
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUpgrading furanic platforms to α-enaminones: tunable continuous flow hydrogenation of bio-based cyclopentenones†
Lídia A. S.
Cavaca
 a,
Jaime A. S.
Coelho
b,
Susana D.
Lucas
c,
Rui M. S.
Loureiro
c,
Rafael F. A.
Gomes
*a and
Carlos A. M.
Afonso
*a
a,
Jaime A. S.
Coelho
b,
Susana D.
Lucas
c,
Rui M. S.
Loureiro
c,
Rafael F. A.
Gomes
*a and
Carlos A. M.
Afonso
*a
aResearch Institute for Medicines (iMed.ULisboa), Faculty of Farmacy, Universidade de Lisboa, Avenida Professor Gama Pinto, 1649-003 Lisbon, Portugal. E-mail: rafael.gomes@campus.ul.pt; carlosafonso@ff.ulisboa.pt
bCentro de Química Estrutural, Institute of Molecular Sciences, Faculty of Sciences, Universidade de Lisboa, Campo Grande, 1749-016 Lisbon, Portugal
cHovione FarmaCiencia SA, Sete Casas, 2674-506 Loures, Portugal
First published on 24th November 2022
Abstract
Here we describe a tunable continuous flow hydrogenation of trans-4,5-diamino cyclopentenones (DCPs) allowing the selective stepwise synthesis of novel bifunctionalized cyclopentanones (up to 96% yield and >92% selectivity). Stability studies of the diamino cyclopentanone motif led to the development of an elimination procedure yielding the corresponding α-enaminones in good yields. Deuteration of the cyclopentene scaffold can also be performed using D2O as a source of D2. A sequential process for the synthesis of α-enaminones directly from furfural is also described. The methodology allows the preparation of a previously reported ATP-sensitive potassium channel agonist in 71% yield.
Introduction
α-Enaminones are versatile synthons exhibiting dual electronic properties.1 The enamine nucleophilic character and enone electrophilic nature of these compounds attracted particular interest as intermediates for the construction of heterocyclic compounds (e.g. tetrahydro-1H-carbazol-1-ones,2 pyrroles,3 quinolones4). Examples of pharmaceutically relevant α-enaminones are dehydro-benzoxazine and dehydro-benzothiazine derivatives, which exhibit activity as ATP-sensitive potassium (KATP) channel agonists (Fig. 1a)5–9 and precursors of imino 1,2,3,4-tetrahydrocyclopent[b]indoles, known to inhibit acetylcholinesterase and monoamine oxidase (Fig. 1a).10 | ||
| Fig. 1 a) Importance of the α-enaminone core in biological active compounds and precursors; b) reported methods for the synthesis of α-enaminones; and c) proposed work towards the preparation of α-enaminones from furfural applying a more sustainable continuous flow hydrogenation of DCPs. | ||
The extensive utility of this synthon promotes the development of new methods for the assembly of α-enaminones, constituting a topic of ongoing interest.
Over the years, different protocols have been developed for the synthesis of α-enaminones (Fig. 1b): (1) the conventional multistep synthesis from diketones;11–16 (2) cyclic ketone oxidative coupling with anilines or O-benzoylhydroxylamines under mild conditions, including electrochemical methods;17–21 more recently, (3) a three-component gold-catalysed coupling/cyclization reaction.22
Despite all the progress, these elegant methods have some limitations, as they require pre-functionalised amination reagents (i.e. o-benzoylhydroxylamines) and use of stoichiometric amounts of additives and/or oxidants and are often limited to the use of aromatic amines.
In view of these limitations, the design of new protocols that improve the sustainability of the existing methods bypassing the use of high amounts of additives/oxidants and tolerating a broad range of amination reagents would be highly desirable.
Following the “12 Principles of Green Chemistry”, we are contributing to sustainability, where the use of renewable feedstocks (i.e. biomass) is a prevalent trend to reduce the chemical impact of a process.23,24 In this sense, furfural is a renewable raw material included in the U.S. Department of Energy top “10 + 4” list of bio-based materials.25 The discovery of novel methods to prepare added value compounds from bio-based synthons, including furanic platforms such as furfural and 5-hydroxymethylfurfural (HMF), has attracted the attention of researchers over the years.26,27
Our group has been involved in the valorisation of furfurals,28,29 in particular for the preparation of functionalized cyclopentenones.11,30,31 Previous reports show that biomass derived trans-4,5-diamino-cyclopentenones (DCPs) undergo base catalysed β-amine elimination upon thiol 1,4-addition, yielding the corresponding enaminone.11,32,33 Hence we envisioned that hydrogenation of the DCP olefin may lead to similar behaviour under basic conditions, therefore forming the desired scaffold under mild conditions from cheap biomass furanic platforms. Furthermore, the use of continuous flow conditions will allow fine-tuning of the reaction selectivity to obtain either the cyclopentanone or the enaminone, facilitate scale-up/scale-out processes and reduce the reaction waste by reusing heterogeneous catalysts.
