
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reinforcement learning optimization of reaction routes on the basis of large, hybrid organic chemistry–synthetic biological, reaction network data†
Chonghuan
Zhang
 a and
Alexei A.
Lapkin
*ab
a and
Alexei A.
Lapkin
*ab
aDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge CB3 0AS, UK. E-mail: aal35@cam.ac.uk
bCambridge Centre for Advanced Research and Education in Singapore, CARES Ltd, 1 CREATE Way, CREATE Tower #05-05, 138602 Singapore
First published on 5th July 2023
Abstract
Computer-assisted synthesis planning (CASP) accelerates the development of organic synthesis routes of complex functional molecules. CASP tools are generally developed on the basis of rules or data of synthetic chemistry, which include some enzymatic reactions. However, synthetic biology offers a new degree of freedom through the potential to engineer new synthetic steps. In this work, we present a method to hybridize conventional organic synthetic and synthetic biological reaction datasets to guide synthesis planning. A section of organic reactions from the Reaxys® database was combined with metabolic reactions from the KEGG database to create a hybrid dataset. The combined dataset was used to assemble synthetic pathways from multiple building blocks to a target molecule. The route assembly was performed using reinforcement learning, which was adapted to ‘learn the values’ of molecular structures in synthesis planning and to develop a value network to suggest near-optimal multi-step synthesis route choices from the pool of the available reactions. To quantify the added value of synthetic biological reaction transformations in the hybrid routes, three value network ‘decision-makers’ were developed from the organic, biological and hybrid reaction pools. The near-optimal synthetic routes planned from the three reaction pools were evaluated and compared to discuss the benefits of the hybrid synthetic chemical plus synthetic biological reaction decision space in reaction route optimization.
Introduction
Retrosynthetic analysis1 plans the synthetic routes of pharmaceuticals and industrial chemicals by transforming a target molecule into simpler precursors until available building block molecules are reached. The use of computer-assisted synthesis planning (CASP) allows accelerating the development of retrosynthetic routes.2,3 The data-driven CASP methods are using the capacity to mine data from historical literature and patents, thus accessing a large knowledge base of chemistry.4 Recent successes in this field include (i) generation of reaction networks based on graph theory,5 using vertices to represent molecules and directed edges to represent reactions from reactants to products to understand connectivity among molecules,6–11 (ii) manual curation12–14 and algorithmic extraction15,16 of reaction rules and templates from historical reactions to predict functional group transformations, and (iii) template-free deep learning methods to learn from historical reactions and plan new transformations.17–19A seemingly parallel development is that of synthetic biology, in which cellular metabolism is engineered to produce target molecules. Biochemical transformations may potentially allow significant gains in synthesis efficiency for three reasons: (i) it can improve routes' redox efficiency by finding metabolic shortcuts for the key synthetic steps – several synthetic steps with poor efficiencies could be replaced by a biochemical step; (ii) most enzymatic reactions are highly selective;20,21 and (iii) biochemical reactions are performed under mild operating conditions and usually with benign solvents, which may lower operational costs and reduce the life cycle impact of syntheses.22,23 Similar to the reaction network of organic synthesis, a map to visualise metabolic production of bio-based chemicals has been summarised to guide the biosynthetic planning.24 A number of pharmaceutical ingredients and bulk chemicals have been produced economically through (hybrid) metabolic engineering approaches.25,26 CASP tools have also been developed for biocatalytic reactions. For example, Finnigan et al.23 developed RetroBioCat, a reaction-rule-based tool to build biocatalytic pathways and identify enzymes for target molecules, and Probst et al.27 generalised molecular transformer,19 a deep learning reaction prediction transformer inspired by natural language processing (NLP), to predict biocatalytic reaction outcomes and build pathways. These tools indicate the feasibility of integrating biosynthetic reactions and machine learning methods into CASP.
With the knowledge of molecular transformations from organic chemistry and synthetic biology, retrosynthesis relies on multi-step decision-making to select optimal reaction routes among all feasible molecular transformations based on criteria such as exergetic efficiency, E-factor, etc.4,6,28 In linear reaction routes, which include only one-to-one (reactant(s)–product only) ‘wiring’ (using the network's jargon) of the reactions, the decision-making could be done through exhaustive search of all possible reaction routes and ranking of the routes based on a set of predefined criteria. However, in topological-tree-styled reaction routes, which include multiple-to-multiple wiring of reactions (including co-reactants and by-products), the number of options increases exponentially with the increase in the number of synthetic branches and depth; exhaustive search becomes computationally expensive.29 In order to improve the efficiency of route design, the machine learning method of reinforcement learning (RL) has been proposed for application in synthesis planning.18,29–31
Reinforcement learning mimics how an intelligent ‘decision-maker’ takes multi-step actions within a specific problem environment to maximise/minimise the cumulative rewards/penalties of the actions.32 In synthetic planning, the selection of each reaction step within a path is a decision-making step. With the given rules and criteria costs, a ‘decision-maker’ starts synthetic planning by trial and error and algorithmically learns from the simulated experience to perform better in the next iteration (an ‘episode’ within the RL jargon). For example, Schreck et al.29 trained a value network to understand the potential costs of candidate reactions computed from reaction templates at a certain synthetic depth and to select retrosynthetic pathways based on costs. The method was compared with a decomposition heuristic method2 to prove its ability for synthetic planning. Similarly, in metabolic engineering, Koch et al.30 presented a code named RetroPath RL, which uses Monte Carlo tree search (MCTS) reinforcement learning to rank metabolic reaction rules to enable the development of biosynthetic routes. These reinforcement learning tools mainly made decisions from pre-generated reaction rules to compute synthesis pathways, and none of these are focusing on historical reaction networks, since a complex reaction network makes reinforcement learning harder to converge.
Despite the interest in chemoinformatics to combine multiple datasets to have a comprehensive understanding of the chemical space,3,33 to the best of our knowledge, there is only one explicit analysis of the benefits of combining organic chemistry and synthetic biological reaction databases in retrosynthesis planning. Levin et al.34 provided a comprehensive approach of the potential of merging enzymatic and synthetic chemistry with CASP and highlighted the importance of multidisciplinary approaches in advancing synthetic chemistry. This approach was based on enzymatic and synthetic chemistry extracted reaction rules. We foresaw the future of this field and developed our own approach based on historical published data.
In this work, we mined a section of historical reactions from the Reaxys® database35 and all metabolic reactions from Kyoto Encyclopedia of Genes and Genomes (KEGG),36 which is an open-source manually curated bioinformatic library. We compared the influence of the presence of organic synthesis and synthetic biology past reaction data in a dataset used for identification of retrosynthesis pathways of a curated set of drug molecules, which were believed to be difficult to synthesise. To evaluate the identified routes we used atom economy, the number of reaction steps and price of molecular building blocks as key quantifiable performance criteria. The reinforcement learning method from Schreck et al.29 was adapted to build value networks to guide the search for retrosynthesis pathways. Different from other CASP tools, the synthetic pathways from this method were not assembled from the predicted reactions (i.e., using reaction templates15 or the algorithm-generated reactions17) but used historical published data. This reduced the propagated uncertainty from the reaction templates over the identified paths to enable us to focus on the key research questions of this work – how much added value could synthetic biological reactions bring to synthetic organic chemistry in multi-step syntheses? Our secondary research question is whether a reinforcement learning method would converge with the large reaction networks as the reaction sources?
