
Evaluation of amine-based solid adsorbents for direct air capture: a critical review
Debashis
Panda†
 ,
Vaishnavi
Kulkarni†
and
Sanjay Kumar
Singh
*
,
Vaishnavi
Kulkarni†
and
Sanjay Kumar
Singh
*
Department of Chemistry, Indian Institute of Technology Indore, Simrol, Khandwa Road, Indore 453552, Madhya Pradesh, India. E-mail: sksingh@iiti.ac.in
First published on 22nd November 2022
Abstract
Direct air capture (DAC) emerges as a new technology that can contribute to “negative carbon emission”. Recent progress in surface chemistry and material synthesis has allowed a new generation of CO2 adsorbents that can drive the future of DAC and its wide-ranging deployment. This review is intended to shed light on the recent developments in porous amine-based solid sorbents for direct air CO2 capture, adsorbent preparation and characterization, CO2 capture under dry and humid conditions, CO2 adsorption kinetics, adsorption thermodynamics, sorbent regeneration, cyclic stability, essential regeneration techniques, and techno-economic analysis for CO2 capture from air.
 Debashis Panda | Debashis Panda obtained his Ph.D. in Mechanical Engineering from the Indian Institute of Technology Indore, India under the joint supervision of Prof. Sanjay Kumar Singh and Prof. E. Anil Kumar. His research mainly focuses on the synthesis and characterization of solid adsorbents for CO2 capture from various sources including flue gas, biogas, and ambient air. He has also focused on the scalability of adsorbent and explored suitable separation techniques including thermal swing adsorption for direct air CO2 capture applications. His research activity is highly interdisciplinary lying between the areas of engineering and chemistry. |
 Vaishnavi Kulkarni | Vaishnavi Kulkarni received her M.Sc. in Chemistry in 2017 from Savitribai Phule Pune University, India. Currently, she is a Ph.D. student in the Department of Chemistry, Indian Institute of Technology Indore, India under the supervision of Prof. Sanjay Kumar Singh. Her research expertise involves the synthesis of mesoporous/microporous as well as hierarchical materials for CO2 capture. She is currently working on the synthesis of hierarchical silica, metal–organic frameworks, and their further modification for direct air CO2 capture applications. |
 Sanjay Kumar Singh | Sanjay Kumar Singh obtained his PhD in Chemistry from A.P.S. University, Rewa, India. He had consecutive postdoctoral stays at AIST, Osaka, Japan (as a JSPS postdoctoral fellow, and later as AIST postdoctoral scientist with Prof. Qiang Xu) and at Karlsruhe Institute of Technology, Karlsruhe, Germany (as Alexander von Humboldt postdoctoral fellow with Prof. Peter W. Roesky). In 2012, he joined the Indian Institute of Technology Indore, India, as an Assistant Professor in Chemistry, and since Feb. 2022 he is a Professor at the same institution. His research focuses on the design and development of new and efficient catalysts and materials for CO2 capture and utilization, hydrogen production and storage, and biomass transformation to value-added chemicals and fuels. |
1 Introduction
The pursuit of socio-economic growth for the development of human civilization has imparted huge dependence on conventional fossil fuels. This has led to massive greenhouse gas (GHG) emissions such as CO2, causing global climate change.1 Besides the combustion of fossil fuels, a considerable amount of CO2 is also generated during various processes such as cement production, bio/natural gas sweetening, and electricity generation from geothermal vents.2 As a result, the atmospheric CO2 concentration has risen dramatically from 278 ppm at the start of the Industrial Revolution to >400 ppm today.3,4 In fact more than 2000 Gt CO2 has been emitted by humans since the Industrial Revolution.5 This has increased the earth's average temperature by 0.6 to 1 °C. Without any intermediate strategy for negative carbon emissions, the current global GHG emission is expected to increase by about 11% by 2030, compared to 2010 levels.6 The global mean temperature has risen by 1 °C after the pre-industrialization period.7 Therefore, 10 Gt CO2 per year must be removed from the atmosphere each year until 2050, and then 20 Gt CO2 per year from 2050 to 2100 to keep warming below 1.5 °C (Fig. 1).8 In this context, various carbon capture storage (CCS) technologies such as forestation, enhanced weathering, bioenergy and carbon capture and storage (BECCS), pre and post-combustion CO2 capture, and direct air CO2 capture have been deployed on a large scale, which could address both carbon emission problems as well as generation of renewable fuel from captured CO2 to fulfill energy demands.9–11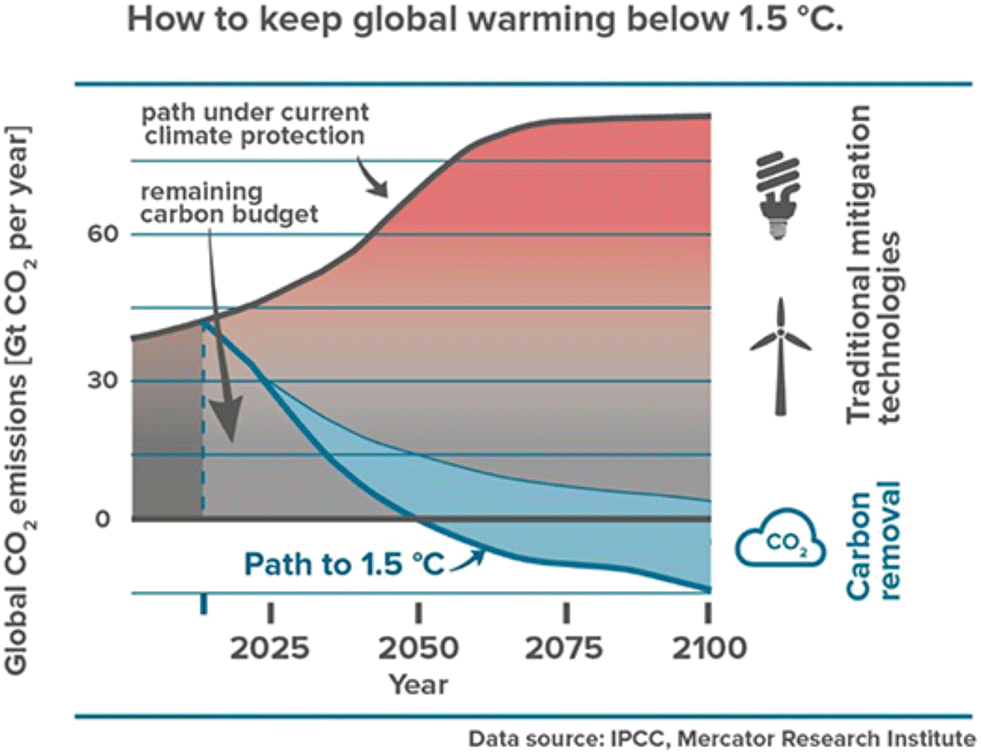 | ||
| Fig. 1 Atmospheric CO2 concentration and CO2 mitigation timeline. Blue shading represents the amount of negative emissions likely needed in this scenario. Reproduced with permission.10 Copyright 2019, Frontiers in Climate. | ||
DAC is recently introduced as modern CCS, a complementary approach to capture CO2 directly from the air at a low energy penalty and convert it into CO2-neutral liquid or gaseous hydrocarbon fuels.12 DAC is geographically agnostic (i.e. located anywhere) and essential for achieving net-zero and thus is now accepted by the world leaders meet at Glasgow (for COP 26).5 Planting trees and rewilding are not enough to reduce the CO2 content in the air up to a permissible limit. Instead, developing cost-effective DAC technology and reducing fossil fuel usage may be a solution to achieve net-zero.13 Nearly 40 direct air capture plants established by companies such as Climeworks and Carbon Engineering are in service around the world, capturing over 9000 tons of CO2 per year directly from the air using various technologies.14,15 To have a meaningful impact of carbon removal on climate, DAC needs to be economical and practical to capture CO2 at gigatons level. With a carbon capture capacity of 1 Mt CO2 per year, Ozkan et al. estimated that 1250 DAC plants would be required to remove 25 Gt of CO2 by 2030.5 In this context, selecting a suitable CO2 capture technology for DAC is tricky, as there is always a trade-off between performance and capture cost. The most important parameter is the low concentration of CO2 in the air (0.04%) compared to point sources (such as flue gas ∼10–15%). Therefore, the kinetics and thermodynamics of gas separation from the air are less favorable.15 In general, both absorption and adsorption processes are widely used in DAC.16 Adsorption processes, using solid sorbents capable of capturing CO2 from any stationary sources or atmosphere have shown many potential advantages,16 compared to other conventional CO2 capture techniques using aqueous amine solvents, such as CO2 scrubbing using aqueous N-methyl ethanolamine (MEA) solution.17 The high heat capacities of these solutions and the solvent losses due to evaporation18,19 make the regeneration process highly energy-intensive and expensive. Significantly, the CO2 scrubbing solutions such as the aqueous MEA solution, which has been used in chemical industries for several years, might not be the best choice for CO2 absorption from the air, because of a large volume of gas (about ∼300 times)20 to be treated and the subsequent loss of water and low-boiling MEA from the system. A recent study claimed that to capture 1 ton of CO2 from the air using a conventional monoethanolamine (MEA) solution, the overall cost of $1690 would be required.21 In this context, various studies were carried out to reduce the regeneration energy during the desorption of CO2 from MEA solution. For instance, Zhang et al.22 synthesized three distinct metal oxide-modified KIT-6 catalysts to catalyze the regeneration process of CO2-loaded MEA solution. The group found that KIT-6-based catalysts can reduce the relative regeneration energy consumption by 20.2–33.4% while increasing the CO2 desorption capacity by 31.0–49.3%. In line with this, Hu et al.23 reported that the trio-amine blends MEA-1-dimethylamino-2-propanol/piperazine (1DMA2P–PZ) with different 1DMA2P/PZ mole ratios were formulated and applied for the post-combustion CO2 capture. In addition, the blend-3 containing (MEA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1DMA2P:PZ = 3 M:2.5 M:0.5 M) demonstrated outstanding performance in terms of relative heat duty and reduced the energy penalty by 29.4–55.4% when compared to the 5 M MEA solution. Chen et al.24 investigated the absorption and desorption performance of three different aqueous MEA/MDEA/AMP (2-aminoethanol/N-methyldiethanolamine/2-amino-2-methyl-1-propanol) blends, and blend-2 (5 M, 1/2/2) showed a desorption rate up to 5 times higher, a 58.5% lower heat duty and a 57.6% lower relative cost compared to 5 M aqueous MEA. Similarly, Zhang et al.25 reported the CO2 absorption heat of tertiary amine 2-(dimethylamino)-2-methyl-1-propanol (2DMA2M1P), found to be 36.3 kJ mol−1, which was quite lower than MEA and thus can be an efficient amine solvent for CO2 capture. Despite significant progress in MEA-based CO2 capture, liquid-based hydroxide solution (NaOH or KOH) is the only current commercial absorption technology capable of capturing CO2 from the atmosphere to form water and carbonate. Later, carbonates are treated with lime (Ca(OH)2) to precipitate CaCO3, which is then heated at about 900 °C to release a high purity CO2 gas for further sequestration and utilization.26 However, the footprint of a liquid-based hydroxide solution adsorption system is quite bulky, and the regeneration process is also highly energy-intensive and expensive.27,28 According to the American Physical Society (APS), the capture cost for a realistic design employing liquid hydroxide solution DAC is roughly 600 $ per tCO2.29
:1DMA2P:PZ = 3 M:2.5 M:0.5 M) demonstrated outstanding performance in terms of relative heat duty and reduced the energy penalty by 29.4–55.4% when compared to the 5 M MEA solution. Chen et al.24 investigated the absorption and desorption performance of three different aqueous MEA/MDEA/AMP (2-aminoethanol/N-methyldiethanolamine/2-amino-2-methyl-1-propanol) blends, and blend-2 (5 M, 1/2/2) showed a desorption rate up to 5 times higher, a 58.5% lower heat duty and a 57.6% lower relative cost compared to 5 M aqueous MEA. Similarly, Zhang et al.25 reported the CO2 absorption heat of tertiary amine 2-(dimethylamino)-2-methyl-1-propanol (2DMA2M1P), found to be 36.3 kJ mol−1, which was quite lower than MEA and thus can be an efficient amine solvent for CO2 capture. Despite significant progress in MEA-based CO2 capture, liquid-based hydroxide solution (NaOH or KOH) is the only current commercial absorption technology capable of capturing CO2 from the atmosphere to form water and carbonate. Later, carbonates are treated with lime (Ca(OH)2) to precipitate CaCO3, which is then heated at about 900 °C to release a high purity CO2 gas for further sequestration and utilization.26 However, the footprint of a liquid-based hydroxide solution adsorption system is quite bulky, and the regeneration process is also highly energy-intensive and expensive.27,28 According to the American Physical Society (APS), the capture cost for a realistic design employing liquid hydroxide solution DAC is roughly 600 $ per tCO2.29
In this context, amine-based solid adsorbents are considered to be technically suitable and feasible for DAC due to their ability to capture CO2 at low concentrations and even in the presence of moisture.30–32 The improved performance characteristics such as easy handling, applicability to microgravity environments, and regenerability in either pressure swing (PSA) or temperature swing (TSA) modes expanded the application of amine-based adsorbents for CO2 capture ranging from alkali fuel cell (AFC) to space life support applications.33,34 Notably, such DAC processes with multiple pilot plants are already being developed and commercialized by two established startups: Climeworks and Global Thermostat.35 Since 2006, there has been a significant increase in the number of publications in the area of DAC (Fig. 2), focusing mainly on the development of various porous materials and energy-efficient separation techniques for CO2 capture from air.
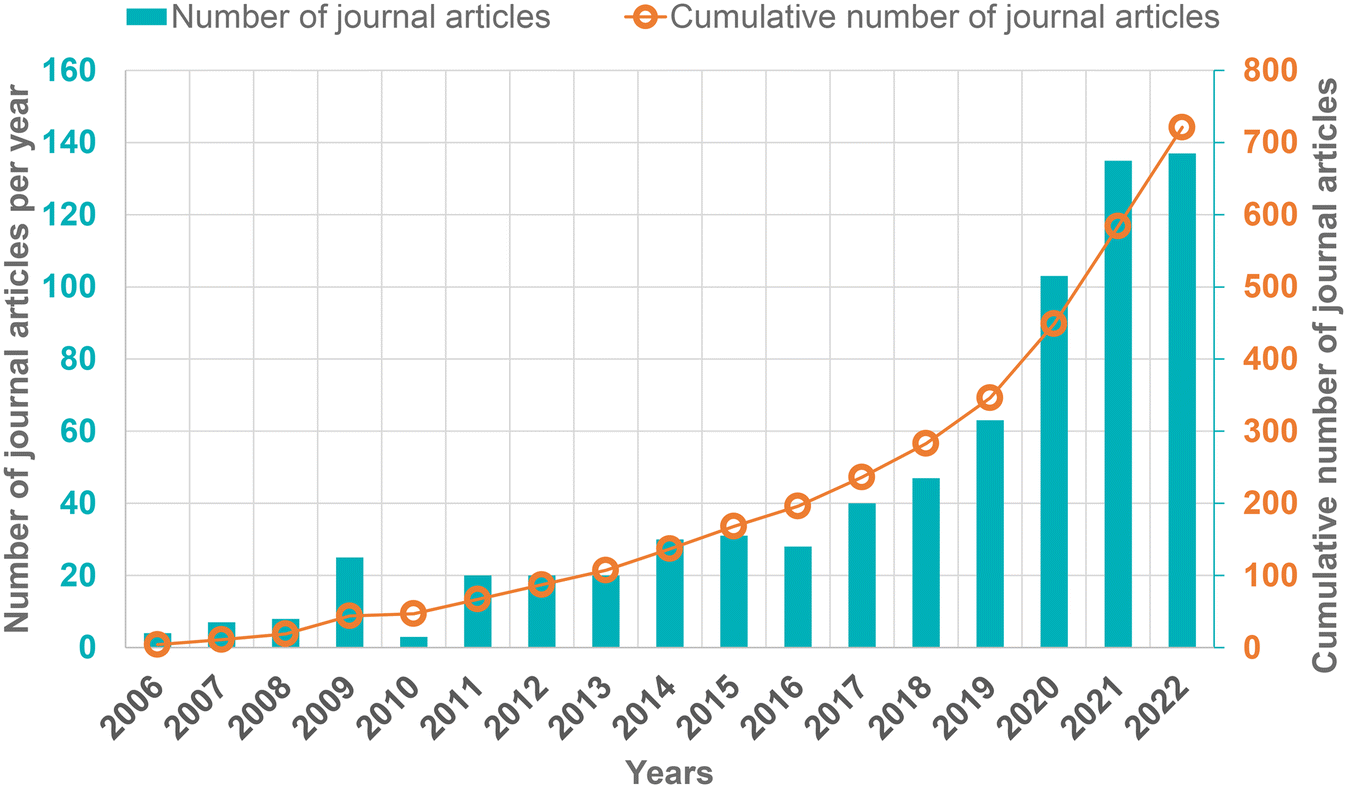 | ||
| Fig. 2 Number of journal articles published from 2006 to 2022 in the area of DAC (data collected from Scopus using the keywords for search: ‘DAC’, ‘direct air capture’, ‘direct air CO2 capture’ and ‘CO2 capture from air’). | ||
A general representation of amine-based adsorbents for direct air CO2 capture is illustrated in Fig. 3, where the amine-based adsorbents capture CO2 from the air at ambient conditions. Subsequently, after CO2 saturation, the sorbents are evacuated at a pressure in the range of 20–400 mbar and heated at a low temperature of 80–130 °C to desorb CO2.36,37 Further, the cost of CO2 capture using amine-based sorbents would significantly reduce by regenerating the sorbent by utilizing waste heat or renewable energy sources. For instance, Fasihi et al.35 estimated that the cost of a low-temperature TSA-based DAC (LT DAC) process using amine-based sorbent would reduce to 133 € per tCO2 in 2020, 60 € per tCO2 in 2030, 40 € per tCO2 in 2040 and 32 € per tCO2 in 2050, if powered by hybrid PV–wind–battery systems for Moroccan conditions or using waste heat.