Results and discussion
Continuous flow hydrogenation of DCP using an H-Cube® Mini Plus for the selective formation of cyclopentanones 2
Aiming at cascade hydrogenation/elimination of DCP, 1a underwent hydrogenation under batch conditions. However, the reaction yielded a mixture of products corresponding to the hydrogenated cyclopentanone 2a and the desired enaminone 3a (see ESI,† Scheme S1). Highlighting the challenges of preparing 4,5-substituted cyclopentanones, we optimised the hydrogenation selectivity towards the formation of cyclopentanone 2a which we could also use to explore the elimination conditions towards enaminones.An emerging strategy to increase reaction selectivity, hence decreasing the formation of side products, is the use of continuous flow. This methodology tends to increase the selectivity due to the higher catalyst/substrate ratio and shorter reaction times.34 Focusing on the hydrogenated product 2a, the reaction was performed under continuous flow using H-Cube® Mini Plus apparatus in order to increase both the selectivity and safety of the hydrogenation reaction. After screening several parameters (see ESI,† Scheme S2) the optimal conditions were selected as 10 bar of H2 pressure, 0.5 mL min−1 flow rate, 7.8 seconds residence time, 65 μL reactor volume, 25 °C and 35 mM substrate concentration.
The scope of the hydrogenation reaction to cyclopentanones 2 was extended to a variety of previously described DCPs,30,35 promoted by three different catalysts. Cyclopentanones 2a–2f and 2h–2k were obtained in high yield and selectivity, with minor formation of the α-enaminone product (Scheme 1). All the three palladium catalysts (5% Pd/C, 20% Pd(OH)2/C and 10% Pd/Al2O3) tested were suitable for this transformation. Each cartridge allowed the running of more than 40 reactions and tolerated different substrates without cross-contamination or loss of efficiency. Overall, 5% Pd/C afforded higher yields (up to 96%) and selectivity (>92%).
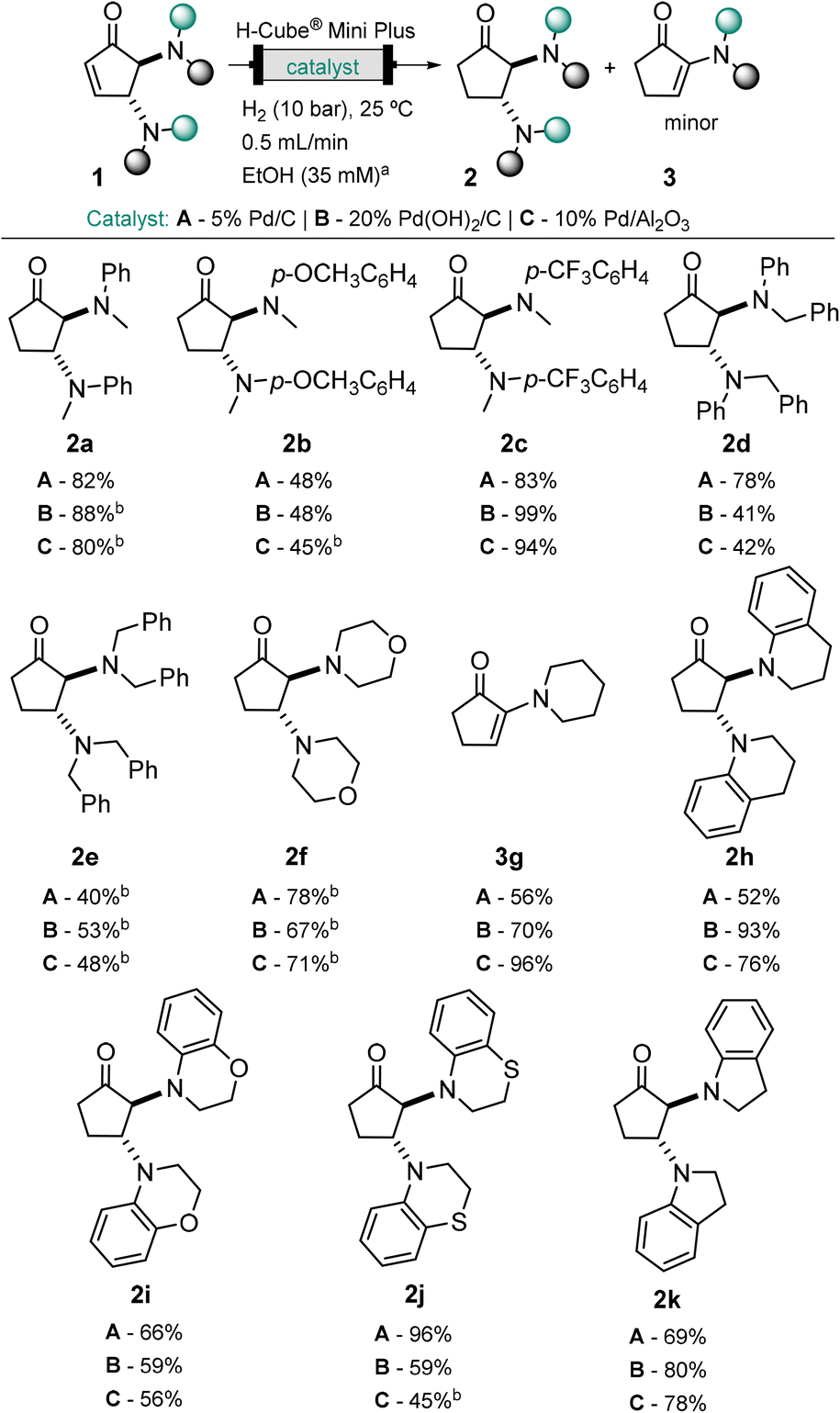 | ||
| Scheme 1 Scope of functionalized cyclopentanones 2a–2k from continuous flow hydrogenation of DCPs 1a–1k. DCPs (20 mg, 1 equiv.) in EtOH (35 mM), 5% Pd/C, 20% Pd(OH)2/C or 10% Pd/Al2O3, H2 (10 bar), 25 °C, 0.5 mL min−1. aConcentration adjusted according to the type of substrate. bObtained as a mixture of DCPs containing α-enaminone as a minor product (<10%, see ESI,† Table S1). | ||