Methods
Building blocks
The molecular building blocks of a target molecule are the smallest precursors required to build up the structure of the target molecule. To make a collection of building blocks and their prices for synthetic planning, here we defined building blocks to be commercially or naturally available small molecules. The collection of commercially available building blocks came from ‘buyable’ molecules crawled from ChemSpace,37 which is an online catalogue of small molecules. For most buyable molecules listed in ChemSpace, more than one price is given from multiple suppliers. We chose only the lowest price available for these molecules.The naturally available building blocks are freely available cofactor metabolites from cell organisms in metabolic reactions, such as ATP and NADPH, and a list of such molecules curated by Blaβ et al.38 was used for the naturally available building blocks. The price for these naturally available molecules is zero. In enzyme-based industrial processes, although the naturally occurring molecules are free to acquire, some of these molecules, specifically cofactors, are difficult to recover and recycle, which makes them economically unviable.39 The common industrial solutions include stoichiometric design that balances each cofactor occurring in the total pathway,40 or integrating multiple pathways to link the generation/degradation of cofactors.21,39 In the current approach, we disregarded the requirement for such cofactors in the analysis of the reactions. As a result, we fully appreciate that some of the suggested reactions may be energetically and economically unfavourable once the cofactor requirement is included. On the one hand, this is a significant handicap of the present implementation. On the other hand, since the purpose of our overall methodology is to supplement the intuition of synthetic chemists with new ideas, and since there is a significant interest in developing both the cofactor recycling strategies and non-native enzymatic reactions, we consider that there is a value in including biochemical reactions in retrosynthesis planning even at such an early stage of development of the methodology. Further work on implicit analysis of cofactor requirements in retrosynthesis planning is planned. Hence, for simplicity, in this work, we consider only the acquisition price of the building blocks to demonstrate the overall approach; the cost of separation and other process ‘costs’ of the syntheses are deliberately left out from the current study.
Fig. 1 shows the price distribution of all building block molecules. The price of approximately 1/10th of all molecules from ChemSpace ranges from 103 to 106 USD per g, which is unreasonably expensive. Therefore, these molecules were removed from the set of building blocks to lower the costs of the potential synthetic routes. In total, we selected 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 282 commercially and 451 naturally available building blocks. A full list of building block molecules can be found in the ESI.†
282 commercially and 451 naturally available building blocks. A full list of building block molecules can be found in the ESI.†
 | ||
| Fig. 1 The price distribution of building block molecules; vertical axis shown in log-scale to distinguish the sharp corner at 103 USD per g. | ||
Mining and merging of Reaxys and KEGG datasets
A section of reactions was mined from Reaxys®. This is the same section as the molecules and reactions to generate the network of organic chemistry (NOC) from our previous studies.6–10 This reaction dataset is comprehensive and covers most major reaction classes.3 However, a disadvantage of this database is that it misses stoichiometry and, sometimes, side products. With the lack of stoichiometry, we do not know the true mass flow of the predicted reaction route, and our predictions would potentially result in low carbon efficiency. However, this could not be avoided at this stage, where the lack of stoichiometry is a general problem for other popular open-source organic reaction databases, such as USPTO41 and Pistachio.42All metabolic reactions were mined from the KEGG reaction database. The KEGG reaction database defines the binary relations of reactions, and directionality of these reactions is not clarified. Since in most cases enzymes bind substrates and catalyse metabolic reactions in both directions, all reactions were assumed to be reversible. Therefore, for all metabolic reactions, both directions were recorded in the local biological dataset. Most metabolic reactions would need energy sources such as ATP to be provided. There are differences in the requirements for cofactors between the forward and the reverse reactions. This is true for most redox reactions, ligases, decarboxylations, phosphorylations and many others. Our assumption ignores the differences in the requirements with regard to cofactors between the forward and the reverse reactions. Since energy criteria were not considered in the reaction assessment scores to select candidate reactions for synthetic planning (see the following section), the use of reverse counterparts of the original reactions might result in energy ineffectiveness of the reaction routes. However, as indicated from the later sections, the biological dataset added only 0.36% of reaction data into the chemical dataset. With the sparse biological reaction data, all forms and possibilities of metabolic transformations including the reverse counterparts were valued to contribute to novel synthesis planning, and therefore we insisted to include the reverse reactions. For practical implementation of the novel synthetic routes, energy integration needs to be carefully considered to industrialize the pathway.
From the two datasets, all molecules and reactions were recorded with their own identification numbers. Due to the use of different identifiers, to merge both datasets we used the RDKit package43 to pairwise compare molecular canonical SMILES strings for all molecules, and reaction SMARTS for all reactions in both datasets. By excluding free metabolites and cofactors, the KEGG reaction dataset includes only reactions with main reactants and main products. These reactions were canonicalised in terms of their molecule SMILES and compared with the canonicalised reactions in the Reaxys dataset. All KEGG molecules and reactions found in Reaxys were renamed with a Reaxys identifier in the local datasets. If the reactants and products were identical for two reaction entries, these two reactions are considered as overlapped reactions. All canonical reaction SMILES from Reaxys and SMILES were converted from their MolFiles. Therefore, no standardization of tautomers and removal of atom mapping were required. Charges were not neutralised since a lot of metabolic reactions need to occur with charge requirements. The statistics of both datasets are shown in Fig. 2a.
 | ||
| Fig. 2 Visualisation of the datasets used in this study. (a) A Venn diagram of molecules and reactions in Reaxys and KEGG datasets. The number of KEGG reactions is the number of reactions from both directions. (b) Visualisation of the hybrid reaction network in ARF layout; red nodes indicate molecules and black edges indicate reactions. | ||
Data from KEGG are significantly more sparse compared with the dataset of reactions mined from Reaxys. Among KEGG data, a proportion of molecules and reactions overlap with the Reaxys data, since Reaxys includes mined reaction data regardless of whether they are from organic synthetic or bio-synthetic sources. To compare the optimal reaction routes computed from candidate reactions from different sources, three local reaction pools were created: reactions from KEGG were labelled as a biological reaction pool (green + brown in Fig. 2a), those from Reaxys and excluding the intersection between Reaxys and KEGG were labeled as chemical reaction pool (pink), whilst the union of the two sets became the hybrid reaction pool (pink, green + brown). The visualisation of the hybrid reaction network in node and edge representation is shown in Fig. 2b.
Since KEGG reactions were manually curated, by no means could the reactions cover the entire synthetic biological reaction space. In reality, the intersection between Reaxys and KEGG may be larger than the overlapping area in Fig. 2a. Thus, the chemical reaction pool (pink) very likely still includes metabolic reactions. However, this is the best we can do in terms of defining the boundary between the chemical and the biological datasets.
Molecular representation in SMILES, especially canonical SMILES, is able to characterize most three-dimensional molecular structure differences, for example cis/trans isomers and enantiomers. In most cases, one SMILES string corresponds to only one chemical structure. However, due to noises or higher-order structure differences (high-level stereoisomers, etc.), in Reaxys, multiple molecules may share one identical canonical SMILES string. One example -of such high-level stereoisomer molecules is shown in the ESI.† Stereochemistry is, frequently, the key reason for using enzyme-catalyzed biochemical reactions; enzymes bind specific substrates to produce specific three-dimensional structural molecules. Failure to detect such a difference in SMILES representations would result in the use of wrong enzymes to catalyse the substrate, which would eventually fail the reaction pathway. In the present implementation of retrosynthesis search we have ignored this potential error due to lack of stereoselectivity representation of molecules in SMILES. As there is a large amount of work ongoing currently to resolve this challenge in molecular representation, and since our approach will be amenable to the use of other molecular representations, we look forward to adopting the more accurate molecular representations in further methodology updates.
In the intersection of the two datasets, one KEGG molecule may have multiple counterparts in Reaxys. The statistics in the intersection of the Venn diagram were counted based on the data from the KEGG-extracted dataset to avoid this issue.
