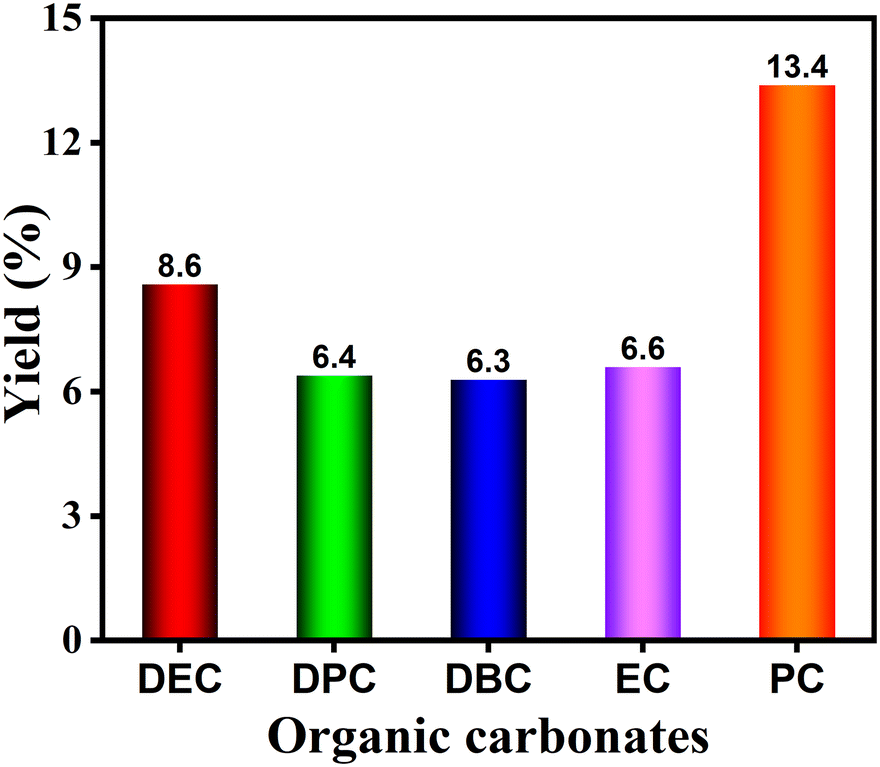 | ||
| Fig. 6 Expandability of electrosynthesis of diverse organic carbonates from urea and corresponding alcohols in an undivided cell. Reaction conditions: Pt plate anode, Ti foil cathode, urea (0.05 mol L−1), NaBr (0.15 mol L−1), alcohol (20 mL), current density = 20 mA cm−2, 288 K, 12 h, 1 atm. | ||
4. Conclusions
In summary, a novel, efficient, and green protocol for the synthesis of DMC by electrochemical conversion of urea and methanol was developed under mild conditions. Apart from the MC intermediate and DMC product, a new compound of methyl formate (MF) was also formed in the electrosynthesis reaction. A high DMC yield of 50.4% and DMC selectivity of 100% were obtained over the best-performance Pt (+)|Ti (−) electrode system, with NaBr as the most efficient supporting electrolyte. NaBr was verified to play a significant role in the electrosynthesis of DMC, which could provide sufficient conductivity, generate Br−/Br2 as mediators, and increase the electrosynthesis reaction rate. In addition, methoxide anion derived from the electro-activation of methanol was confirmed, which could efficiently react with urea to form MC and continued reacting with MC to synthesize the final DMC product under mild electrosynthesis conditions. The mild electrosynthesis method is also employed to synthesize DEC, DPC, DBC, EC, and PC. This work provides a novel and promising approach for the electrochemical conversion of CO2-derived urea to organic carbonates under mild conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 21706162), Guangdong Basic and Applied Basic Research Foundation (No. 2021A1515010150), Science and Technology Program of Guangdong Province (No. 2019B090904004), and China Postdoctoral Science Foundation (No. 2017M622790).References
- M. Zhang, Y. H. Xu, B. L. Williams, M. Xiao, S. J. Wang, D. M. Han, L. Y. Sun and Y. Z. Meng, J. Cleaner Prod., 2021, 279, 123344 CrossRef CAS.
- P. Kumar, V. C. Srivastava, U. L. Štangar, B. Mušič, I. M. Mishra and Y. Meng, Catal. Rev.: Sci. Eng., 2021, 63, 363–421 CrossRef CAS.