It is noteworthy that despite the 10 bar system pressure, the deprotection of benzylamines in DCPs 1d and 1e was not observed. This selectivity for the hydrogenation of the enone may be due to the short residence time (7.8 s). In most cases, amine elimination with formation of α-enaminone was not observed when arylamine-DCPs were employed. The small extent of elimination observed for 2a explains the sensitivity of these compounds to temperature (40 °C) during solvent evaporation. In the case of alkyl amines (2e–2g), we observed the elimination to enaminones 3 to some extent, most pronounced in the case of piperidine DCP 1g, which afforded solely the α-enaminone product 3g.
To better understand the α-enaminone product 3a formation, the stability of cyclopentanone form 2a was initially assessed by exposing the reaction crude mixture to different deuterated solvents (see ESI,† Fig. S2) and additives with acidic or basic properties (see ESI,† Fig. S3 and S4). The studies suggested that β-amine elimination was inhibited under slightly acidic conditions (i.e. in non-neutralized CDCl3 and with NaH2PO4). On the other hand, a base catalysed elimination of the amine in the presence of K2CO3 and possibility of an autocatalytic mechanism by N-methyl aniline were observed.
Continuous flow hydrogenation of DCPs using an H-Cube® Mini Plus for the selective formation of α-enaminones 3
Based on these results, we envisioned that a simple treatment of the crude reaction mixture, rich in cyclopentanone 2, with K2CO3 would promote the elimination, therefore converting the cyclopentenones 1 to the corresponding α-enaminones 3. We started the elimination studies by i) employing a sequential HPLC column filled with K2CO3 connected to the H-Cube® apparatus outlet; ii) stirring the output of the hydrogenation reaction with a K2CO3 saturated aqueous solution; iii) stirring with solid K2CO3 under heterogeneous conditions (see ESI,† section 6). The simple use of solid K2CO3 afforded the desired product in high yields and the base could be easily removed by filtration. By using this methodology, α-enaminone 3a was obtained in 73% yield using 5% Pd/C, with the recovery of the free N-methyl aniline by column chromatography in neutral alumina (81% yield).The scope of α-enaminones was extended to various DCPs by selecting the most appropriate palladium catalyst. The desired compounds were obtained in up to 83% isolated yield (Scheme 2). Despite most DCPs affording the desired product at room temperature, dibenzylamine cyclopentanone derivative 2e was an exception, requiring higher temperatures (60 °C) and 2 equivalents of K2CO3 to yield 42% of 3e.