Reaction assessment scores
In reinforcement learning multi-step decision-making, each decision-making step is associated with a reward/penalty, whilst the whole multi-step decision-making process is associated with an expected return/cost, which is the accumulation of rewards/penalties of all decision-making steps. For each of the decision-making steps having multiple options, a well-trained value network predicts expected returns/costs of the options by foreseeing the cumulative return/cost of the following steps from the current options, and then selects the option with the maximised expected return or the minimised expected cost. In the synthetic problem, each decision-making step is to select a reaction option from the reaction pool for the current molecule, and the whole multi-step decision-making process is to assemble the multi-step reaction pathway. Here, to assess the reaction options, we chose the penalty-expected cost evaluation system and designed reaction assessment scores to represent the penalty to select the candidate reactions. The scores would later be also used to quantify the performance of synthesis planning from the three different reaction pools.Various criteria could be applied to evaluate candidate reactions, subject to the optimization objectives and data availability.6 In this work, our objective was to find efficient reaction routes whilst maintaining environmental efficiency. It is expected to improve the route search in the implementation of the overall system with yield, selectivity and reaction conditions included. However, the larger part of the dataset, Reaxys, provides such attributes only for a small portion of literature-excerpted reactions, and this information cannot be found at all from the KEGG dataset or other commonly used biological databases. Therefore, only global criteria determined from the data available in both Reaxys and KEGG databases were used to design the assessment scores. After trials to avoid failure of computation and biases, the global criteria were designed to include atom economy of reaction steps and price of building blocks to consider both route efficiency and operational costs. Of course, changes in global criteria would significantly alter the optimization results. Here we chose a minimum set of criteria to demonstrate the overall approach. Nevertheless, for the purpose of ideation and generating leads for further chemical and biological work, this analysis is still valid. The analysis and evaluation of routes based on only-Reaxys data published previously6 also suggest that the results of route analysis, specifically the ideas that synthetic chemists generated on the basis of the suggested reactions, were always appreciated much more than the absolute ranking from the reaction scores.
We considered factors from Jacob et al.6 and Schreck et al.29 to design global criteria related to penalty scores. The factors considered in the penalty scores were not as many as those in the work by Jacob et al.,6 which were only feasible to use in an investigation on a specific reaction pathway. However, these are more comprehensive than the ones used in Schreck et al.,29 which used simple numerical values to judge the number of reactions and molecules in the pathway.
For a reaction pathway, penalties were added to the reactions and the building block molecules. For any reaction or building block molecule in the pathway, the penalty was designed to be lower than 1. The penalty of a reaction is shown in eqn (1).
| penaltyr = 1 − AEi | (1) |
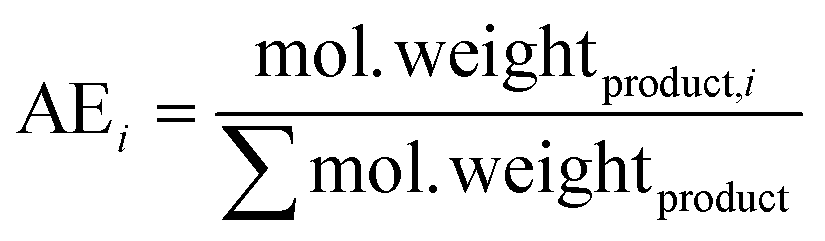 | (2) |
Apart from building blocks, the reaction pathway may also terminate at a ‘dead-end molecule’, which means no other reaction link with the molecule, or a ‘maximum-depth molecule’, which means the end-point molecule reaches the maximum allowed route depth from the target molecule, which was set to be 10 synthesis steps. The ‘decision-maker’ fails to find a proper pathway in these cases, and therefore, adapted from Schreck et al.,29 the penalty for a dead-end molecule is 100, and the penalty for a maximum-depth molecule is 10.
The expected cost of a molecule in the reaction pathway is the cumulative penalties of all reactions and end-point molecules from the sub-pathway from the molecule as target molecule to its sub-branches (shown in eqn (3)). The expected cost of a molecule is also equal to the penalty of the reaction linked with the molecule as a product, plus expected costs of all reactants in the reaction. For example, in Fig. 4, the expected cost of m2 is the sum of the penalty of a building block m6 and a max-length molecule m10 plus the penalty of reactions r1 and r3.
 | (3) |
Workflow of reaction route optimization
A reinforcement learning approach adapted from Schreck et al.29 was applied to create value network ‘decision-makers’ towards identification of the optimal reaction routes. The optimization workflow shown in Fig. 3 was conducted in 20 iterations to optimise the decision-making process and provide promising value network models. The value network was later used to suggest near-optimal reaction routes for target molecules from the reaction pool. To compare the impacts of chemical, biological and hybrid reaction pools, the same workflow was run three times to train and produce three value network models from the three reaction pools. | ||
| Fig. 3 Workflow to collect reaction data and train reinforcement learning value network for reaction route optimization. | ||
Reinforcement learning decision-making always starts from defining the decision space and defining how the ‘decision-maker’ interacts with the decision space (defining ‘environment’ within RL jargon). The workflow starts with defining a synthesis planning environment, which includes (i) a reaction pool comprising all molecules and reactions for the ‘decision-maker’ to choose from, (ii) evaluation score functions to assess reactions and synthetic routes, and (iii) a set of molecules as target molecules to initialise retrosynthesis planning.
In Schreck et al.,29 the decision space for the decision-maker were the candidate reaction rules to break down the target molecule into simpler precursors, using reaction templates developed from Coley et al.'s method.15 Whilst Coley et al.15 extracted templates from the open-source USPTO reaction dataset41 with a much smaller reaction space and used the templates to predict reaction products, Schreck et al.29 extracted templates from the more comprehensive Reaxys and used the templates to predict reaction reactants for CASP. Nevertheless, the top-one accuracy reported for one-step reaction major product prediction from the current reaction template method has reached only 71.8%.15 With a reaction prioritiser,44 reaction templates most relevant to the given molecules were selected, which weakly improved the accuracy. In synthesis planning, with the same reaction template method used to predict reaction reactants, uncertainty was still carried in the one-step reaction predictions, and this uncertainty would be significantly propagated in multi-step synthesis planning. This is within the context of organic synthesis. With metabolic reactions, the more complex, stereo-specific reaction mechanisms are expected to be harder to extract from reaction templates and are prone to have lower accuracy. This means biochemical retrosynthesis from reaction templates has a great chance of synthesis failure. Therefore, we used known reactions to propose confident pathways, and with this, we could highlight the key research question – benefits of including synthetic biological reaction decision space into the reaction route optimization.
For the target molecule set, molecules were filtered to be in the SMILES string length of 20 to 400. This was to maintain the target molecules from different datasets with fair synthetic difficulty. The aim was to include only 100000 molecules to maintain reasonable computational costs. This was the case for the chemical and hybrid reaction pools. 100000 molecules (excluding the molecular building blocks) were randomly selected from the molecule set as targets. Also, in each iteration of the optimization, the target molecule set was reshuffled to increase randomness. However, since the biological dataset records only approximately 30000 molecules, all molecules with a SMILES string length of 20 to 400 (building blocks exclusive), i.e. 12281 molecules, were included as the set of target molecules to compute synthesis planning.
For each target molecule, to compute its retrosynthesis route, all reactions in the reaction pool using the target molecule as one of the reaction products were marked as possible reaction options. If no reaction was found from the dataset, the molecule was marked as a dead-end molecule, as no synthesis step could be further added to the molecule. A dead-end molecule in the pathway is highly disfavoured by the ‘decision-maker’.
A ‘decision-maker’ selected one of the reaction options as the next synthesis step for the target molecule. For each reactant in the selected reaction, as shown in Fig. 4, if the reactant was a building block or a dead-end molecule, no further synthesis step is required. If not, the reactant became the next step target molecule. The same procedure was repeated to add the next reaction to the retrosynthesis route until all end-point molecules at all branches (resulting from multiple reactant reactions in the route) were building blocks, dead-end molecules, or maximum-depth molecules, where the maximum allowed depth was set to be 10 synthesis steps from the target molecule, which is also highly disfavoured.