- Z. Yang, L. Liu, H. An, C. Li, Z. Zhang, W. Fang, F. Xu and S. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 9968–9979 CrossRef CAS.
- B. Puértolas, M. Rellán-Piñeiro, J. L. Núñez-Rico, A. P. Amrute, A. Vidal-Ferran, N. López, J. Pérez-Ramírez and S. Wershofen, ACS Catal., 2019, 9, 7708–7720 CrossRef.
- T. Nienałtowski, P. Szczepanik, P. Małecki, D. Czajkowska-Szczykowska, S. Czarnocki, J. Pawłowska, A. Kajetanowicz and K. Grela, Chem. – Eur. J., 2020, 26, 15708–15717 CrossRef PubMed.
- J. H. Chan, A. Tsolakis, J. M. Herreros, K. X. Kallis, C. Hergueta, S. Sittichompoo and M. Bogarra, Fuel, 2020, 263, 116742 CrossRef CAS.
- M. Pan, W. Qian, Z. Zheng, R. Huang, X. Zhou, H. Huang and M. Li, Fuel, 2019, 257, 115920 CrossRef CAS.
- Y. Jin, P. M. L. Le, P. Gao, Y. Xu, B. Xiao, M. H. Engelhard, X. Cao, T. D. Vo, J. Hu, L. Zhong, B. E. Matthews, R. Yi, C. Wang, X. Li, J. Liu and J.-G. Zhang, Nat. Energy, 2022, 7, 718–725 CrossRef CAS.
- J. Hou, L. Lu, L. Wang, A. Ohma, D. Ren, X. Feng, Y. Li, Y. Li, I. Ootani, X. Han, W. Ren, X. He, Y. Nitta and M. Ouyang, Nat. Commun., 2020, 11, 5100 CrossRef CAS PubMed.
- N. Dubouis, T. Marchandier, G. Rousse, F. Marchini, F. Fauth, M. Avdeev, A. Iadecola, B. Porcheron, M. Deschamps, J.-M. Tarascon and A. Grimaud, Nat. Mater., 2021, 20, 1545–1550 CrossRef CAS PubMed.
- H.-Z. Tan, Z.-Q. Wang, Z.-N. Xu, J. Sun, Y.-P. Xu, Q.-S. Chen, Y. Chen and G.-C. Guo, Catal. Today, 2018, 316, 2–12 CrossRef CAS.
- I. E. Muskat; and F. Strain, US Pat., 2379250, 1941 Search PubMed.
- K. Nishihira, K. Mizutare and S. Tanaka, EP Pat., 425197, 1991 Search PubMed.
- U. Romano, R. Tesei, G. Cipriani and L. Micucci, US Pat., 4218391, 1980 Search PubMed.
- Z. Zhang, S. Liu, L. Zhang, S. Yin, G. Yang and B. Han, Chem. Commun., 2018, 54, 4410–4412 RSC.
- Z.-Q. Wang, M.-J. Zhang, X.-B. Hu, V. P. Dravid, Z.-N. Xu and G.-C. Guo, Chem. Commun., 2020, 56, 403–406 RSC.
- Y. Liao, F. Li, X. Dai, N. Zhao and F. Xiao, Chin. J. Catal., 2017, 38, 1860–1869 CrossRef CAS.
- Y. Song, X. He, B. Yu, H.-R. Li and L.-N. He, Chin. Chem. Lett., 2020, 31, 667–672 CrossRef CAS.
- M. Tojo and H. Miyaji, US Pat., 8148566B2, 2012 Search PubMed.
- K. Shukla and V. C. Srivastava, Catal. Rev.: Sci. Eng., 2017, 59, 1–43 CrossRef CAS.
- A. Sánchez, L. M. Gil and M. Martín, J. CO2 Util., 2019, 33, 521–531 CrossRef.
- H. Yang, H. Sun, W. Zeng, F. Dai, Y. Duan, M. Shi, Z. Hua, X. Yang and B. Zhang, React. Chem. Eng., 2022, 7, 2399–2409 RSC.
- A. H. Tamboli, A. A. Chaugule and H. Kim, Chem. Eng. J., 2017, 323, 530–544 CrossRef CAS.
- J. Artz, T. E. Muller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- K. Xuan, S. Chen, Y. Pu, Y. Guo, Y. Guo, Y. Li, C. Pu, N. Zhao and F. Xiao, J. CO2 Util., 2022, 59, 101960 CrossRef CAS.