 | ||
Scheme 2 Scope of α-enaminones 3a–3m obtained after basic treatment of the continuous flow hydrogenation crude mixture obtained using the corresponding Pd catalyst A, B or C. aConcentration adjusted according to the type of substrate (see ESI† for detailed information). bObtained only by treatment with 2 equivalents of K2CO3 at 60 °C. cIsolated yield in gram scale reaction (1g). dObtained directly from hydrogenation reaction, without requiring treatment with K2CO3; the yield without chromatographic purification is given in parentheses. eObtained using D2O as a source of D2 under reported conditions and followed by in acetone-d6/D2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and K2CO3 after 3 days. :1) and K2CO3 after 3 days. | ||
A single cartridge of 5% Pd/C containing 150 mg of catalyst allowed full conversion in gram scale reaction using 1a for the preparation of 3a. To this end, solutions of DCP (200 mg, 35 mM) were fed to the reactor in a series of 4 sequential injections (each injection corresponds to an average of 310 reactor volumes). The yields were consistent in all cycles (see ESI,† Fig. S5 and Table S2) and overall, the desired α-enaminone was obtained in 93% isolated yield and a productivity of 1.7 g L−1 h−1. To further improve the metrics of the reaction, significantly more concentrated solutions (173 mM) were fed to the reactor in a series of 3 sequential injections which allowed the production of a similar amount of product with only a fraction of the solvent, equivalent to 231 reactor volumes (see ESI,† Fig. S6 and Table S3). The productivity was significantly increased at this concentration, achieving 53.6 g L−1 h−1, despite a decrease in yield to 76%.
The importance of labelling scaffolds with hydrogen isotopes, in conjunction with the ease of forming D2 using the H-Cube® Mini Plus apparatus, led us to study the incorporation of deuterium in our cyclopentane scaffolds. Compounds labelled with hydrogen isotopes (i.e. deuterium and tritium) are of extreme utility in mechanistic, spectroscopic and tracer studies.36 Isotope labelled compounds prove to have additional uses in medicinal chemistry, such as slowing the metabolism of drug candidates.37 The insertion of deuterium at C4 and at enolizable C5 positions was possible affording α-enaminone 3m in 83% yield (Scheme 2 and ESI,† Scheme S3).
Sequential continuous flow process by tandem hydrogenation/elimination
At this stage, α-enaminones were obtained from furfural in a stepwise manner. In order to avoid intermediate isolation and purification, we developed a sequential continuous flow process to obtain α-enaminones directly from furfural.A previously described continuous flow system for the preparation of DCPs from furfural38 was coupled with the herein described hydrogenation setup (see ESI,† Scheme S4). The sequential reaction was able to afford α-enaminone 3a directly from furfural in 1 g L−1 h−1 productivity (Scheme 3).
 | ||
| Scheme 3 Sequential continuous flow process for the direct synthesis of α-enaminone 3a from furfural, integrating the in situ preparation of DCP 1a and the herein described hydrogenation setup. | ||
Catalyst leaching measurement of Cu and Pd was conducted through ICP-AES analyses using the crude sample obtained from this flow process (see ESI,† section 10). The results indicated leaching of Cu in 679 ppm mg−1 of sample, while the amount of Pd was below the limit of detection (<0.16 ppm for hydrogenation reaction). The amount of Cu detected in the sample was consistent with previous work,38 although a simple washing of the crude sample with water after redissolution in EtOAc was enough to reduce the amount of Cu to 83.5 ppm mg−1 of sample.
Synthesis of ATP-sensitive potassium channel agonist 3n
To showcase the versatility of the method, a model ATP-sensitive potassium (KATP) channel agonist was selected as the target molecule. The active dehydro-benzoxazine α-enaminone scaffold has been previously reported, and the selected derivative exhibits IC50 in the μM range.9The original total synthesis of this compound comprises two steps from cyclopentanone, affording the desired α-enaminone 3n in 50% global yield. Despite the straightforward process, the procedure requires harsh conditions and sensitive reagents.9
In order to improve the existing method in terms of sustainability and easier handling of materials, the synthesis of α-enaminone 3n is proposed applying the described continuous flow process.