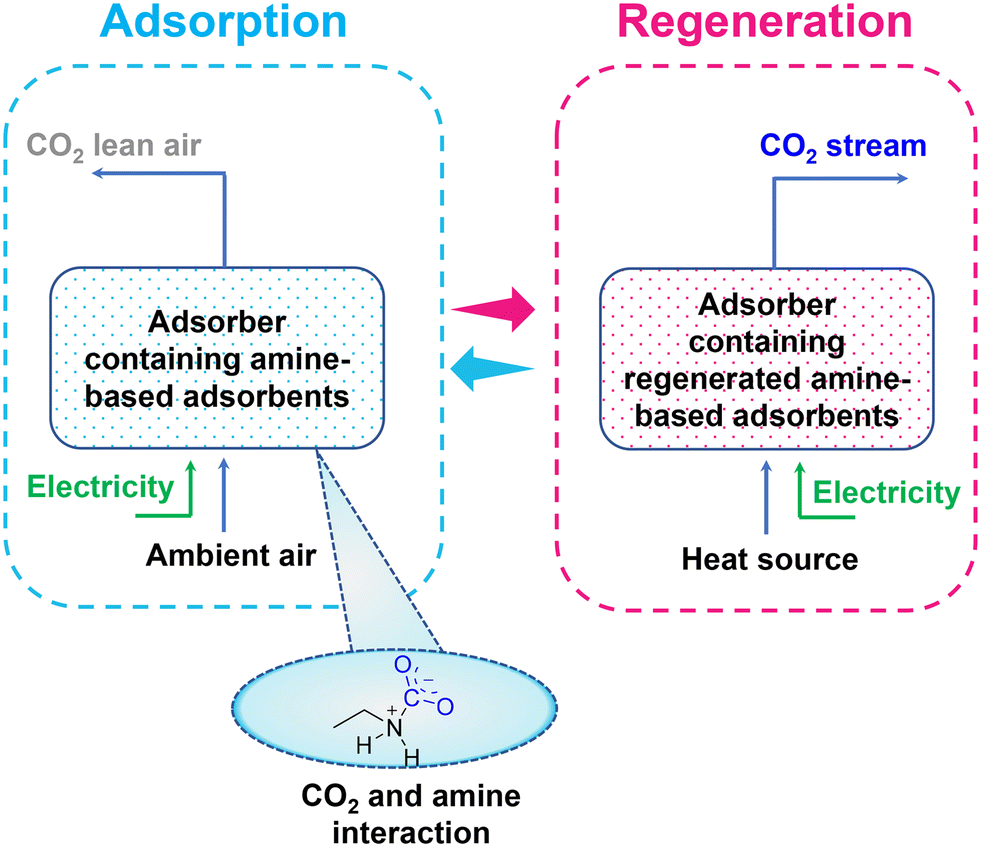 | ||
| Fig. 3 Schematic representation of amine-based adsorbent for direct air CO2 capture. | ||
Amine-based solid adsorbents, consisting of a mixture of amine and porous solid support, exhibit better CO2 capture performance due to the mutual benefit of CO2 chemisorption by amines and CO2 physisorption by the remaining solid support. In this regard, physisorbents such as zeolites,38 silica,39–41 and MOFs,42,43 are typically used as solid support to produce amine-based solid adsorbents. The interaction mechanism of CO2 with amine (especially MEA) is well explored in the literature,44–46 for instance by Caplow et al.47 and Danckwerts et al.48 Typically, reaction mechanism for CO2 adsorption over amine based adsorbents can be categorized into three types (as shown in eqn (1)–(6)).44–46
Zwitterion mechanism (type I): in the zwitterion mechanism, amine reacts with CO2 to form a zwitterion (eqn (1)) in the first step,47–49 and then the zwitterion is deprotonated by a free base (such as amine, OH−, or H2O) to form carbamate (in dry condition),50 and/or bicarbonate (under humid condition).51
| CO2 + RNH2 ⇌ RNH2+COO− (Ea = ∼40–50 kcal mol−1) | (1) |
| RNH2+COO− + RNH2 ⇌ RNHCOO− + RNH3+ (Ea = ∼13.2 kcal mol−1) (anhydrous condition) | (2) |
| CO2 + RNH2 + H2O ⇌ RNH3+ + HCO3− (Ea = ∼21 kcal mol−1) (hydrous condition) | (3) |
Single-step mechanism (type II): in the single-step termolecular mechanism, proposed by Da Silva and Svendsen,52 amine reacts with CO2 and a base simultaneously without the formation of any stable isolable zwitterion species. In this mechanism, instead of amine, water molecules act as a proton acceptors.49,53
| CO2 + RNH2⋯B ⇌ RNHCOO−⋯BH+ | (4) |
Carbamic acid mechanism (type III): carbamic acid mechanism also follows a two-step reaction pathway, wherein, initially amine reacts with CO2 to form carbamic acid, and then carbamic acid reacts (proton transfer) with another amine to form carbamate.54,55
| CO2 + RNH2 ⇌ RNHCOOH | (5) |
| RNHCOOH + RNH2 ⇌ RNHCOO− + RNH3+ | (6) |
Among these three mechanisms, the zwitterion mechanism is mostly referred to in research articles44 while addressing the CO2 capture by amines whether in solutions or solid adsorbents under dry or humid conditions.51 Similar to liquid amine, species such as carbamic acid, alkylammonium carbamate, and/or bicarbonate are also identified during CO2 adsorption over amine-based adsorbent, depending upon the amine loading and adsorption conditions56 Kortunov et al.57 reported that over the adsorbent surface, the nucleophilic attack of amine (Lewis base) on CO2 carbon resulted in the formation of zwitterion RH2N+–COO− (referred as 1,3-zwitterion, where 1 and 3 indicate the positive and negative centers, respectively). Further, according to the working conditions, 1,3-zwitterion may transform to ammonium carbamate via intermolecular proton transfer with amine (Brønsted base) or afford carbamic acid via intramolecular hydrogen transfer. Notably, under anhydrous conditions, two primary amines reacts with CO2 to yield ammonium carbamate.58 For the reaction takes place in the presence of water; water acts as a nucleophile, reacting with electrophilic CO2 to form carbonic acid57,59 and which further produce carbonate/bicarbonate by reacting with amine.57,60 In addition, water can hydrate carbamate anion to form carbonate or bicarbonate by releasing the amine molecule for further reaction with CO2.57,61 Hence, the maximum theoretical amine efficiency (molar CO2/N ratio) is 0.5 for anhydrous and 1.0 for hydrous conditions.62 Primary and secondary amines can react with CO2 in both dry and humid conditions, whereas, tertiary amine reacts with CO2 only under humid conditions.63 Further, the formation of carbamic acid or carbamate depends upon amine loading (or amine surface density),58,64 when there is a large amine loading on the substrate, alkylammonium carbamate predominates,64–67 while carbamic acid or hydronium carbamate is favored under low amine loading.58,64,66 Under the humid condition, water can also assist the nucleophilic attack of CO2 with the amine to generate hydronium carbamate, or it can act as a nucleophile that amines assist in producing ammonium bicarbonate at expense of carbamate.51 For highly hindered amine or tertiary amine, water becomes a more competitive Lewis base, resulting in the production of ammonium bicarbonate in comparison to carbamate.51
Various probing techniques such as Fourier transform infrared (FTIR),65,66,68–7013C solid-state NMR,64,67,71–7315N NMR,74,75 and X-ray diffraction76 are used to characterize the species form during CO2 adsorption on amine-based adsorbents. However, the mechanisms of CO2 interaction with amines, present in solution form or inside the adsorbent are still unclear. For instance, the zwitterion intermediates have been repeatedly reported in literature but experimentally not detected.45,51 Various researchers have tried to elucidate the amine–CO2 plausible reaction mechanism in terms of the energy barrier of 1,3-zwitterions. The activation energy barrier comes in the order of 40–50 kcal mol−1 for a 1:1 reaction between CO2 and amines.54,66,69 For instance Said et al.51 reported the energy barrier (Gibbs activation energy resp.) Ea (ΔrG* resp.) of 1,3-zwitterions to be 42.8 (44.8 resp.), and 40.0 (41.0 resp.) kcal mol−1 for CO2 interaction with primary and secondary amine (gaseous stage) respectively through a four-membered mechanism (Fig. 4). Activation barrier reduces further for the interaction of CO2 and amine diluted with a suitable solvent such as CCl4 (41.8 versus 44.8 kcal mol−1 in the gas state) or water (31.3 kcal mol−1). On the other hand, when CO2 reacts with a diamine such as N-methylmethanamine in the gaseous state, catalytic assistance of a Brønsted base facilitates the reaction by decreasing the energy barrier (26.5 kcal mol−1). Similarly, activation barrier of 21.0 kcal mol−1 is associated with the formation of bicarbonate and carbonate by the reaction of CO2 and water in the presence of aqueous amine. Hence, Said et al.51 proposed that the formation of 1,3-zwitterions may not prefer a four-member mechanism due to the high activation barrier (>30 kcal mol−1) rather may follow a unified six-membered mechanism. The six-membered mechanism involves the nucleophilic attack of CO2 by a Lewis base (amines or water) with hydrogen transfer or exchange assisted by a Brønsted base (amines or water). The plausible CO2–amine interaction mechanism and associated activation energy, and possible reaction mechanism for CO2–amine–water interactions are shown in Fig. 4.51
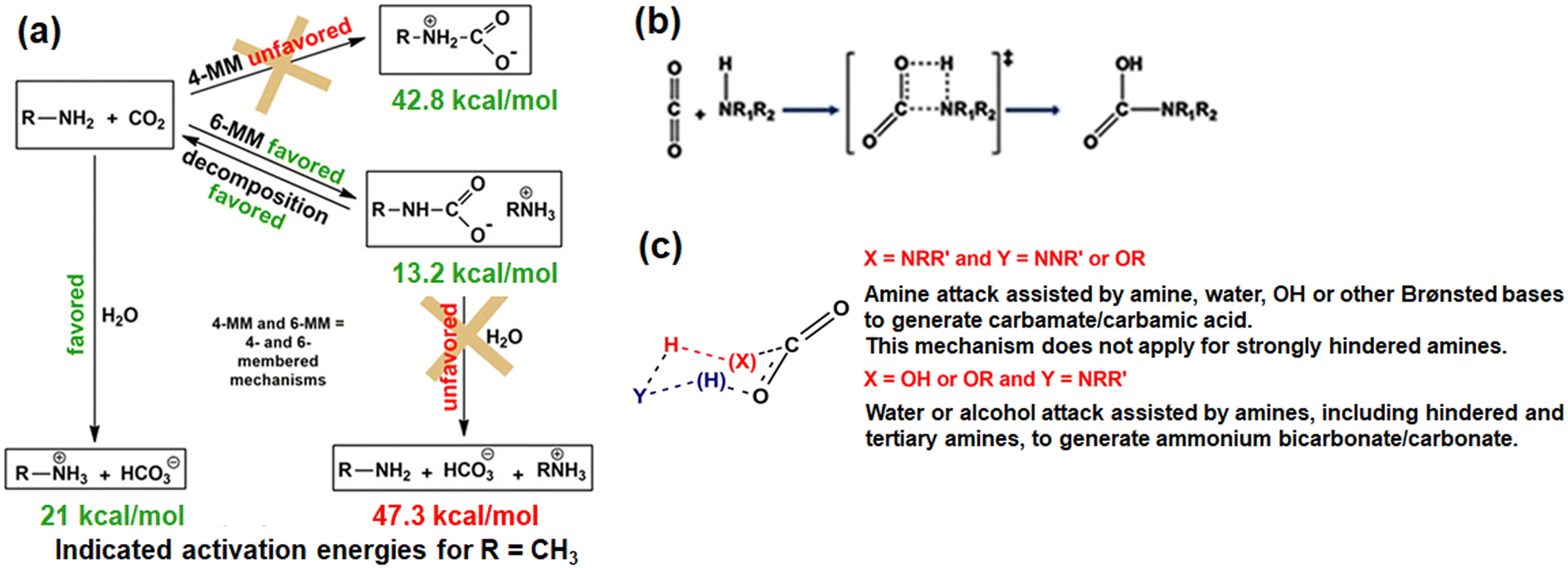 | ||
| Fig. 4 (a) Illustrative CO2–amine interaction pathways, involving (b) four-membered and (c) six-membered transition states. Reproduced with permission.51 Copyright 2020 American Chemical Society. | ||
Although CO2 adsorption on amine-based adsorbents is a well-established technique with extensive research, particularly on capturing high-concentration CO2 from point sources such as flue gas and the cement industry, the material design for the DAC application, where CO2 concentration is very low (0.04%), is still trivial. One needs to be more careful while choosing the best adsorbent and the evaluation parameters which will basically decide the overall CO2 capture performance. Readers can find some interesting reviews on material development and techno-economic analysis of multiple techniques used for direct air capture.15,16,26,32,77–80 However, only limited focused work appeared on exploring the evaluation parameters for amine-based adsorbents in DAC applications.16 This review addresses the recent developments in amine-based porous solid sorbents for CO2 capture, adsorbent preparation, and characterization, evaluation parameters such as CO2 capture under dry and humid conditions, CO2 adsorption kinetics, adsorption thermodynamics, sorbent regeneration, cyclic stability, essential techniques and economic analysis for CO2 capture from air.
2 Synthesis of amine-based solid adsorbent for direct air CO2 capture
DAC via amine-based adsorbent has gained significant interest nowadays due to their high CO2 adsorption performance at ultra-dilute CO2 concentrations and even in the presence of moisture.30,39,41,81,82 However, the performance of amine-based adsorbent predominantly depends upon its synthetic procedure and process parameters. The amine-based adsorbents used for DAC are typically synthesized in four ways: (i) physical impregnation (class I adsorbents), (ii) chemical grafting (class II adsorbents), (iii) in situ polymerization (class III adsorbents) and (iv) combination of both chemical grafting and physical impregnation (class IV adsorbents)16 as displayed in Fig. 5.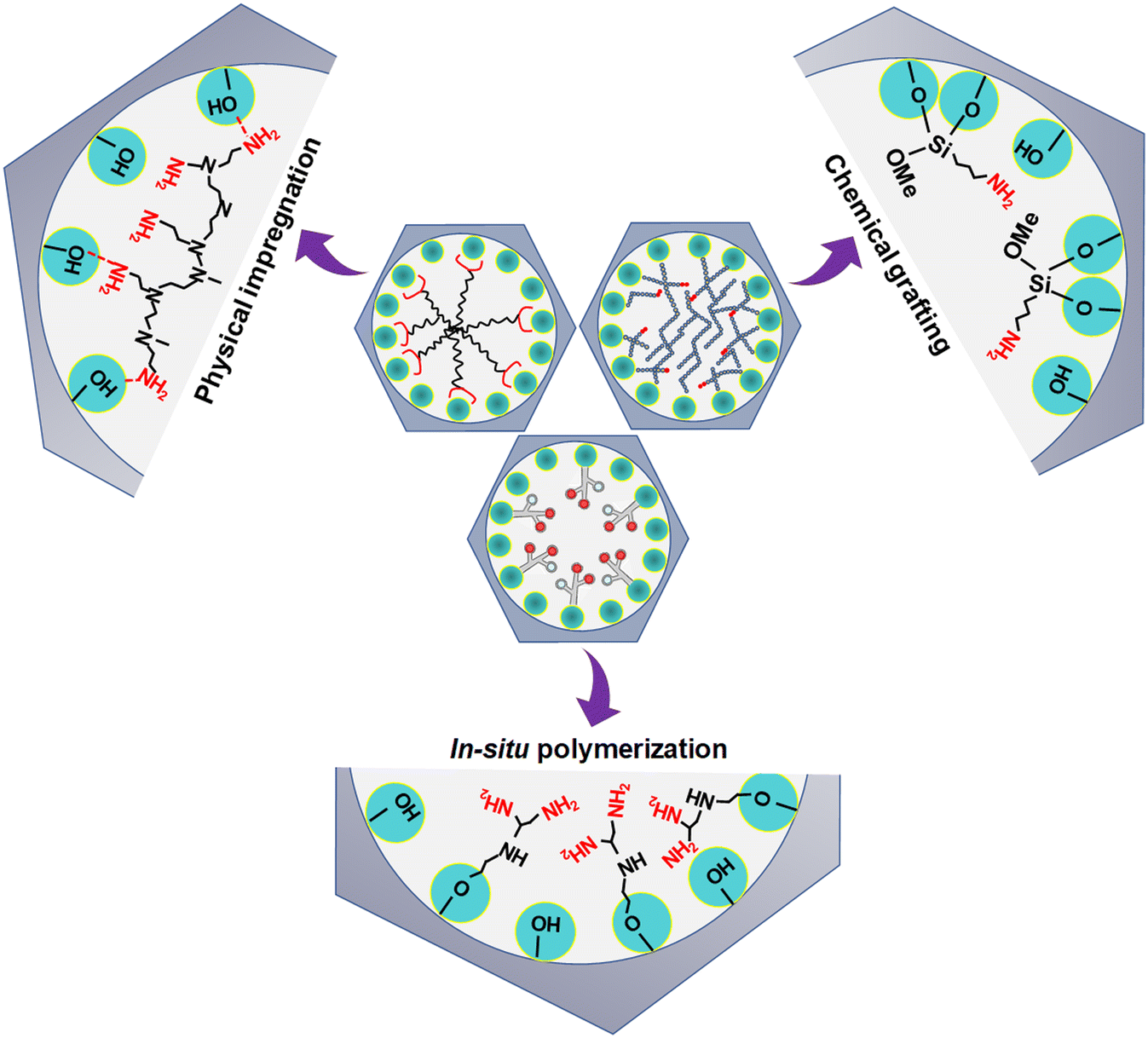 | ||
| Fig. 5 Schematic representation of amine-based porous solid adsorbent. Adapted with permission from ref. 100. Copyright 2015 American Chemical Society. | ||
Typically, in the physical impregnation method, the selected amine is first added to a solvent (such as water or any organic solvents e.g., methanol or ethanol) and uniformly dispersed by mechanical or magnetic stirring. Later, the desired quantity of porous solid support is added to the amine solution, followed by continuous stirring (for a specific duration) to disperse amine into the pores of the support. The resultant solution is finally filtered and the solvent is removed under pressure to obtain the amine-based solid adsorbent, where amines are expected not to have any direct chemical bonding with the support.83 The loading of amine inside the pores of the support depends upon the available pore volume of the support and the amine density. The amine viscosity greatly influences its dispersion in the porous support system and the resulting adsorbent's degree of saturation. Further, the pore size and morphology restrict the molecular and kinetic diffusion of an amine into the pores of the support.39,84 Mesopore and macropore volume (larger voids) are majorly responsible for containing the amine inside the sorbent pores/voids.39 The voids present inside the solid support may contain amine; however, the residual amine species will agglomerate on the support surface once the percolation limit has been reached. The benefit of the physical impregnation method lies in its simple and facile synthesis process and the ability to accommodate large quantities of amine in the adsorbent having a high pore volume. However, low amine stability due to amine degradation and amine evaporation are the significant drawbacks that impede the adsorbent application in DAC. Amine-impregnated adsorbents are also called class 1 adsorbents16 and among them small amine molecules, such as tetraethylenepentamine (TEPA),82,85–87 or amino polymers (e.g. polyethylenimine, PEI)39,88–90 within robust porous support largely used for CO2 capture from air. In the chemical grafting method, amine molecules containing the silane group are grafted on the surface of porous solid support via covalent bonding. Grafting occurs through a condensation reaction between the aminosilane species (such as 3-aminopropyltriethoxysilane, APTES) and the hydroxyl group present on the support surface. In general, the synthesis involves mixing the desired amount of porous solid support in a solvent (in general anhydrous toluene) and aminosilane, followed by vigorous stirring and reflux.9 The resulting solution is eventually filtered after completion of the reaction and repeatedly washed to remove any unreacted amine molecules from the sorbent.83 Typically, amine-grafted adsorbents are called class 2 adsorbents, which are more resilient than class 1 adsorbents. However, their CO2 uptake capacity is limited as the surface becomes saturated with aminosilane species.91 Combining the advantages of impregnation and grafting, a new class of amine-based adsorbents is explored, known as class 3 amine-based adsorbents.16 Class 3 adsorbents are fabricated by in situ ring-opening polymerization of amine-containing monomers such as aziridine in the pores of support; thus, the resulting aminopolymers are covalently bound to the support, leading to the high stability feature of these materials.92 However, the rapid reaction rate of aziridine polymerization makes it challenging to control the polymer formed, and the toxic nature of aziridine makes this approach unappealing on the laboratory scale. Hyperbranched aminosilicas93–95 and melamine dendrimer-mesoporous silica hybrids,96 polysilsesquioxanes97,98 are a few class 3 adsorbents widely explored in literature for CO2 capture application. Another type of amine-based adsorbent is called class 4 adsorbent.99 They are synthesized by combining both grafting and impregnation techniques, typically known as double-functionalized methods, wherein previously grafted amine-based sorbents are impregnated with aminosilanes to increase their adsorption sites by occulting more amine species to them. However, very limited work has been carried out on class 4 adsorbents for DAC application compared to flue gas CO2 capture.99
3 Evaluation parameters and CO2 adsorption performance of amine-based solid adsorbent under DAC condition
The selection of suitable adsorbent properties for DAC is cumbersome, as several operational parameters affect the CO2 capture performance of a sorbent while working in practical environmental conditions. The following sections describe in detail the technical and economic factors affecting the performance of the adsorbent for CO2 capture from the air.3.1 CO2 adsorption performance of amine-based solid adsorbent for DAC
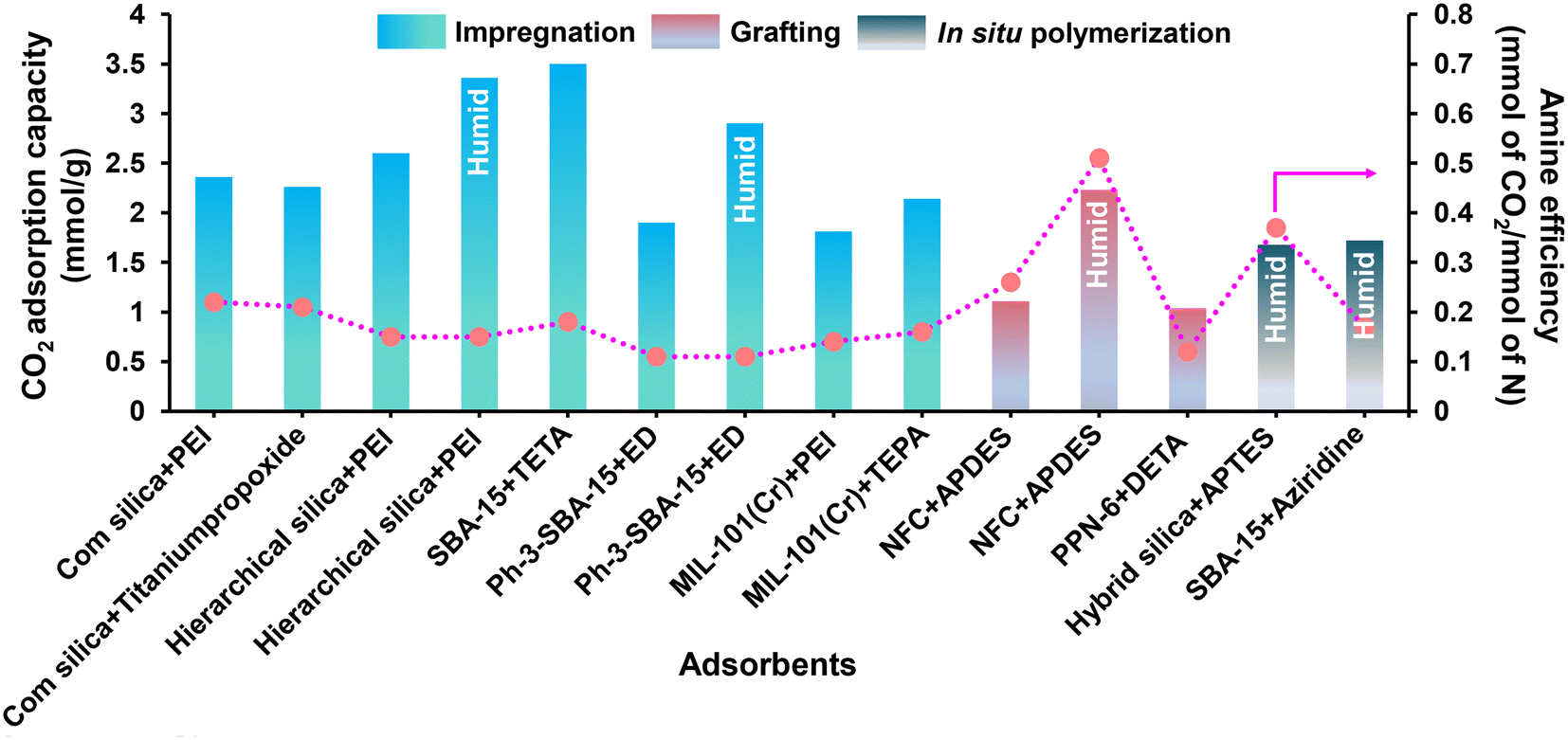
000 Da into sorbents maximizes the effective content of amine in the adsorbent.81 Notably, the literature reported that adsorbents with branched PEI (Mw of 800 Da) achieved the highest CO2 adsorption capacity (Table 1).81,88,39 Further, the vacuum level during the drying step is vital to efficiently migrate the amine into the pore network and create favorable amino polymer distributions. Kwon et al.39 reported that a mild vacuum (33.33 mbar) would be better for superior amine loading and amine migration into the pore network. It was seen that the amine efficiency increases with amine content as more free amines are added to the system; however, the amine efficiency will reach a maximum, typically at a loading of 1.6 g PEI per g SIO2 (corresponds to ∼40 nm aminopolymer film thickness), as the pores eventually become occluded and CO2 diffusion is hindered. Therefore, a delicate balance between the presence of adsorption sites and amine loading is created to maximize the CO2 uptake capacity with faster kinetics.104 To address the thermal stability issue of PEI-modified adsorbents, Choi et al.88 studied a 3-aminopropyltrimethoxysilane (APTES) modified PEI silica sorbent (A-PEI) and titanium(IV) propoxide, which enhanced the kinetics, CO2 uptake capacity (>2 mmol g−1) along with the thermal stability as compared to the pristine PEI modified adsorbents (CO2 capacity: 1–2 mmol g−1). Modification of the chemical structure of PEI by Ti additives differs in its accessibility or mass transport through the pores. Further, the mesoporous framework is also an important aspect influencing the performance of amine-modified adsorbents for CO2 capture at very low pressures. Chen et al.105 reported a PEI-impregnated resin (HP20/PEI-50) with an excellent CO2 uptake capacity of 2.26 mmol g−1 and almost stable amine efficiency of ∼0.19–0.20 mmol of CO2 per mmol of N in the PEI content range of 40–50% under DAC conditions. The presence of mesopores (43–68 nm) in the adsorbent facilitated the high amine loading (up to 50 wt%), resulting into enhanced CO2 diffusion and increased CO2 adsorption capacity. Similarly, Wang et al.106 reported a PEI impregnated mesoporous carbon (MC) adsorbent, where the MC framework can accommodate high PEI content owing to the large mesoporous volume and area. The interconnecting channels also serve as a rapid CO2 route, boosting the sorption/desorption kinetics. In addition, they also enhanced the kinetic inhibition of CO2 internal diffusion within the PEI films by adding polymer-based surfactant such as sorbitan monooleate (Span 80). This transforms the PEI film from a homogenous solid block to a polymer network, resulting in high CO2 diffusion rates. Sehaqui et al.107 reported a highly porous bio-based sorbent composed of oxidised nanofibrillated cellulose (NFC) and 44 wt% polyethylenimine (PEI) synthesized by freeze-drying method to achieve a CO2 capacity of 2.22 mmol g−1 with stability over five cycles, and exceptionally low adsorption half time of 10.6 min under atmospheric conditions (moist air with ∼400 ppm of CO2) and at 80% RH. Further, temperature and pressure/concentration also have a greater role in CO2 adsorption capacity and amine mobility inside the porous adsorbents. Pang et al.108 explored the mechanism of CO2 adsorption in amine-based adsorbents by comparing the isotherm at various temperatures and pressures in a dry environment. The group reported a transition of CO2 sorption isotherm from a transport-limited regime to a thermodynamically limited one. In the case of linear poly(propylenimine) based SBA-15 sorbents, at low partial pressures relevant to direct air capture (PCO2 = 0.3 Torr), the CO2 uptake capacity and amine efficiency decreased with the increase in adsorption temperature, in accordance with a thermodynamically limited exothermic adsorption process. However, as the pressure increases (∼1 bar), the CO2 uptake capacity and amine efficiency of PEI/SBA-15 increased with increasing temperature (cross-over), which may be attributed to the increased flexibility and mobility of the PEI molecules, thus helping greater penetration of CO2 into the amine sites. Furthermore, when the temperature rises, the films produced by the crosslink between CO2 and PEI (formation of ammonium carbamate ion pairs) become less dominating over CO2 penetration, resulting in increased capacity.109 In the similar context, recently Miao et al.86 reported the favourable desorption temperature for PEI and TEPA based SBA-15 adsorbent for DAC application. They found that, a temperature interval of 90–110 °C can be considered as the optimal desorption temperature of PEI/SBA-15 and 90 °C for TEPA/SBA-15 composite (50% amine loading), which could help to minimise amine volatilization. Notably, TEPA/SBA-15 composite exhibited better desorption kinetics than PEI/SBA-15 at the same degassing conditions, evidenced its suitability for DAC.