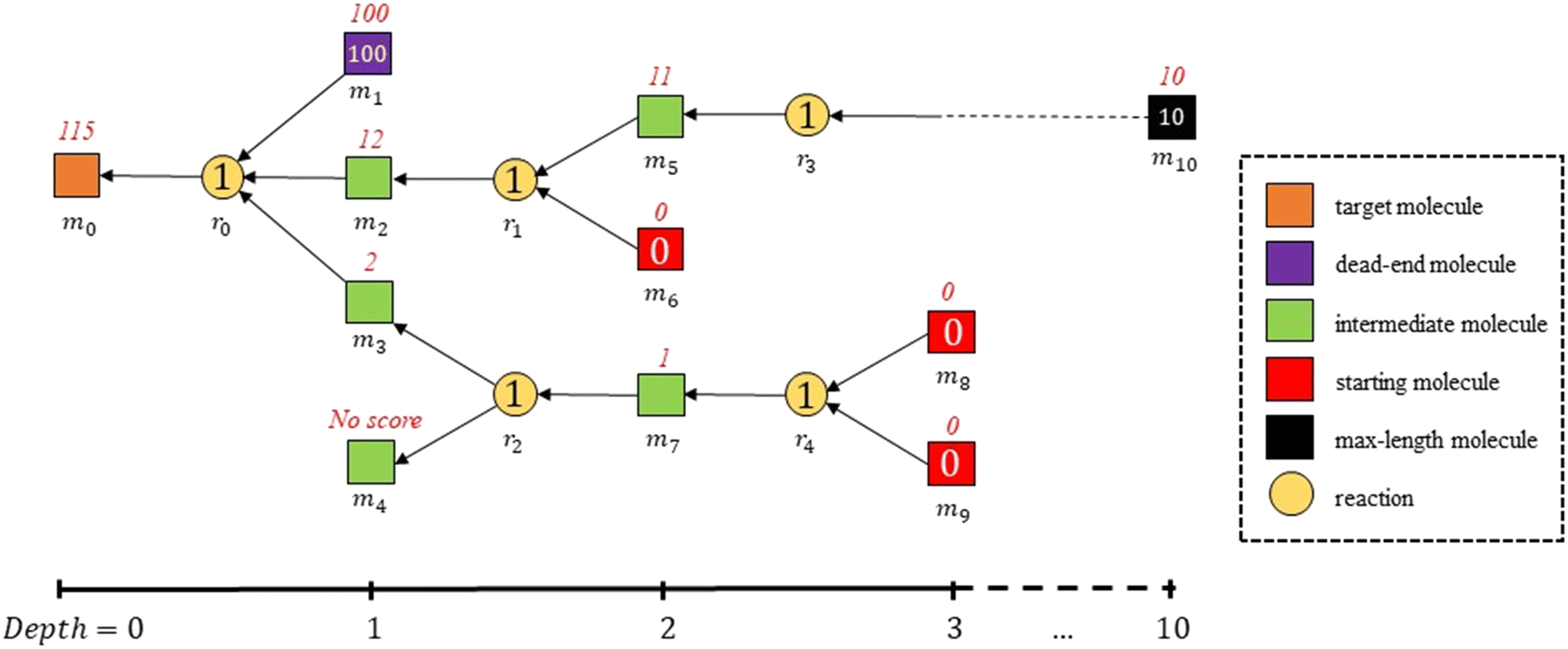 | ||
| Fig. 4 Schematics of retrosynthesis planning of a target molecule m0, with labelled penalties and expected costs in the planned route. In the schematics, molecules are marked as mi and reactions as ri. The axis below represents the depth from the target molecule m0, where the maximum allowed depth is 10. All ri are selected by a ‘decision-maker’ (a random selector or a value network model) from the available reactions in the reaction network. The penalties of reactions and end-point molecules are shown inside the nodes, and the expected costs of molecules are shown in red on top of the nodes. In the schematics, all reactions have penalties of 1 and all building blocks have no penalty only for simplification. However, in most cases, reactions and building blocks always have penalties ranging from 0 to 1. | ||
The ‘decision-maker’ came from either random sampling of the candidate reactions, or a trained value network (discussed below), and the possibility of random sampling follows the noise-level distribution in Fig. 3 and eqn (4). The possibility of the trained ‘decision-maker’ is one minus that of random sampling.
 | (4) |
 is the expected costs of reactant molecules in their specific residual depths. Essentially, this means the value network would understand the expected costs of molecular structures after exploring the reaction space and always point to the molecule structures that are easier to synthesise as the following steps.
is the expected costs of reactant molecules in their specific residual depths. Essentially, this means the value network would understand the expected costs of molecular structures after exploring the reaction space and always point to the molecule structures that are easier to synthesise as the following steps. | (5) |
Not only does the expected cost of a molecule depend on the molecule itself but also on the depth of the molecule in the pathway. If the molecule requires a long synthesis pathway, whether the pathway of a molecule reaches maximum-depth molecules or building-block molecules relies on its residual depth, i.e. the maximum allowed depth (10 steps) subtracted from the current depth from the target molecule. To learn from the simulated experience, the residual depth and the corresponding expected costs of all molecules in the pathway were collected following the designed penalty rules. This did not include the side-product molecules in the reactions, since the side products have no expected costs. However, the effects of side products were included in the reaction penalty, which counts for the atom economy of the reaction.
The same procedure was repeated for all target molecules to collect residual depths and expected costs of all simulated molecules. For the former 10 iterations, the simulation of each target molecule was repeated 10 times to add randomness to the built pathways. However, for the latter 10 iterations, since all pathways were built by the trained value networks, the repeated simulation results were identical, and thus only one simulation was required for each target molecule. In each iteration (in order to compute retrosynthesis once for each of the 100000 sampled target molecules), the ‘decision-maker’ chose candidate reactions from the large decision space, and approximately a million expected cost values of molecules at their corresponding residual depths were collected. The multiple expected costs of the same molecule at the same residual depth were averaged to count into the training data.
As shown in Fig. 3, at each iteration, the trained multi-layered perception (MLP) (discussed below) was eventually updated as the value network ‘decision-maker’ for the next iteration. The optimization was terminated after 20 iterations, and the value network at the last iteration became the final ‘decision-maker’ to predict expected costs of molecules and select reactions based on eqn (5) to build retrosynthesis pathways.
Machine learning to learn from molecules
Following Schreck et al.,29 machine learning models mimic the mathematical relationship between the molecules and their residual depths as inputs and the corresponding expected costs as output. To digitise molecules into mathematical models, extended-connectivity fingerprint (ECFP)45 was applied, which is a topological fingerprint to convert the circular structure of the neighborhood of each non-hydrogen atom into bytes. In this work, the radius of the fingerprint was four (ECFP4), which detects the multiple layers of the neighborhoods from the molecule centre, and all molecules were converted into a 2048 fixed-length bit string. Overall, the input has 2049 features, from which 2048 are binary variables from ECFP, and one from the residual depth.MLP neural network was used as the machine learning model to learn from the data, and this was conducted by using the deep learning API Keras.46 Although over one million data points were obtained from each iteration, the structure of the MLP was simple to avoid data overfitting, especially from the 2048 binary variables. The MLP includes an input layer of 2049 nodes, followed by a batch normalization layer to standardize the inputs. Three hidden layers of 30, 15 and 5 nodes using the exponential linear unit (elu) activation function were added, and right after each hidden layer, three dropout layers, with a dropout rate of 0.3, 0.2 and 0.1, were added to randomly reduce the size of hidden nodes to avoid overfitting. This was eventually followed by an output layer of one node, also with the elu activation function, which approximates the molecular expected cost. For specification, MLP used a learning rate of 0.002 to slowly learn from data, ‘mean square error’ as the loss function, and ‘adam’ as the optimiser. At each iteration, the collected data were split into training data and test data at the ratio of 4:1 and digitalised into 2049 inputs and one output to fit the specified MLP model. With a slow learning rate, we set 50 epochs for the MLP to learn from the training data.