The hydrogenation conditions described above were employed to yield a different product than the expected. Preliminary characterization by 1H NMR and HRMS suggests the formation of an α-enaminone resulting from additional reduction of the aromatic nitro group (see ESI,† Scheme S5). Adjustment of the original conditions to 2 mL min−1 flow rate and change to ACN/EtOH (1:1) to increase the substrate concentration inhibited the reduction of the nitro group, hence yielding the desired product 3n in 71% yield from the corresponding cyclopentenone (Scheme 4).
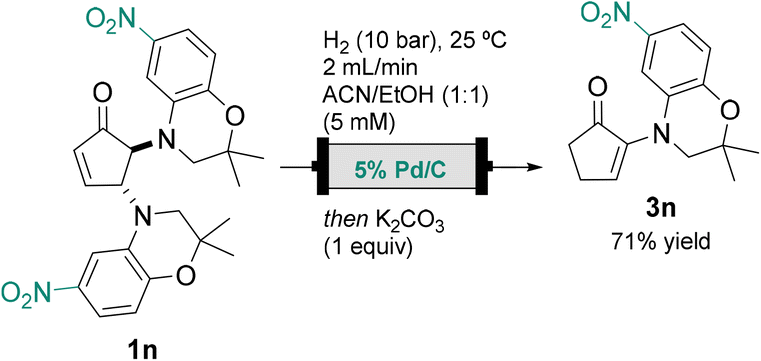 | ||
| Scheme 4 Synthesis of biologically active α-enaminone 3n applying the presented continuous flow hydrogenation methodology. | ||
This new approach overcomes price, raw material availability and sustainability limitations of the original total synthesis. The sustainability of the process was enhanced not only by the use of furfural but also by the implementation of a continuous flow process that allowed catalyst reuse. The increased safety and the use of milder conditions in all steps are also advantages.
Quantitative and qualitative green metrics of the process
We compared the quantitative and qualitative green metrics of the continuous flow hydrogenation/elimination of DCP 1a and reported batch procedures in the preparation of α-enaminones according to the CHEM21 toolkit (Table 1).39 The reported procedures were selected in a way to encompass current different approaches, including 1) an electrochemical procedure (method 1),18 2) a method using hydroxylamines as amine sources (method 2)20 and 3) a procedure using TEMPO+ PF6− as an oxidant (method 3).17 Since it was not possible to compare substrate 1a to methods 2 and 3, the calculations were conducted with similar derivatives (i.e. N-benzylaniline for method 2 and 4-chloro-N-methylaniline for method 3). The yields and the “reaction mass efficiency (RME)” are comparable between the procedures, except for the hydroxylamine method that presents a low RME value. Regarding the “optimum efficiency (OE)”, it is significantly better in the hydrogenation/elimination process. Although the “atom economy (AE)” is expectedly lower than that of the other procedures due to free amine elimination, the AE metric value is compensated by RME.| Metric | This work | This work, from furfural | Method 1 | Method 2 | Method 3 |
|---|---|---|---|---|---|
| a PMI values not counting chromatographic procedures.“Reaction mass efficiency (RME)” is the division of the product and reactant mass. “Optimum efficiency (OE)” is the ratio of RME and “atom economy (AE)”. | |||||
| Yield (%) | 76 | 76 | 79 | 71 | 62 |
| RME | 49 | 46 | 54 | 22 | 51 |
| OE | 76 | 50 | 55 | 34 | 52 |
| PMI | 386 (36a) | 388 (38a) | 56a | 61a | 403 (67a) |
The “process mass intensity” (PMI) values were calculated taking into account the workup and chromatographic procedures whenever experimental information was available. The herein described method has a PMI value of 386, significantly lower than the value of 403 observed for method 3. It is noteworthy that methods 1–3 use commercially available cyclopentanone as the substrate, known to be prepared from furfural upon the Piancatelli rearrangement and hydrogenation.40,41 Despite DCPs not being commercially available, they can be easily obtained in a single step from furfural in quantitative yields.30 Nevertheless, the quantitative green metrics of the process starting from furfural were assessed (Table 1). For that, the synthesis of DCPs from furfural 1a30 was accounted in the overall process, including work-up and chromatographic solvents (see ESI,† Table S11). No significant changes were observed in the PMI value of 388.