| Support | Amine type | Temp (°C) | CO2 concentration (ppm) | Amine loading (mmol of N per g of the adsorbent) | CO2 adsorption capacity (mmol g−1) | Amine efficiency (mmol of CO2 per mmol of N) | Method | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Dry CO2 | Humid CO2 | ||||||||
|
a 67% relative humidity.
b 19% relative humidity.
c
ca. 0.9–1.4 vol% relative humidity.
d 50% relative humidity.
e 30% relative humidity.
f 85% relative humidity.
g 80% relative humidity.
h 32% relative humidity, TGA, thermogravimetric analysis; IR, infrared analyzer; TPD, temperature-programmed desorption; GC, gas chromatography, and vol., volumetric. Com silica, commercial silica; APTES, (3-aminopropyl)triethoxysilane; PEI, branched polyethylenimine (MW = 800 Da); PEI-A, branched polyethylenimine (MW = 600–1000 Da); PEI-M, branched PEI (MW = 1800 Da); PEI-U, branched polyethylenimine (MW = 2000 Da); PEI-H, branched PEI (MW = 25000 Da); PEI-ln, linear PEI (Mn = 423 Da); PEI-lnH, linear PEI (MW = 2500 Da); TEPA, tetraethylenepentamine; PAA, poly(allylamine); PEG, poly(ethylene glycol) (Mw ∼ 200); ED, ethylenediamine; PPI, linear poly(propylenimine); PEG poly(ethylene glycol); PGA, poly(glycidyl) amine, (Mn = 50); PEHA, pentaethylenehexamine; PO, propylene oxide; BO, α-butylene oxide; TETA, triethylenetetramine; CTMA+, cetyltrimethylammonium cations; NOHM-I-PEI, polyethylenimine tethered silica nanoparticles with ionic bond; SIP, solvent impregnated polymers; TTE, triglycidyl trimethylolpropane ether; CA, cellulose acetate. |
|||||||||
| Com silica | PEI | 25 | 400 | 10.5 | 2.36 | — | 0.22 | TGA | 88 |
| Com silica | PEI + APTES | 25 | 400 | 10.7 | 2.26 | — | 0.21 | TGA | 88 |
| Com silica | PEI + titanium(IV) propoxide | 25 | 400 | 10.5 | 2.19 | — | 0.21 | TGA | 88 |
| Com silica | TEPA | 35 | 400 | 10.1 | 2.50 | 0.25 | TGA | 87 | |
| Fumed silica | PEI-H | 25 | 420 | 33 wt% | 1.18 | 1.77a | — | IR | 123 |
| Fumed silica | PEI-H | 25 | 420 | 50 wt% | 1.71 | 1.41a | — | IR | 123 |
| Fumed silica | PEI-ln | 25 | 400 | 50 wt% | 2.34 | — | — | IR | 81 |
| Fumed silica | PEI | 25 | 400 | 50 wt% | 2.44 | — | — | IR | 81 |
| Fumed silica | PEI-M | 25 | 400 | 50 wt% | 1.69 | — | — | IR | 81 |
| Fumed silica | PEI-H | 25 | 400 | 50 wt% | 1.67 | — | — | IR | 81 |
| Hierarchical silica | PEI | 30 | 400 | 2.62 g g−1 | 2.6 | 3.36b | 0.15 | TGA | 39 |
| Mesocellular silica foam | PAA | 25 | 400 | 7.24 | 0.86 | — | 0.12 | TGA | 89 |
| Mesocellular silica foam | PEI | 25 | 400 | 10.7 | 1.74 | — | 0.16 | TGA | 89 |
| Mesocellular silica foam | PEI-lnH | 25 | 400 | 11.4 | 1.05 | — | 0.09 | TGA | 89 |
| Mesoporous silica | PEI-M | 20 | 393.5 | 40 wt% | — | 1.66c | — | IR | 124 |
| γ-Alumina | PEI | 25 | 400 | 11.2 | 1.74 | 0.16 | TGA | 125 | |
| γ-Alumina | PEI | 30 | 400 | 7.95 | — | 1.96d | 0.25 | IR | 126 |
| PME/CTMA+ | PEI | 25 | 400 | 40 wt% | 2.18 | 2.92 | — | TGA | 90 |
| SBA-15 | PEI | 25 | 400 | 9.23 | 1.05 | — | 0.11 | TGA | 125 |
| SBA-15 | PEI-ln | 75 | 400 | 50 wt% | 0.51 | — | — | TPD | 127 |
| SBA-15 | PEI + PEG | 30 | 400 | 5.75 | 0.79 | — | 0.14 | TGA | 128 |
| SBA-15 | PPI | 35 | 400 | 13.8 | 1.25 | — | 0.16 | TGA and vol. | 108 |
| SBA-15 | PEI | 25 | 400 | 75 wt% | 1.90 | — | — | TGA | 86 |
| SBA-15 | TEPA | 25 | 400 | 75 wt% | 3.44 | — | — | TGA | 86 |
| SBA-15 | PPI | 35 | 400 | 8.17 | 0.31 | — | 0.04 | TGA | 129 |
| SBA-15 | PGA | 35 | 400 | 10.2 | 0.6 | — | 0.11 | TGA and vol. | 109 |
| SBA-15 | PEI | 30 | 400 | 30 wt% | 0.65 | — | 0.10 | TGA | 130 |
| SBA-15 | PPI (linear) | 30 | 400 | 14 | 1.85 | — | 0.13 | TGA | 131 |
| SBA-15 | TETA | 35 | 400 | 19 | 3.50 | — | 0.18 | TGA | 132 |
| Zr-SBA-15 | PEI | 25 | 400 | 8.3 | 0.85 | 0.10 | TGA | 133, 134 | |
| Ph-3-SBA-15 | ED | 35 | 400 | 16.7 | 1.9 | 2.9e | 0.11 | TGA | 41 |
| CA-SiO2 | PEI | 35 | 395 | 16.1 | 0.59 | 1.60f | — | TGA | 101 |
| Sipernat 50S | PEHA-PO-1-2 | 25 | 400 | — | 1.25 | — | — | TGA | 40 |
| Sipernat 50S | PEHA-BO-1-2 | 25 | 400 | — | 1.11 | — | — | TGA | 40 |
| Sipernat 50S | TEPA-PO-1-2 | 25 | 400 | — | 1.34 | — | — | TGA | 40 |
| Resin HP20 | PEI | 25 | 400 | 50 wt% | 2.26 | — | — | — | 105 |
| Mesoporous carbon | PEI | 25 | 400 | 55 wt% | 2.25 | 2.58g | — | GC | 106 |
| MIL-101(Cr) | PEI | 25 | 400 | 1.76 | 1.25 | 1.26h | 0.10 | TGA | 135 |
| MIL-101(Cr) | PEI | 25 | 400 | 50 wt% | 1.81 | — | 0.14 | vol. | 43 |
| MIL-101(Cr) | TEPA | 25 | 400 | 50 wt% | 2.14 | — | 0.16 | vol. | 43 |
| TTE | PEI-H | 20 | 400 | 30 wt% | — | 1.14 | — | Isotopic analyzer | 136 |
| Nanofibrillated cellulose (NFC) | PEI-A | 25 | 400 | 44 wt% | — | 2.22 | 0.22 | IR | 107 |
| NOHM-I-PEI in SIP film | PEI-U | 25 | 400 | 50 wt% | 1.05 | 1.66 | — | TGA & IR | 110 |
Several next-generation encapsulation techniques, such as micro-encapsulated carbon sorbents (MECS) and solvent-impregnated polymers (SIPs), have been recently explored to develop hybrid CO2 capture sorbents that combine the benefits of solid sorbents and liquid solvents. The kinetics of CO2 adsorption in a liquid solvent is usually slow. As a result, the liquid solvent is kept inside the polymer matrix, which has a high CO2 permeability, allowing for a synergistic effect on CO2 sorption behaviors, increased mass transfer, and rapid CO2 capture. The advantage of SIPs over MECS is that it does not require a microfluidic device to create sorbent particles, making them more straightforward and having a better potential for scale-up. Recently, Rim et al.110 synthesized liquid-like polyethylenimine tethered silica nanoparticles with an ionic bond (NOHM-I-PEI), incorporated into a shell material and UV-cured to yield gas-permeable solid sorbents with uniform NOHMs loading (NPEI-SIPs) as shown in Fig. 6. NPEI-SIPs had a CO2 adsorption capacity of 1.05 mmol g−1 at 400 ppm, 25 °C at 50% PEI loading, with rapid CO2 adsorption kinetics (4.16 μmol g−1 min−1) and claimed to be a well-suited system for DAC application.
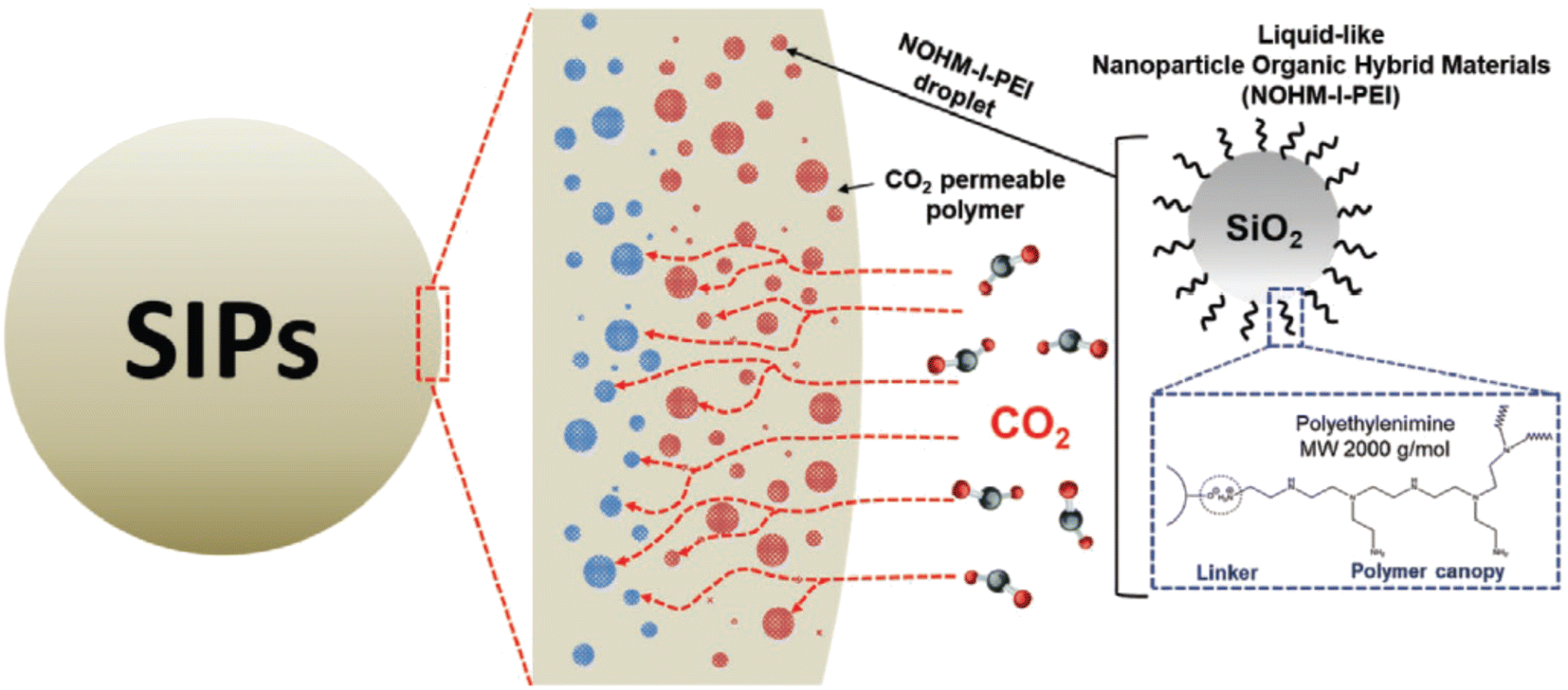 | ||
| Fig. 6 Structure of liquid-like nanoparticle organic hybrid materials with ionically tethered polyethylenimine (NOHM-I-PEI) and the schematic of microdroplets of NOHM-I-PEI in solvent impregnated polymers (SIPs). Reproduced with permission.110 Copyright 2021, Wiley-VCH GmbH. | ||
Recently, amine-appended MOFs have been explored to show remarkable performance in DAC owing to their extraordinary surface area, giant pore volume, and tunable pore architecture, allowing amines to immobilize into the pore wall and interact with the CO2 at ultra-dilute concentration. For instance, Lee et al.115 prepared an amine-appended metal–organic framework (en-Mg2(dobpdc)) (where H4-dobpdc = 4,4′-dihydroxy-(1,1′-biphenyl)-3,3′-dicarboxylic acid) functionalized with en (ethylene diamine). This material exhibited a superior CO2 adsorption capacity of 2.83 mmol g−1 from ambient air (pCO2 = 0.4 mbar) owing to carbamic acid (not a carbamate) formation by the chemical reaction associated with the free amine group and CO2. However, the CO2 sorbent shown by Lee et al. could not achieve saturation at 400 ppm CO2 in air, which was attributed to strong intramolecular interactions between the neighboring amine groups, which can be surpassed at higher pressure CO2.116,117 As a consequence, the CO2 uptake of en-Mg2(dobpdc) drops sharply to 0.120 mmol g−1 at a somewhat higher temperature of 50 °C at 0.4 mbar. To overcome such a problem, Liao et al.118 substituted the ‘ethylenediamine (en)’ molecule with an even shorter diamine named ‘hydrazine (N2H4)’; where with an overall amine loading of 6.01 mmol of N per g of the adsorbent, a very high CO2 adsorption capacity of 3.89 mmol g−1 amongst other MOFs was achieved (Table 2). In addition, Gebald et al.119 reported a CO2 adsorption capacity of 1.11 (dry CO2), and 2.13 (humid CO2, 91% relative humidity) over APDES–NFC (APDES: 3-aminopropylmethyldiethoxysilane, NFC: nano fibrillated cellulose) at 0.4 mbar. The remarkable CO2 adsorption performance of APDES–NFC was presumably attributed firstly to the interaction of mesopores of APDES–NFC with CO2via physisorption and hence increased amine efficiency (0.54 mmol of CO2 per mmol of N) is achieved. Secondly, the aminosilane dimer molecule for each hydroxyl group at the cellulose surface and unreacted silanol groups presumably catalyzed the interaction of CO2 molecules with the amine groups, enhancing the amine efficiency beyond the theoretical limit of carbamate formation. Lu et al.120 examined the impact of amine-grafted PPN-6-CH2DETA (PPN: porous polymer networks; DETA: diethylenetriamine) on CO2 capture from the air. High CO2 selectivity and uptake capacity (1.04 mmol g−1) was observed due to the highly porous three-dimensional carbon scaffolds. Notably, tertiary amine DETA (diethylenetriamine) has a 6% higher CO2 loading capacity with low heat of adsorption (53.8 vs. 67.3 kJ mol−1) as compared to the amine-modified pore expanded silica (TRI-PE-MCM-41) at 400 ppm (0.98 mol kg−1).
| Support | Amine type | Temp. (°C) | CO2 concentration (ppm) | Amine loading (mmol of N per g of the adsorbent) | CO2 adsorption capacity (mmol g−1) | Amine efficiency (mmol of CO2 per mmol of N) | Method | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Dry CO2 | Humid CO2 | ||||||||
| Mesocellular silica foam | APTMS | 25 | 400 | 2.70 | 0.54 | — | 0.20 | TGA | 84 |
| Mesocellular silica foam | APTMS | 25 | 400 | 3.75 | 1.0 | — | 0.30 | TGA | 84 |
| Mesocellular silica foam | MAPS | 25 | 400 | 2.41 | 0.17 | — | 0.07 | TGA | 84 |
| O-Al2O3 | APTES | 30 | 400 | 5.43 | 0.76 | — | 0.14 | TGA | 91 |
| D-Al2O3 | APTES | 30 | 400 | 4.13 | 0.62 | — | 0.15 | TGA | 91 |
| Silica gel | AEATPMS | 25 | 400–440 | 2.48 | 0.40 | 0.44a | Dry: 0.16 | TGA | 112 |
| Humid: 0.18 | |||||||||
| Nano fibrillated cellulose | AEAPDMS | 25 | 506 | 4.9 | — | 1.39a | 0.28 | IR | 113 |
| Nano fibrillated cellulose | APDES | 23 | 400 | — | 1.11 | 2.13b | Dry: 0.26 | IR | 119 |
| Humid: 0.51 | |||||||||
| PPN-6 | DETA | 22 | 400 | 8.5 | 1.04 | 0.12 | vol. | 120 | |
| RFAS | APTES | 25 | 400 | 8.07 | 1.71c | 0.21 | IR | 137 | |
| SBA-15 | APTMS | 25 | 395 | — | 0.14 | — | TGA | 138 | |
| SBA-15 pellet | APTMS | 25 | 395 | — | 0.09 | 0.13d | — | TGA | 138 |
| SBA-15 | Alkyl halide + ammonia | 30 | 400 | 1.62 | 0.07 | — | 0.043 | TGA | 75 |
| SBA-15 | TEPA | 23.4 | 400 | 12.2 | 3.59e | — | TPD | 82 | |
| Pore-expanded MCM-41 | TRI | 30 | 400 | — | 0.64 | 0.55f | — | TGA | 103 |
| Pore-expanded MCM-41 | TRI | 25 | 400 | 7.9 | 0.98 | — | 0.12 | TGA | 33 |
| Mg2(dobpdc) | Ethylenediamine | 25 | 390 | — | 2.83 | — | — | vol. | 115 |
| Mg2(dobdc) | Hydrazine | 25 | 400 | 6.01 | 3.89 | — | — | TGA | 118 |
| Mg-MOF-74 | Ethylenediamine | 25 | 400 | 1.51 | — | — | TGA | 139 | |
| Mg2(dobpdc) | mmen | 25 | 390 | — | 2.0 | — | — | vol. | 42 |
| Cr-MIL-101-SO3H | TAEA | 20 | 400 | 1.12 | — | — | TGA | 140 | |
| In situ polymerization | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| a 40% humidity. b 91% relative humidity. c 4% relative humidity. d 80% relative humidity. e 49% relative humidity. f 73% relative humidity. g 60% relative humidity. h Fully humidified condition; TGA, thermogravimetric analysis; IR, infrared analyzer; TPD, temperature-programmed desorption; GC, gas chromatography, and vol., volumetric. APTES, (3-aminopropyl)triethoxysilane; MAPS, N-methylaminopropyl-trimethoxysilane; AEAPDMS, [N-(2-aminoethyl)-3-aminopropylmethyl]dimethoxysilane; APDES, 3-aminopropylmethyldiethoxysilane; APTMS, (3-aminopropyl)trimethoxysilane; AEATPMS, [N-(2-aminoethyl)-3-aminopropyl]trimethoxysilane; DETA, diethylenetriamine; mmen, N,N′-dimethylethylenediamine; TAEA, tris(2-aminoethyl)amine; TRI, 2-[2-(3-trimethoxysilylpropylamino)ethylamino]ethylamine, AHTSA, amine hybrid titania/silsesquioxane composite aerogel. | |||||||||
| SBA-15 | Z-L-Lysine + APTMS | 25 | 400 | 5.18 | 0.60 | — | 0.12 | TGA | 92 |
| Hybrid silica | APTES | 30 | 400 | 4.5 | — | 1.68g | 0.37 | TGA | 111 |
| SBA-15 | Aziridine | 25 | 400 | 9.9 | — | 1.72h | 0.17 | MS | 95 |
| AHTSA | APTES | 30 | 400 | 8.47 | 1.64 | — | 0.19 | IR | 114 |
| Macroporous silica | L-Alanine | 50 | 400 | 10.98 | 2.65 | — | 0.24 | TGA | 141 |
3.2 CO2 adsorption kinetics of amine-based solid adsorbent for DAC
Adsorption kinetics is a time-dependent calculation of adsorption at constant pressure or concentration, which provides the necessary information about the adsorbate diffusion into the adsorbent's pores. Since CO2 concentration in air is very low (400 ppm), the CO2 adsorption kinetics (capture-release cycle) of the amine-based adsorbents is worth exploring. The adsorption kinetics also affects the working capacity of the adsorbent, and the working capacity can be the same as the equilibrium adsorption capacity of the sorbent possesses high adsorption kinetics.88 Despite the significant importance of adsorption kinetics, the number of publications reporting the kinetic data of CO2 during direct air capture is currently limited. Only a few articles describe the sorbent's kinetic behavior in terms of adsorption half time95,111,113,114,134,142 or the breakthrough time.143 Adsorption half time is the time to reach half of the pseudo-equilibrium adsorption capacity, typically calculated from pure component isotherm either by volumetric or gravimetric measurement.95 Compared to the flue gas environment (10–15% CO2), the adsorption kinetics of CO2 in DAC (0.04% CO2) typically take a longer time for amine-based adsorbents.39 This behavior is primarily affected by various factors such as CO2 diffusion to bulk materials (boundary layer diffusion), mass transfer in and out of the pores, structure and density of the grafted amines, accessibility to amine moieties, intrinsic chemical reaction rates, and others.39,94For instance, Kwon et al.39 compared the kinetics of conventional mesoporous material-supported amine sorbents (class 1, PEI-based sorbents) during CO2 adsorption from the simulated air and flue gases (400 ppm and 10% CO2) at a fixed temperature. At higher CO2 concentration, 2.6 g PEI per g sorbent exhibited 30–40% higher CO2 uptake at the initial stage but it lasted only for a few minutes, whereas the same material showed 70–80% of the total CO2 uptake capacities at a slow initial rate for 400 ppm CO2 concentration. The observed trend can be attributed to a higher driving force for adsorption under flue gas conditions causing an enhanced surface diffusion barrier during the initial stage of the adsorption process. In contrast, for 400 ppm CO2, a low driving force favoured diffusion of CO2 relatively deeper into the aminopolymer film; hence, adsorption occurred until saturation was achieved. For instance, PEI-H-SiO2 displayed a high uptake capacity of 2.6 mmol CO2 per g at 400 ppm dry CO2 at 50 °C.39 Though the temperature-induced adsorption capacity (kinetic limitation) was observed to be improved at 400 ppm CO2, it was not accompanied by increased adsorption kinetics as the sorption rates decreased with the increase in temperature, conversely to the case of the 10% CO2 conditions.