Results and discussion
Reaction route optimization
The logic of the value network ‘decision-maker’ is as follows: it determines the costs of molecules based on their functional structures and previous synthetic performance and minimises the costs of synthetic planning by selecting the overall low-cost molecules. The expected costs of the molecules were learned through the proposed neural network. Generally at each iteration, after 50 epochs of learning, the test data outputs usually show approximately 45% scaled root-mean-square error (RMSE) and 65% Pearson correlation coefficient (correlation) from the test data approximations. The RMSE and correlation equations and results for all three environments and all 20 iterations are shown in the ESI.† The RMSEs are high since we tried to learn from 2049 features out of millions of molecules, and by no means could an MLP with three hidden layers fit all the costs of molecular structures using such a simple model structure at once. Also, we did not expect the MLP to grasp all details from the observations, since a large portion was from trial-and-error noise, which would eventually cause overfitting. However, approximately 70% correlation means that the model learned the overall relationship among molecular structure, retrosynthetic depth and the expected costs, which was promising for overall predictions. With the convergence of the reinforcement learning optimization after 20 iterations, we have proven that the above method is a useful strategy. With a dense and noisy reaction network, a large decision-making domain is provided; an underfitted MLP structure would help the reinforcement learning optimization to learn from uncertainty and explore the decision-making domain through optimization iterations.With well-trained value network models, the optimization results improved over the iterations. The statistics of the expected costs of molecules from the biological reaction pool over the 20 iterations is shown in Fig. 5, and the chemical and hybrid reaction pathways show similar optimization trends (shown in Fig. 6). At iteration 0, the median of expected costs for all target molecules reaches approximately 100, which means that in most cases, the random sampling ‘decision-maker’ picks dead-end molecules to build reaction routes for the target molecules. For a great portion of the outliers, the ‘decision-maker’ selects multiple dead-end molecules, which approaches the expected costs of multiple hundreds. By learning from trial-and-error results, the value network reduces the expected costs of most target molecules, with median expected costs being stabilised below 10 in the last five iterations and finalised at 5.2 at the last iteration. Along the 20 iterations, although the portion of outliers also reduces, there are still outliers that reach costs over 200 in the last five iterations. These are large protein molecules which usually have molar weights over 500 and are believed to be hard to synthesize, which include C16-KDO2-lipid A, UDP-4-amino-4-deoxy-L-arabinose, etc. The situation of target molecule ferricytochrome c has not been improved over the 20 iterations, which stabilises at the expected costs of 704 in the biological reaction pathway in Fig. 5.
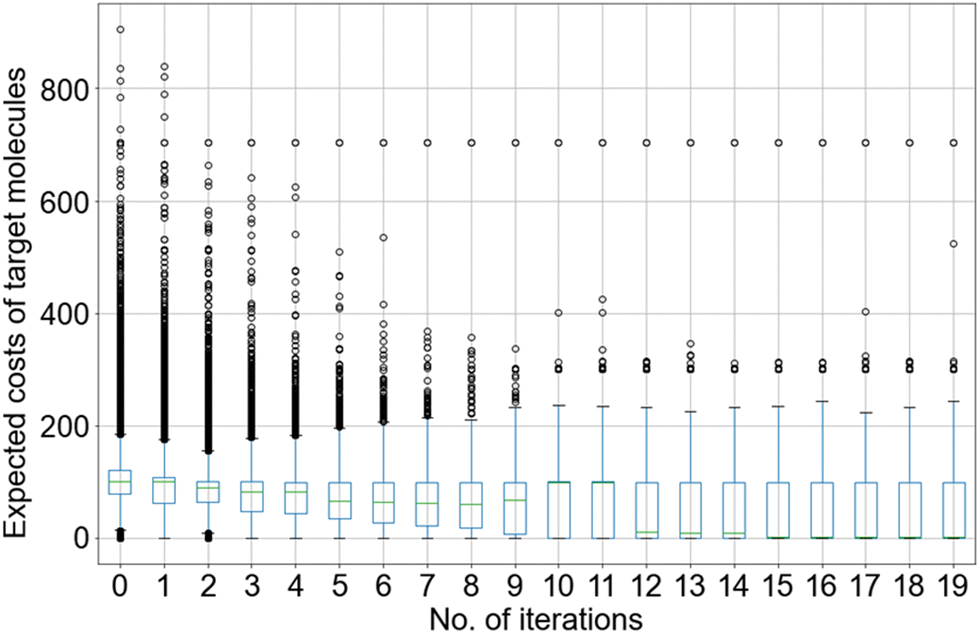 | ||
| Fig. 5 The statistic of expected costs of all target molecules in the biological pathway optimised along with the number of iterations, shown in the boxplot. | ||
 | ||
| Fig. 6 The optimization results of median expected costs of target molecules from the chemical, biological and hybrid environments, shown in the log-scale vertical axis to distinguish the tails at the last five iterations. | ||
Comparison of the optimization results from the three datasets
The expected costs of target molecules rely on the molecule synthetic difficulty and the quality of the decision-maker to reach a near-optimal synthetic route. Since we used large target molecule pools and fix the molecule synthetic difficulty by filtering the molecule SMILES string length from 20 to 400, the synthetic difficulty was fair for the chemical, biological and hybrid reaction pools. Hence, we use the median expected costs of target molecules to judge the optimization results from the three reaction pools. With the value network ‘decision-makers’ being trained and optimised, whilst the median expected cost of the target molecules from the biological reaction pool has a significant jump at iteration 12, those from the chemical and the hybrid reaction pools both reduce gradually over the 20 iterations (shown in Fig. 6). The three curves all tend to be stable in the last five iterations, which means that they all approach optimization limits. This suggests that the reinforcement learning method is able to converge within 20 iterations, although we provided three large reaction pools as decision-making domains.At the last iteration, the medians are 4.3, 5.2, and 4.15, respectively, for the three reaction pools. This can be interpreted such that in most cases, the molecule synthetic difficulty reduces in the hybrid reaction pool compared with the organic synthesis or synthetic biology ones alone. It also suggests that although the addition of the biological dataset only adds 0.36% data into the chemical dataset (Fig. 2a, in terms of the number of reactions), overall it adds value by 3.4% to the organic synthesis to reach better synthetic results (by comparing the expected costs of molecule medians of 4.3 and 4.15 in organic and hybrid synthesis, respectively): it is able to improve the redox efficiency and find more opportunities for synthetic shortcuts among molecules via hybridising the reaction pools. The value 3.4% relates to the synthesis of all target molecules on average within the reaction space. The improvement on heavier drug molecules is much more significant than that, as we discuss in the following section. Moreover, with the lack of biological data, at this stage, the total number of reactions increases by only 0.36% by hybridizing the biological data. The increase of 3.4% added value in synthesis proves the feasibility of the hybrid method. Further improvements will arise from increasing the range of biological transformations.
We focus on the conceptual assembly of hybrid reaction pathways, whilst the ability of biological transformations to substitute some key catalytic steps is highly valued. However, we acknowledge that this interpretation is specific to the used assessment criteria and penalty scores. Other advantages of biological reactions such as greenness and close-to-ambient reaction conditions have not been covered by the current methodology. We also did not implement any quantification of the drawbacks of biological reactions. For example, it is common for biological reactions to be highly dependent on the rest of the cellular metabolic network, which increases the operational costs of reactions. We also did not consider product separation for any of the reactions in the current implementation. Although correlation equations and machine learning tools have emerged to correlate these scores, they are not yet highly accurate. To industrialize the hybrid pathway, the best solution for now would be to manually assess the final suggested routes.