The PMI values for comparison with methods 1 and 2 are given in parenthesis (Table 1). This is due to the lack of information concerning the chromatographic procedure of the reported methods. Thus, the PMI used for comparison was calculated after excluding the chromatographic solvents parameter. The PMI values of 56 and 61, respectively, are slightly higher than the corresponding values calculated for our method, including for the synthesis from furfural.
Qualitative analysis of the processes was performed based on the aforementioned toolkit, and to each criterion was attributed a colored flag concerning the sustainability of the process (Table 2). Despite these methodologies receiving a green flag for being catalytic and conducted at room temperature, only the new procedure receives a green flag for being performed under flow conditions. A single red flag is attributed to our method regarding the use of chromatographic purification, which is common to all methods. Concerning solvents, ethanol is considered a preferred solvent, conferring an advantage compared to methods 2 and 3 which were attributed with a red flag for using hazardous solvents. All the four methods used a critical element in the process. Nevertheless, a yellow flag is attributed to Pd, as well as to Cu and Sc used in method 2. Ag, Pt and Sb are considered very critical elements receiving a red flag.
| Green flag represents “preferred”. Yellow flag represents “acceptable”. Red flag represents “undesirable”. |
|---|

|
In a global consideration, the described method is comparable to recent reported procedures in terms of quantitative metrics. Regarding qualitative measures, the most advantageous criterion is the use of continuous flow. A clear advantage of this method is the use of biomass feedstocks such as furfural and bioethanol which is in accordance with the 12 principles of green chemistry.
NMR kinetic studies and DFT calculations for mechanism elucidation
To elucidate the elimination reaction mechanism, both quantitative 1H-NMR kinetics and computation studies were performed.Firstly, the relevance of electronic effects of the aryl amine substituent on the elimination reaction was evaluated (see ESI,† section 13). To this end, competitive experiments were conducted using cyclopentanones 2a, 2b and 2c. Stoichiometric mixtures of 2a/2b and 2a/2c were treated with K2CO3 and the profile of elimination was followed over time by 1H NMR experiments in CDCl3 (see ESI,† Fig. S10 and S11). These results suggest a high dependence of the elimination reaction with the electronic character of the arylamine substituent. The order of stability is then proposed to be 2b (OMe) > 2a (H)> 2c (CF3).
Finally, quantitative 1H NMR kinetic experiments were performed to study the reaction kinetics (see ESI,† section 14). The elimination of cyclopentanone 2a in the corresponding α-enaminone 3a was assessed by 1H NMR experiments over time in methanol-d4, promoted by K2CO3. A pseudo zero-order kinetics in solution was observed (see ESI,† Fig. S12 and S13).
To further understand the mechanism of the elimination reaction, density functional theory (DFT) calculations were performed at the ωB97X-D/Def2-TZVPP/PCM(SMD, ethanol)//B3LYP/6-31G(d) level of theory. Aniline-substituted cyclopentanones 2a, 2b and 2c and bis-dimethylamino-cyclopentanone were selected as model substrates (see ESI,† section 15). Calculations suggest that the β-amine elimination reaction proceeds through the enolate intermediate via protonation by EtOH (solvent of the reaction, see ESI,† Fig. S14 and S15). Calculations also support that a second molecule of the substrate in its enol form could participate as a proton donor (see ESI,† Fig. S16), explaining the occurrence of the neat reaction.
The calculated activation barriers corroborate the observed competitive experiments where the elimination rate follows the following trend: dialkylamine > aniline (p-CF3 > H > p-OMe).
Conclusions
A continuous flow approach is described for the stepwise synthesis of a series of previously inaccessible 2,3-diamino-cyclopentanones in high yield and selectivity through a tunable hydrogenation of 4,5-trans-diamino-cyclopent-2-enones under mild conditions. Studies on the stability of diamino cyclopentanones led to the development of a procedure to selectively obtain α-enaminones under basic conditions. Preparation of α-enaminones directly from biomass derived furfural was accomplished by a sequential continuous flow process that first yielded the corresponding diamino cyclopentenone followed by tandem hydrogenation/elimination. Incorporation of deuterium in high extent on the diamino cyclopentenone C4 and C5 was accomplished by employing deuterated water on the H-Cube® Mini Plus apparatus. The flow process was used to prepare a previously reported ATP-sensitive potassium channel agonist.Mechanistic highlights revealed the influence of the electronic features of aniline substituents as well as a pseudo zero-order reaction kinetics in solution.