The CO2 adsorption kinetics greatly depends upon the synthesis methodology of amine loaded into the adsorbent. It has been seen that compared to amine-impregnated adsorbents, amine-grafted and in situ polymerized amine-based adsorbents exhibit better CO2 adsorption kinetics in DAC conditions except for NFC–PEI (made by a freeze-drying process) (Fig. 8, Table 3). For instance, Choi et al.95 reported the adsorption half time of various amine-modified hyperbranched aminosilica (HAS) adsorbents and found the CO2 adsorption capacity of amine-modified HAS adsorbents was increased to 1.72 mmol g−1 by increasing the amine loading to 9.9 mmol g−1 with an adsorption half-time of ∼167 min. Later, the same group also compared the adsorption kinetics of various amine-based adsorbents (class 1 and class 3) for CO2 capture from air.88 They found that the conventional class 1 amine-based adsorbent (PEI/silica) also displayed the highest CO2 uptake of 2.36 mmol g−1 when exposed to CO2 (400 ppm) in Ar, due to the presence of a larger amount of polymeric amines inside the pores (10.5 mmol N per g of adsorbent). However, it led to a longer adsorption half-time, whereas, after mixing the additive with PEI (tetrapropylorthotitanate and (3-aminopropyl)triethoxysilane (APTES)), the adsorption half time of the resultant mixture was reduced without significant change in CO2 uptake. The adsorption half time and corresponding average CO2 adsorption rate was 309 min and 3.8 μmol g−1 min−1 for PEI/silica, 196 min and 5.8 μmol g−1 min−1 for A-PEI/silica, 210 min and 5.2 μmol g−1 min−1 for T-PEI/silica and 167 min and 5.2 μmol g−1 min−1 for HAS6 respectively.88 The reduced CO2 adsorption rate was probably due to a change in the chemical structure of PEI by mixing with additives, causing tuned amine accessibility and mass transport through the pores. A study of air capture using TRI-PE-MCM-41 (triamine-grafted pore-expanded mesoporous silica) recorded a breakthrough time of 167 min and an adsorption capacity of 0.98 mmol g−1.143 In another study using MAHSM (mono amine-based hybrid silica material), the CO2 adsorption capacity of 1.68 mmol g−1 was obtained with the adsorption half time of 50 min. A rate of 17.9 μmol g−1 min−1 during CO2 capture from the simulated air (containing 400 ppm CO2, 60% RH) was achieved at 30 °C with a flow rate of 100 ml min−1 in 24 h.111 Gebald et al. reported the adsorption half time of AEAPDMS-NFC-FD (aminosilane modified nanofibrillated cellulose aerogel) was 92 min, corresponding to an average CO2 adsorption rate of 7.6 μmol g−1 min−1, which is quite less than PEI.113 On the other hand, Sehaqui et al.107 reported a porous sheet-like biocomposite (PEI modified nanofibrillated cellulose) adsorbent, synthesized by freeze-drying process, exhibiting exceptionally low adsorption half time of 10.6 min with a faster uptake rate (105 μmol g−1 min−1) due to the higher porosity and micro meter range pores available in these sorbents.
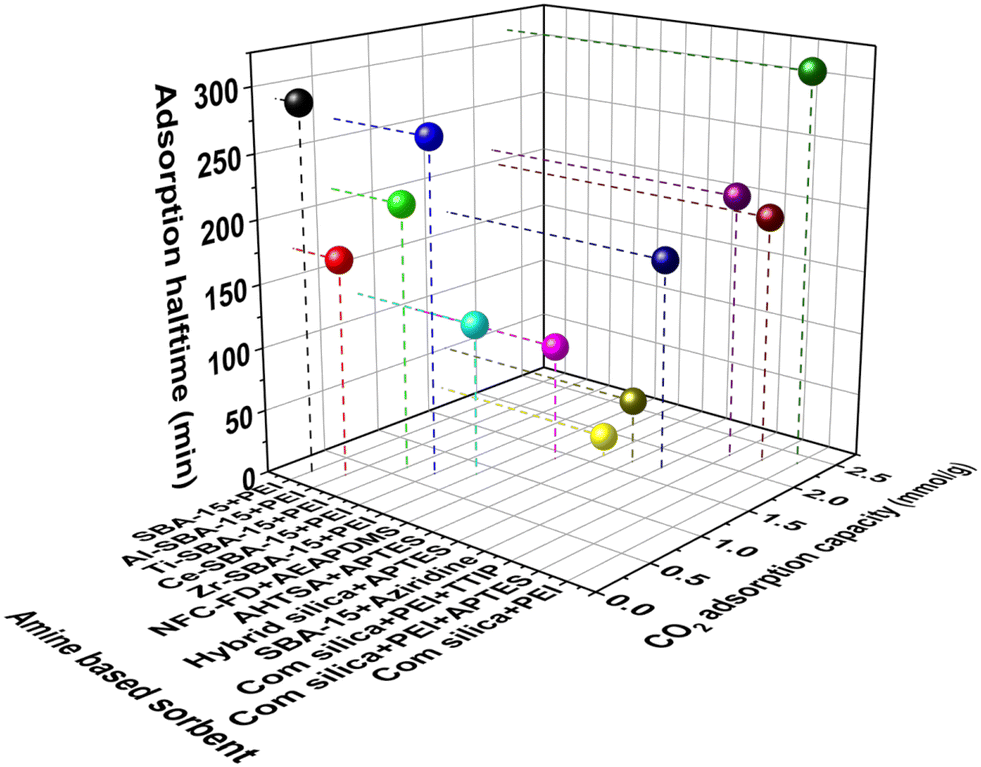 | ||
| Fig. 8 Comparative CO2 adsorption kinetics of amine-based adsorbents (based on Table 3). | ||
| Amine based composite | CO2 adsorption capacity (mmol g−1) | Adsorption halftime (min) | Adsorption rate (μmol g−1 min−1) | Method | Ref. |
|---|---|---|---|---|---|
| a Data retrieved from data from graph software (version 1.0); Com silica, commercial silica. | |||||
| Com silica + PEI | 2.36 | 309 | 3.8 | TGA | 88 |
| Com silica + PEI + APTES | 2.26 | 196 | 5.8 | TGA | 88 |
| Com silica + PEI + titanium(IV) propoxide | 2.19 | 210 | 5.2 | TGA | 88 |
| SBA-15 + aziridine | 1.72 | 167 | — | MS | 95 |
| Hybrid silica + APTES | 1.68 | 50 | 17.9 | TGA | 111 |
| Zr-SBA-15 + PEIa | 0.85 | 185 | — | TGA | 134 |
| Ce-SBA-15 + PEIa | 0.68 | 264 | — | TGA | 134 |
| Ti-SBA-15 + PEIa | 0.64 | 210 | — | TGA | 134 |
| Al-SBA-15 + PEIa | 0.29 | 171 | — | TGA | 134 |
| SBA-15 + PEIa | 0.19 | 288 | — | TGA | 134 |
| NFC + PEI | 2.22 | 10.6 | 105 | IR | 107 |
| NFC-FD + AEAPDMS | 1.39 | 92 | 7.6 | IR | 113 |
| AHTSA + APTES | 1.64 | 15.88 | — | IR | 114 |
| RFAS + APTES | 1.78 | 233 | — | IR | 137 |
Though amine-impregnated adsorbents are not the best in terms of CO2 adsorption kinetics, exceptions are the amine-impregnated MOFs, where the CO2 adsorption kinetics readily depend on the textural properties of MOFs, the presence of the open metal site and associated amine ligands. For instance, in the case of mmen-Mg2(dobpdc) (where mmem is N,N′-dimethylethylenediamine and dobpdc is 4,4′-dihydroxy-(1,1′-biphenyl)-3,3′-dicarboxylate),42 CO2 adsorption kinetics of mmen-Mg2(dobpdc) tend to be slightly faster than other amine-based adsorbents, where amines are deposited on the adsorbents by evaporation or polymerization method. Notably, mmen-Mg2(dobpdc) took only 60 min to achieve the 4.6% equilibrium CO2 adsorption capacity compared to the benchmark impregnated polyethylenimine silica gel under DAC condition,88 which took nearly 210 min to reach the same equilibrium adsorption capacity. The readily accessible amines in mmen considerably improve adsorption rates, hence facilitating the rapid adsorption–desorption cycles. A similar relationship of CO2 adsorption kinetics with the textural properties and molecular weight of amine can be seen in silica-based adsorbents. Although both the composites (PPI/MCF and PPI/SBA-15) (where, PPI is poly(propylene imine), MCF is a mesocellular silica foam and SBA-15 is Santa Barbara Amorphous-15, a type of mesoporous silica) adsorb roughly the same amount of CO2, PPI/MCF, having a larger pore volume, has resulted in more rapid uptake under the adsorption conditions of 400 ppm CO2/N2 at 35 °C.108 A rapid tailing out for secondary amines is reported, while primary amines displayed CO2 adsorption for a prolonged time at a faster initial rate. To study the effect of the molecular weight of adsorbents and their CO2 adsorption, PPI polymers of different molecular weights were prepared by altering the polymerization time. The lower composite molecular weight oligomer showed faster CO2 adsorption kinetics than the higher molecular weight polymer composite and vice versa. This is due to the relative degree of amine branching that affects the CO2 affinity and not just the diffusion properties of the gas.129 After all, the CO2 adsorption kinetics of amine-based adsorbents also depend upon the design of the adsorbent bed (fluidized bed or small-scale fixed bed). For instance, Zhang et al.124 used a bubbling fluidized bed reactor for direct air CO2 capture on PEI–silica adsorbent and found that almost 100% equilibrium CO2 adsorption capacity can be accomplished with 7.5 s contact time between the adsorbent and the ambient air. This might be due to the adsorbent's rapid reaction kinetics, more turbulent gas flow to the adsorbent in the reactor, and highly efficient gas–solid contact in the whole riser of the reactor.
3.3 Stability of amine-based solid adsorbents for DAC
The stability of CO2 adsorbents is critical for practical DAC application, as the lifetime of adsorbents has a significant influence on the entire process cost.92 The degree of change in adsorption capacities across repeated adsorption/desorption cycles is described as the regenerability of CO2 adsorbents.88 A fully regenerable sorbent can resist several adsorption/desorption cycles without losing its CO2 adsorption capacity, reducing the frequency of sorbent replacement and hence improving the DAC economics. From the practical point of view, amine-based adsorbents are typically subjected to thermal, moisture, and oxidative stability issues, which are briefly discussed in this section. The sorbent regeneration and cyclic CO2 adsorption–desorption stability of amine-based solid adsorbents are described in Table 4.| Support | Amine type | Sorbent regeneration condition | Stability performance | Ref. |
|---|---|---|---|---|
| a Data retrieved from data from graph software (version 1.0). | ||||
| Silica | PEI | 110 °C for 3 h under Ar flow at 100 mL min−1 | ∼30% capacity loss in 4 cyclic runs | 88 |
| Silica | A-PEI | 110 °C for 3 h under Ar flow at 100 mL min−1 | ∼9% capacity loss in 4 cyclic runs | 88 |
| Silica | T-PEI | 110 °C for 3 h under Ar flow at 100 mL min−1 | ∼1.3% capacity loss in 4 cyclic runs | 88 |
| Fumed silica | PEI | 85 °C for 3 h under vacuum | ∼3.5% capacity loss in 4 cyclic runs | 123 |
| Fumed silica | PEI | 85 °C for 3 h under airflow at 335 mL min−1 | No apparent loss of capacity in 4 cyclic runs | 81 |
| Hierarchical silica | PEI | 110 °C for 6 h under He flow at 100 mL min−1 | 16% capacity loss in 5 cyclic runs in humid conditions, whereas no appreciable change in CO2 adsorption capacity in dry condition | 39 |
| Mesoporous silica | PEI | 130 °C for 2 h under N2 flow (moisture content 8.8 vol%) at 8000 mL min−1 | No apparent loss of capacity in 4 cyclic runs | 124 |
| SBA-15 | PEI | 110 °C for 9 h under Ar flow at 100 mL min−1 | Stable over short multicycle operations (3 cyclic runs) | 125 |
| SBA-15 | PPI | 110 °C for 10 min under N2 flow | No apparent loss of capacity in 50 cyclic runs | 108 |
| SBA-15 | LPPI | 110 °C for 30 min under He flow at flow rate of 90 mL min−1 | No apparent loss of capacity in 20 cyclic runs | 131 |
| Zr-SBA-15 | PEI | 110 °C for 3 h under Ar flow at 100 mL min−1 | 2% capacity loss in 4 cyclic runs | 134 |
| Ph-3-ED/SBA-15 | Ethylenediamine | 90 °C for 10 min under He flow at flow rate of 90 mL min−1 | No apparent loss of capacity in 25 cyclic runs | 41 |
| CA-SiO2 | PEI | 110 °C for 2 h under He flow | ∼9% pseudo-equilibrium capacity and 16% breakthrough capacity loss in 20 cyclic runs | 101 |
| Resin-HP-20 | PEI | 100 °C for 3 h under N2 flow at 100 mL min−1 | ∼1.5% capacity loss in 5 cyclic runs | 105 |
| Mesoporous carbon | PEI | 110 °C under N2 flow at 50 mL min−1 | 3% capacity loss in 10 cyclic runs | 106 |
| Mg/(dobdc) | Ethylenediamine | 110 °C for 3 h under Ar flow | No apparent loss of capacity in 4 cyclic runs | 139 |
| Mg2(dobpdc) | Ethylenediamine | 150 °C for 2 h under simulated air (0.39 mbar CO2) purge at 60 mL min−1 | 6% capacity loss in 5 cyclic runs | 115 |
| Mg2(dobpdc) | mmen | 150 °C for 30 min under N2 flow at 25 mL min−1 | No apparent loss of capacity in 10 cyclic runs | 42 |
| MIL-101-Cr | PEI | 110 °C for 3 h under He flow at 90 mL min−1 | CO2 uptake dropped 2.7% and 1.9% after the first and second cycles, respectively (total 3 cyclic runs) | 135 |
| Cr-MIL-101-SO3H | TAEA | 80 °C for 60 min at high vacuum | No apparent loss of capacity in 15 cyclic runs | 140 |
| TTE | PEI | 120 °C for 30 min by water steam at 2000 mL min−1 | No apparent loss of capacity in 10 cyclic runs | 136 |
| Nano fibrillated cellulose | AEAPDMS | 90 °C and 40% relative humidity under Ar flow at 800 mL min−1 | Stable over 20 cyclic runs | 113 |
| Silica gel | AEATPMS | 90 °C under vacuum with a sufficient humid environment | Good repeatability over 40 consecutive cyclic runs | 112 |
| Pore-expanded MCM-41 | TRI | 100 °C for 3 h by synthetic air at 10 mL min−1 | ∼24% capacity loss in 4 cyclic runsa | 103 |
| SBA-15 | Aziridine | 110 °C for 3 h under Ar flow at 100 mL min−1 | Relatively stable for 4 cyclic runs | 95 |
| Hybrid silica | APTES | 80 °C for 15 min under He flow at 70 mL min−1 | Stable over 50 cyclic runs | 111 |
| AHTSA | APTES | 90 °C for 30 min under N2 flow at 300 mL min−1 | No apparent loss of capacity in 15 cyclic runs | 114 |
| SBA-15 | Z-L-Lysine, APTMS | 110 °C for 3 h under a pure argon purge | Stable over short multicycle operations (3 cyclic runs) | 92 |
000 g mol−1) exhibited no sign of leaching.81 However, aminopolymer containing primary amines is more susceptible to urea formation than secondary or tertiary amines. Such aminopolymers may get deactivated rapidly in the presence of CO2 atmosphere through the intermediacy of isocyanate followed by a reaction with primary or secondary amine into di- and tri-substituted open-chain urea or N-substituted imidazolidinones.70
The literature reports inferred that incorporating additives to polyamines during adsorbent synthesis and desorption of amine-based adsorbent under humid conditions (as also mentioned in section 3.3.3) is a promising way to prevent urea formation.72 Choi et al. reported a comparative study on cyclic CO2 stability of low molecular weight PEI-impregnated silica before and after incorporating additives titanium(IV) propoxide (tetrapropyl orthotitanate) and (3-aminopropyl)triethoxysilane.88 It was observed that after tetrapropyl orthotitanate (C12H28O4Ti) modification, the amine-based adsorbent exhibited only 1.36% loss in CO2 cyclic stability compared to PEI-based silica (30% loss). Goeppert et al. also reported the prolonged 50 thermo-cyclic stability of polyamines (PEHA and TEPA) supported Sipernat 50S silica after adding an epoxide stabilizing agent (propylene oxide).40 The group reported that compared to TEPA-PO-1-2/50S, PEHA-PO-1-2/50S based adsorbents are more thermally stable, even up to 50 cycles due to the presence of more one –CH2–CH2–NH– group than TEPA. A desorption time of 5 min at 85 °C was sufficient to desorb all the CO2 from the adsorbent with no noticeable changes over time. Abhilash et al.111 synthesized monoamine-based hybrid silica material using a combination of 3-aminopropyl triethoxysilane (APTES) and vinyl triethoxysilane (VS) and carried out a CO2 adsorption experiment under a 400 ppm CO2 environment at 60% RH.111 Fifty cycles were performed at 30 °C in ambient air followed by desorption at 80 °C under He flow, and the shifting of carbamate carbonyl peak (164.5 ppm) was verified using 13C CP-MAS NMR. The adsorbent exhibited excellent cyclic stability with no shifting of carbamate carbonyl peak, indicating no urea formation. Interestingly, the studied adsorbent is highly stable and can regenerate quickly by desorbing CO2 at a relatively low temperature (80 °C) within a few minutes (10 min).
Various researchers have attempted to enhance the oxidative stability of amine-based sorbents by functionalizing or blending the aminopolymers with certain additives. Chuang et al. used poly(ethylene glycol) (PEG) as an additive to reduce the oxidation of supported TEPA by facilitating hydrogen bonding between PEG and TEPA.152,153 By restricting amine sites from access to oxygen, these hydrogen-bonding interactions between amines and OH groups boosted TEPA dispersion and reduced amine oxidation to imide species.152,153 Choi et al.154 hybridized branched PEI with 1,2-epoxybutane, enabling the availability of hydrogen bonding groups directly onto the aminopolymer to suppress urea formation and oxidative degradation of amines simultaneously by reducing the fraction of primary amine. The oxidative stability of another aminopolymer, namely poly(glycidyl amine) (PGA) containing both ethylene oxide and primary amine groups in each repeating unit, was studied by Sujan et al.109 The sample was exposed to 21% O2 with a balance of N2 at 110 °C, resulted in the enhancement in the stability of the sorbent, and hence the sorbent could retain 50% of its CO2 adsorption capacity.109
In a study by Goeppert et al. under oxidative conditions, the stability of tetraethylenepentamine (TEPA) and pentaethylenehexamine (PEHA) modified sorbents were found to be significantly enhanced by introducing propylene oxide (PO) as intramolecular alcohol functionalities.40 The oxidative stability experiment conducted in the aerobic atmosphere at 100 °C for 24 h revealed that the CO2 adsorption capacity of PO containing TEPA sorbent remains stable at 25 °C but decreased by 16% at 55 °C and 18% at 85 °C. For PEHA-PO, comparable results were obtained, indicating the increased oxidative stability of PO-modified aminopolymer sorbents.
Another approach to enhance oxidative stability included using small molecule poly(propylenimine) (PPI) in linear and branched aminopolymer forms. The linear PPI, namely tripropylenetetramine (TPTA) (188 g mol−1) containing primary as well as secondary amines, showed only a 20% loss in amine efficiency after oxidative treatment in air at 110 °C for 24 h.132 Pang et al.108,132 critically analyzed the oxidative stability of small and large molecules of amine introduced to the porous support. Switching from a PEI to a poly(propylenimine) (PPI) (without secondary amine) increases the spacing of the amines in the backbones. It significantly improves the stability of small molecule aminopolymers against oxidative degradation. In oxidizing conditions, small-molecule amines such as ethylenimines can undergo chain rearrangement reactions to form six-membered rings that quickly oxidize to piperazinones. Propylenimines, on the other hand, form four- or eight-membered rings, which are more stable toward deactivation. Further, PPI having a lower molecular weight of 700 g mol−1 and 1000 g mol−1 maintained about 65% of its amine efficiency. On the other hand, samples with higher molecular weight PPI (6700 g mol−1 and 36000 g mol−1) retained over 83% efficiency in the presence of an oxidative environment.108 This is due to the inaccessibility of oxygen to penetrate the polymer block to carry out the oxidative reaction. Sarazen et al. demonstrated the use of branched PPI along with acid initiators such as HBr, HClO4, HCl, and CH3SO3H, where sorbents with HBr retained 76% of their CO2 adsorption capacity after 12 h of oxidation in air at 110 °C.129 Recently, Rosu et al.131 evaluated the effect of extended aging (∼2 years) of linear PPI and observed that the linear PPI with a molecular weight of 700 g mol−1 retained 68% of its CO2 adsorption capacity after oxidation for 12 h in 21% O2/He stream under 400 ppm CO2. Also, the same sample could retain 66% of its adsorption capacity during short TSA cycles (6 cycles, 1 h adsorption at 35 °C, and 30 min desorption at 110 °C).
Recently, Kumar et al.41 developed alkyl-amine rich small molecules modified with ethylenediamine/propane-1,3-diamine for the DAC process. The accelerated oxidation stability experiments at high temperatures (90 and 110 °C) under 21% O2 for 24 h revealed that the ethylenediamine-modified sorbent retained 35% and 20% CO2 adsorption capacity and 39% and 23% amine efficiency at 90 and 110 °C, respectively. On the other hand, propane-1,3-diamine impregnated sorbent maintained 22% and 13% CO2 adsorption capacity and 20% and 10% amine efficiency at 90 and 110 °C, respectively. They concluded that the aromatic cores could change the distances between the oligomeric alkyl amine chains and hinder the electronics of the impregnated amine molecules and thus show lesser oxidative stability compared to the PPI-based sorbents.
Steam or moisture-containing non-adsorbing gases, such as CO2, can be employed as a purge gas for amine-based adsorbents, enabling pure CO2 to be extracted.156 Sayari et al. reported the mechanism behind the stability of amine-based adsorbents to humidity.72 Monoamine, triamine-grafted pore-expanded MCM-41 mesoporous silica (PE-MCM-41), and polyethylenimine impregnated PE-MCM-41 were studied to adsorb pure CO2 and desorbed CO2 in the presence of moisture (20 °C as dew point). It was found that when desorption was carried out under dry conditions (moderate to high temperature), the cycle stability dropped by 14–45% after 40 cycles due to the formation of urea as a byproduct or reaction between amine and CO2. However, in the studied amine-based adsorbent, when desorbed in the presence of moisture, the cyclic stability was retained for up to 750 cycles without the formation of urea. The mechanism of urea formation was probed by 13C CP MAS NMR and DRIFT study. The MAS NMR peak at 164.6 ppm (for carbamate) and 1600 cm−1 (NH2 scissoring) was seen in amine-modified PE-MCM-41 when they underwent desorption under humid conditions. However, carbamate decomposes to produce urea in a dry desorption environment, which is shown by the new peak that appeared at 160.5 ppm in MAS NMR and two new bands (1658 and 1560 cm−1) in the IR pattern as shown in Fig. 9.