Optimization results for drug molecules
To test the performance of the final value network ‘decision-makers’ and also investigate the added value of biological data in the synthesis of larger molecules of interest, we compared the final reaction routes computed for these molecules from the three reaction pools. The KEGG drug database47 gives a list of drug molecules as active pharmaceutical ingredients, and molecules crawled from the database were used as larger molecules of interest to compute reaction pathways to them. Since the reaction pathways were compared by reaction pools from organic chemistry, biological and hybrid databases, only 3821 drug molecules coexisting in the chemical and biological datasets were used as the target drug molecules to compare the optimal reaction pathways from the three datasets. Here we also filtered these molecules into a set of 3746 to contain only molecules with SMILES string length greater than 10 to increase the synthetic planning complexity; see further details in the ESI.†The results from the three reaction pools are shown in Fig. 7. Different from other random selected target molecules with shorter SMILES string lengths, it is more difficult to find synthetic routes for these drug molecules. Whilst the target molecules are usually being synthesised within five steps, the cost to make drug molecules reach a median of 100 for the three datasets, which means that the routes always point to a dead-end molecule. This indicates that due to the molecular complexity, the majority of drug molecules cannot be synthesised using the demonstrated method and datasets. One of the reasons for the difficulty to synthesize these molecules is that a large number of drug molecules were originally from partial or pure natural extracts. For these molecules, the possibility of biochemical synthesis from precursor molecules is still unclear. Moreover, synthesis of these molecules would be easier if we enlarge the building blocks dataset. We set the building blocks to be, to the best of our knowledge, most commercially available small molecules (which fall into Reaxys and KEGG databases), whilst in industry, the synthesis of drug molecules sometimes starts from middle-sized simpler drug molecules. Longer pathways would be a solution to find synthesis routes of some drug molecules. However, to increase the reaction steps in the pathway, the number of trial-and-error experiments needs to be significantly increased to initiate reinforcement learning, which would increase the computational burden. This would also increase the model error represented as RMSE. In this way, the predicted reaction pathways from the model decision-maker would have an increased uncertainty. For further work, we could use partial reactions to predict functional transformations. In this way, more possible solutions could be given to the synthetic routes. However, it is also seen that the hybrid environment exhibits a heavier tail towards lower costs to make the molecules. This means that the method opens possibilities to synthesise a significant proportion of drug molecules, making use of the full set of chemistry combining organic synthetic and synthetic biological reactions.
 | ||
| Fig. 7 Statistics of costs to make drug molecules determined from the final value network ‘decision-maker’ using the biological, chemical and hybrid reaction pools. Data for this figure can be found in the ESI.† | ||
An example of these successfully synthesised molecules is glucosinolate, an active pharmaceutical ingredient of multiple Chinese medicines, which are antibacterial, antioxidant, anticarcinogenic, etc.48 We illustrate the following 7-step synthetic route of glucosinolate in Scheme 1, suggested by the value network. The cost of making glucosinolate by this route is 3.66, with five building blocks used, three illustrated in Scheme 1, and another two are cofactors 3′-phosphoadenyl sulfate in enzymatic reaction 0 and UDP-glucose in enzymatic reaction 1 circulating over cell organisms, or in this, over the in vitro bioreactor. The depth of the longest branch is six steps. The route uses four organic chemical reactions and four synthetic biological reactions. Excluding free metabolites and cofactors such as oxygen, etc., the route produces in total two side products – pyruvate in organic reaction 0 and carbapen-2-em-3-carboxylate in enzymatic reaction 3. To compare, there is no entire organic chemical route to synthesise this molecule, and the cost of the purely biological route is 7.15.
 | ||
| Scheme 1 The proposed synthetic route of glucosinolate by the final hybrid value network ‘decision-maker’, with glucosinolate shown as target, and organic chemical, synthetic biological reactions coloured in red and green respectively. Free metabolites and cofactors are excluded from the scheme. | ||
Another example of a hybrid route is the synthesis of atropine, an anticholinergic medication to treat nerve agent poisoning and slow heart rate as well as to decrease production of saliva in surgery.49 It is mainly naturally extracted. However, we illustrate a 7-step synthetic route of atropine in Scheme 2. The cost of making atropine by this route is 2.16, with five building blocks used, three illustrated in Scheme 2, and another two are cofactors, NADPH used in enzymatic reaction 0 and S-adenosylmethionine used in enzymatic reaction 1. The cost of making atropine is cheaper than that of the latter example since most reactions are one-to-one wiring, and most steps in the retrosynthesis pathway reduce the complexity of the intermediate molecule, which increases carbon flow efficiency. The route uses five organic chemical reactions and two synthetic biological reactions. There is no chemical route to synthesise this molecule, since one of the key steps, reduction of tropinone (enzymatic reaction 0) does not exist in the current chemical reaction pool, whilst this is feasible via enzymatic catalysis by tropinone reductase. It does not have a biological route since starting from the building blocks, most transformations take place via synthetic chemistry.
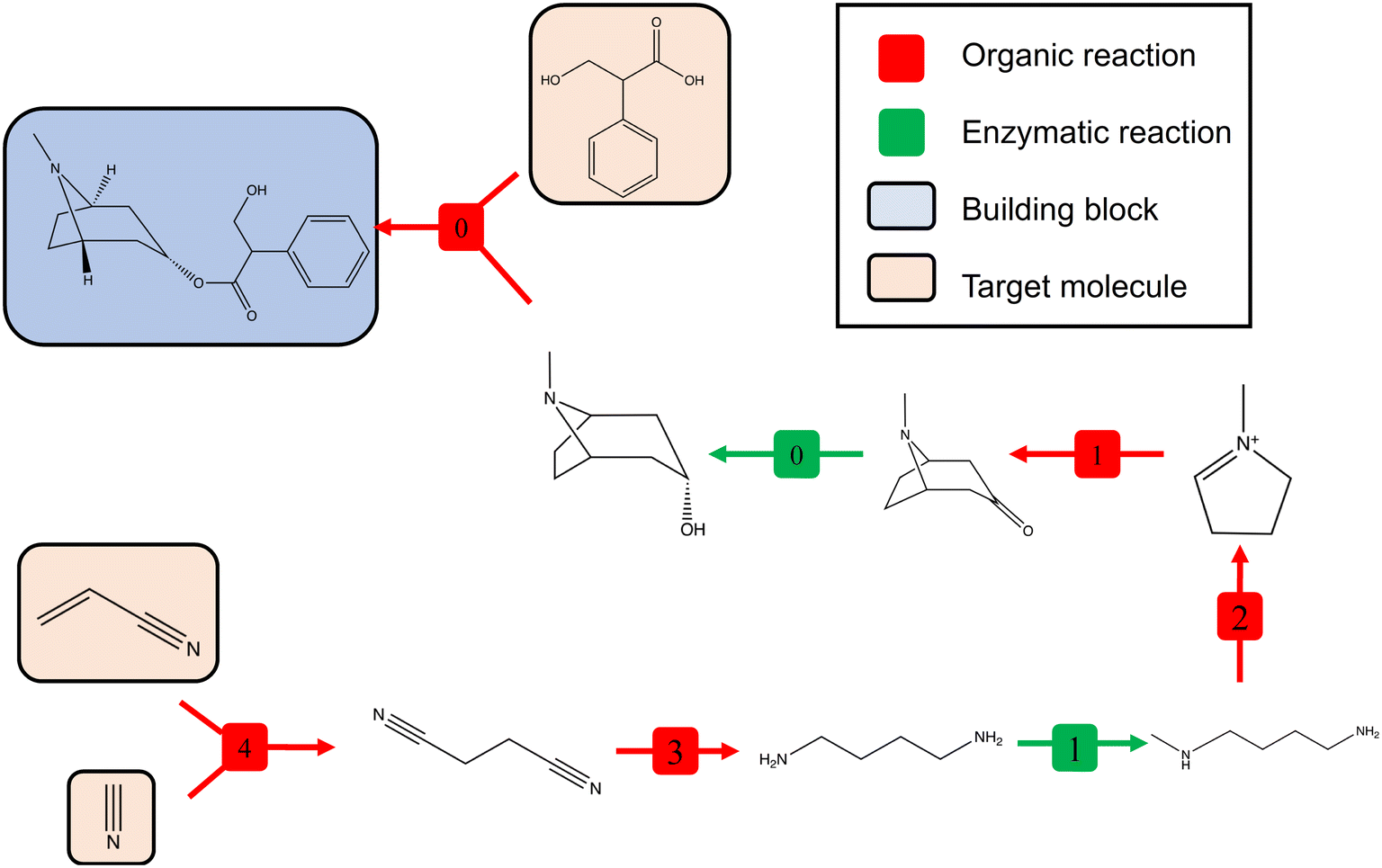 | ||
| Scheme 2 The proposed synthetic route of atropine by the final hybrid value network ‘decision-maker’. Free metabolites and cofactors are excluded from the scheme. | ||
The proposed routes for the above examples indicate the feasibility of the method, especially for larger functional molecules. In the proposed routes all reactions are existing historical literature examples. Compared with the CASP tools based on predicted reactions, this gives confidence to actually perform each reaction step in the reaction routes. However, since the method did not consider yield, selectivity, greenness and other reaction/process parameters due to current lack of data, the routes proposed for target molecules always need to be further investigated using more conventional approaches. Since these routes would include intermediate isolation and purification, decoupling of rather different synthesis conditions and process configurations that exist in the biochemical and synthetic organic chemistry processes can be done. Certainly, these case studies are proofs of concept that demonstrate that it is feasible to address the challenge of designing a ‘better’ (by some metric) route through a combination of biochemical and organic chemistry synthesis options which are identified by means of reinforcement learning retrosynthesis and using data from large reaction databases.