On a final note, this method is a new green methodology for the synthesis of α-enaminones. It encompasses several principles among the “12 Principles of Green Chemistry”, by making use of biorenewable raw materials, avoiding the use of oxidants and additives, allowing catalyst reuse, and easy free amine recovery.
Experimental
Materials and methods
All solvents were of analytical grade and distilled prior to use. Unless otherwise stated, all reagents were used as received from commercial suppliers. Continuous flow hydrogenation reactions were conducted in an H-Cube® Mini Plus from ThalesNano® using ThalesNano CatCart® cartridge systems (30 mm L, 0.226 mL dead volume). NMR spectra were recorded in a Bruker Fourier 300 spectrometer. High resolution mass spectrometry (HRMS) results were recorded in a Thermo Scientific Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (Thermo Scientific™ Q Exactive™ Plus). Infrared spectra were recorded in a Bruker Alpha II FT-IR spectrometer. Inductively coupled-plasma atomic emission spectroscopy (ICP-AES) analyses were carried out on a Horiba Jobin Yvon ULTIMA sequential ICP, using the Horiba Jobin Yvon ICP Analyst 5.4 software. A monochromator with a Czerny Turner spectrometer was used. The gas used was argon.Synthetic procedures
:1).
:1). α-Enaminone 3a was obtained as a yellow oil (9 mg, 65% yield). N-Methyl aniline was recovered (6.8 mg, 81% yield).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Fundação para a Ciência e Tecnologia (FCT) for financial support (PD/BD/143127/2019, PTDC/QUI-QOR/32008/2017, UIDB/04138/2020, UIDP/04138/2020 UIDB/00100/2020, UIDP/00100/2020 and LA/P/0056/2020). J. A. S. C. thanks the FCT for Scientific Employment Stimulus 2020/02383/CEECIND. The project leading to this application has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 951996. The NMR spectrometers are part of the National NMR Network (PTNMR) partially supported by Infrastructure Project No. 022161 (co-financed by FEDER through COMPETE 2020, POCI and PORL and FCT through PIDDAC).Notes and references
- A.-Z. A. Elassar and A. A. El-Khair, Tetrahedron, 2003, 59, 8463–8480 CrossRef CAS.
- H. Shi, T. Guo, D. Zhang-Negrerie, Y. Du and K. Zhao, Tetrahedron, 2014, 70, 2753–2760 CrossRef CAS.
- J. Cossy, C. Poitevin, L. Sallé and D. G. Pardo, Tetrahedron Lett., 1996, 37, 6709–6710 CrossRef CAS.
- D. Lankri, G. Albarghouti, M. Mahameed and D. Tsvelikhovsky, J. Org. Chem., 2017, 82, 7101–7113 CrossRef CAS PubMed.
- V. Calderone, R. Spogli, A. Martelli, G. Manfroni, L. Testai, S. Sabatini, O. Tabarrini and V. Cecchetti, J. Med. Chem., 2018, 51, 5085–5092 CrossRef PubMed.
- A. Martelli, G. Manfroni, P. Sabbatini, M. L. Barreca, L. Testai, M. Novelli, S. Sabatini, S. Massari, O. Tabarrini, P. Masiello, V. Calderone and V. Cecchetti, J. Med. Chem., 2013, 56, 4718–4728 CrossRef CAS PubMed.
- E. Carosati, H. Lemoine, R. Spogli, D. Grittner, R. Mannhold, O. Tabarrini, S. Sabatini and V. Cecchetti, Bioorg. Med. Chem., 2005, 13, 5581–5591 CrossRef CAS.
- V. Cecchetti, V. Calderone, O. Tabarrini, S. Sabatini, E. Filipponi, L. Testai, R. Spogli, E. Martinotti and A. Fravolini, J. Med. Chem., 2003, 46, 3670–3679 CrossRef CAS.
- Y. Matsumoto, R. Tsuzuki, K. Takayama, T. Yoden, W. Uchida, M. Asano, S. Fujita, I. Yanagisawa and T. Fujikura, Chem. Pharm. Bull., 1996, 44, 103–114 CrossRef CAS PubMed.
- D. M. Fink, M. G. Palermo, G. M. Bores, F. P. Huger, B. E. Kurys, M. C. Merriman, G. E. Olsen, W. Petko and G. O'Malley, Bioorg. Med. Chem., 1996, 6, 625–630 CrossRef CAS.
- J. P. M. Nunes, C. A. M. Afonso and S. Caddick, RSC Adv., 2013, 3, 14975 RSC.
- J. Tamariz, R. Bautista and A. Jerezano, Synthesis, 2012, 44, 3327–3336 CrossRef.