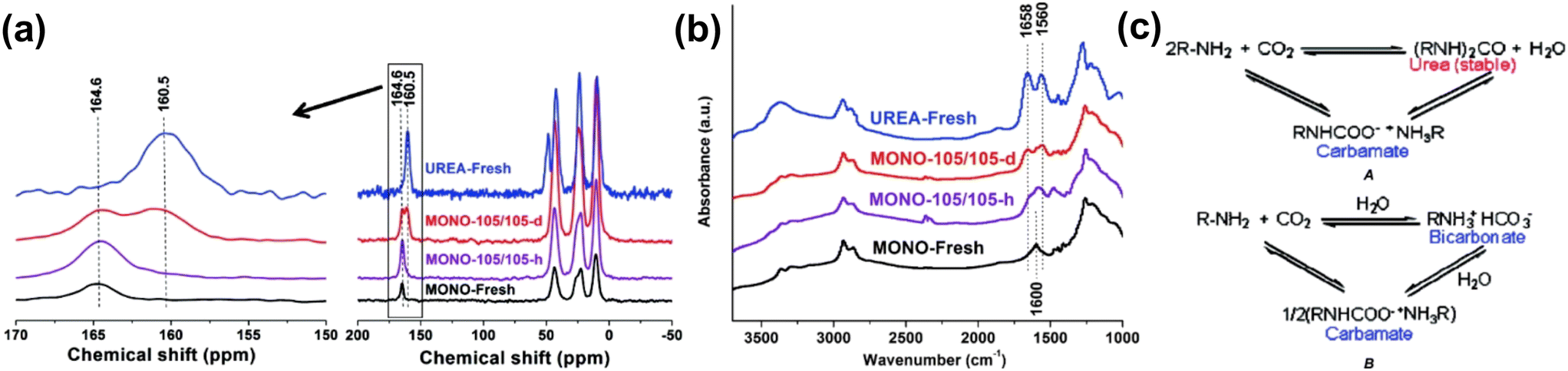 | ||
| Fig. 9 (a) 13C CP MAS NMR spectra, (b) DRIFT spectra for fresh urea, fresh mono-MCM-41 and after cyclic desorption in dry and humid conditions, and (c) reaction mechanism of amine and CO2 to form urea as well as a carbamate. Reproduced with permission.72 Copyright 2010, American Chemical Society. | ||
The author also reported that it is also possible to deactivate the strongly formed urea groups in the cyclic adsorbent via hydrolysis at 200 °C under a flow of nitrogen-containing as little as 0.15% RH (dew point at 20 °C) for 24 h. AEATPMS-modified silica gel and AEAPDMS-modified nano-fibrillated cellulose have been reported as one of the most stable adsorbents under thermal swing regeneration (in a humid environment), preventing the formation of urea.112,113
Furthermore, MOFs in the DAC process require partial or complete drying of the gas streams. Some MOFs are hydrophilic and can end up adsorbing more H2O than CO2.157 However, amine-modified MOFs prove to be more stable under humid conditions of the DAC process. In the case of PEI/MIL-101-Cr,135 and N2H4/Mg2(dobdc)118 the measured CO2 adsorption capacity before and after the exposure to humidity of 32% RH and under 82% RH, respectively, showed little change in the adsorption capacity, suggesting the material is stable under DAC working conditions.
3.4 Adsorption thermodynamics of amine-based solid adsorbent for DAC
Adsorption thermodynamics describes the energetic interaction between adsorbate and the adsorbent's surface as a function of state variables (pressure, temperature, chemical composition, and others).158 To design an adsorption-based system, it is essential to understand the thermodynamic properties, especially the enthalpy of adsorption.159 The enthalpy of adsorption is defined as the heat released when adsorbate binds to an adsorbent. Experimentally, the heat (enthalpy) of adsorption (ΔHads) is evaluated by either direct or indirect methods.160 In the direct method (usually called the calorimetric method), the heat released upon the contact between adsorbate and adsorbent is measured using a calorimeter (differential scanning calorimetry or microcalorimetry). In the indirect method, the enthalpy of adsorption is estimated from the adsorption isotherm data at multiple temperatures by fitting adsorption isotherms using Langmuir, Toth, or related thermodynamic models and then applying the Clausius–Clapeyron equation.160 The enthalpy of adsorption can also be expressed as isosteric heat of adsorption (Qst) at particular coverage. The isosteric heat of adsorption has the same magnitude as the enthalpy of adsorption but with an opposite sign (ΔHads = −Qst).161 The isosteric heat of adsorption provides necessary information about the nature of the adsorption process (exothermic or endothermic) it allows quantifying the energy needed to desorb the adsorbate from the adsorbent.162 Among the two measurement methods, the isosteric heat measurement by the Clausius–Clapeyron equation is widely used compared to the calorimetric measurement.160 However, the heat value calculated from the indirect method sometimes underestimates the actual heat value due to the inconsistency in adsorption fitting. Hence, for practical DAC application, the heat of desorption and the heat of regeneration, along with the heat of adsorption, are also calculated using calorimetry.163The isosteric heat of adsorption for CO2 capture over a few reported amine-based adsorbents under DAC conditions comes under 49–130 kJ mol−1 (Fig. 10, Table 5). These values depend on the amine–CO2 interaction, type, basicity, and amount of amine loaded in adsorbent/amine support.163 Among primary, secondary, and tertiary amines, the adsorbents loaded with primary amine exhibit high heat of CO2 adsorption, even at very low CO2 concentrations due to their aggressive interaction with CO2.163 For instance, the bare adsorbent such as silica exhibited heat of CO2 adsorption typically in the range of 20 to 40 kJ mol−1.164 However, introducing primary amine such as 3-aminopropyl silane to the silica substrate greatly enhanced its heat of CO2 adsorption in the range of 65 to 130 kJ mol−1 depending on the density of amine sites.163,165 With the increase in amine density in the substrate (>1.2 mmol N per g of adsorbent), the heat of CO2 adsorption at zero coverage was observed to show an increasing trend, primarily due to the formation of strong alkylammonium carbamate species (92 kJ mol−1) in dry conditions by allowing multiple amines to interact with one CO2 molecule. Further, at a higher amine loading, the heat of adsorption at zero coverage increased significantly once the average amine spacing reached a value below ca. 11–12 Å, assuming all sites are equally spaced. On the other hand, at low amine density in the substrate, the heat of CO2 adsorption was decreased to 40–60 kJ mol−1 similar to bare SBA-15 support because of the less interaction of CO2 with the fewer isolated amine species present in the substrate. In such a case, carbamic acid will be preferred over carbamate.163,164 Similar observation was found by Potter et al.91 by revealing the heat of CO2 adsorption (using microcalorimetry) of different species formed (using in situ FTIR) during the interaction of CO2 and 3-aminopropyltriethoxysilane (APTES) grafted mesoporous alumina (D-Al2O3 and O-Al2O3). Lower APTES loading into alumina resulted in the formation of carbamic acid (due to more isolated amine species), which is generally less energetically stable and exhibits low heat of CO2 adsorption (45–65 kJ mol−1) in alumina pores as compared to alkylammonium carbamate (98 kJ mol−1). Further, the same group also reported that the resultant heat of CO2 adsorption of APTES modified alumina also depends upon the initial basicity of alumina surface. Alumina D-Al2O3 is more basic than O-Al2O3 due to presence of hydroxyl groups and O2− group (heat of CO2 adsorption: 79 kJ mol−1vs. 52 kJ mol−1) and it was believed that the heat of adsorption would further increase after amine loading on D-Al2O3. However, after the same equivalent APTES loading, the initial heat of CO2 adsorption in D-Al2O3 was notably lower than O-Al2O3 due to the restriction in interaction of CO2 with strong binding sites of alumina by the APTES grafting process. Therefore, D-Al2O3 overcame a larger energy barrier to interact with CO2.91
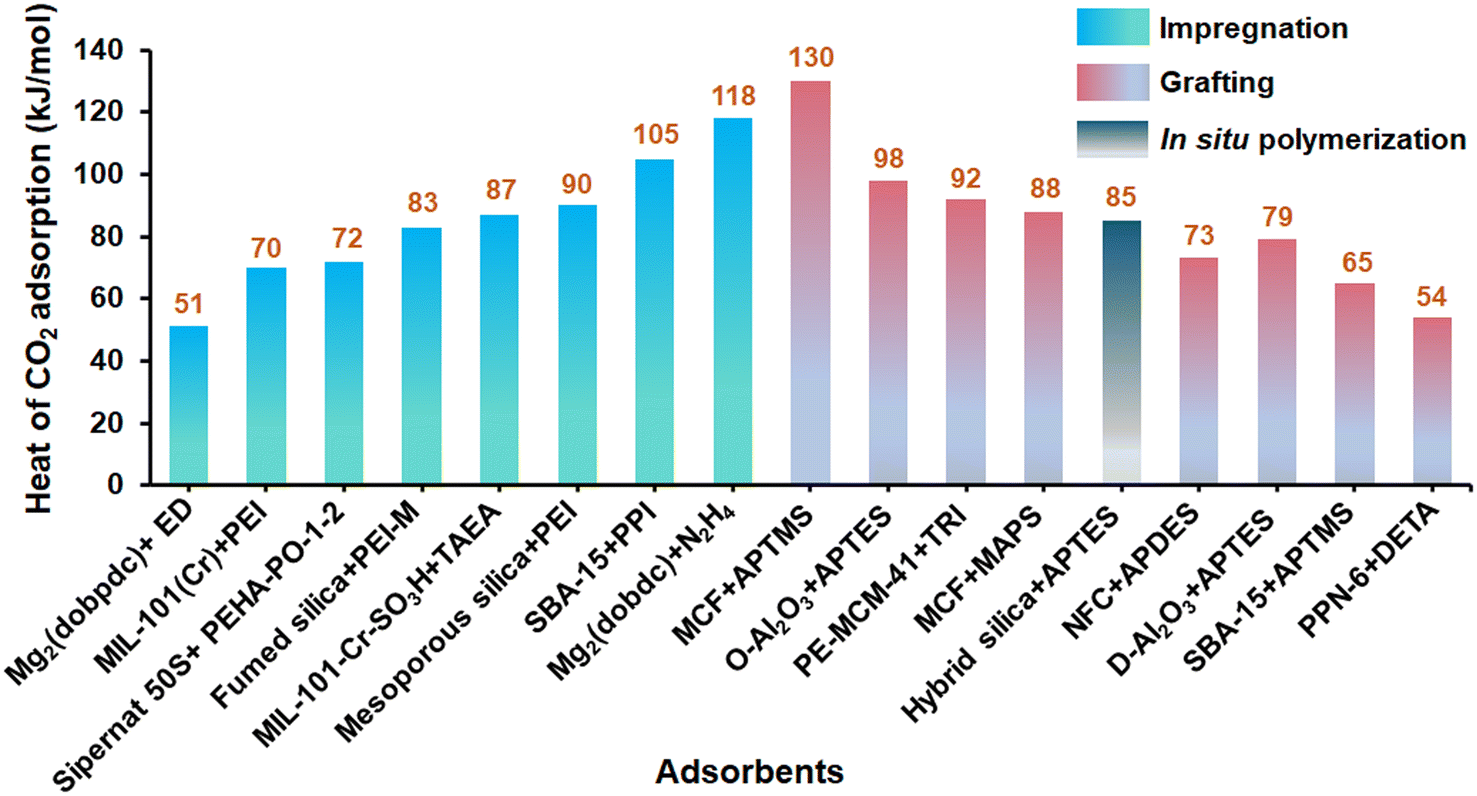 | ||
| Fig. 10 Heat of adsorption for CO2 capture over literature-reported amine-based adsorbents (based on Table 5). | ||
| Amine based composite | Heat of adsorption (kJ mol−1) | Method | Ref. |
|---|---|---|---|
| a Isosteric heat of CO2 adsorption at zero coverage obtained by the Clausius–Clapeyron equation and isotherms fitted by the respective thermodynamic models like virial, Toth, etc. b Isosteric heat of CO2 adsorption at very low surface coverage obtained by the Clausius–Clapeyron equation using the experimental isotherm data. | |||
| Fumed silica + PEI-M | 83 | Calorimetry | 81 |
| Mesoporous silica + PEI | 90 | Calorimetry | 124 |
| SBA-15 + PPI | 105a | Toth isotherm | 108 |
| PEHA-PO-1-2-Sipernat 50S | 72 | Calorimetry | 40 |
| Mg2(dobpdc) + ed | 49–51a | Dual-site Langmuir–Freundlich | 115 |
| Mg2(dobdc) + hydrazine | 118a | Langmuir–Freundlich | 118 |
| Mg2(dobdc) + mmem | 71a | Dual-site Langmuir–Freundlich | 42 |
| MIL-101-Cr + PEI | 70b | Directly from isotherm data | 135 |
| Cr-MIL-101-SO3H + TAEA | 87a | Triple-site Langmuir | 140 |
| O-Al2O3 + APTES | 98 | Calorimetry | 91 |
| D-Al2O3 + APTES | 69 | Calorimetry | 91 |
| MCF + APTMS | 130b | Directly from isotherm data | 84 |
| MCF + MAPS | 88b | Directly from isotherm data | 84 |
| Nano fibrillated cellulose + APDES | 73b | Directly from isotherm data | 119 |
| SBA-15 + APTMS | 65a | Langmuir | 138 |
| Pore-expanded MCM-41 + TRI | 92b | Directly from isotherm data | 33 |
| PPN-6 + DETA | 54a | Triple-site Langmuir | 120 |
| Hybrid silica + APTES | 85 ± 5 | Calorimetry | 111 |
Adsorbent modified with secondary amine (associated with methylaminopropyl silane, MAPS) exhibited heat of CO2 adsorption similar to that of primary amine at a similar amine density but is less effective for CO2 capture from air.84,163 For example, Didas et al.84 reported the high heat of CO2 adsorption (130 kJ mol−1) for APTMS grafted mesoporous cellular form (MCF) as compared to those grafted with MAPS (88 kJ mol−1). Notably, tertiary amines such as dimethylaminopropyl silane (DMAPS) are inefficient for DAC application due to their poor interaction with CO2. On the other hand, if an adsorbent is loaded with amines of all types, including primary, secondary, and tertiary, the net heat of CO2 adsorption of the adsorbent is not equal to the sum of the heat of CO2 adsorption of individual amine species of the adsorbent, but rather depends on the individual content of amines and their reactivity toward CO2. For instance, commercially available low-molecular-weight branched PEI has a carbon:nitrogen molar ratio of 2:1 [(C2H5N)n, linear form] with a distribution of amine sites as 44% primary, 33% secondary, and 22% tertiary amines.163 Primary, secondary, and tertiary amines exhibit typically heat of CO2 adsorption values around 84 kJ mol−1, 72 kJ mol−1, and 48 kJ mol−1, respectively.34,166 Therefore, the heat of CO2 adsorption of the highest PEI-loaded adsorbent (PEI-7.5/SBA-15) is found to be 93 kJ mol−1, which is less than the summation of the heat of reaction of its stand-alone constituent amines. This observation can be attributed primarily to the fact that primary, and a few secondary amines present in PEI are majorly responsible for the adsorption of CO2 even at a low concentration of CO2. It is demonstrated that PEI-SBA-15 exhibited optimal heat of CO2 adsorption with better CO2 uptake despite the low concentration of CO2, and hence is perfect for DAC applications.163 In a similar work, Pang et al.108 reported that compared to PEI–silica composite (50–80 kJ mol−1), the isosteric heat of CO2 adsorption at zero surface coverage on PPI–silica composite (105 kJ mol−1) is considerably greater, presumably due to the higher basicity of propylenimines than ethylenimines, which resulted in strong adsorption of CO2 on PPI.
Although high enthalpy of adsorption for DAC is the prerequisite criteria for CO2 capture from air, it imposes a high parasitic energy load on the plant. Therefore, researchers focus on incorporating smaller chain amine into adsorbents without compromising the CO2 adsorption capacity.40,167 Secondary amino groups have been reported to bind CO2 less strongly than primary amino groups.30 This is advantageous during the adsorbent's endothermic regeneration step when CO2 has to be released. Therefore, the amount of energy required for the desorption could be lowered by employing polyamines mainly containing secondary amino groups. Goeppert et al.81 estimated the heat of CO2 adsorption, the heat of CO2 desorption, and the heat of CO2 regeneration respectively as 83 kJ mol−1 (at 85 °C), 82 kJ mol−1, and 170 kJ mol−1 (from 25 to 110 °C) for branched PEI based fumed silica using calorimetry. On the contrary, when the fumed silica is modified with linear PEI, which consists of secondary amine, the associated heat of CO2 adsorption (42 to 53 kJ mol−1) and desorption (40 to 49 kJ mol−1) decreased significantly.150 The estimated heat of regeneration is almost half of the heat of regeneration when considering a 20% aqueous solution of MEA (330 kJ mol−1, considering an absorption of 0.4 mol of CO2 for 1 mol of MEA) and is therefore energetically favorable. To further reduce the associated thermal energy for CO2 regeneration without compromising its uptake, the same group, Goeppert et al.40 estimated the heat of CO2 adsorption of polyamines (PEHA and TEPA) supported Sipernat 50S silica with the addition of stabilizing agent propylene oxide (PO) on direct isothermal calorimetry. With the addition of PO, the heat of CO2 reaction for PEHA-PO-1-2/50S was decreased to 72 kJ mol−1, which is 10 kJ mol−1 less than PEHA/50S.
Considering humid conditions, although the CO2 adsorption capacity of amine-based adsorbents rises, especially at ultra-diluted CO2 pressures, the H2O content must be kept as low as possible.168 For DAC, temperature swing desorption or temperature-vacuum swing desorption is preferentially used to recover the adsorbents during the repetitive adsorption–desorption cycles. High moisture sorption in the adsorbent leads to higher parasitic energy requirements during the regeneration of a sorbent. It has been observed that, during the regeneration of adsorbent under TVSA, if the H2O loading in the air increases from 20 to 60 RH% at 30 °C, the thermal energy demand increased from 532 to 586 kJ mol−1 CO2.168 This is because the total energy required to regenerate an adsorbent during the co-adsorption of CO2 and H2O in the DAC condition is the sum of the sensible heat required to bring the material to desorption temperature and the heat input for the desorption enthalpy of H2O and CO2. Typically, during CO2 adsorption under dry conditions, the enthalpy change of reaction values were reported as 50 to 118 kJ mol−1 and 56 kJ mol−1 for hydrous conditions, additionally with 47 to 53 kJ mol−1 for H2O adsorption.34,113 Since moisture in the air is inevitable, the net thermal regeneration energy for any practical DAC process could be decreased significantly by increasing the CO2 adsorption capacity of the material beyond 2 mmol g−1 to nullify the sensible heat effect.168
3.5 CO2 over other gases selectivity of amine-based solid adsorbents for DAC
The high selective adsorption of CO2 from the other bulk components of the air (78% N2, 21% O2, and 1–2% H2O) is the most essential and desirable criteria to be achieved for any amine-based adsorbent. Usually, for DAC the selectivity for CO2 is calculated based on the ratio of CO2 uptake capacity (at 0.4 mbar) to that of the other components (for example, N2 or O2) according to their partial pressure in the air.169 The adsorbent having high selectivity for CO2 has three crucial significances in DAC, including (i) production of ultra-high pure (UHP) CO2, (ii) reduction of cost associated with the CO2 capture process, and (iii) requirement of less energy for transportation and compression. Since air contains a large concentration of N2 and O2, it is crucial to develop amine-based adsorbents that ideally would not react with either N2 or O2 at room temperature and atmospheric pressure. Further, the CO2 purity should be greater than 90% to meet the department of energy (DOE) demand to be utilized for further value-added applications such as underground storage.138Belmabkhout et al.33,143 reported a novel adsorbent (triamine-grafted pore-expanded mesoporous silica TRI-PE-MCM-41) that exhibited excellent CO2/N2 and CO2/O2 selectivity (∼ infinity) with high CO2 uptake (0.9 ± 0.09 mmol g−1) when exposed to the simulated air containing CO2 (0.03%) and N2 (79.98%) and O2 (19.99%) at 25 °C and 1 bar. The experiment was conducted in a breakthrough setup, where both O2 and N2 appeared in the column downstream almost immediately after the process started, indicative of a minimal adsorption capacity for N2 and O2. Further, the same group also reported the adsorption performance of TRI-PE-MCM-41 in the presence of simulated air containing CO2:N2 = 0.04%:99.96% in a dry condition as well as in presence of moisture 27% and 64% relative humidity (RH), at 25 °C and 1 bar.143 TRI-PE-MCM-41 excelled in high CO2–amine interaction as indicated by a steep rise in a breakthrough curve, with high CO2 adsorption capacity and H2O uptake of 4.7 mmol g−1 and 6.29 mmol g−1 at 27 and 64% RH respectively, due to the partial formation of bicarbonate by the reaction of amine molecules with CO2.143 The corresponding CO2 adsorption capacity was 32 and 56% higher than the capacity under dry conditions. Advantageously, even in the presence of moisture, TRI-PE-MCM-41 exhibited high CO2/N2 comparable to that observed under dry conditions (Fig. 11).
 | ||
| Fig. 11 (a) Column-breakthrough curves for CO2:carbon-free air = 0.03:99.97% mixture at 25 °C and 1 bar in dry condition. (b) Breakthrough curves for CO2:N2 = 0.04:99.96 mixture at 25 °C and 1 bar with and without the presence of moisture (27% and 64% RH). Reproduced with permission.33 Copyright 2009, ACS. | ||
Lu et al.120 investigated the CO2 adsorption performance of ethylenediamine and diethylenetriamine (DETA) modified porous polymer networks PPN-6-CH2DETA in the condition similar to air, considering the partial pressure of the gases present in the air. PPN-6-CH2DETA exhibited an extraordinarily high CO2/N2 selectivity (3.8 × 1010), CO2/(N2 + O2) selectivity (3.6 × 1010) as calculated using ideal adsorption solution theory (IAST), CO2 adsorption capacity (1.04 mmol g−1), high stability towards moisture, and excellent CO2 purity (99.999%). Compared to ethylenediamine (which has only short diamine chains), DETA which has also a third amine available for interaction with the CO2 molecule can be loaded at a higher loading into PPN-6 without any pore blockage. The high loading capacity and chemical interactions with CO2 boost the CO2 selectivity of DETA by 7 orders of magnitude than the corresponding ethylenediamine-modified PPN-6-CH2. The group also marked that O2 has minimal interference in competing for adsorption sites against CO2 when PPN-6-CH2DETA is subjected to air due to its strong interaction with CO2–DETA, highlighting the suitability of PPN-6-CH2DETA for DAC application.
Similarly, Lee et al.115 reported the CO2/N2 selectivity of 70000 and CO2 purity of 97% in the simulated air [390 ppm CO2 and 21% O2 balanced with N2, and Ar (99.999%)] for en-Mg2(dobpdc) MOF (en: ethylenediamine), which is higher than mmen-Mg2(dobpdc), (mmen; N,N-dimethylethylenediamine) (corresponding selectivity 49000 and purity 96%).42 The high CO2/N2 selectivity was attributed to carbamate species (–NHCOOH) formation by the specific interactions between the amine groups of en and CO2. Apart from the preferential interaction of CO2 with amine-based adsorbent, CO2 purity also depends upon the desorption process cycle in the practical application. Compared to TSA, TVSA would be sufficient for CO2 purification. The vacuum degasses the amine-based adsorbent more suitably, ensuring minimum loss of CO2 at a shorter cycle time. The analogous observation was reported by Stuckert et al.,138 where SBA-15-APTMS exhibited ∼99% CO2 purity under TVSA (0.01 bar and 95 °C) conditions as compared to 9% CO2 purity observed in TSA (1 bar, and 120 °C) under dry DAC condition (dry ambient air containing 395 ppm CO2).