Conclusions
We presented an efficient method to suggest near-optimal biochemical synthesis routes via data mining from organic chemistry and synthetic biology reaction datasets and reinforcement learning decision-making. With this method, we proved that by providing historical literature reaction sources as large decision-making spaces, the reinforcement learning model has learned from the connectivity and values of molecules in the reaction data and converged in 20 iterations. It becomes an efficient method for synthesis planning optimization.We also proved that with atom economy, numbers of reaction steps, and price of building blocks as key criteria to quantify retrosynthesis performance, overall, biological reactions offer substantial cost savings for organic synthesis, and hybridising chemical and biological reactions to plan synthetic routes is better than conventional organic synthesis alone by 3.4% on average, with respect to the synthesis of all target molecules in the molecular space, due to the advantages of synthetic biological reactions, such as improving reaction redox efficiency and enabling synthetic shortcuts. This is a significant value since the biological data only comprise 0.36% of the total reaction data. We expect the value to be dramatically increased as more biochemical reactions are available for reaction network optimization, for example, through design of non-native enzymes.
With respect to drug molecules, we could especially benefit from the well-trained value network to plan their synthetic routes. The case studies of making glucosinolate and atropine molecules using our method indicate that these two syntheses would be significantly eased with the help of synthetic biology reactions. The example of atropine also proves the possibility of synthesis of a natural product. This methodology could be further extended to mine more comprehensive reaction data to further understand the true costs of using biological reactions, which would make it possible to plan reaction routes with reduced uncertainty.
Data availability
Reaxys molecule and reaction data are accessible to users via Elsevier. KEGG reaction and molecule data are available via KEGG APIs. All other data are shared via the ESI.†Conflicts of interest
AAL is a founder of Chemical Data Intelligence (CDI) Pte Ltd (https://cdi-sg.com), which was set up to commercially exploit the chemical data networks.Acknowledgements
CZ is grateful to Cambridge Trust CSC Scholarship for funding his PhD study. We gratefully acknowledge the collaboration with RELX Intellectual Properties SA and their technical support, which enabled us to mine Reaxys. Copyright© 2020 Elsevier Limited except certain content provided by third parties. Reaxys is a trademark of Elsevier Limited. Reaxys data were made accessible to our research project via the Elsevier R&D Collaboration Network.References
- E. J. Corey, Robert Robinson Lecture, Retrosynthetic thinking—essentials and examples, Chem. Soc. Rev., 1988, 17(0), 111–133 RSC.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Computer-Assisted Synthetic Planning: The End of the Beginning, Angew. Chem., Int. Ed., 2016, 55(20), 5904–5937 CrossRef PubMed.
- A. Thakkar, T. Kogej, J.-L. Reymond, O. Engkvist and E. J. Bjerrum, Datasets and their influence on the development of computer assisted synthesis planning tools in the pharmaceutical domain, Chem. Sci., 2020, 11(1), 154–168 RSC.
- J. M. Weber, Z. Guo, C. Zhang, A. M. Schweidtmann and A. A. Lapkin, Chemical data intelligence for sustainable chemistry, Chem. Soc. Rev., 2021, 50(21), 12013–12036 RSC.
- R. J. Wilson, Introduction to graph theory, John Wiley & Sons, Inc., 1986 Search PubMed.
- P. M. Jacob, P. Yamin, C. Perez-Storey, M. Hopgood and A. A. Lapkin, Towards automation of chemical process route selection based on data mining, Green Chem., 2017, 19(1), 140–152 RSC.
- A. A. Lapkin, P. K. Heer, P. M. Jacob, M. Hutchby, W. Cunningham, S. D. Bull and M. G. Davidson, Automation of route identification and optimisation based on data-mining and chemical intuition, Faraday Discuss., 2017, 202(0), 483–496 RSC.
- P.-M. Jacob and A. Lapkin, Statistics of the network of organic chemistry, React. Chem. Eng., 2018, 3(1), 102–118 RSC.
- J. M. Weber, P. Lió and A. A. Lapkin, Identification of strategic molecules for future circular supply chains using large reaction networks, React. Chem. Eng., 2019, 4(11), 1969–1981 RSC.
- J. M. Weber, A. M. Schweidtmann, E. Nolasco and A. A. Lapkin, Modelling Circular Structures in Reaction Networks: Petri Nets and Reaction Network Flux Analysis, in Computer Aided Chemical Engineering, ed. S. Pierucci, F. Manenti, G. L. Bozzano and D. Manca, Elsevier, 2020, vol. 48, pp. 1843–1848 Search PubMed.
- M. Fialkowski, K. J. M. Bishop, V. A. Chubukov, C. J. Campbell and B. A. Grzybowski, Architecture and Evolution of Organic Chemistry, Angew. Chem., Int. Ed., 2005, 44(44), 7263–7269 CrossRef CAS PubMed.
- C. M. Gothard, S. Soh, N. A. Gothard, B. Kowalczyk, Y. Wei, B. Baytekin and B. A. Grzybowski, Rewiring Chemistry: Algorithmic Discovery and Experimental Validation of One-Pot Reactions in the Network of Organic Chemistry, Angew. Chem., Int. Ed., 2012, 51(32), 7922–7927 CrossRef CAS PubMed.
- B. A. Grzybowski, S. Szymkuć, E. P. Gajewska, K. Molga, P. Dittwald, A. Wołos and T. Klucznik, Chematica: A Story of Computer Code That Started to Think like a Chemist, Chem, 2018, 4(3), 390–398 CAS.