- R. Saada, S. Kellici, T. Heil, D. Morgan and B. Saha, Appl. Catal., B, 2015, 168–169, 353–362 CrossRef CAS.
- Z. Song, B. Subramaniam and R. V. Chaudhari, React. Chem. Eng., 2020, 5, 101–111 RSC.
- Y. He, H. Cheng, Z. Pan and F. Cheng, React. Chem. Eng., 2021, 6, 2170–2180 RSC.
- F. Wang, Y. Pu, J. Yang, T. Wang, L. Chen, N. Zhao and F. Xiao, Chin. J. Chem. Eng., 2019, 27, 1879–1887 CrossRef CAS.
- B. Jia, X. Sun, M. Chen, J. Jian, K. You, H. A. Luo, Y. Huang, X. Luo, B. Jin, N. Wang and Z. Liang, React. Chem. Eng., 2021, 6, 1854–1868 RSC.
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- D. S. P. Cardoso, B. Šljukić, D. M. F. Santos and C. A. C. Sequeira, Org. Process Res. Dev., 2017, 21, 1213–1226 CrossRef CAS.
- D. Anastasiadou, E. J. M. Hensen and M. C. Figueiredo, Chem. Commun., 2020, 56, 13082–13092 RSC.
- Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed.
- X. Yuan, B. Lu, J. Liu, X. You, J. Zhao and Q. Cai, J. Electrochem. Soc., 2012, 159, E183–E186 CrossRef CAS.
- F. J. Li, L. G. Wang, S. Xu, S. T. Liang and N. N. Zhang, RSC Adv., 2021, 11, 15477–15485 RSC.
- F.-Y. Chen, L. Xiao, C.-X. Yang and L. Zhuang, Acta Phys.-Chim. Sin., 2015, 31, 2310–2315 CAS.
- L. G. Wang, H. Q. Li, S. M. Xin and F. J. Li, Catal. Commun., 2014, 50, 49–53 CrossRef CAS.
- S. M. Xin, L. G. Wang, H. Q. Li, K. L. Huang and F. J. Li, Fuel Process. Technol., 2014, 126, 453–459 CrossRef CAS.
- R. Kishi, H. Ogihara, M. Yoshida-Hirahara, K. Shibanuma, I. Yamanaka and H. Kurokawa, ACS Sustainable Chem. Eng., 2020, 8, 11532–11540 CrossRef CAS.
- Y. Zhou, R. Ganganahalli, S. Verma, H. R. Tan and B. S. Yeo, Angew. Chem., Int. Ed., 2022, 61, e202202859 CAS.
- C. Yan, B. Lu, X. Wang, J. Zhao and Q. Cai, J. Chem. Technol. Biotechnol., 2011, 86, 1413–1417 CrossRef CAS.
- M. C. Figueiredo, V. Trieu, S. Eiden and M. T. M. Koper, J. Am. Chem. Soc., 2017, 139, 14693–14698 CrossRef CAS PubMed.
- M. Tariq, Z. Phys. Chem., 2020, 234, 295–312 CrossRef CAS.
- K. M. Lee, J. H. Jang, M. Balamurugan, J. E. Kim, Y. I. Jo and K. T. Nam, Nat. Energy, 2021, 6, 733–741 CrossRef CAS.
- L. Li and S. Luo, Org. Lett., 2018, 20, 1324–1327 CrossRef CAS PubMed.
- D. Horii, M. Atobe, T. Fuchigami and F. Marken, Electrochem. Commun., 2005, 7, 35–39 CrossRef CAS.
- G. Jia, W. Zhang, Z. C. Jin, W. D. An, Y. F. Gao, X. Zhang and J. R. Liu, Electrochim. Acta, 2014, 144, 1–6 CrossRef CAS.
- Y. T. Yu, X. H. Liu, W. Zhang, Y. Zhang, L. J. Li, Z. Z. Cao, Z. H. Guo, H. Wang, G. Jia, Y. S. Pan and Y. F. Gao, Ind. Eng. Chem. Res., 2013, 52, 6901–6907 CrossRef CAS.
- A. Funakawa, I. Yamanaka and K. Otsuka, J. Electrochem. Soc., 2006, 153, D68 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00480a |
| This journal is © The Royal Society of Chemistry 2023 |