3.6 Shaping and structuring of amine-based solid adsorbent for DAC
Amine-based adsorbents for carbon capture can satisfy CO2 capture needs while consuming the least energy. However, this results in a significant pressure drop and issues related to the mass transfer and attrition loss,170 particularly when the adsorbents are used in powdered form in a cyclic adsorption process, including PSA and/or TSA. The use of structured monolithic adsorbents has been proposed to address the drawbacks of randomly packed beds, thereby increasing the energy efficiency and economic value of the carbon capture process.171 Recently, additive manufacturing, commonly known as 3D printing, has shown to be a viable platform for shaping porous materials and creating scalable configurations that address these concerns.172 Honeycomb monolith structure has been vastly used for air drying and CO2 capture and separation application,173 as the honeycomb monolith structure has the advantage of low-pressure drop at a high flow rate as well as superior heat and mass transfer compared to the packed bed.174 Typically, mineral-like cordierite is commonly used as construction material for honeycomb monolith synthesis, due to its low thermal expansion. Such monoliths can be wash-coated with high surface area porous material and coated with amine for CO2 capture applications.175 Kulkarni and Sholl utilized a large structured monolith contractor made of amine-functionalized silica adsorbent (TRI-PE-MCM-41) for TSA-based DAC application, using the standard shipping container-sized air capture unit, they could get a daily throughput of ∼1.1 ton CO2 at 85.5% purity.155 Sakwa-Novak et al.176 evaluated the material characteristics and performance of alumina-supported PEI sorbents in their powdered and honeycomb monolith forms. Powder sorbents made by grinding unfunctionalized monolith and then functionalizing with PEI were compared to the monolithic alumina modified with PEI. The monolithic adsorbents showed a CO2 adsorption capacity of 0.75 mmol g−1 compared to the powdered adsorbents (0.7 mmol g−1) at 30 wt% amine loading. The slightly higher adsorption capacity was attributed to the high amine loading (higher volume of PEI content) of monolithic sorbents, implying that some PEI was deposited in the macropores of the structure (Fig. 12). After being exposed to running steam for varied durations, the CO2 adsorption capabilities of PEI-impregnated extruded alumina monolith was stable over five cycles of CO2 adsorption and has shown a volumetric capacity of 350 mol CO2 per m3 with a faster (350 min) equilibration time under 400 ppm CO2/N2, 30 °C, and 0.4 m s−1 gas flow conditions. On the other hand, Wijesiri et al.177 evaluated a PEI impregnated mesocellular foam silica pellet for capturing CO2 from 420 ppm CO2 in N2 under both dry and humid conditions. At a temperature of 46 °C, the maximum capacity of 2.52 mmol g−1 was achieved at a moisture content of 2% mol H2O and 1.94 mmol g−1 under dry conditions. | ||
| Fig. 12 Alumina-supported honeycomb monolith and SEM image of its square channel cross-section, impregnated with PEI for DAC application, adapted with permission.176 Copyright 2016, WILEY-VCH Verlag GmbH & Co. | ||
3.7 Moisture swing sorption: a new emerging CO2 regeneration technology for DAC
K. S. Lackner178 came up with the distinctive concept of moisture swing sorption for DAC to limit the exorbitant expense of amine-based chemisorbent regeneration. The idea of moisture swing adsorption is very similar to green leaves, which capture net CO2 in the presence of sunlight and release it at night. Moisture swing CO2 sorbent act as an “artificial leaf” to bind CO2 in nearly dry conditions and release it in humid environments by changing the moisture content of the surrounding air. Therefore, the interconversion of carbonate and bicarbonate moieties proposes the adsorption and desorption process.179 Due to their minimal environmental and economic cost by avoiding expensive heating/cooling cycles180 or significant changes in the pressure over the sorbent, these materials are promising for reversible CO2 capture through humidity swing and appealing for large-scale applications.181 The fact that the humidity swing process needs to operate in a dry area, and the limited adsorption capacity are the few potential downsides of a humidity swing method. Further, a large-scale DAC plant based on moisture swing requires significant water to operate. It is estimated that about 10 gigatons of CO2 could use as much as 100 km3 of water,9 compared to a world water consumption of 3600 km3.182 The reaction pathway of CO2 sorption/desorption on a moisture-swing absorbent reported by Shi et al.181 is shown in Fig. 13 and eqn (7)–(10).| H2O ⇌ H+ + OH− | (7) |
| CO32− + H+ ⇌ HCO3− | (8) |
| OH− + CO2 ⇌ HCO3− | (9) |
| HCO3− + HCO3− ⇌ CO32− + CO2 + H2O | (10) |
| CO32−·nH2O ⇌ HCO3−·m1H2O + OH−·m2H2O + (n − m1 − m2 − 1)H2O | (11) |
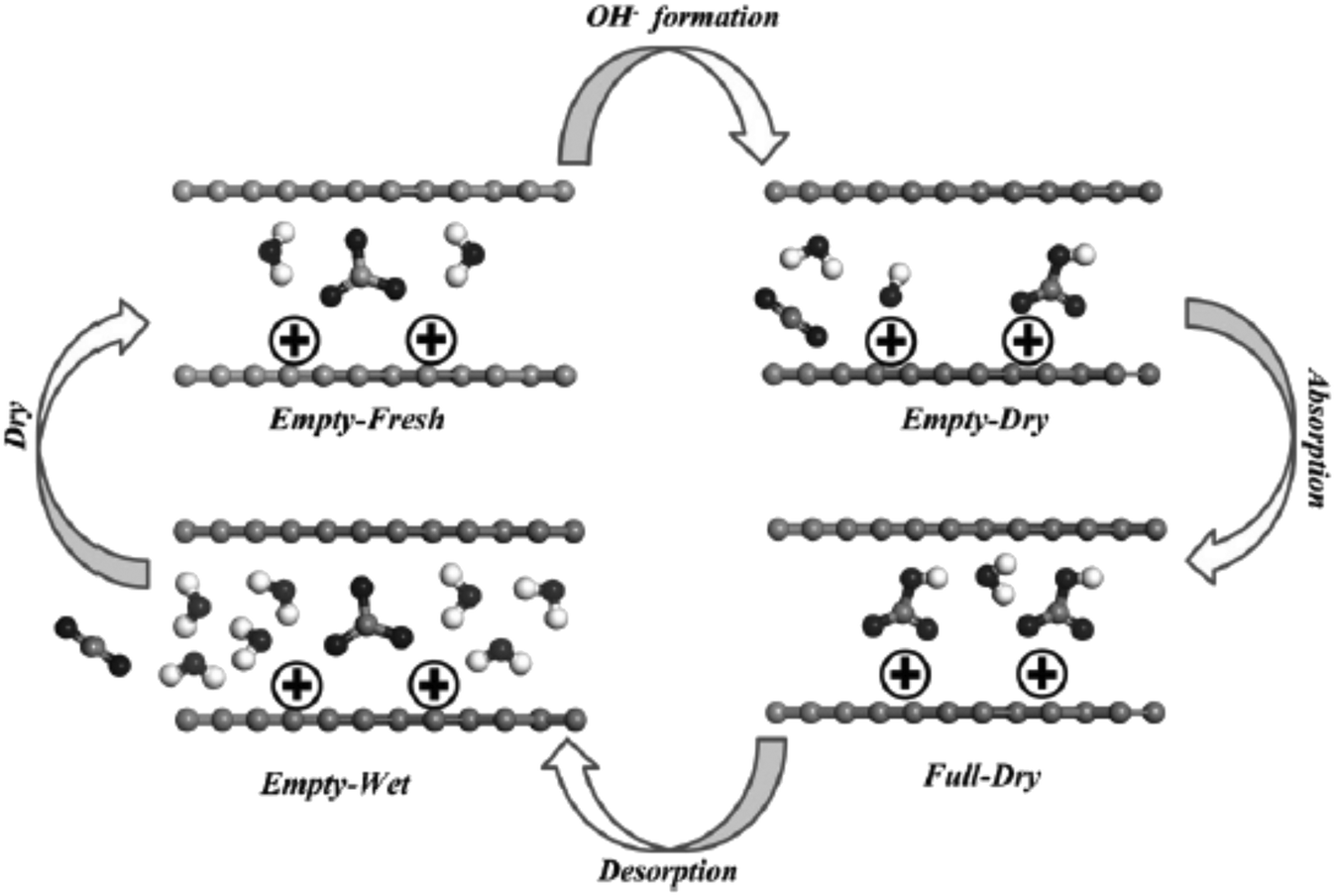 | ||
| Fig. 13 Reaction pathway of CO2 absorption/desorption on a moisture-swing absorbent. The empty-fresh state is the sorbent in dry condition with a few water molecules in the surrounding. The empty-dry state is when H2O splits into H+ ion and OH− ion, which is ready to absorb CO2, and H+ ion is combined with CO32− forming HCO3− ion [eqn (7) and (8)]. The full-dry state is the fully loaded sorbent in the dry condition [eqn (9)], eqn (7)–(9) present the absorption process. The empty-wet state is the regenerating absorbent releases CO2 in the wet condition (desorption [eqn (10)]) reproduced with permission.181 Copyright 2016, Wiley. | ||
Typically, ion hydration/dehydration mechanism in ion-exchange resin plays a greater role in the moisture-swing sorption system. The eqn (11) belongs to the hydration of water in the moisture-swing adsorbent, representing the equilibrium between carbonate and bicarbonate ions. The lower is water molecules (n) in dry conditions, the higher is the OH− ions, and hydrated bicarbonate generation, resulting in larger CO2 capture in the sorbent. However, at higher water molecules in wet conditions, carbonate ion hydration occurs, resulting increase in hydration Gibbs free energy and the release of CO2. As evidenced from eqn (11), Shi et al.181 reported that with an increase in 8 to 60 water molecules, the difference in hydration Gibbs free energy increases quickly from a negative to a positive value, then becomes stable at a plateau of 15 kcal mol−1 in bulk water. Many moisture-swing sorbents consist of quaternary ammonium ions (NR4+), which are strong bases in aqueous solutions similar to Na+ ions. The cations in the matrix or support are replaced by anions such as carbonate, bicarbonate, and hydroxide ions during CO2 adsorption. Their relative abundance depends on the CO2 loading of the resin and the quantity of moisture present.9 For moisture swing adsorption, the reference material is Excellion membrane (I-200), a commercially available extruded polyethylene membrane containing crosslinked chloromethylated polystyrene resin with quaternary ammonium hydroxide groups (NMe3+–MS OH−).179 This membrane has been utilized to capture CO2 due to humidity changes. However, the Excellion membrane displayed slow desorption and absorption profiles with an overall rate of 2.1 × 10−3 mmol g−1 min−1 and a moderate swing size of 1.3 × 10−1 mmol g−1 (Table 6, entry 1).179 Further, the Excellion membrane required over 2 h to capture and release 0.13 mmol of CO2 per 1 g of material to complete a cycle. To overcome the slow rate of adsorption/desorption, new materials were explored where ammonium ions lacked abstractable β-hydrogens, and the material did not contain ester or amide bonds that could be hydrolysed.179 He et al.179 designed a templated p(NMe3+–MS OH−) carbon black sorbent containing three methyl substituents and one benzyl substituent which links the ammonium group to the polymer backbone. The material outperformed the other four materials (Table 6). The overall rate of CO2 capture and release for CB-g-p(NMe3+–MS OH−) was found to be 3 times higher than the Excellion membrane. Moreover, the swing size was 3.8 times larger owing to its surface area of 6.2 m2 g−1 and a highly porous structure with an average pore size of 0.83 mm. On the other hand, compared to the materials which were made by conventional high internal phase emulsion (HIPE) polymerization, the increased pore size found in porous polymers generated using Pickering HIPE polymerization improved CO2 capture ability.179 The biggest swing size was 7.2 × 10 −1 mmol g−1 (Table 6, entry 6) and the quickest overall rate was 2.5 × 10−2 mmol g−1 (Table 6, entry 4), both of which were 5.5 and 11.9 times higher than those achieved with Excellion membrane.184 The ideal parameters of humidity swing sorbents for traditional point-source capture and those for air capture were found to be significantly different. Micro-/mesoporous materials with a high surface area are preferred for point source capture, while larger pore sizes and better interconnectivity pores (as seen in colloidal crystal templated material and the HIPE-based material) are better for direct air CO2 capture using a humidity swing process.179 Further, considering the energy consumption, the moisture swing adsorbents need much less energy for CO2 desorption (∼50 kJ mol−1)178 compared to other conventional adsorbents, which makes it economical for large-scale CO2 capture from the air and further utilization. Notably, the free energy released by water evaporation is utilized in moisture-induced cycle while avoiding the use of heat for sorbent regeneration.183
| Material | CO2 concentration (ppm) | Relative humidity (RH%) cycle | Swing size or CO2 sorption capacity (mmol g−1) | Absorption rate (mmol g−1 min−1) | Desorption rate (mmol g−1 min−1) | Overall rateb (mmol g−1 min−1) | Ref. |
|---|---|---|---|---|---|---|---|
| a Adsorption half time (minute). b Overall rate = 1/{(1/absorption rate) + (1/desorption rate)}. | |||||||
| Excellion membrane | Pure CO2 (90%) | 20% | 0.13 | 4.0 × 10−3 | 4.5 × 10−3 | 2.1 × 10−3 | 179 |
| CB-g-p(NMe3+–MS OH−) | 400 | 20% to 95% | 0.14 | 1.8 × 10−2 | 1.2 × 10−2 | 7.2 × 10−3 | 179 |
| Colloidal crystal templated p(NMe3+–MS OH−) | 400 | 20% to 95% | 0.37 | 2.8 × 10−2 | 1.8 × 10−2 | 1.1 × 10−2 | 179 |
| HIPE templated p(NMe3+–MS OH−) | 400 | 20% to 95% | 0.49 | 1.1 × 10−1 | 3.3 × 10−2 | 2.5 × 10−2 | 179 |
| PolyHIPE (1.5% crosslinking) | 400 | 20% to 95% | 0.23 | 3.2 × 10−2 | 3.0 × 10−2 | 1.5 × 10−2 | 184 |
| Pickering HIPE polymer | 400 | 20% to 95% | 0.72 | 3.7 × 10−2 | 2.7 × 10−2 | 1.6 × 10−2 | 184 |
| CB-g-xPCMS-OH | 400 | 20% to 95% | 0.14 | 2.0 × 10−2 | 1.5 × 10−2 | 8.6 × 10−3 | 185 |
| PS-CC-film/VBTMACl/PMVPMACl/M/20% | 400 | 20% to 95% | 0.57 | 6.4 × 10−3 | 1.4 × 10−2 | 4.4 × 10−3 | 186 |
| Polypropylene and a resin – composite sheet comprising quaternary ammonium functional groups | 400 | 0.5% | 0.82 | — | — | 187 | |
| Q-cellulose | 400 | 20% to 90% | 0.18 | 9.8 (25 °C)a | 188 | ||
| (QCS)/poly(vinyl alcohol)-hybrid aerogels | 400 | 3% to 95% | 0.18 | 10.72 (20 °C)a | 189 | ||
3.8 Techno-economic analysis of amine-based adsorbents for DAC
The techno-economic analysis is carried out to predict the economic viability of the DAC technology using the energy and cost estimation of the overall process.15 In this review, the techno-economic analysis of DAC based on amine-based adsorbents is briefly discussed. Besides the adsorbent's properties, the cost of CO2 capture using amine-based adsorbents for DAC is highly dependent on the design of the adsorbent contactor and the regeneration process.15,190 Since a high airflow rate over the adsorbent contractor is required for DAC, air compression may not be a good alternative, leaving temperature and vacuum the only viable regeneration drivers. As a result, the vacuum-thermal swing adsorption (TVSA) cycle is used in most sorbent-based DAC systems.36 DAC companies such as Climeworks and Global Thermostat, use the contractor of air-filter-like structures and honeycomb monoliths, respectively, and a TVSA process for CO2 capture from air.36Sabatino et al.36 recently reported a sorbent-based DAC system using the TVSA process, where the process comprises (i) adsorption, (ii) purge, (iii) regeneration, and (iv) repressurization steps. The adsorption step was carried out at ambient conditions (Tads = 20 °C, CO2 concentration: 400 ppm), and the preheating and regeneration step (Tdes = 100–120 °C) were carried out with the virtue of simultaneous heating and vacuum (0.1 bar) to remove N2 from void space and recover the captured CO2 from the adsorbent respectively. The group estimated the energy, and cost of DAC using the VTSA process on four amine-based adsorbents namely APDES–NFC, TRI-PE-MCM-41, MIL-101(Cr)-PEI-800, and Lewatit VP OC 106. In all cases, the CO2 purity was found to be in the range of 94% to 99%, with water being the main impurity. The overall CO2 capture cost was estimated to be 149–427 $ per tCO2. For these four amine-based sorbent processes, the total energy demand varied in a range of 4.9–13.3 MJ kg−1 CO2, where the thermal energy had the most significant contribution (4–11.8 MJ kg−1), whereas the electrical energy accounted for 0.8–1.8 MJ kg−1, which indicated that the results were in line with the Climeworks and Global Thermostat report.35,191,192 The process developed by Climeworks uses special cellulose fiber supported by amines in a solid form requiring 0.7–1.0 MJ kg−1 (electrical) and 5.4–7.2 MJ kg−1 (thermal) energy.35 An entire DAC cycle of Climeworks runs about 4–6 h and produces 99.9% pure CO2, using waste heat as a thermal source for regeneration.35 Global Thermostat, a USA-based company, used amine-polymer adsorbent for a TVSA-based DAC system. The adsorption is carried out under ambient conditions, and the desorption is accompanied by vacuum and saturated steam at 85–95 °C, producing CO2 purity >98.5%. The total cycle time of the overall process is below 30 min, and the net thermal and electrical energy demands were 4.2–5.0 MJ kg−1, and 0.54–0.94 MJ kg−1, respectively.193
A similar observation was also found by Sinha et al.194 using MIL-101(Cr)-PEI-800 and mmen-Mg2(dobpdc) amine append MOFs for TVSA run DAC system, where Tads = 20 °C, CO2 concentration: 400 ppm, Tdes = 100 °C at 1 atm pressure. A study showed that sorbent would require a thermal energy demand of about 5.11 MJ kg−1 for the MIL-101(Cr)-PEI-800 and 3.6 MJ kg−1 for the mmen-Mg2(dobpdc). Their work showed that the net cost of CO2 removal would be in the range of 75–140 $ per tCO2 and 60–190 $ per tCO2 for the former and later sorbents respectively, considering the lifetime of adsorbent between 1 and 3 years. However, the CO2 capture cost of MIL-101(Cr)-PEI-800 and mmen-Mg2(dobpdc) using the same TVSA process could be reduced to below 75 $ per tCO2 and 82 $ per tCO2 if the sorbent cost was lowered to 15 $ per kg and 50 $ per kg respectively.195 Sadiq et al.196 also demonstrated a pilot-scale version of a mobile DAC machine using MOF–polymer nanocomposite using the TVSA process. The adsorption was carried out at ambient conditions by flowing the air over the adsorbent bed at a high flow rate (50 m3 h−1) with the help of a blower. The adsorbent was coated as a thin layer onto long resistive heating sheets. The desorption was done by heating the adsorbent bed to 80 °C under a vacuum (0.037 bar). The plant exhibited the lowest energy consumption for regeneration (5.76 MJ kg−1), moderate CO2 purity (70–80%), and an operational cost of 35–350 $ per tCO2.
In the same context, Kulkarni and Sholl used the TRI-PE-MCM-41 monolith and evaluated the energy and cost of the TSA-based DAC unit. The net energy demand was 0.79 MJ kg−1 (electrical energy) and 6.0 MJ kg−1 (thermal energy). Advantageously, the cost of capture was estimated to be ∼100 $ per tCO2.155 Bajamundi et al.197 reported a TVSA-based DAC 8-bed system using amine-functionalized polystyrene. The resultant thermal energy demand was 27.4 MJ kg−1 and the electrical energy demand was ∼5.0–26.3 MJ kg−1. The DAC module was able to produce CO2 purity up to 95%. Adsorption took place at ambient conditions (10–30 °C) and desorption was accomplished by heating the bed to around 80 °C coupled with a vacuum (<0.15 bar). Finally, McQueen et al.79 also evaluated TVSA-based DAC using monolith contactors made of amine-modified adsorbents. The design (using the 5-step TVSA process) and calculation are similar to Sinha et al.,194 but the cost of CO2 capture is evaluated based on the regeneration using different modes of renewable and conventional energy sources. According to their research, the cost of CO2 capture for DAC systems powered by geothermal, natural gas, and nuclear power plants was 205 $ per tCO2, 223 $ per tCO2, and 233 $ per tCO2, respectively. The energy and cost associated with the DAC system of various amine-based adsorbents are listed in Table 7.
| Sorbent | CO2 conc. (ppm) | Operating conditions | Energy demand (MJ kg−1) | Regen. source | CO2 purity (%) | CO2 production | DAC cost $ per tCO2 | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Adsorption | Desorption | ||||||||||
| T ads (°C) | T des (°C) | P des (bar) | E electrical | E themal | |||||||
| T ads, adsorption temperature; Tdes, desorption temperature; Pdes, desorption pressure; HT, high temperature; Elec., electrical. | |||||||||||
| Amine-based cellulose (Climeworks) | 400 | Ambient | 100 | 0.2 | 0.7–1.0 | 5.4–7.2 | Waste heat | 99.9 | 0.135 t CO2 per day | 75 | 35, 192, 197 |
| Amino-polymer (Global Thermostat) | 400 | Ambient | 85–95 | 0.5–0.9 | 0.5–0.9 | 4.2–5.0 | Steam | >98.5 | — | 113 | 193 |
| Amine-polystyrene | 400 | Ambient | 80 | <0.15 | 5.0–26.3 | 27.4 | Heating fluid | >95 | 3.4 kg CO2 per cycle | — | 197 |
| PE-MCM-41 + TRI | 400 | Ambient | 110 | 1.4 | 0.78 | 6 | Steam | 88 | 1.1 tCO2 per day | ∼100 | 155 |
| PE-MCM-41 + TRI | 400 | Ambient | 100–120 | 0.1 | 0.8–1.0 | 7.1–7.7 | Heating fluid | >94 | 3.8–10.6 kg m−3 h−1 | 149–427 | 36 |
| NFC + APDES | 400 | Ambient | 100–120 | 0.1 | 1.1–1.3 | 8.6–10.1 | Heating fluid | >94 | 3.8–10.6 kg m−3 h−1 | 149–427 | 36 |
| Lewatit | 400 | Ambient | 100–120 | 0.1 | 0.6–1.3 | 10.1–11.8 | Heating fluid | >94 | 3.8–10.6 kg m−3 h−1 | 149–427 | 36 |
| Mesoporous silica + PEI | 395.5 | Ambient | 130 | 1 | 3.4 | 3.2 | Elec. | 40 tCO2 per day | 108–152 | 124 | |
| MIL-101(Cr) + PEI-800 | 400 | Ambient | 135–480 | 1 | 5.1 | HT steam | — | — | 75–140 | 194 | |
| mmen-Mg2(dobpdc) | 400 | Ambient | 135–480 | 1 | 3.6 | HT stream | — | — | 60–190 | 194 | |
| MOF–polymer | 400 | Ambient | 80 | 0.037 | 2.4 | 5.7 | Heating sheets | >70 | 6 kg CO2 per day | 35–350 | 196 |
3.9 Comparison of evaluation parameters of amine-based solid adsorbent for DAC: a perspective
Since the existing CO2 market is too small compared to the net CO2 emitted every year (250 million tons per year vs. over 40 gigatons),198 the installation of large numbers of DAC plants at affordable prices is gaining interest globally. In such a case, the specific focus must be on the material design, particularly on the amine-based solid adsorbent for DAC. It is also required to compare all of their major evaluation parameters on a single platform. Patel et al.198 reported a critical assessment of evaluation parameters of the CO2 adsorbent materials such as CO2 adsorption capacity, CO2/other gas selectivity, CO2 kinetics, isosteric heat of adsorption, cost (adsorbent cost and CO2 capture cost) and economic feasibility, and regenerability or cyclic stability of adsorbents along with their targeted limit. Although the evaluation parameters were initially designed for CO2 capture from a point source, the same evaluation parameters could also be helpful for screening adsorbents for DAC with some modifications, as mentioned in Table 8.| Checkpoints | Evaluation criteria | Preferable desired limit |
|---|---|---|
| 1 | CO2 adsorption capacity | >2 mmol of CO2 per g of sorbent |
| 2 | CO2/other gas selectivity | ≥100 |
| 3 | CO2 adsorption kinetics | 80% uptake within 2 min (rate: 1 mmol g−1 min−1) |
| 4 | Heat of adsorption | 35–50 kJ mol−1 |
| 5 | Cyclic stability | >1000 cycles |
| 6 | Regeneration temperature | 80–120 °C |
| 7 | Thermal and mechanical stability | Adsorbent should be stable at >150 °C, with high mechanical strength and low attrition index (AI) |
| 8 | Sorbent cost | <$10 per kg |
However, while considering the amine-based adsorbents, none of them could satisfy all of the desired limits, making it challenging to design an optimum adsorbent for CO2 capture. Considering few of the typical evaluation parameters (CO2 uptake, heat of adsorption, and stability) of amine-based adsorbents akin to DAC conditions are compared and shown in Fig. 14. Though Mg2dobpdc + ED and Mg2dobdc + mmen appeared promising for DAC applications (Fig. 14) due of their high CO2 uptake (>2 mmol g−1), prolonged thermo-cyclic stability (∼100%), and marginal heat of adsorption (50–70 kJ mol−1), rigorous process modeling and adsorbent evaluation metrics are required to screen the adsorbents for practical DAC application.199,200 Nevertheless, a few of the amine-based adsorbents, such as MIL-101(Cr) + PEI, NFC + APDES, TRI-PE-MCM-41, and mmen-Mg2(dobpdc) have already drawn the attention of numerous researchers, and few of them have already been employed in commercial or pilot scale TVSA based DAC processes, as also discussed in the previous section (refer section 3.8).