- B. Mikulak-Klucznik, P. Gołębiowska, A. A. Bayly, O. Popik, T. Klucznik, S. Szymkuć, E. P. Gajewska, P. Dittwald, O. Staszewska-Krajewska, W. Beker, T. Badowski, K. A. Scheidt, K. Molga, J. Mlynarski, M. Mrksich and B. A. Grzybowski, Computational planning of the synthesis of complex natural products, Nature, 2020, 588(7836), 83–88 CrossRef CAS.
- C. W. Coley, R. Barzilay, T. S. Jaakkola, W. H. Green and K. F. Jensen, Prediction of Organic Reaction Outcomes Using Machine Learning, ACS Cent. Sci., 2017, 3(5), 434–443 CrossRef CAS PubMed.
- C. W. Coley, W. H. Green and K. F. Jensen, RDChiral: An RDKit Wrapper for Handling Stereochemistry in Retrosynthetic Template Extraction and Application, J. Chem. Inf. Model., 2019, 59(6), 2529–2537 CrossRef CAS.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction, ACS Cent. Sci., 2019, 5(9), 1572–1583 CrossRef CAS PubMed.
- M. H. S. Segler, M. Preuss and M. P. Waller, Planning chemical syntheses with deep neural networks and symbolic AI, Nature, 2018, 555(7698), 604–610 CrossRef CAS PubMed.
- P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano and T. Laino, Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy, Chem. Sci., 2020, 11(12), 3316–3325 RSC.
- J. M. Woodley and N. J. Turner, New Frontiers in Biocatalysis, in Handbook of Green Chemistry, 2019, pp. 73–86 Search PubMed.
- R. A. Sheldon and J. M. Woodley, Role of Biocatalysis in Sustainable Chemistry, Chem. Rev., 2018, 118(2), 801–838 CrossRef CAS PubMed.
- Y.-S. Ko, J. W. Kim, J. A. Lee, T. Han, G. B. Kim, J. E. Park and S. Y. Lee, Tools and strategies of systems metabolic engineering for the development of microbial cell factories for chemical production, Chem. Soc. Rev., 2020, 49(14), 4615–4636 RSC.
- W. Finnigan, L. J. Hepworth, S. L. Flitsch and N. J. Turner, RetroBioCat as a computer-aided synthesis planning tool for biocatalytic reactions and cascades, Nat. Catal., 2021, 4(2), 98–104 CrossRef CAS PubMed.
- S. Y. Lee, H. U. Kim, T. U. Chae, J. S. Cho, J. W. Kim, J. H. Shin, D. I. Kim, Y.-S. Ko, W. D. Jang and Y.-S. Jang, A comprehensive metabolic map for production of bio-based chemicals, Nat. Catal., 2019, 2(1), 18–33 CrossRef CAS.
- V. E. Balderas-Hernández, A. Sabido-Ramos, P. Silva, N. Cabrera-Valladares, G. Hernández-Chávez, J. L. Báez-Viveros, A. Martínez, F. Bolívar and G. Gosset, Metabolic engineering for improving anthranilate synthesis from glucose in Escherichia coli, Microb. Cell Fact., 2009, 8(1), 19 CrossRef PubMed.
- K. C. Thomas and W. M. Ingledew, Production of 21%(v/v) ethanol by fermentation of very high gravity(VHG) wheat mashes, J. Ind. Microbiol., 1992, 10(1), 61–68 CrossRef CAS.
- D. Probst, M. Manica, Y. G. Nana Teukam, A. Castrogiovanni, F. Paratore and T. Laino, Biocatalysed synthesis planning using data-driven learning, Nat. Commun., 2022, 13(1), 964 CrossRef CAS PubMed.
- A. Voll and W. Marquardt, Reaction network flux analysis: Optimization-based evaluation of reaction pathways for biorenewables processing, AIChE J., 2012, 58(6), 1788–1801 CrossRef CAS.
- J. S. Schreck, C. W. Coley and K. J. M. Bishop, Learning Retrosynthetic Planning through Simulated Experience, ACS Cent. Sci., 2019, 5(6), 970–981 CrossRef CAS PubMed.
- M. Koch, T. Duigou and J.-L. Faulon, Reinforcement Learning for Bioretrosynthesis, ACS Synth. Biol., 2020, 9(1), 157–168 CrossRef CAS PubMed.
- A. Khan and A. Lapkin, Searching for optimal process routes: A reinforcement learning approach, Comput. Chem. Eng., 2020, 141, 107027 CrossRef CAS.
- M. van Otterlo and M. Wiering, Reinforcement Learning and Markov Decision Processes, in Reinforcement Learning: State-of-the-Art, ed. M. Wiering and M. van Otterlo, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 3–42 Search PubMed.
- C. W. Coley, Defining and Exploring Chemical Spaces, Trends Chem., 2021, 3(2), 133–145 CrossRef CAS.
- I. Levin, M. Liu, C. A. Voigt and C. W. Coley, Merging enzymatic and synthetic chemistry with computational synthesis planning, Nat. Commun., 2022, 13(1), 7747 CrossRef CAS PubMed.
- Elsevier Reaxys, https://www.reaxys.com/ (accessed 6 Feb 2023) Search PubMed.
- M. Kanehisa and S. Goto, KEGG: Kyoto Encyclopedia of Genes and Genomes, Nucleic Acids Res., 2000, 28(1), 27–30 CrossRef CAS PubMed.
- ChemSpace, https://chem-space.com (accessed 21 Feb 2023) Search PubMed.
- L. K. Blaß, C. Weyler and E. Heinzle, Network design and analysis for multi-enzyme biocatalysis, BMC Bioinf., 2017, 18(1), 366 CrossRef PubMed.
- J. U. Bowie, S. Sherkhanov, T. P. Korman, M. A. Valliere, P. H. Opgenorth and H. Liu, Synthetic Biochemistry: The Bio-inspired Cell-Free Approach to Commodity Chemical Production, Trends Biotechnol., 2020, 38(7), 766–778 CrossRef CAS PubMed.
- T. Shi, P. Han, C. You and Y.-H. P. J. Zhang, An in vitro synthetic biology platform for emerging industrial biomanufacturing: Bottom-up pathway design, Synth. Syst. Biotechnol., 2018, 3(3), 186–195 CrossRef PubMed.
- D. Lowe, Chemical reactions from US patents, https://figshare.com/articles/dataset/Chemical_reactions_from_US_patents_1976-Sep2016_/5104873 (accessed 2 May 2023) Search PubMed.
- J. Mayfield, D. Lowe and R. Sayle, Pistachio. 3.0 edn, 2019 Search PubMed.
- RDKit: Open-source cheminformatics, https://www.rdkit.org (accessed 6 Feb 2023) Search PubMed.
- M. H. S. Segler and M. P. Waller, Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction, Chem. – Eur. J., 2017, 23(25), 5966–5971 CrossRef CAS PubMed.
- D. Rogers and M. Hahn, Extended-Connectivity Fingerprints, J. Chem. Inf. Model., 2010, 50(5), 742–754 CrossRef CAS PubMed.
- F. Chollet, Keras, https://github.com/fchollet/keras (accessed 17 July 2022) Search PubMed.
- KEGG DRUG Database, https://www.genome.jp/kegg/drug/ (accessed 10 Dec 2022) Search PubMed.
- A. P. Vig, G. Rampal, T. S. Thind and S. Arora, Bio-protective effects of glucosinolates – A review, LWT–Food Sci. Technol., 2009, 42(10), 1561–1572 CrossRef CAS.
- A. Lofton, Atropine, in Encyclopedia of Toxicology (Second Edition), ed. P. Wexler, Elsevier, New York, 2005, pp. 190–192 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00406b |
| This journal is © The Royal Society of Chemistry 2023 |