- C.-M. Ho and T.-C. Lau, New J. Chem., 2000, 24, 859–863 RSC.
- P. G. Gassman, T. J. van Bergen, D. P. Gilbert and B. W. Cue Jr, J. Am. Chem. Soc., 1974, 96, 5495–5508 CrossRef CAS.
- K. Sato, S. Inoue, D. P. Gilbert and B. W. Cue Jr, J. Org. Chem., 1973, 38, 551–554 CrossRef CAS.
- M. A. Tobias, J. G. Strong and R. P. Napier, J. Org. Chem., 1970, 35, 1709–1711 CrossRef CAS.
- B. Xu, Y. Shang, X. Jie, X. Zhang, J. Kan, S. L. Yedage and W. Su, Green Chem., 2020, 22, 1827–1831 RSC.
- K. Hu, P. Qian, J. H. Su, Z. Li, J. Wang, Z. Zha and Z. Wang, J. Org. Chem., 2019, 84, 1647–1653 CrossRef CAS PubMed.
- Y.-J. Li, L. Zhang, N. Yan, X.-H. Meng and Y.-L. Zhao, Adv. Synth. Catal., 2018, 360, 455–461 CrossRef CAS.
- Y. Li, R. Zhang, X. Bi and J. Fu, Org. Lett., 2018, 20, 1207–1211 CrossRef CAS.
- Y. Li, H. Gao, Z. Zhang, P. Qian, M. Bi, Z. Zha and Z. Wang, Chem. Commun., 2016, 52, 8600–8603 RSC.
- J. Li, Y. Xu, X. Hu, S. Zhu and L. Liu, Org. Lett., 2020, 22, 9478–9483 CrossRef CAS PubMed.
- G. T. Whiteker, Org. Process Res. Dev., 2019, 23, 2109–2121 CrossRef CAS.
- A. Ivanković, Int. J. Sustainable Green Energy, 2017, 6, 39–48 CrossRef.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- G. Shen, B. Andrioletti and Y. Queneau, Curr. Opin. Green Sustainable Chem., 2020, 26, 100384 CrossRef.
- R. C. Saxena, D. K. Adhikari and H. B. Goyal, Renewable Sustainable Energy Rev., 2009, 13, 167–178 CrossRef.
- J. P. M. António, R. F. M. Frade, F. M. F. Santos, J. A. S. Coelho, C. A. M. Afonso, P. M. P. Gois and A. F. Trindade, RSC Adv., 2014, 4, 29352–29356 RSC.
- S. Subbiah, S. P. Simeonov, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Green Chem., 2013, 15, 2849 RSC.
- R. F. A. Gomes, N. R. Esteves, J. A. S. Coelho and C. A. M. Afonso, J. Org. Chem., 2018, 83, 7509–7513 CrossRef CAS.
- M. S. Estevão and C. A. M. Afonso, Tetrahedron Lett., 2017, 58, 302–304 CrossRef.
- M. L. Di Gioia, M. Nardi, P. Costanzo, A. De Nino, L. Maiuolo, M. Oliverio and A. Procopio, Molecules, 2018, 23, 1891 CrossRef PubMed.
- M. Nardi, P. Costanzo, A. De Nino, M. L. Di Gioia, F. Olivito, G. Sindona and A. Procopio, Green Chem., 2017, 19, 5403–5411 RSC.
- C. Wiles and P. Watts, Green Chem., 2014, 16, 55–62 RSC.
- S. W. Li and R. A. Batey, Chem. Commun., 2007, 3759–3761, 10.1039/b709337n.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS PubMed.
- C. S. Elmore and R. A. Bragg, Bioorg. Med. Chem. Lett., 2015, 25, 167–171 CrossRef CAS.
- R. F. A. Gomes, L. A. S. Cavaca, J. M. Gonçalves, R. Ramos, A. F. Peixoto, B. I. Arias-Serrano and C. A. M. Afonso, ACS Sustainable Chem. Eng., 2021, 9, 16038–16043 CrossRef CAS.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- P. Jia, X. Lan, X. Li and T. Wang, ACS Sustainable Chem. Eng., 2019, 7, 15221–15229 CrossRef CAS.
- T. Shen, R. Hu, C. Zhu, M. Li, W. Zhuang, C. Tang and H. Ying, RSC Adv., 2018, 8, 37993–38001 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00292b |
| This journal is © The Royal Society of Chemistry 2023 |