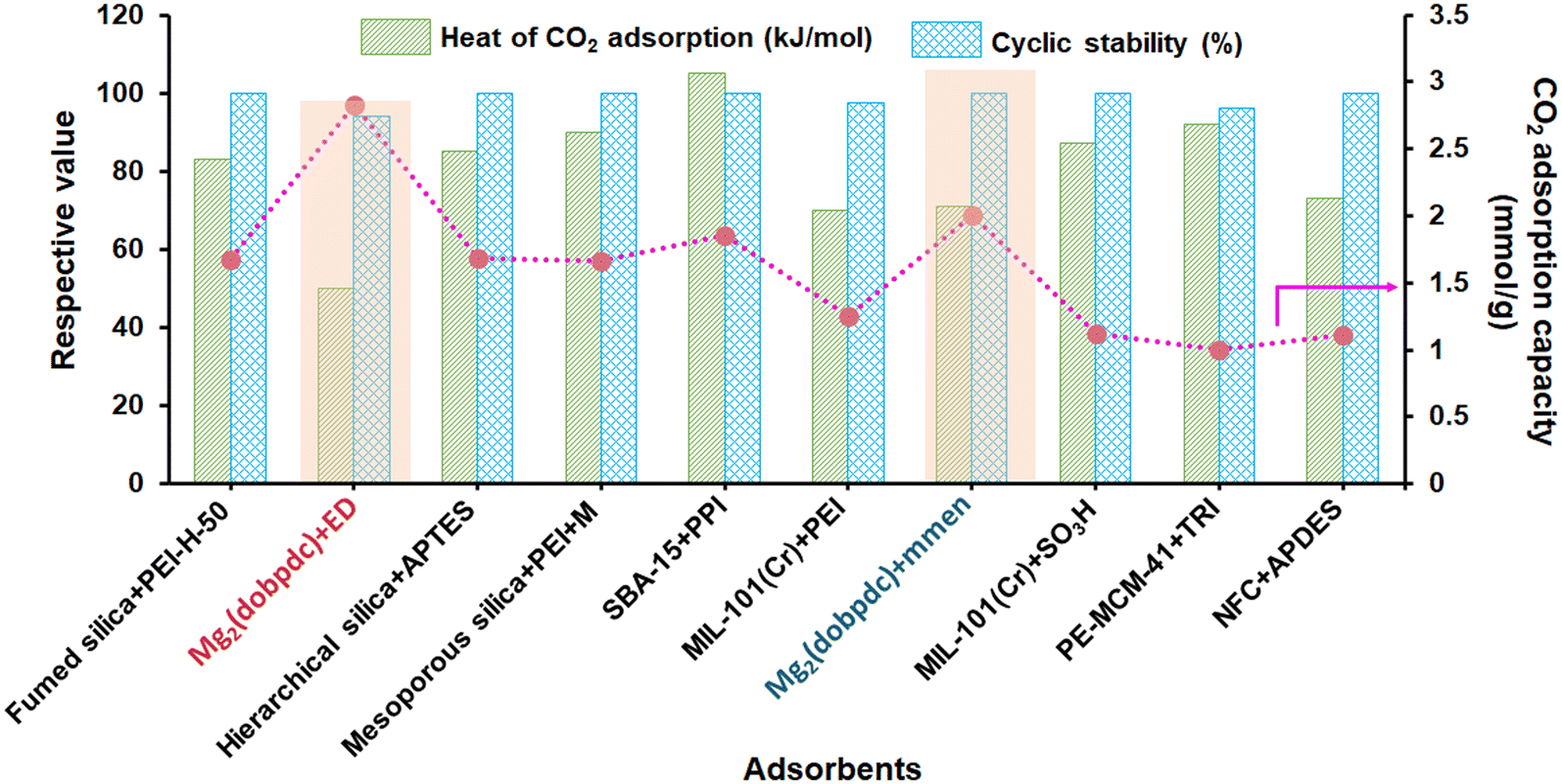 | ||
| Fig. 14 Comparison of evaluation parameters of various amine-based adsorbents (data was chosen from Tables 2, 3 and 5). | ||
In actual practice, the utilization of MOFs in DAC is limited due to their limited bulk-scale production and high capital cost. Despite the vast potential, the high cost associated with the precursor chemicals and machinery for the production of MOFs in large quantities retards its viability at present.201 However, the use of MOF on polymer support or MOF–polymer nanocomposite could decrease the overall cost of the adsorbent.194 The porous polymer support with low specific heat would be beneficial in transferring heat away from the material, thereby aiding in desorption.34 Satyapal et al.34 developed an HSC+ supported pressure swing adsorption (PSA) machine to remove CO2 in space shuttle applications. The device was able to run hundreds of cycles and capture CO2 up to 4% from dry air at ambient pressure with regeneration at a 1.33 mbar vacuum. After all, the field of DAC utilizing amine-based adsorbents is still in its infancy, and there is a lot of room for improvement in terms of sorbents and processes, of which only a handful are highlighted here.
(i) From the synthesis point of view, the focus should be on the cost-effective, facile, and faster synthesis of amine-grafted adsorbents, which typically consume several hours for amine condensation reaction.83 Further, limited research articles are available on type 3 adsorbents and type 4 adsorbents (double-functionalized amine-based adsorbents) for DAC applications.
(ii) Since thermal swing or vacuum-thermal swing regeneration is suitable for DAC, especially for amine-based adsorbents, the adsorbent design mainly influences the thermodynamics and kinetics of CO2 adsorption. The focus needs to be devoted towards developing large-scale structured adsorbents using emerging technology such as 3D printing to decrease the pressure drop along the adsorption column and better CO2 kinetics without affecting the CO2 adsorption performance as compared to the powdered form.
(iii) For a commercial DAC plant, the amine-based adsorbents must be stable for thousands of cycles. Though amine-impregnated adsorbents are suitable for high amine loading and hence excellent CO2 uptake, they faced severe issues in cyclic adsorption–desorption stability due to amine leaching. Therefore, amine-grafted adsorbents could be the preferred option for DAC due to their high thermocyclic, moisture, and oxidative stability compared to the amine-impregnated adsorbents. However, a greater focus still needs to be devoted to increasing the CO2 adsorption capacity of amine-grafted adsorbents.
(iv) The CO2 desorption process needs to be less energy-intensive and low-cost. Moreover, renewable energy resources need to be explored more for the desorption process to make the DAC process carbon neutral and sustainable. Selecting suitable amine-based adsorbents, which can be regenerated at low temperatures (80–120 °C), may greatly reduce the cost of CO2 capture.202 The techno-economic analysis of Fasihi et al.35 inferred that the utilization of waste heat for CO2 desorption can further reduce the CO2 capture cost by about 40% compared to the lack of free waste heat. The heat required for regeneration of sorbents, can come from a wide range of cost-effective thermal energy supply alternatives, such as heat pumps, geothermal and waste heat generated from some industrial plants such as combined heat and power plants, waste incinerators, electrolysers, or from the synthetic fuel production process.202,203 The combination of temperature and vacuum swing desorption (TVSD) can be used in practice to produce high purity CO2,204 which is well commercialized by the Climeworks plant at Hinwil,191 where combined heating and vacuum are used for CO2 desorption at <100 °C using waste heat developed from municipal waste incineration. Moreover, moisture swing adsorbents such as amine-based ion exchange resin, as reported by Lackner et al., can also be considered as a promising candidate due to its facile low temperature regeneration (45 °C).178
(v) Further, the selectivity of CO2 to other bulk components (such as moisture, O2, and N2) of amine-based adsorbents needs to be high, so that low parasitic energy205 is required for moisture or N2 regeneration during the desorption of CO2 and to produce high purity grade CO2 (99%) for pipeline transportation (pressure of 10–15 MPa is typically chosen).206
(vi) For moisture-swing adsorption, the used adsorbent, such as ion-exchange resin, can be manufactured with a greater charge density to improve the capacity of moisture-swing sorbents, and materials with varied cation distances can be employed under different humidity conditions.9
(vii) Finally, the most critical aspect is the cost of CO2 capture, which depends upon all the above-discussed milestones. A high sorbent CO2 uptake capacity and high CO2/other gas selectivity can reduce the cost of CO2 capture by reducing the amount of sorbent required.198 Research should be focused on gathering reliable data on the techno-economic and life-cycle performance of DAC under the actual conditions to predict the environmental and economic viability.15 Furthermore, incentives for CO2 storage should be encouraged, such as the 45Q tax credit in the US, which will stimulate learning and cost reductions of DAC at this early stage of its development.207
4 Summary and outlook
CO2 capture from the air is the need of the hour to combat anthropogenic CO2 emissions from small and distributed sources, including commercial/domestic fuel-based thermal appliances and the transportation sector. The ability of DAC to harvest CO2 directly from the atmosphere and sequester it onboard without any need for sophisticated infrastructure and prolonged transportation to recycling sites creates a new era for suitable material and process development. Amine-based materials have shown promising characteristics to be used as suitable adsorbents for DAC, owing to their high CO2 adsorption capacity at ultra-dilute CO2 concentration, excellent CO2 over other components selectivity, and high adsorption under humid conditions, and lower regeneration energy corresponding to liquid amines. Moreover, amine-based adsorbents can be synthesized in a variety of processed including impregnation, grafting, in situ polymerization, and double functionalization techniques. Advantageously, the textural properties and nature of the amine-based adsorbents have been extensively optimized and established to define their suitability for CO2 captures, such as their giant surface area (for better dispersion amine without clogging), large pore volume (to accommodate more amine moieties), hierarchal pore structure with interconnected pore opening (to facilitate better amine mobility without steric restrictions and faster CO2 kinetics), enriched surface silanol group (for occluding more amine species by chemical grafting) and so on.Further, DAC performance depends on the amine loading, amine type, amine density, and its basicity. Impregnating more reactive amine species (primary amine) into the pore improves CO2 uptake until the percolation is achieved. The preliminary evaluation parameters for optimal sorbent design and their future outlooks have been discussed in this review. Exploration of amine-modified MOF-based adsorbents via in situ polymerization and double functionalization techniques, and their techno-economic analyses needs more attention. Furthermore, to reduce the heat and mass transfer limitation, the regeneration enthalpy of CO2 of amine-based adsorbents, and the cost of CO2 capture, innovative regeneration techniques such as moisture swing, electro swing, and magnetic swing need to be further explored and implemented at commercial level. Overall, sorbent design is a crucial factor in DAC, which still needs to be explored further to govern the cost of the DAC process. In the best-case scenario, with extensive and focused efforts, the long-term cost of CO2 removal using the DAC technique can be reduced from its existing price to 40-80 $ per tCO2. Aggressive research and innovation in DAC would undoubtedly slow down catastrophic global climate change, resulting in a better living environment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank IIT Indore and SERB New Delhi for the funding support.References
- O. Edenhofer, Y. Sokona, J. C. Minx, E. Farahani, S. Kadner, K. Seyboth, A. Adler, I. Baum, S. Brunner, B. Kriemann, J. Savolainen, S. Schlömer, C. von Stechow and T. Zwickel, Climate Change 2014 Mitigation of Climate Change Working Group III Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change Edited by, 2014 Search PubMed.
- O. T. Qazvini and S. G. Telfer, J. Mater. Chem. A, 2020, 8, 12028–12034 RSC.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2016, 16, 16–22 CrossRef PubMed.
- Global Monitoring Laboratory - Carbon Cycle Greenhouse Gases, https://gml.noaa.gov/ccgg/trends/, (accessed 28 October 2022).
- M. Ozkan, S. P. Nayak, A. D. Ruiz and W. Jiang, iScience, 2022, 25, 103990 CrossRef CAS PubMed.
- Net Zero Coalition|United Nations, https://www.un.org/en/climatechange/net-zero-coalition, (accessed 28 October 2022).
- U.S. Global Change Research Program, U.S. Glob. Chang. Res. Progr., 2018, 1, p. 470 Search PubMed.
- National Academies of Sciences, Engineering, and Medicine, Negative Emissions Technologies and Reliable Sequestration: A Research Agenda, The National Academies Press, Washington, DC, 2019, DOI:10.17226/25259.
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed.
- C. Beuttler, L. Charles and J. Wurzbacher, Front. Clim., 2019, 1, 10 CrossRef.
- D. Goud, R. Gupta, R. Maligal-Ganesh and S. C. Peter, ACS Catal., 2020, 10, 14258–14282 CrossRef CAS.
- D. W. Keith, M. Ha-Duong and J. K. Stolaroff, Clim. Change, 2006, 74, 17–45 CrossRef CAS.
- Can carbon capture facilities reverse climate change?|Climate Crisis News|Al Jazeera, https://www.aljazeera.com/news/2021/11/1/cop, (accessed 11 December 2021).
- Seizing the Moment – Why Carbon Capture is so Important for COP26 - Green Recruitment Company, https://www.greenrecruitmentcompany.com/blog/2021/11/seizing-the-moment-why-carbon-capture-is-so-important-for-cop26, (accessed 11 December 2021).
- M. Erans, E. S. Sanz-Pérez, D. P. Hanak, Z. Clulow, D. M. Reiner and G. A. Mutch, Energy Environ. Sci., 2022, 15, 1360–1405 RSC.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- P. Luis, Desalination, 2016, 380, 93–99 CrossRef CAS.
- B. R. Strazisar, R. R. Anderson and C. M. White, Energy Fuels, 2003, 17, 1034–1039 CrossRef CAS.
- A. Chakma and A. Meisen, J. Chromatogr. A, 1988, 457, 287–297 CrossRef CAS.
- K. Z. House, A. C. Baclig, M. Ranjan, E. A. Van Nierop, J. Wilcox and H. J. Herzog, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20428–20433 CrossRef CAS.
- A. Kiani, K. Jiang and P. Feron, Front. Energy Res., 2020, 8, 92 CrossRef.
- R. Zhang, T. Li, Y. Zhang, J. Ha, Y. Xiao, C. Li, X. Zhang and H. Luo, Sep. Purif. Technol., 2022, 300, 121702 CrossRef CAS.
- X. Hu, J. Huang, X. He, Q. Luo, C. Li, C. Zhou and R. Zhang, Fuel, 2022, 316, 123216 CrossRef CAS.
- G. Chen, G. Chen, M. Peruzzini, R. Zhang and F. Barzagli, Sep. Purif. Technol., 2022, 291, 120939 CrossRef CAS.
- R. Zhang, X. He, T. Liu, C. Li, M. Xiao, H. Ling, X. Hu, X. Zhang, F. Tang and H. Luo, Sep. Purif. Technol., 2022, 295, 121292 CrossRef CAS.
- S. Shayegh, V. Bosetti and M. Tavoni, Front. Clim., 2021, 3, 46 Search PubMed.
- F. Zeman, Environ. Sci. Technol., 2007, 41, 7558–7563 CrossRef CAS PubMed.
- A. Bandi, M. Specht, T. Weimer and K. Schaber, Energy Convers. Manage., 1995, 36, 899–902 CrossRef CAS.
- R. Socolow, M. Desmond, R. Aines, J. Blackstock, O. Bolland, T. Kaarsberg, N. Lewis, M. Mazzotti, A. Pfeffer, K. Sawyer, J. Siirola, B. Smit and J. Wilcox, Direct Air Capture of CO2 with Chemicals: A Technology Assessment for the APS Panel on Public Affairs, American Physical Society Search PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- E. E. Ünveren, B. Ö. Monkul, Ş. Sarıoğlan, N. Karademir and E. Alper, Petroleum, 2017, 3, 37–50 CrossRef.
- A. Cherevotan, J. Raj and S. C. Peter, J. Mater. Chem. A, 2021, 9, 27271–27303 RSC.
- Y. Belmabkhout, R. Serna-Guerrero and A. Sayari, Ind. Eng. Chem. Res., 2010, 49, 359–365 CrossRef CAS.
- S. Satyapal, T. Filburn, J. Trela and J. Strange, Energy Fuels, 2001, 15, 250–255 CrossRef CAS.
- M. Fasihi, O. Efimova and C. Breyer, J. Cleaner Prod., 2019, 224, 957–980 CrossRef CAS.
- F. Sabatino, A. Grimm, F. Gallucci, M. van Sint Annaland, G. J. Kramer and M. Gazzani, Joule, 2021, 5, 2047–2076 CrossRef CAS.
- A. Gambhir and M. Tavoni, One Earth, 2019, 1, 405–409 CrossRef.
- H. Thakkar, A. Issa, A. A. Rownaghi and F. Rezaei, Chem. Eng. Technol., 2017, 40, 1999–2007 CrossRef CAS.
- H. T. Kwon, M. A. Sakwa-Novak, S. H. Pang, A. R. Sujan, E. W. Ping and C. W. Jones, Chem. Mater., 2019, 31, 5229–5237 CrossRef CAS.
- A. Goeppert, H. Zhang, R. Sen, H. Dang and G. K. S. Prakash, ChemSusChem, 2019, 12, 1712–1723 CrossRef CAS PubMed.
- D. R. Kumar, C. Rosu, A. R. Sujan, M. A. Sakwa-Novak, E. W. Ping and C. W. Jones, ACS Sustainable Chem. Eng., 2020, 8, 10971–10982 CAS.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- G. Rim, F. Kong, M. Song, C. Rosu, P. Priyadarshini, R. P. Lively and C. W. Jones, JACS Au, 2022, 2, 380–393 CrossRef CAS PubMed.
- B. Lv, B. Guo, Z. Zhou and G. Jing, Environ. Sci. Technol., 2015, 49, 10728–10735 CrossRef CAS PubMed.
- S. Kim, H. Shi and J. Y. Lee, Int. J. Greenhouse Gas Control, 2016, 45, 181–188 CrossRef CAS.
- H. Bin Xie, Y. Zhou, Y. Zhang and J. K. Johnson, J. Phys. Chem. A, 2010, 114, 11844–11852 CrossRef PubMed.
- M. Caplow, J. Am. Chem. Soc., 1968, 90, 6795–6803 CrossRef CAS.
- P. V. Danckwerts, Chem. Eng. Sci., 1979, 34, 443–446 CrossRef CAS.
- A. Hartono, E. F. da Silva and H. F. Svendsen, Chem. Eng. Sci., 2009, 64, 3205–3213 CrossRef CAS.
- P. M. M. Blauwhoff, G. F. Versteeg and W. P. M. Van Swaaij, Chem. Eng. Sci., 1984, 39, 207–225 CrossRef CAS.
- R. B. Said, J. M. Kolle, K. Essalah, B. Tangour and A. Sayari, ACS Omega, 2020, 5, 26125–26133 CrossRef CAS PubMed.
- E. F. Da Silva and H. F. Svendsen, Ind. Eng. Chem. Res., 2004, 43, 3413–3418 CrossRef CAS.
- P. D. Vaidya and E. Y. Kenig, Chem. Eng. Technol., 2010, 33, 1577–1581 CrossRef CAS.
- B. Arstad, R. Blom and O. Swang, J. Phys. Chem. A, 2007, 111, 1222–1228 CrossRef CAS PubMed.
- G. F. Versteeg, L. A. J. Van Dijck and W. P. M. Van Swaaij, Chem. Eng. Commun., 1996, 144, 113–158 CrossRef CAS.
- R. Afonso, M. Sardo, L. Mafra and J. R. B. Gomes, Environ. Sci. Technol., 2019, 53, 2758–2767 CrossRef CAS PubMed.
- P. V. Kortunov, M. Siskin, L. S. Baugh and D. C. Calabro, Energy Fuels, 2015, 29, 5919–5939 CrossRef CAS.
- S. A. Didas, M. A. Sakwa-Novak, G. S. Foo, C. Sievers and C. W. Jones, J. Phys. Chem. Lett., 2014, 5, 4194–4200 CrossRef CAS PubMed.
- P. V. Kortunov, L. S. Baugh, M. Siskin and D. C. Calabro, Energy Fuels, 2015, 29, 5967–5989 CrossRef CAS.
- P. V. Kortunov, M. Siskin, M. Paccagnini and H. Thomann, Energy Fuels, 2016, 30, 1223–1236 CAS.
- P. V. Kortunov, M. Siskin, L. S. Baugh and D. C. Calabro, Energy Fuels, 2015, 29, 5940–5966 CrossRef CAS.
- Y. G. Ko, S. S. Shin and U. S. Choi, J. Colloid Interface Sci., 2011, 361, 594–602 CrossRef CAS PubMed.
- X. Shen, H. Du, R. H. Mullins and R. R. Kommalapati, Energy Technol., 2017, 5, 822–833 CrossRef CAS.
- T. Čendak, L. Sequeira, M. Sardo, A. Valente, M. L. Pinto and L. Mafra, Chem. – Eur. J., 2018, 24, 10136–10145 CrossRef PubMed.
- M. W. Hahn, M. Steib, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2015, 119, 4126–4135 CrossRef CAS.
- M. W. Hahn, J. Jelic, E. Berger, K. Reuter, A. Jentys and J. A. Lercher, J. Phys. Chem. B, 2016, 120, 1988–1995 CrossRef CAS PubMed.
- L. Mafra, T. Čendak, S. Schneider, P. V. Wiper, J. Pires, J. R. B. Gomes and M. L. Pinto, J. Am. Chem. Soc., 2017, 139, 389–408 CrossRef CAS PubMed.
- Y. Zhai and S. S. C. Chuang, Energy Technol., 2017, 5, 510–519 CrossRef CAS.
- S. A. Didas, R. Zhu, N. A. Brunelli, D. S. Sholl and C. W. Jones, J. Phys. Chem. C, 2014, 118, 12302–12311 CrossRef CAS.
- A. Sayari, A. Heydari-Gorji and Y. Yang, J. Am. Chem. Soc., 2012, 134, 13834–13842 CrossRef CAS PubMed.
- M. L. Pinto, L. Mafra, J. M. Guil, J. Pires and J. Rocha, Chem. Mater., 2011, 23, 1387–1395 CrossRef CAS.
- A. Sayari and Y. Belmabkhout, J. Am. Chem. Soc., 2010, 132, 6312–6314 CrossRef CAS PubMed.
- C. H. Chen, D. Shimon, J. J. Lee, F. Mentink-Vigier, I. Hung, C. Sievers, C. W. Jones and S. E. Hayes, J. Am. Chem. Soc., 2018, 140, 8648–8651 CrossRef CAS PubMed.
- R. W. Flaig, T. M. Osborn Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef CAS PubMed.
- E. G. Moschetta, M. A. Sakwa-Novak, J. L. Greenfield and C. W. Jones, Langmuir, 2015, 31, 2218–2227 CrossRef CAS PubMed.
- F. Inagaki, C. Matsumoto, T. Iwata and C. Mukai, J. Am. Chem. Soc., 2017, 139, 4639–4642 CrossRef CAS PubMed.
- M. Yang, C. Ma, M. Xu, S. Wang and L. Xu, Curr. Pollut. Rep., 2019, 5, 272–293 CrossRef CAS.
- Y. Deng, J. Li, Y. Miao and D. Izikowitz, Energy Rep., 2021, 7, 3506–3516 CrossRef.
- N. McQueen, K. V. Gomes, C. McCormick, K. Blumanthal, M. Pisciotta and J. Wilcox, Prog. Energy, 2021, 3, 032001 CrossRef.
- F. Kong, G. Rim, M. G. Song, C. Rosu, P. Priyadarshini, R. P. Lively, M. J. Realff and C. W. Jones, Korean J. Chem. Eng., 2022, 39, 1–19 CrossRef CAS.
- A. Goeppert, H. Zhang, M. Czaun, R. B. May, G. K. S. Prakash, G. A. Olah and S. R. Narayanan, ChemSusChem, 2014, 7, 1386–1397 CrossRef CAS PubMed.
- A. Kumar, D. G. Madden, M. Lusi, K. J. Chen, E. A. Daniels, T. Curtin, J. J. Perry and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed.
- C. Chen, S. Zhang, K. H. Row and W. S. Ahn, J. Energy Chem., 2017, 26, 868–880 CrossRef CAS.
- S. A. Didas, A. R. Kulkarni, D. S. Sholl and C. W. Jones, ChemSusChem, 2012, 5, 2058–2064 CrossRef CAS PubMed.
- A. Goeppert, H. Zhang, R. Sen, H. Dang and G. K. S. Prakash, ChemSusChem, 2019, 12, 1712–1723 CrossRef CAS PubMed.
- Y. Miao, Z. He, X. Zhu, D. Izikowitz and J. Li, Chem. Eng. J., 2021, 426, 131875 CrossRef CAS.
- D. W. F. Brilman and R. Veneman, Energy Procedia, 2013, 37, 6070–6078 CrossRef CAS.
- S. Choi, M. L. Gray and C. W. Jones, ChemSusChem, 2011, 4, 628–635 CrossRef CAS PubMed.
- W. Chaikittisilp, R. Khunsupat, T. T. Chen and C. W. Jones, Ind. Eng. Chem. Res., 2011, 50, 14203–14210 CrossRef CAS.
- A. Sayari, Q. Liu and P. Mishra, ChemSusChem, 2016, 9, 2796–2803 CrossRef CAS PubMed.
- M. E. Potter, K. M. Cho, J. J. Lee and C. W. Jones, ChemSusChem, 2017, 10, 2192–2201 CrossRef CAS PubMed.
- W. Chaikittisilp, J. D. Lunn, D. F. Shantz and C. W. Jones, Chem. – Eur. J., 2011, 17, 10556–10561 CrossRef CAS PubMed.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS PubMed.
- J. H. Drese, S. Choi, R. P. Lively, W. J. Koros, D. J. Fauth, M. L. Gray and C. W. Jones, Adv. Funct. Mater., 2009, 19, 3821–3832 CrossRef CAS.
- S. Choi, J. H. Drese, P. M. Eisenberger and C. W. Jones, Environ. Sci. Technol., 2011, 45, 2420–2427 CrossRef CAS PubMed.
- E. J. Acosta, C. S. Carr, E. E. Simanek and D. F. Shantz, Adv. Mater., 2004, 16, 985–989 CrossRef CAS.
- G. Qi, L. Fu, X. Duan, B. H. Choi, M. Abraham and E. P. Giannelis, Greenhouse Gases: Sci. Technol., 2011, 1, 278–284 CAS.
- N. Shanmugam, K. T. Lee, W. Y. Cheng and S. Y. Lu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2521–2526 CrossRef CAS.
- X. Wang, L. Chen and Q. Guo, Chem. Eng. J., 2015, 260, 573–581 CrossRef CAS.
- S. A. Didas, S. Choi, W. Chaikittisilp and C. W. Jones, Acc. Chem. Res., 2015, 48, 2680–2687 CrossRef CAS PubMed.
- A. R. Sujan, S. H. Pang, G. Zhu, C. W. Jones and R. P. Lively, ACS Sustainable Chem. Eng., 2019, 7, 5264–5273 CrossRef CAS.
- A. Heydari-Gorji and A. Sayari, Ind. Eng. Chem. Res., 2012, 51, 6887–6894 CrossRef CAS.
- A. Wagner, B. Steen, G. Johansson, E. Zanghellini, P. Jacobsson and P. Johansson, Int. J. Spectrosc., 2013, 2013, 1–8 CrossRef.
- W. J. Son, J. S. Choi and W. S. Ahn, Microporous Mesoporous Mater., 2008, 113, 31–40 CrossRef CAS.
- Z. Chen, S. Deng, H. Wei, B. Wang, J. Huang and G. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 6937–6945 CrossRef CAS PubMed.
- J. Wang, H. Huang, M. Wang, L. Yao, W. Qiao, D. Long and L. Ling, Ind. Eng. Chem. Res., 2015, 54, 5319–5327 CrossRef CAS.
- H. Sehaqui, M. E. Gálvez, V. Becatinni, Y. Cheng Ng, A. Steinfeld, T. Zimmermann and P. Tingaut, Environ. Sci. Technol., 2015, 49, 3167–3174 CrossRef CAS PubMed.
- S. H. Pang, R. P. Lively and C. W. Jones, ChemSusChem, 2018, 11, 2628–2637 CrossRef CAS PubMed.
- A. R. Sujan, D. R. Kumar, M. Sakwa-Novak, E. W. Ping, B. Hu, S. J. Park and C. W. Jones, ACS Appl. Polym. Mater., 2019, 1, 3137–3147 CrossRef CAS.
- G. Rim, T. G. Feric, T. Moore and A. H. A. Park, Adv. Funct. Mater., 2021, 31, 2010047 CrossRef CAS.
- K. A. S. Abhilash, T. Deepthi, R. A. Sadhana and K. G. Benny, ACS Appl. Mater. Interfaces, 2015, 7, 17969–17976 CrossRef CAS PubMed.
- J. A. Wurzbacher, C. Gebald and A. Steinfeld, Energy Environ. Sci., 2011, 4, 3584–3592 RSC.
- C. Gebald, J. A. Wurzbacher, P. Tingaut, T. Zimmermann and A. Steinfeld, Environ. Sci. Technol., 2011, 45, 9101–9108 CrossRef CAS PubMed.
- Y. Kong, G. Jiang, Y. Wu, S. Cui and X. Shen, Chem. Eng. J., 2016, 306, 362–368 CrossRef CAS.
- W. R. Lee, S. Y. Hwang, D. W. Ryu, K. S. Lim, S. S. Han, D. Moon, J. Choi and C. S. Hong, Energy Environ. Sci., 2014, 7, 744–751 RSC.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- N. Planas, A. L. Dzubak, R. Poloni, L. C. Lin, A. McManus, T. M. McDonald, J. B. Neaton, J. R. Long, B. Smit and L. Gagliardi, J. Am. Chem. Soc., 2013, 135, 7402–7405 CrossRef CAS PubMed.
- P. Q. Liao, X. W. Chen, S. Y. Liu, X. Y. Li, Y. T. Xu, M. Tang, Z. Rui, H. Ji, J. P. Zhang and X. M. Chen, Chem. Sci., 2016, 7, 6528–6533 RSC.
- C. Gebald, J. A. Wurzbacher, A. Borgschulte, T. Zimmermann and A. Steinfeld, Environ. Sci. Technol., 2014, 48, 2497–2504 CrossRef CAS PubMed.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna and H. C. Zhou, J. Phys. Chem. C, 2013, 117, 4057–4061 CrossRef CAS.
- G. Qi, L. Fu and E. P. Giannelis, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- H. Lyu, H. Li, N. Hanikel, K. Wang and O. M. Yaghi, J. Am. Chem. Soc., 2022, 2022, 12989–12995 CrossRef PubMed.
- A. Goeppert, M. Czaun, R. B. May, G. K. S. Prakash, G. A. Olah and S. R. Narayanan, J. Am. Chem. Soc., 2011, 133, 20164–20167 CrossRef CAS PubMed.
- W. Zhang, H. Liu, C. Sun, T. C. Drage and C. E. Snape, Chem. Eng. Sci., 2014, 116, 306–316 CrossRef CAS.
- W. Chaikittisilp, H. J. Kim and C. W. Jones, Energy Fuels, 2011, 25, 5528–5537 CrossRef CAS.
- M. A. Sakwa-Novak and C. W. Jones, ACS Appl. Mater. Interfaces, 2014, 6, 9245–9255 CrossRef CAS PubMed.
- X. Wang, X. Ma, V. Schwartz, J. C. Clark, S. H. Overbury, S. Zhao, X. Xu and C. Song, Phys. Chem. Chem. Phys., 2012, 14, 1485–1492 RSC.
- M. A. Sakwa-Novak, S. Tan and C. W. Jones, ACS Appl. Mater. Interfaces, 2015, 7, 24748–24759 CrossRef CAS PubMed.
- M. L. Sarazen, M. A. Sakwa-Novak, E. W. Ping and C. W. Jones, ACS Sustainable Chem. Eng., 2019, 7, 7338–7345 CrossRef CAS.
- A. Holewinski, M. A. Sakwa-Novak and C. W. Jones, J. Am. Chem. Soc., 2015, 137, 11749–11759 CrossRef CAS PubMed.
- C. Rosu, S. H. Pang, A. R. Sujan, M. A. Sakwa-Novak, E. W. Ping and C. W. Jones, ACS Appl. Mater. Interfaces, 2020, 12, 38085–38097 CrossRef CAS PubMed.
- S. H. Pang, L. C. Lee, M. A. Sakwa-Novak, R. P. Lively and C. W. Jones, J. Am. Chem. Soc., 2017, 139, 3627–3630 CrossRef CAS PubMed.
- Y. Kuwahara, D. Y. Kang, J. R. Copeland, N. A. Brunelli, S. A. Didas, P. Bollini, C. Sievers, T. Kamegawa, H. Yamashita and C. W. Jones, J. Am. Chem. Soc., 2012, 134, 10757–10760 CrossRef CAS PubMed.
- Y. Kuwahara, D.-Y. Kang, J. R. Copeland, P. Bollini, C. Sievers, T. Kamegawa, H. Yamashita and C. W. Jones, Chem. – Eur. J., 2012, 18, 16649–16664 CrossRef CAS PubMed.
- L. A. Darunte, A. D. Oetomo, K. S. Walton, D. S. Sholl and C. W. Jones, ACS Sustainable Chem. Eng., 2016, 4, 5761–5768 CrossRef CAS.
- X. Xu, M. B. Myers, F. G. Versteeg, B. Pejcic, C. Heath and C. D. Wood, Chem. Commun., 2020, 56, 7151–7154 RSC.
- L. He, M. Fan, B. Dutcher, S. Cui, X. Dong Shen, Y. Kong, A. G. Russell and P. McCurdy, Chem. Eng. J., 2012, 189–190, 13–23 CrossRef CAS.
- N. R. Stuckert and R. T. Yang, Environ. Sci. Technol., 2011, 45, 10257–10264 CrossRef CAS PubMed.
- S. Choi, T. Watanabe, T. H. Bae, D. S. Sholl and C. W. Jones, J. Phys. Chem. Lett., 2012, 3, 1136–1141 CrossRef CAS PubMed.
- H. Li, K. Wang, D. Feng, Y. P. Chen, W. Verdegaal and H. C. Zhou, ChemSusChem, 2016, 9, 2832–2840 CrossRef CAS PubMed.
- F. Q. Liu, L. Wang, Z. G. Huang, C. Q. Li, W. Li, R. X. Li and W. H. Li, ACS Appl. Mater. Interfaces, 2014, 6, 4371–4381 CrossRef CAS PubMed.
- V. Nikulshina, C. Gebald and A. Steinfeld, Chem. Eng. J., 2009, 146, 244–248 CrossRef CAS.
- Y. Belmabkhout, R. Serna-Guerrero and A. Sayari, Chem. Eng. Sci., 2010, 65, 3695–3698 CrossRef CAS.
- A. K. Voice and G. T. Rochelle, Ind. Eng. Chem. Res., 2014, 53, 16222–16228 CrossRef CAS.
- T. Wang and K. J. Jens, Energy Procedia, 2012, 23, 102–110 CrossRef CAS.
- S. Chi and G. T. Rochelle, Ind. Eng. Chem. Res., 2002, 41, 4178–4186 CrossRef CAS.
- M. Jahandar Lashaki, S. Khiavi and A. Sayari, Chem. Soc. Rev., 2019, 48, 3320–3405 RSC.
- A. Ahmadalinezhad and A. Sayari, Phys. Chem. Chem. Phys., 2013, 16, 1529–1535 RSC.
- A. Heydari-Gorji, Y. Belmabkhout and A. Sayari, Microporous Mesoporous Mater., 2011, 145, 146–149 CrossRef CAS.
- H. Zhang, A. Goeppert, G. K. S. Prakash and G. Olah, RSC Adv., 2015, 5, 52550–52562 RSC.
- S. Bali, T. T. Chen, W. Chaikittisilp and C. W. Jones, Energy Fuels, 2013, 27, 1547–1554 CrossRef CAS.
- C. S. Srikanth and S. S. C. Chuang, ChemSusChem, 2012, 5, 1435–1442 CrossRef CAS PubMed.
- C. S. Srikanth and S. S. C. Chuang, J. Phys. Chem. C, 2013, 117, 9196–9205 CrossRef CAS.
- W. Choi, K. Min, C. Kim, Y. S. Ko, J. W. Jeon, H. Seo, Y.-K. Park and M. Choi, Nat. Commun., 2016, 7, 12640 CrossRef CAS PubMed.
- A. R. Kulkarni and D. S. Sholl, Ind. Eng. Chem. Res., 2012, 51, 8631–8645 CrossRef CAS.
- L. A. Darunte, K. S. Walton, D. S. Sholl and C. W. Jones, Curr. Opin. Chem. Eng., 2016, 12, 82–90 CrossRef.
- S. Keskin, T. M. van Heest and D. S. Sholl, ChemSusChem, 2010, 3, 879–891 CrossRef CAS PubMed.
- P. Saha and S. Chowdhury, Insights Into Thermodynamics, ed. M. Tadashi, INTECH Open Acess Publisher, 2011, ch. 16, pp. 349–364 Search PubMed.
- A. L. Myers, AIChE J., 2002, 48, 145–160 CrossRef CAS.
- J. C. Moreno-Piraján and L. Giraldo, J. Chem. Eng. Data, 2020, 65, 3130–3145 CrossRef.
- A. Nuhnen and C. Janiak, Dalton Trans., 2020, 49, 10295–10307 RSC.
- D. Panda, E. A. Kumar and S. K. Singh, J. CO2 Util., 2020, 40, 101223 CrossRef CAS.
- M. E. Potter, S. H. Pang and C. W. Jones, Langmuir, 2016, 33, 117–124 CrossRef PubMed.
- M. A. Alkhabbaz, P. Bollini, G. S. Foo, C. Sievers and C. W. Jones, J. Am. Chem. Soc., 2014, 136, 13170–13173 CrossRef CAS PubMed.
- C.-J. Yoo, L.-C. Lee and C. W. Jones, Langmuir, 2015, 31, 13350–13360 CrossRef CAS PubMed.
- A. L. Kohl and R. B. Nielsen, Alkanolamines for Hydrogen Sulfide and Carbon Dioxide Removal, ed. A. L. Kohl and R. B. Nielsen, Gulf Professional Publishing, Houston, 1997, ch. 2, pp. 40–186 Search PubMed.
- A. Goeppert, S. Meth, G. K. S. Prakash and G. A. Olah, Energy Environ. Sci., 2010, 3, 1949–1960 RSC.
- J. A. Wurzbacher, C. Gebald, N. Piatkowski and A. Steinfeld, Environ. Sci. Technol., 2012, 46, 9191–9198 CrossRef CAS PubMed.
- A. A. Al-Absi, M. Mohamedali, A. Domin, A. M. Benneker and N. Mahinpey, Chem. Eng. J., 2022, 447, 137465 CrossRef CAS.
- B. Yeskendir, J. P. Dacquin, Y. Lorgouilloux, C. Courtois, S. Royer and J. Dhainaut, Mater. Adv., 2021, 2, 7139–7186 RSC.
- F. Rezaei and P. Webley, Sep. Purif. Technol., 2010, 70, 243–256 CrossRef CAS.
- S. Lawson, X. Li, H. Thakkar, A. A. Rownaghi and F. Rezaei, Chem. Rev., 2021, 121, 6246–6291 CrossRef CAS PubMed.
- H. Yang, X. Fang, Z. Li, H. Sun and H. Chen, ACS Sustainable Chem. Eng., 2022, 10, 1759–1764 CrossRef CAS.
- R. M. Heck, S. Gulati and R. J. Farrauto, Chem. Eng. J., 2001, 82, 149–156 CrossRef CAS.
- L. A. Darunte, Y. Terada, C. R. Murdock, K. S. Walton, D. S. Sholl and C. W. Jones, ACS Appl. Mater. Interfaces, 2017, 9, 17042–17050 CrossRef CAS PubMed.
- M. A. Sakwa-Novak, C. J. Yoo, S. Tan, F. Rashidi and C. W. Jones, ChemSusChem, 2016, 9, 1859–1868 CrossRef CAS PubMed.
- R. P. Wijesiri, G. P. Knowles, H. Yeasmin, A. F. A. Hoadley and A. L. Chaffee, Ind. Eng. Chem. Res., 2019, 58, 3293–3303 CrossRef CAS.
- K. S. Lackner, Eur. Phys. J.: Spec. Top., 2009, 176, 93–106 Search PubMed.
- H. He, W. Li, M. Zhong, D. Konkolewicz, D. Wu, K. Yaccato, T. Rappold, G. Sugar, N. E. David and K. Matyjaszewski, Energy Environ. Sci., 2013, 6, 488–493 RSC.
- T. Wang, J. Liu, M. Fang and Z. Luo, Energy Procedia, 2013, 37, 6096–6104 CrossRef CAS.
- X. Shi, H. Xiao, K. S. Lackner and X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4026–4029 CrossRef CAS PubMed.
- S. Rost, D. Gerten, A. Bondeau, W. Lucht, J. Rohwer and S. Schaphoff, Water Resour. Res., 2008, 44, 9405 CrossRef.
- T. Wang, K. S. Lackner and A. B. Wright, Phys. Chem. Chem. Phys., 2013, 15, 504–514 RSC.
- H. He, W. Li, M. Lamson, M. Zhong, D. Konkolewicz, C. M. Hui, K. Yaccato, T. Rappold, G. Sugar, N. E. David, K. Damodaran, S. Natesakhawat, H. Nulwala and K. Matyjaszewski, Polymer, 2014, 55, 385–394 CrossRef CAS.
- H. He, M. Zhong, D. Konkolewicz, K. Yacatto, T. Rappold, G. Sugar, N. E. David and K. Matyjaszewski, J. Mater. Chem. A, 2013, 1, 6810–6821 RSC.
- H. He, M. Zhong, D. Konkolewicz, K. Yacatto, T. Rappold, G. Sugar, N. E. David, J. Gelb, N. Kotwal, A. Merkle and K. Matyjaszewski, Adv. Funct. Mater., 2013, 23, 4720–4728 CrossRef CAS.
- T. Wang, K. S. Lackner and A. Wright, Environ. Sci. Technol., 2011, 45, 6670–6675 CrossRef CAS PubMed.
- C. Hou, Y. Wu, T. Wang, X. Wang and X. Gao, Energy Fuels, 2018, 33, 1745–1752 CrossRef.
- J. Song, J. Liu, W. Zhao, Y. Chen, H. Xiao, X. Shi, Y. Liu and X. Chen, Ind. Eng. Chem. Res., 2018, 57, 4941–4948 CrossRef CAS.
- G. Holmes and D. W. Keith, Philos. Trans. R. Soc., A, 2012, 370, 4380–4403 CrossRef CAS PubMed.
- S. Deutz and A. Bardow, Nat. Energy, 2021, 6, 203–213 CrossRef CAS.
- Direct air capture technology and carbon removal, https://climeworks.com/co2-removal, (accessed 7 May 2022).
- E. Ping, M. Sakwa-Novak and P. Eisenberger, in International Conference on Negative CO2 Emissions, Gothenburg, 2018 Search PubMed.
- A. Sinha, L. A. Darunte, C. W. Jones, M. J. Realff and Y. Kawajiri, Ind. Eng. Chem. Res., 2017, 56, 750–764 CrossRef CAS.
- H. Azarabadi and K. S. Lackner, Appl. Energy, 2019, 250, 959–975 CrossRef.
- M. M. Sadiq, M. P. Batten, X. Mulet, C. Freeman, K. Konstas, J. I. Mardel, J. Tanner, D. Ng, X. Wang, S. Howard, M. R. Hill and A. W. Thornton, Adv. Sustainable Syst., 2020, 4, 2000101 CrossRef CAS.
- C. J. E. Bajamundi, J. Koponen, V. Ruuskanen, J. Elfving, A. Kosonen, J. Kauppinen and J. Ahola, J. CO2 Util., 2019, 30, 232–239 CrossRef.
- H. A. Patel, J. Byun and C. T. Yavuz, ChemSusChem, 2017, 10, 1303–1317 CrossRef CAS PubMed.
- J. Park, H. O. Rubiera Landa, Y. Kawajiri, M. J. Realff, R. P. Lively and D. S. Sholl, Ind. Eng. Chem. Res., 2020, 59, 7097–7108 CrossRef CAS.
- Y. S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS.
- E. S. M. El-Sayed and D. Yuan, Green Chem., 2020, 22, 4082–4104 RSC.
- Y. Qiu, P. Lamers, V. Daioglou, N. McQueen, H. S. de Boer, M. Harmsen, J. Wilcox, A. Bardow and S. Suh, Nat. Commun., 2022, 13, 1–13 Search PubMed.
- C. Breyer, M. Fasihi, C. Bajamundi and F. Creutzig, Joule, 2019, 3, 2053–2057 CrossRef.
- J. Elfving, C. Bajamundi, J. Kauppinen and T. Sainio, J. CO2 Util., 2017, 22, 270–277 CrossRef CAS.
- M. Oschatz and M. Antonietti, Energy Environ. Sci., 2018, 11, 57–70 RSC.
- M. Anheden, A. Andersson, C. Bernstone, S. Eriksson, J. Yan, S. Liljemark and C. Wall, Greenh. Gas Control Technol., 2005, pp. 2559–2564 Search PubMed.
- G. F. Nemet, M. W. Callaghan, F. Creutzig, S. Fuss, J. Hartmann, J. Hilaire, W. F. Lamb, J. C. Minx, S. Rogers and P. Smith, Environ. Res. Lett., 2018, 13, 063003 CrossRef.
Footnote |
| † Authors with equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |