DOI:
10.1039/D3RA07416A
(Paper)
RSC Adv., 2023,
13, 34670-34680
Discovery, synthesis, and cytotoxic evaluation of isoquinolinequinones produced by Streptomyces albidoflavus derived from lichen†
Received
31st October 2023
, Accepted 22nd November 2023
First published on 28th November 2023
Abstract
Four isoquinolinequinones (1–4) were isolated from the fermentation broth of Streptomyces albidoflavus which were derived from lichens. Among them, mansouramycin H (1) was identified as a new isoquinolinequinone by comprehensive spectroscopic data analysis. The mansouramycins from S. albidoflavus presented broad cytotoxic activities, especially against MDA-MB-231, but the SAR and mechanism were still unclear. The total synthesis of mansouramycin H (1) and its twenty-three derivatives were completed and their cytotoxic activities against MDA-MB-231 were evaluated in vitro. Primary SAR revealed that the piperazine moieties introduced into the amino group at C-7 could improve the activities of mansouramycins. Benzoyl and phenylacetyl groups on piperazine fragments had better activities than those of benzyl substitution; the alkyl substituent on piperazine exhibited optimal activity. Among them, compound 1g showed the strongest cytotoxicity against MBA-MB-231 cells with an IC50 value of 5.12 ± 0.11 μM. Mechanistic studies revealed that 1g induced apoptosis in MBA-MB-231 cells through down-regulating the protein expression of Bcl-2, up-regulating the protein expression of bax, and, meanwhile, activating the cleavage of caspase-3 and caspase-9. 1g caused S phase cell cycle arrest in MBA-MB-231 cells by reducing the protein expression of CDK2 and cyclin A2 and increasing the protein levels of p21.
Introduction
Isoquinolinequinones with a cytotoxic cyclohexadiene diketone unit possess a highly effective and broad spectrum anti-tumor effect.1–3 Quinone was involved in the Michael addition reaction with DNA and/or essential proteins, which caused cell cycle arrest in tumor cells. In addition, it also disrupted the intracellular redox balance and promoted the production of reactive oxygen species (ROS), thereby triggering cell apoptosis.1–4 Since the first isoquinolinequinone, mimosamycin, was reported in 1976,5 more than fifty members of this class of alkaloids were isolated from natural sources. These isoquinolinequinones were mainly isolated from marine organisms, including sponges (e.g. mimosamycins from Reniera sp.6), bryozoans (e.g. caulibugulones from Caulibugula intermis7) and nudibranchs (e.g. fennebricins from Jorunna funebris8). In addition, mansouramycins with an isoquinolinequinone scaffold were also obtained from streptomycete.9,10 Most of the isoquinolinequinones exhibited excellent cytotoxic activity. For example, mimosamycin inhibited multiple tumour cells with IC50 values ranging from 0.04–10 μg mL−1,11 and mansouramycins presented strong cytotoxicity against a panel of up to 36 tumor cell lines.9,10 However, a few isoquinolinequinones did not show the expected activity or showed weak activity, indicating that the different substituents had a great influence on the toxicity of the compounds.
Lichens are commonly considered as reciprocal symbiosis organisms between fungi and cyanobacteria and/or green algae, and are an important source of secondary metabolites with potential anti-tumor activity.12–14 In searching for new potential cytotoxic isoquinolinequinones, we investigated the secondary metabolites of Streptomyces albidoflavus (YIM 131469), which was isolated from a lichen sample Parmotrema tinctorum collected on stone surfaces in Shangri-La, China. Four isoquinolinequinone alkaloids, mansouramycin H (1), mansouramycin F (2),10 3-methyl-7-(methylamino)-5,8-isoquinolinedione (3),9 and mansouramycin A (4),9 were identified from ethyl acetate extracts of a large-scale (120 L) fermentation broth of S. albidoflavus (Fig. 1). Nevertheless, the lack of structural diversity and the quality of isoquinolinequinones from S. albidoflavus limited our further research on their cytotoxic activities and the structure–activity relationship (SAR). To resolve the above questions, we achieved the total synthesis of mansouramycin H (1). And taking it as the lead compound, twenty-three derivatives (1a–1w) were synthesized using an analogous approach. The cytotoxic activity of all synthetic analogs on MBA-MB-231 cells were investigated. The compound 1g with the most optimal activity showed the effect of promoting apoptosis and cycle arrest in MBA-MB-231 cells.
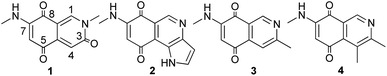 |
| | Fig. 1 Structures of compounds 1–4. | |
Results and discussion
Isolation and structural elucidation
The 120 L large-scale fermentation broth of strain YIM 131469 was collected and centrifuged to produce supernatant and mycelium. The supernatant was extracted with ethyl acetate and the organic phase was concentrated. The EtOAc extract (62 g) was isolated using various separation methods including silica gel, sephadex LH-20, and ODS to yield pure compounds 1–4.
Compound 1 was isolated as a bright yellow solid. Its molecular formula was determined to be C11H10N2O3 based on HRESIMS at m/z [M+H]+ 219.0763 and 13C NMR data. The IR spectrum revealed the characteristics of amino (3355 cm−1) and carbonyl groups (1734 cm−1). The 1H NMR data of 1 presented the presence of three singlet aromatic hydrogens at δH 8.70, 6.68, and 5.66, an amino hydrogen at δH 7.94 and two methyl groups at δH 3.57 and 2.79. The 13C NMR data of 1 along with the help of its HSQC experiment suggested eleven carbon signals, comprising three carbonyl carbons at δC 178.4, 177.1, and 162.5, six aromatic carbons at δC 151.3, 144.4, 141.0, 113.7, 110.1 and 100.8, and two methyls at δC 37.5 and 29.0. According to the analysis of the 1H and 13C NMR data of 1 and the comparison with the data of the known mimosamycin,15 the parent nucleus of 1 was determined to be isoquinoline-3,5,8-trione. Compound 1 differs from mimosamycin in that the methyl signal at position C-6 disappeared and the methoxy signal at position C-7 was replaced by an aminomethyl signal. COSY correlations from NH (δH 7.94) to CH3 (δH 2.79) and HMBC correlations from 7-NHCH3 (δH 2.79) to C-7 (δC 151.3), from H-6 (δH 5.66) to C-5 (δC 177.1) and C-4a (δC 141.0) aids corroborate this notion. Meanwhile, HMBC correlations observed from H-1 (δH 8.70) to C-3 (δC 162.5), C-4a (δC 141.0) and C-8 (δC 177.1), from H-4 (δH 6.68) to C-5 (δC 178.4), C-8a (δC 110.1), from 2-NCH3 (δH 3.57) to C-1 (δC 144.4), C-3 (δC 162.5) further confirmed the structure of 1. Therefore, 1 was confirmed as 2-methyl-7-(methylamino)isoquinoline-3,5,8(2H)-trione, named mansouramycin H.
Moreover, three known mansouramycins 2–4 were identified by comparing their spectroscopic data in the literature.9,10 The cytotoxic activity of isolated alkaloids 1–4 against six human cancer cell lines was investigated. The experimental results were shown in Table 1, compounds 1–4 exhibited different degrees of cytotoxic activities against a variety of tumor cells. Notably, MDA-MB-231 breast cancer cells were most sensitive to mansouramycins from YIM 131469. However, the low biomass of mansouramycins limited the further exploration of their activities. To further explore their SAR and molecular mechanism, the total synthesis and derivatization of mansouramycin H (1) were performed in the following study.
Table 1 Cytotoxic (IC50 in μM) activities of compound 1–4
| Compound |
A-549 |
BGC-823 |
HCT-116 |
Hepg2 |
Ishikawa |
MDA-MB-231 |
| 1 |
167.41 ± 4.89 |
>200 |
185.26 ± 0.78 |
191.23 ± 1.55 |
163.17 ± 5.93 |
122.20 ± 3.27 |
| 2 |
24.15 ± 1.63 |
29.90 ± 2.17 |
17.84 ± 3.05 |
18.79 ± 1.22 |
27.62 ± 3.34 |
14.41 ± 0.86 |
| 3 |
35.82 ± 1.88 |
23.01 ± 1.23 |
16.62 ± 0.11 |
19.54 ± 0.82 |
51.37 ± 3.00 |
22.82 ± 0.74 |
| 4 |
>200 |
>200 |
176.01 ± 4.87 |
164.53 ± 6.35 |
>200 |
101.71 ± 5.61 |
| Adriamycin |
1.31 ± 0.38 |
2.12 ± 0.12 |
0.63 ± 0.05 |
1.14 ± 0.21 |
1.74 ± 0.13 |
3.49 ± 0.50 |
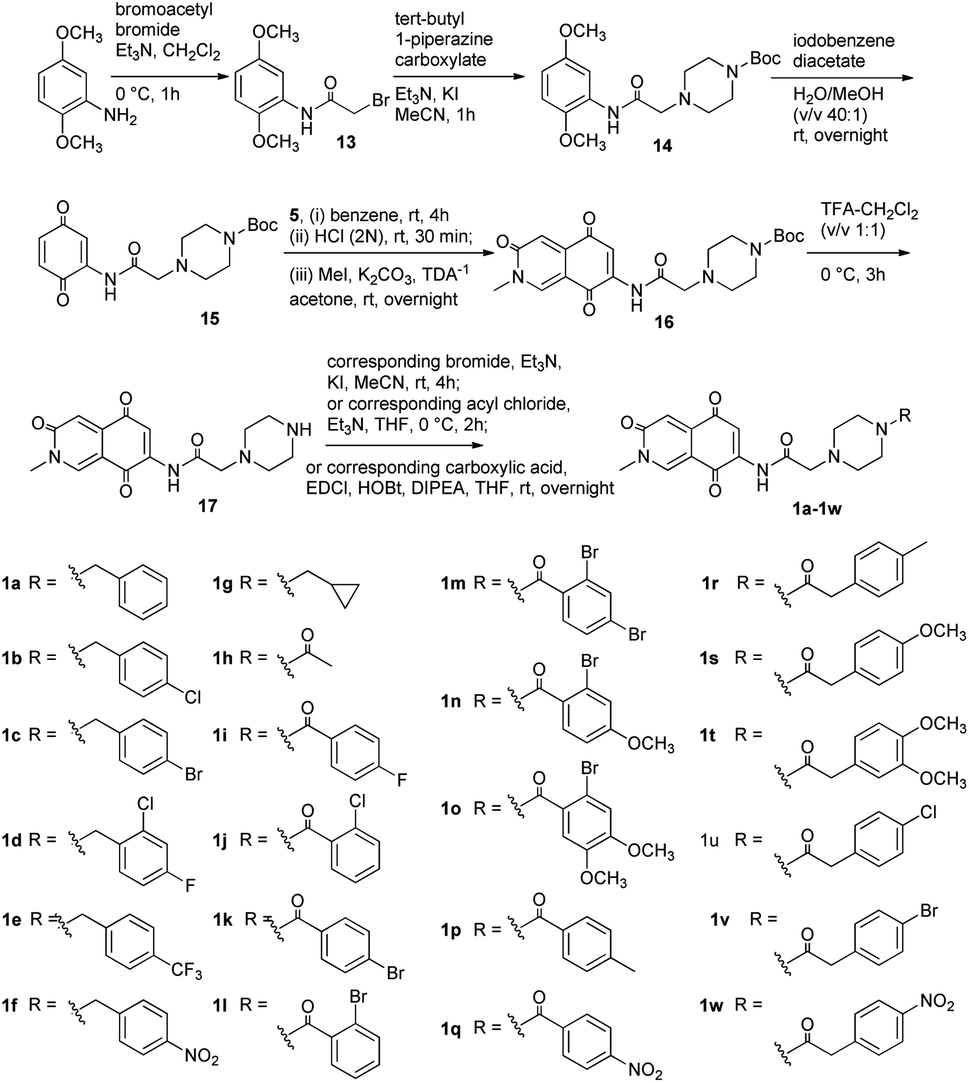
Synthesis of 1 and its derivatives
The retrosynthetic analysis of 1 was revealed in Scheme 1. The isoquinoline-3,5,8-trione skeleton was constructed by Diels–Alder reaction of compound 6 as a dienphile with diene 5. At the same time, 6 serves as a key intermediate molecule, which could be obtained from 8 through amino protection and oxidation.
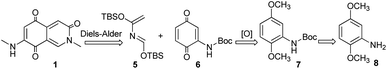 |
| | Scheme 1 Retrosynthetic analysis of compound 1. | |
According to the foregoing retrosynthetic strategy, our work began with the synthesis of dienophile 6 from commercially available 2,5-dimethoxyaniline (8). As outlined in Scheme 2, 8 was reacted with (Boc)2O to give the N-Boc-aniline 7 and subsequently oxidized to obtain 2-N-amino-p-benzenedione 6. Following the literature protocol,16 acetyl chloride was slowly dropped into the refluxing formamide to acquire N-formylacetamide 9, and then treated with tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf) to get 2-aza-1,3-butadiene 5. The Diels–Alder reaction between 6 and 5 was followed by acidification to successfully construct isoquinoline 3,5,8-trione. It undergone N-methylation reaction on nucleophilic N sites and boc protection removal in turn to obtain the target compound 1.
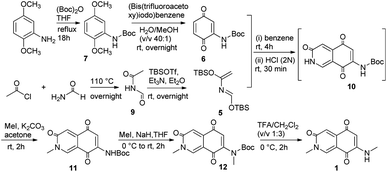 |
| | Scheme 2 Syntheses of compound 1. | |
It was found that the cytotoxic activity of compound 1 was not as good as expected (Table 1). We speculated that 1 contained an insoluble heterocyclic structure, which made it not easy to penetrate the cell membrane to play its biological role. Piperazine moiety was of great significance in the design of anti-tumor drugs due to its excellent pharmacokinetic properties. It could improve the lipid–water partition coefficient of compound and stimulate their bioactive potential.17,18 Therefore, we designed a series of derivatives 1a–1w by introducing the different piperazinyl acetic acid fragments on the amino group at C-7 of scaffold (Scheme 3). Using a similar approach of synthesizing compound 1, analogs 1a–1w were obtained. The synthesis of analogs was initiated with the preparation of compound 13 from commercial 2,5-dimethoxyaniline and bromoacetyl bromide.19 Then amide 14 was obtained by N-alkylation reaction of 13 and N-Boc-piperazine. The intermediate 14 was treated with iodobenzene diacetate to obtain the dienophile 15. After that, using the same method of preparing 11, the key intermediate 16 was obtained from 15 and diene 5 through Diels–Alder reaction and N-methylation in one pot. Finally, 16 was deprotected and treated with haloalkanes, substituted benzoic acids or phenylacetic acids to afford analogs 1a–1w.
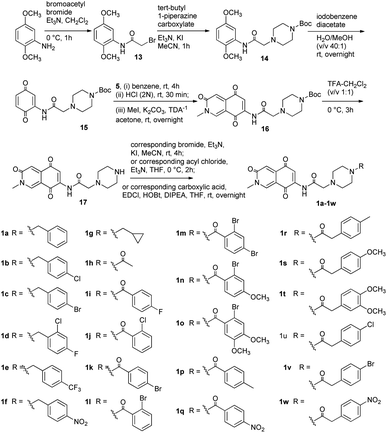 |
| | Scheme 3 Syntheses of compounds 1a–1w. | |
Cytotoxic activity and structure–activity relationships analyses
In order to search for more potent bioactive analogs, the cytotoxic activities of the synthesized derivatives were screened using MBA-MB-231 cells by MTT method. As shown in Table 2, most of the derivatives were more active than compound 1, indicating that introduction of the piperazine moieties could obviously enhance their cytotoxic activities. At the same time, the substituents at piperazine ring affected the activities of mansouramycins. The acyl substitution on piperazine can improve the biological activity of the compound (1h–1w > 1), and the phenylacetyl group was preferred over the benzoyl group (1r–1w > 1i–1o, 1q). Moreover, the acyl group was more beneficial than the benzyl group to improve the cytotoxic activity of the compound (1i–1p and 1r–1w > 1a–1f). However, the saturated alkyl substitution on the piperazine fragment was most effective in enhancing the cytotoxic activity (1g > 1a–1f and 1h–1w). As the most potential candidate for cytotoxic lead compound, cytotoxic spectrum of 1g was investigated. It showed broad cytotoxic activities against MDA-MB-231, HCT-116, BGC-823, A-549, HepG2, ishikawa cells, 293T cells and huvce cells, with IC50 values of 5.12, 7.24, 9.57, 7.50, 6.12, 8.45, 15.98 and 17.31 μM, respectively. Compound 1g exhibited selectivity towards various tumor cells (293T cells, SI: 1.67–3.12; Huvce cells, SI: 1.81–3.38). Especially, 1g displayed an optimal selectivity to MDA-MB-231 cells (SI 293T/MDA-MB-231: 3.12; Huvce/MDA-MB-231: 3.38). To understand the active mechanism of 1g, the effect of 1g on cell cycle and apoptosis in MDA-MB-231 cells were further explored in the following experiments.
Table 2 Cytotoxic activities of compound 1a–1w in MBA-MB-231 cells
| No. |
IC50 (μM) |
| 1a |
27.08 ± 2.23 |
| 1b |
34.30 ± 1.89 |
| 1c |
41.79 ± 1.52 |
| 1d |
>100 |
| 1e |
>100 |
| 1f |
>100 |
| 1g |
5.12 ± 0.11 |
| 1h |
17.38 ± 0.64 |
| 1i |
16.52 ± 0.41 |
| 1j |
11.54 ± 1.07 |
| 1k |
13.05 ± 1.01 |
| 1l |
12.05 ± 0.66 |
| 1m |
23.37 ± 0.99 |
| 1n |
16.33 ± 0.32 |
| 1o |
16.14 ± 0.74 |
| 1p |
10.34 ± 0.45 |
| 1q |
62.99 ± 3.15 |
| 1r |
6.89 ± 0.16 |
| 1s |
9.04 ± 0.67 |
| 1t |
9.95 ± 1.33 |
| 1u |
10.28 ± 1.18 |
| 1v |
8.88 ± 0.83 |
| 1w |
12.18 ± 1.54 |
1g caused MDA-MB-231 cell S phase arrest
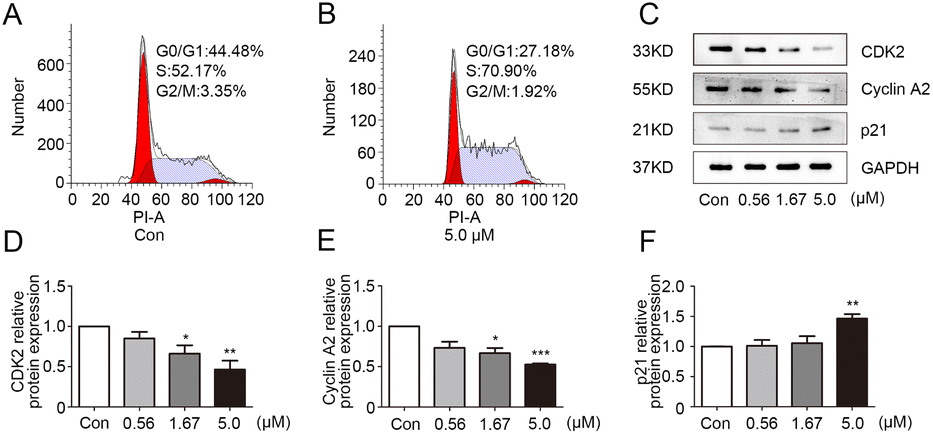
In order to exclude the influence of cell death on the test, the growth curve experiment was carried out. The results (Fig. S1†) showed that the three concentrations of 0.56, 1.67 and 5.0 μM had no obvious influence on the growth and proliferation of MDA-MB-231 cells in 24 h. Therefore, these dosages of 1g were selected for subsequent experiments. The effect of 1g on the cell cycle of MDA-MB-231 was analyzed by flow cytometry. As depicted in Fig. 2A and B, the proportion of MDA-MB-231 cells in the S phase was significantly higher in the 1g treatment group in comparison with vehicle group. The result suggested that 1g caused S phase cell cycle arrest in MDA-MB-231 cells. Cell cycle progression was positively regulated by cyclin-dependent kinases (CDKs) and CDK inhibitors.20 CDK2 can be activated by cyclin A and plays a vital role in cell division throughout S phase.21 As an important CKI, p21 negatively regulated cell cycle development by interacting with CDK2.22 To further explore the mechanism of 1g in S phase arrest, the expression levels of related regulators cyclin A2, CDK2, and p21 were determined by western blot in 1g-treated MDA-MB-231 cells. As shown in Fig. 2C–F, the protein expression levels of cyclin A2 and CDK2 were obviously down-regulated in 1g-treated cells, while the expression level of p21 was up-regulated in a dose-dependent manner. Consequently, these data confirmed that 1g effectively arrested the cell cycle of MDA-MB-231 cells in S phase.
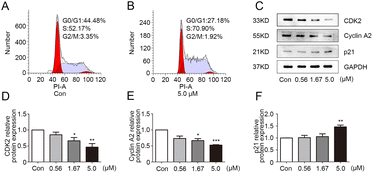 |
| | Fig. 2 Compound 1g caused S phase cell cycle arrest in MBA-MB-231 cells. (A and B) Cell cycle distribution were analyzed by flow cytometry in MBA-MB-231 cells treated with 1g (0 or 5.0 μM). (C) The protein levels of CDK2, cyclin A2 and p21 in MBA-MB-231 cells treated with 1g (0.56, 1.67 and 5.0 μM) were evaluated by western blot. (D–F) The relative quantities analysis of CDK2, cyclin A2 and p21 in MBA-MB-231 cells. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. control. | |
1g induced apoptosis in MDA-MB-231 cells
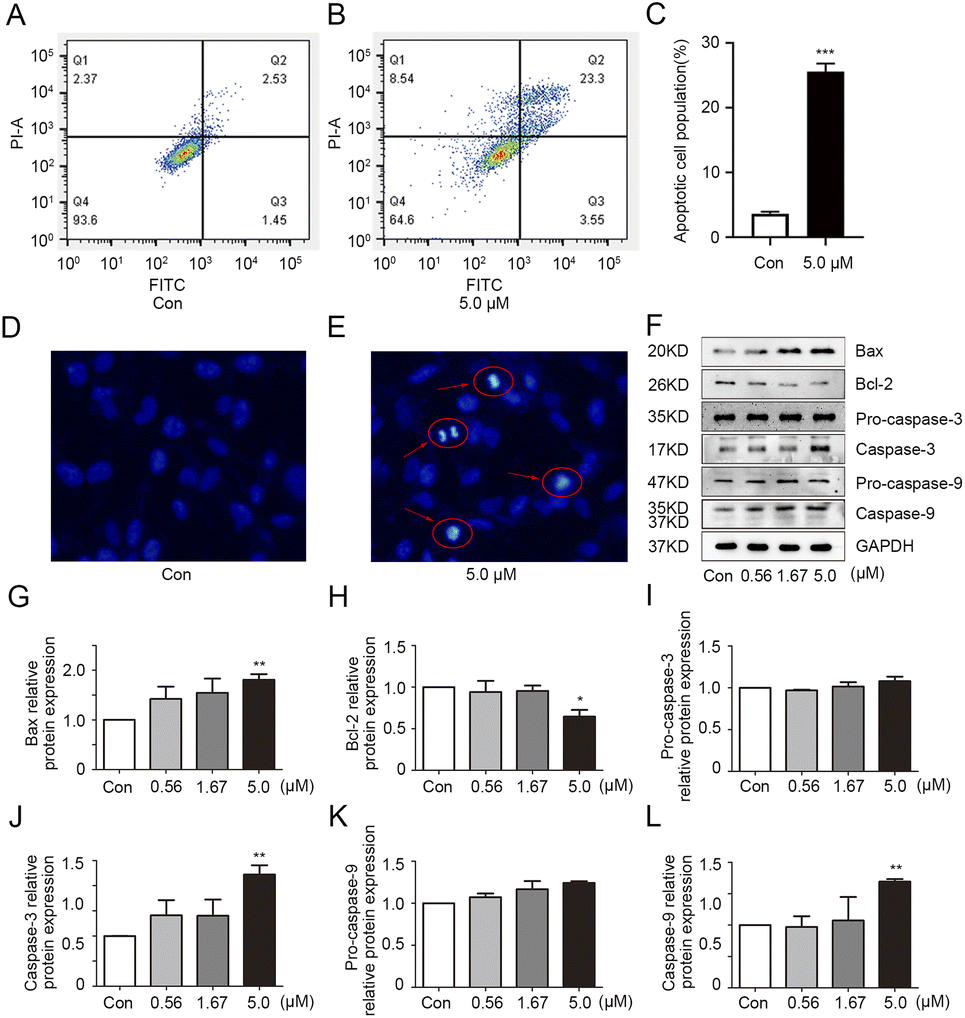
To explore the effect of 1g on apoptosis in MDA-MB-231 cells, apoptosis detection was performed by flow cytometric analysis. As shown in Fig. 3A–C, more apoptotic cell populations were observed in 1g-treated cells compared with the vehicle group. To further confirm above observation, the morphological alterations of apoptotic cells were examined by Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 258 staining method. As expected, the typical morphological characteristics of apoptosis such as cell membrane blistering, chromatin atrophy and the formation of apoptotic bodies were clearly observed in 1g-treated cells (Fig. 3D and E). All the above results revealed that 1g had the ability to induce apoptosis of MDA-MB-231 cells. The increase of the pro-apoptotic protein bax over the anti-apoptotic protein Bcl-2 could promote the release of apoptosis-inducing factors, thus playing a significant role in the process of apoptosis.23 Additionally, caspase-9 and caspase-3 are essential in the apoptotic cascade.24 In the process of execution of the apoptotic cascade, activated caspase-9 could cause the activation of executioner caspase-3 to trigger excessive programmed cell death. As shown in Fig. 3F–L, 1g positively regulated the protein expression of bax and the cleavage of pro-caspase 9 and pro-caspase 3 with a concentration-dependent manner, while inhibiting bcl-2 protein expression in MDA-MB-231 cells. Overall, these observations indicated 1g could induce cell death by increasing the protein ratio of bax/bcl-2 and promoting the activations of pro-caspase 9 and pro-caspase 3.
258 staining method. As expected, the typical morphological characteristics of apoptosis such as cell membrane blistering, chromatin atrophy and the formation of apoptotic bodies were clearly observed in 1g-treated cells (Fig. 3D and E). All the above results revealed that 1g had the ability to induce apoptosis of MDA-MB-231 cells. The increase of the pro-apoptotic protein bax over the anti-apoptotic protein Bcl-2 could promote the release of apoptosis-inducing factors, thus playing a significant role in the process of apoptosis.23 Additionally, caspase-9 and caspase-3 are essential in the apoptotic cascade.24 In the process of execution of the apoptotic cascade, activated caspase-9 could cause the activation of executioner caspase-3 to trigger excessive programmed cell death. As shown in Fig. 3F–L, 1g positively regulated the protein expression of bax and the cleavage of pro-caspase 9 and pro-caspase 3 with a concentration-dependent manner, while inhibiting bcl-2 protein expression in MDA-MB-231 cells. Overall, these observations indicated 1g could induce cell death by increasing the protein ratio of bax/bcl-2 and promoting the activations of pro-caspase 9 and pro-caspase 3.
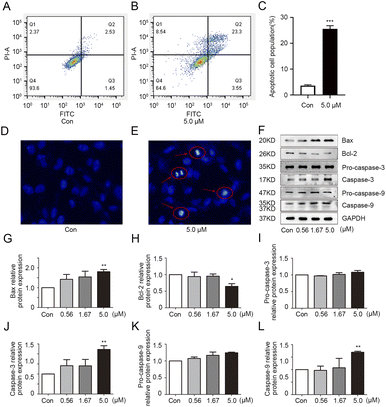 |
| | Fig. 3 Compound 1g induced apoptosis in MBA-MB-231 cells. (A–C) 1g induced apoptosis was analyzed by flow cytometry in MBA-MB-231 cells treated with 1g (0 or 5.0 μM). (D–E) The nuclear morphology alterations of MBA-MB-231 cells were detected by Hoechst 33258 staining. (F) The protein levels of bax, bcl-2, pro-caspase-3, caspase-3, pro-caspase-9 and caspase-9 in MBA-MB-231 cells treated with 1g (0.56, 1.67 and 5.0 μM) were evaluated by western blot. (G–L) The relative quantities analysis of bax, bcl-2, caspase-3, pro-caspase-3, caspase-9 and pro-caspase-9 in MBA-MB-231 cells. *p < 0.05, **p < 0.01, and ***p < 0.001 vs. control. | |
Conclusions
In conclusion, four isoquinolinequinone alkaloids were isolated from Streptomyces albidoflavus, including one novel compound mansouramycin H (1). Meanwhile, we have achieved the first total synthesis of the cytotoxic natural product 1 with 36.8% yield. The similar strategy was adopted to design and synthesize twenty-three derivatives of 1 (1a–1w), and most of them exhibited stronger cytotoxic activities than 1. According to SAR analysis, the introduction of the piperazine units significantly enhanced the cytotoxic activity of the compound in vitro. The substituents on the piperazine unit greatly affected the cytotoxic activity, with the saturated alkane substituent (1g) showing the optimal activity. The in-depth biological studies displayed obvious morphological changes, cell cycle arrest in S phase and apoptosis induction in MBA-MB-231 cells treated with 1g. Therefore, mansouramycins with piperazine unit may be valuable leads for the development of new antitumor drug candidates. However, the key molecular mechanism of mansouramycins and antitumor effect in vivo still needs to be determined.
Experimental section
General information
All reagents and solvents are purchased from commercial suppliers without further purification unless otherwise specified. Ultraviolet spectra were determined by a Beckman Coulter DU 730 nucleic acid/protein analyzer (Beckman Coulter, Brea, CA, USA). IR spectra (film) were obtained from a Tensor 27 FT-IR spectrometer (Bruker, Ettlingen, Germany). ESIMS were measured by using a 1290-6420 Triple Quadrupole LC-MS spectrometer (Agilent, Santa Clara, CA, USA). HRESIMS were recorded on an Agilent G6230 TOF mass spectrometer. 1D and 2D NMR experiments were performed on a Bruker AV-600 spectrometer, δ in ppm relative to TMS, J in Hz. Column chromatography were performed on YMC*GEL ODS-A (S-50 μm, 12 nm) (YMC Co., Ltd, Kyoto, Japan), silica gel (100–200 mesh, 300–400 mesh, 1000–1200 mesh, Qingdao Marine Chemical Ltd, Qingdao, China) and sephadex LH-20 (GE Healthcare Bio-sciences AB, Uppsala, Sweden). MTT assays were determined by a microplate reader (BioTek Synergy H1, BioTek Instruments, Inc., VT, USA). The protein extraction kit were produced by the Beyotime Institute of Biotechnology (Beijing, China). The primary antibodies for cyclin A2, CDK2, p21, bax, bcl-2, pro-caspase-3, caspase-3, pro-caspase-9, caspase-9, GAPDH, and horse-radish peroxidase (HRP)-conjugated secondary antibodies were purchased from Cell Signaling Technology (Dancers, MA, USA).
Microbial material
Strain S. albidoflavus (YIM 131469) was isolated from a lichen sample Parmotrema tinctorum collected on stone surfaces in Shangri-La, Yunnan Province, China. The identification of this strain was based on the phylogenetic analysis of the 16S rRNA gene sequence, and the similarity with Streptomyces albidoflavus (GenBank accession no. AB571553) was 99.69%. The strain (No. YIM 131469) was deposited at the Yunnan Institute of Microbiology, Yunnan University, China.
Fermentation, extraction, and isolation
A slant culture of S. albidoflavus was inoculated into 500 mL Erlenmeyer flasks containing 100 mL of seed medium.25 The pH was 7.2 without adjustment and the flasks were cultivated for 2 days at 28 °C on a rotary shaker at 180 rpm. This subculture was used to inoculate fermentation medium with 10% volume. The large-scale fermentation was carried out in 1000 mL/2000 mL Erlenmeyer flask with 200 mL/400 mL of fermentation medium composed of soluble starch 5 g L−1, beef extract 3 g L−1, yeast extract 5 g L−1, peptone 3 g L−1, glucose 1 g L−1, CaCO3 4 g L−1, with a pH of 7.2 with no adjustment, and the flasks were incubated for 7 days at 28 °C on a rotary shaker at 180 rpm.
The fermentation broth (120 L) was separated by a centrifuge, and the resulting aqueous phase was extracted with ethyl acetate three times. The organic phase was vacuum-dried to obtain 62 g of crude extract. The total extract was subjected to silica gel (100–200 mesh) column chromatography eluting with a CH2Cl2–MeOH solvent system (from 100:1 to 20:1, 10:1, 5:1, 2:1 and 1:1) to yield ten fractions. Fraction 4 was divided into eight parts (Fr.4.1–Fr.4.8) by sephadex LH-20 chromatography (MeOH). Fr.4.2 was further segregated by silica gel column chromatography (CH2Cl2–MeOH, 100:1–20:1) and ODS column chromatography (MeOH–H2O, 40:60) to give compound 3 (12.1 mg). Fr.4.3 was subjected to ODS column chromatography (MeOH–H2O, 40:60) to produce four sections. Fr.4.3.2 was isolated by sephadex LH-20 chromatography (MeOH) to produce 1 (2.4 mg). Fr.4.3.3 was subjected to silica gel column chromatography and eluted with CH2Cl2–MeOH (100:1) to yield 4 (4.7 mg). Fr.4.4 was isolated through silica gel column chromatography (petroleum ether–EtOAc, 3:1) to yield six subfractions, and then Fr.4.4.3 was subjected to silica gel column chromatography (CH2Cl2–MeOH, 100:1) to yield 2 (1.0 mg).
2-Methyl-7-(methylamino)isoquinoline-3,5,8(2H)-trione (1). Yellow solid; UV (MeOH) λmax (log ε) 217 (3.3) nm, 325 (3.2) nm, 358 (3.1) nm; IR (film) νmax 3355, 2923, 2854, 1734, 1688, 1647, 1602, 1543, 1419, 1324, 1058, 624 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 8.70 (1H, s, H-1), 7.94 (1H, brs, 7-NH), 6.68 (1H, s, H-4), 5.66 (1H, s, H-6), 3.57 (3H, s, 2-CH3), 2.79 (3H, d, J = 5.1 Hz, 7-NCH3); 13C NMR (150 MHz, DMSO-d6) δ 178.4 (C, C-5), 177.1 (C, C-8), 162.5 (C, C-3), 151.3 (C, C-7), 144.4 (CH, C-1), 141.0 (C, C-4a), 113.7 (CH, C-4), 110.1 (C, C-8a), 100.8 (CH, C-6), 37.5 (CH3, 2-CH3), 29.0 (CH3, 7-NCH3); ESIMS m/z 219 [M+H]+; HRESIMS m/z 219.0763 [M+H]+ (calcd for C11H11N2O3+, 219.0764).
Synthese of 1 and 1a–1w
The synthesis steps of 1 and 1a–1w were shown in Schemes 2 and 3. The intermediates were prepared as follows.
1,1-Dimethylethyl-N-(2,5-dimethoxyphenyl) carbamate (7). 2,5-dimethoxyaniline (5.00 g, 32.64 mmol) and (Boc)2O (8.55 g, 39.17 mmol) were dissolved in THF (100 mL). The mixture was heated to reflux and stirred for 18 h. After that, the reaction mixture was evaporated under vacuum and the residue was chromatographed with silica gel column (P. ether/EtOAc = 20:1) to yield 7 (7.86 g, 95.0%) as an oil. 1H NMR (600 MHz, CDCl3) δ 7.80 (brs, 1H), 7.11 (d, J = 3.0 Hz, 1H), 6.75 (d, J = 8.8 Hz, 1H), 6.49 (dd, J = 8.8, 3.0 Hz, 1H), 3.81 (s, 3H), 3.78 (s, 3H), 1.52 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 154.2, 152.7, 141.8, 128.9, 110.9, 107.2, 104.3, 80.4, 56.3, 55.9, 28.4 (3 × C).
1,1-Dimethylethyl-N-(3,6-dioxo-1,4-cyclohexa-dien-1-yl) carbamate (6). 7 (6.00 g, 23.69 mmol), MeOH (3.0 mL) and H2O (120 mL) were stirred vigorously at room temperature, then [bis(trifluoroacetoxy)iodine]benzene (25.49 g, 59.27 mmol) was slowly added, and the reaction took place overnight at room temperature. Then, the reaction mixture was extracted three times with CH2Cl2. The combined organic phases were washed with saturated salt water and dried with anhydrous Na2SO4. Then the solvent was removed under reduced pressure, the residue was separated by silica gel column chromatography (P. ether/EtOAc = 50:1) to obtain the target compound 6 (3.98 g, 75.3%) as a yellow solid. 1H NMR (600 MHz, CDCl3) δ 7.39 (brs, 1H), 7.21 (d, J = 2.4 Hz, 1H), 6.74 (d, J = 10.1 Hz, 1H), 6.70 (dd, J = 10.1, 2.4 Hz, 1H), 1.51 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 187.4, 182.4, 151.1, 139.3, 138.2, 133.1, 112.5, 82.7, 28.1 (3 × C).
N-formyl acetamide (9). Formamide (20.00 g, 444.05 mmol) was stirred vigorously at 110 °C and acetyl chloride (80.33 g, 1023.31 mmol) was slowly dropped into it. After that, the reaction mixture was stirred at 110 °C for 6 h, and the reaction courses were monitored by thin-layer chromatography (TLC) on silica gel-precoated F254 Merck plates. When the reaction was completed, the mixture was cooled to room temperature. The mixture was neutralized to pH 8–9 through slowly adding saturated NaHCO3 aqueous solution, and then extracted with EtOAc. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under a vacuum to give a crude product. Silica gel column chromatography (P. ether/EtOAc = 2:1) was used for purification to obtain white solid 9 (24.09 g, 62.3%). 1H NMR (600 MHz, DMSO-d6) δ 11.10 (brs, 1H), 8.96 (d, J = 8.6 Hz, 1H), 2.06 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ 171.8, 163.4, 23.3.
1,1-Dimethylethyl-N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)carbamate (11). 9 (7.00 g, 80.37 mmol) and Et3N (20.34 g, 200.99 mmol) were added into anhydrous Et2O (35 mL), and stirred at room temperature under nitrogen protection for 30 min. Slowly dropped TBSOTf (46.75 g, 176.85 mmol) of anhydrous Et2O (70 mL) solution to the reaction mixture, and stirred overnight at room temperature. After the reaction was completed, the Et2O phase was concentrated under reduced pressure to obtain the crude product 5 (20.39 g). It was used without further purification for the next reaction. Under nitrogen protection, 6 (1.30 g, 5.82 mmol) and 5 (2.76 g) were dissolved in anhydrous benzene (10 mL) and stirred at room temperature for 4 h. Subsequently the mixture was added HCl (2N, 1 mL). After stirring for 30 min, the reaction mixture was diluted with water (10 mL) and extract with EtOAc. The organic layer was washed with saturated salt water and concentrated in vacuo to obtain yellow oil 10. To a solution of 10 in acetone (100 mL), iodomethane (2.48 g, 17.48 mmol), K2CO3 (1.61 g, 11.65 mmol) and TDA-1 (564.91 mg, 1.75 mmol) were added at room temperature. The mixture was stirred overnight, then extracted with ethyl acetate. The organic solution was washed with brine and dried with anhydrous Na2SO4. After the solvent removed in vacuo, the residue was separated by silica gel column chromatography (P. ether/EtOAc = 1:1) to yield yellow solid 11 (1.46 g, 82.6%). 1H NMR (600 MHz, DMSO-d6) δ 8.80 (s, 1H), 8.72 (s, 1H), 7.29 (s, 1H), 6.71 (s, 1H), 3.59 (s, 3H), 1.49 (s, 9H); 13C NMR (150 MHz, DMSO-d6) δ 182.7, 175.8, 162.3, 151.5, 145.4, 144.1, 138.8, 114.8, 114.4, 109.5, 82.2, 37.8, 27.8 (3 × C).
1,1-Dimethylethyl-N-methyl-N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroiso- quinolin-7-yl)carbamate (12). To the suspension of 11 (1.45 g, 4.77 mmol) in anhydrous THF (40 mL) was added slowly NaH (137.22 mg, 5.72 mmol) at 0 °C. After stirring at this temperature for 15 min, the reaction mixture was restored to room temperature and added methyl iodide (811.61 mg, 5.72 mmol), then reacted for 2 h. The reaction was quenched by addition of saturated NH4Cl aqueous solution and the solution was extract with EtOAc. The combined organic phase was dried on Na2SO4 and concentrated. The crude product was purified by silica gel column chromatography (P. ether/EtOAc = 3:1) to get 12 (1.07 g, 70.2%) as a yellow solid. 1H NMR (600 MHz, DMSO-d6) δ 8.79 (s, 1H), 6.91 (s, 1H), 6.74 (s, 1H), 3.61 (s, 3H), 3.15 (s, 3H), 1.36 (s, 9H); 13C NMR (150 MHz, DMSO-d6) δ 183.1, 176.7, 162.0, 153.0, 152.4, 144.8, 138.9, 127.0, 114.3, 110.6, 81.5, 37.8, 36.4, 27.5 (3 × C).
Mansouramycin H (1). To a solution of the 12 (1.00 g, 3.14 mmol) in CH2Cl2, TFA (10 mL) was added dropwise at 0 °C and the reaction was kepted for 2 h. The mixture was worked up by using saturated NaHCO3 aqueous solution (20 mL) and CH2Cl2. The separated CH2Cl2 portion was washed with saturated salt water and dried with anhydrous Na2SO4. The evaporated material was separated by silica gel column chromatography (P. ether/EtOAc = 2:3), gaving the target compound 1 (0.65 g, 88.6%). 1H NMR (600 MHz, DMSO-d6) δ 8.69 (s, 1H), 7.93 (d, J = 5.1 Hz, 1H), 6.68 (s, 1H), 5.66 (s, 1H), 3.57 (s, 3H), 2.79 (d, J = 5.3 Hz, 3H); 13C NMR (150 MHz, DMSO-d6) δ 178.4, 177.1, 162.5, 151.3, 144.4, 141.0, 113.7, 110.2, 100.8, 37.5, 29.0.
2-Bromo-N-(2,5-dimethoxy-phenyl)acetamide (13). The mixture of 2,5-dimethoxyaniline (2.00 g, 13.05 mmol), CH2Cl2 (30 mL) and Et3N (1.98 g, 19.57 mmol) were stirred at 0 °C for 15 min. Then, 2-bromoacetyl bromide (3.16 g, 15.66 mmol) was slowly dropped into the mixture and continue to stir for 1 h. The crude material was diluted with saturated NaHCO3 aqueous solution and extracted with EtOAc. Afterwards, the mixture was dried over Na2SO4 and the volatiles were evaporated in vacuum. The crude product was purified by silica gel column chromatography (P. ether/EtOAc = 10:1) to obtain 13 (2.62 g, 73.2%) as a yellow soild. 1H NMR (600 MHz, CDCl3) δ 8.80 (brs, 1H), 8.04 (d, J = 3.0 Hz, 1H), 6.82 (d, J = 8.9 Hz, 1H), 6.62 (dd, J = 8.9, 3.0 Hz, 1H), 4.02 (s, 2H), 3.87 (s, 3H), 3.78 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 163.2, 153.8, 142.5, 127.6, 110.9, 109.3, 105.9, 56.4, 55.8, 29.7.
1,1-Dimethylethyl-4-[2-[(2,5-dimethoxyphenyl)amino]-2-oxoethyl]-1-pipera-zinecarboxylate (14). The intermediate 13 (6.00 g, 21.89 mmol) was added to a mixture of N-boc piperazine hydrochloride (5.36 g, 24.07 mmol), Et3N (5.54 g, 54.74 mmol) and catalytic amount KI in MeCN (100 mL) and stirred at room temperature for 1 h. After extracted by EtOAc, the combined organic layers were dried over anhydrous Na2SO4 and concentrated in vacuum. Then the solid was purified using silica gel column chromatography with P. ether/EtOAc (5:1) to obtain a white product (14) (7.91 g, 94.9%). 1H NMR (600 MHz, CDCl3) δ 9.73 (brs, 1H), 8.10 (d, J = 2.4 Hz, 1H), 6.80 (dd, J = 8.9, 2.4 Hz, 1H), 6.58 (d, J = 8.9 Hz, 1H), 3.85 (s, 3H), 3.78 (s, 3H), 3.51 (t, J = 4.8 Hz, 4H), 3.16 (s, 2H), 2.57 (t, J = 4.8 Hz, 4H), 1.47 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 167.9, 154.7, 153.9, 142.5, 127.9, 110.8, 108.6, 105.9, 80.0, 62.2, 56.4, 55.8, 53.2 (4 × C), 28.4 (3 × C).
1,1-Dimethylethyl-4-[2-[(3,6-dioxo-1,4-cyclo-hexadien-1-yl)amino]-2-oxo-ethyl]-1-piperazinecarboxylate (15). The solution of 14 (7.00 g, 18.45 mmol) in MeOH (10 mL) and H2O (400 mL) was stirred vigorously at room temperature. To the solution was added slowly iodobenzene diacetate (14.85 g, 46.10 mmol), and the mixture was stirred overnight. After dilution with H2O, the product was extracted using CH2Cl2. The combined organic phases were washed with saturated aqueous NaHCO3, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was then purified by silica gel column chromatography (P. ether/EtOAc = 5:1) to yield desired intermediate 15 (6.07 g, 94.2%) as a yellow soild. 1H NMR (600 MHz, CDCl3) δ 9.99 (brs, 1H), 7.60 (d, J = 2.4 Hz, 1H), 6.78 (d, J = 10.1 Hz, 1H), 6.74 (dd, J = 10.1, 2.4 Hz, 1H), 3.54 (t, J = 4.8 Hz, 4H), 3.19 (s, 2H), 2.55 (t, J = 4.8 Hz, 4H), 1.46 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 187.9, 182.7, 169.9, 154.6, 138.1, 138.0, 133.3, 115.0, 80.1, 62.1, 53.2 (4 × C), 28.4 (3 × C).
1,1-Dimethylethyl-4-[2-[(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)amino]-2-oxoethyl]-1-piperazinecarboxylate (16). A mixture of 15 (4.00 g, 11.45 mmol) and 5 (5.42 g) in anhydrous benzene (12 mL) was stirred at room temperature for 4 h. The mixture was acidified with HCl (2N, 1 mL) and stirred for 30 min. The reaction mixture was extracted with EtOAc and the organic phase was concentrated under reduced pressure. The residue was added into the solution of iodomethane (4.88 g, 34.38 mmol), K2CO3 (3.16 g, 22.87 mmol) and TDA-1 (555.70 mg, 1.72 mmol) in acetone (100 mL). After stirring overnight, the reaction solution was extracted with EtOAc and the organic part was concentrated at reduced pressure. The residue was separated by silica gel column chromatography (CH2Cl2/MeOH = 50:1) to acquire the key intermediate 16 (3.53 g, 71.6%), a yellow solid. 1H NMR (600 MHz, CDCl3) δ 10.39 (brs, 1H), 8.36 (s, 1H), 7.90 (s, 1H), 7.11 (s, 1H), 3.68 (s, 3H), 3.56 (t, J = 4.9 Hz, 4H), 3.23 (s, 2H), 2.57 (t, J = 4.9 Hz, 4H), 1.47 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 182.9, 176.8, 170.0, 162.7, 154.6, 143.2, 141.6, 138.8, 118.2, 117.0, 110.0, 80.1, 62.1, 53.2 (4 × C), 38.7, 28.4 (3 × C).
General procedure for the preparation of 1a–1w. To the solution of 16 (3.00 g, 6.97 mmol) in CH2Cl2 (60 mL) was added dropwise the TFA (20 mL) at 0 °C, and stirred for 3 h. The reaction mixture was diluted by saturated NaHCO3 aqueous solution (20 mL) and extracted by CH2Cl2. The reaction product (17) was concentrated under reduced pressure and used for the next reaction without purification. A mixture of 17 (1.0 equiv.), corresponding halide reagent (1.1 equiv.), Et3N (1.2 equiv.) and KI (a catalytic amount) in MeCN were stirred at room temperature for 4 h. Afterwards, the resulting mixture was extracted using EtOAc and purified to obtain the target compound 1a–1g. The corresponding acyl chloride (1.1 equiv.) was added respectively to a solution of 17 (1.0 equiv.) and Et3N (1.3 equiv.) in anhydrous THF, and the reaction mixture was stirred at 0 °C for 1 h. The resulting solution was extracted, drird and concentrated. The crude product were separated by silica gel column chromatography to give the target compounds 1h and 1k. Additionally, the mixture of carboxylic acid (1.2 equiv.), anhydrous THF, DIPEA (3.0 equiv.), EDCI (1.5 equiv.) and HOBt (1.5 equiv.) were stirred at room temperature for 30 min. Then the intermediate 17 (1.0 equiv.) was added and stirred overnight. The mixture was treated with saturated NaHCO3 aqueous solution and EtOAc, and the organic phases were collected and dried with anhydrous Na2SO4. The solvent was removed, then the residue was isolated to afford compounds 1i–1j and 1l–1w.
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-benzyl-1-piperazine-acetamide (1a). Yellow solid; 39.66 mg (81.3%); IR (film) νmax 3319, 3284, 2949, 2920, 2836, 1650, 1451, 1412, 1113, 1014 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.44 (brs, 1H), 8.39 (s, 1H), 7.93 (s, 1H), 7.34–7.31 (m, 4H), 7.28–7.25 (m, 1H), 7.12 (s, 1H), 3.69 (s, 3H), 3.57 (s, 2H), 3.21 (s, 2H), 2.65–2.60 (m, 8H); 13C NMR (150 MHz, CDCl3) δ 182.9, 176.9, 170.6, 162.7, 143.2, 141.7, 138.8, 129.1, 128.3 (C × 4), 127.2, 118.2, 117.0, 110.1, 62.9, 62.1, 53.5 (C × 2), 53.3 (C × 2), 38.6; MS (ESI) m/z [M+H]+ 421.1, [M+Na]+ 443.0; HRESIMS m/z 443.1688 [M+Na]+ (calcd for C23H24N4O4Na+, 443.1695).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(4-chlorobenzyl)-1-piperazineacetamide (1b). Yellow solid; 39.15 mg (74.2%); IR (film) νmax 3327, 2949, 2838, 1648, 1450, 1410, 1112, 1014 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.38 (s, 1H), 8.88 (s, 1H), 7.60 (s, 1H), 7.39 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.3 Hz, 2H), 6.73 (s, 1H), 3.61 (s, 3H), 3.49 (s, 2H), 3.26 (s, 2H), 2.63–2.63 (m, 4H), 2.50–2.36 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 183.2, 176.3, 170.9, 162.1, 145.5, 142.2, 138.7, 137.3, 131.6, 130.7, 128.2 (C × 3), 115.8, 114.4, 109.3, 61.3, 61.2, 52.9 (C × 2), 52.7 (C × 2), 37.8; MS (ESI) m/z [M+H]+ 455.1, [M+Na]+ 477.1; HRESIMS m/z 477.1300 [M+Na]+ (calcd for C23H23ClN4O4Na+, 477.1306).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(4-bromobenzyl)-1-piperazineacetamide (1c). Yellow solid; 46.92 mg (81.0%); IR (film) νmax 3282, 2954, 2848, 1643, 1452, 1410, 1305, 1113, 1014 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.42 (s, 1H), 8.39 (s, 1H), 7.91 (s, 1H), 7.44 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 8.3 Hz, 2H), 7.10 (s, 1H), 3.68 (s, 3H), 3.51 (s, 2H), 3.21 (s, 2H), 2.69–2.53 (m, 8H); 13C NMR (150 MHz, CDCl3) δ 182.9, 176.9, 170.5, 162.6, 143.2, 141.7, 138.8, 137.1, 131.4 (C × 2), 130.7 (C × 2), 121.0, 118.2, 117.0, 110.1, 62.1, 62.0, 53.5 (C × 2), 53.2 (C × 2), 38.6; MS (ESI) m/z [M+H]+ 499.0, [M+Na]+ 521.1; HRESIMS m/z 499.0975 [M+H]+ (calcd for C23H24BrN4O4+, 499.0981).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(2-chloro-4-fluoro-benzyl)-1-piperazineacetamide (1d). Yellow solid; 42.46 mg (77.4%); IR (film) νmax 3310, 2955, 2918, 2850, 1655, 1492, 1445, 1302, 1261, 1230, 1021 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.44 (brs, 1H), 8.40 (s, 1H), 7.92 (s, 1H), 7.45 (dd, J = 8.6, 6.3 Hz, 1H), 7.14–7.09 (m, 2H), 6.97 (td, J = 8.6, 2.6 Hz, 1H), 3.69 (s, 3H), 3.64 (s, 2H), 3.22 (s, 2H), 2.69–2.62 (m, 8H); 13C NMR (150 MHz, CDCl3) δ 182.9, 176.9, 170.5, 162.7, 162.3, 160.7, 143.2, 141.7, 138.8, 134.8 (d, JC-F = 10.1 Hz), 131.7 (d, JC-F = 3.0 Hz), 131.6 (d, JC-F = 8.4 Hz), 118.2, 117.0, 116.8 (d, JC-F = 23.9 Hz), 113.9 (d, JC-F = 20.4 Hz), 110.1, 62.1, 58.4, 53.5 (C × 2), 53.1 (C × 2), 38.6; MS (ESI) m/z [M+H]+ 473.0, [M+Na]+ 495.1; HRESIMS m/z 473.1394 [M+H]+ (calcd for C23H23ClFN4O4+, 473.1387).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(4-(trifluoromethyl)benzyl)-1-piperazineacetamide (1e). Yellow solid; 39.95 mg (70.5%); IR (film) νmax 3314, 2947, 2836, 1652, 1450, 1412, 1113, 1015 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.43 (s, 1H), 8.39 (s, 1H), 7.92 (s, 1H), 7.58 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.11 (s, 1H), 3.69 (s, 3H), 3.62 (s, 2H), 3.22 (s, 2H), 2.69–2.58 (m, 8H); 13C NMR (150 MHz, CDCl3) δ 182.9, 176.9, 170.5, 162.6, 143.2, 142.4, 141.7, 138.8, 129.6 (q, JC-F = 31.8 Hz), 129.1 (C × 2), 125.3 (q, JC-F = 3.5 Hz, C × 2), 123.3, 118.2, 117.0, 110.1, 62.2, 62.0, 53.4 (C × 2), 53.2 (C × 2), 38.6; MS (ESI) m/z [M+H]+ 489.1, [M+Na]+ 511.2; HRESIMS m/z 489.1742 [M+H]+ (calcd for C24H24F3N4O4+, 489.1745).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(4-nitrobenzyl)-1- piperazineacetamide (1f). Yellow solid; 35.91 mg (66.5%); IR (film) νmax 3282, 2949, 2837, 1648, 1450, 1411, 1113, 1014 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.38 (s, 1H), 8.87 (s, 1H), 8.21 (d, J = 8.5 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 7.60 (s, 1H), 6.73 (s, 1H), 3.65 (s, 2H), 3.61 (s, 3H), 3.27 (s, 2H), 2.66–2.50 (m, 8H); 13C NMR (150 MHz, CDCl3) δ 183.2, 176.3, 170.8, 162.1, 146.7, 146.7, 145.5, 142.2, 138.7, 129.8 (C × 2), 123.5 (C × 2), 115.8, 114.4, 109.3, 61.3, 61.1, 53.0 (C × 2), 52.7 (C × 2), 37.8; MS (ESI) m/z [M+H]+ 466.1, [M+Na]+ 488.2; HRESIMS m/z 466.1735 [M+H]+ (calcd for C23H24N5O6+, 466.1722).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-cyclopropylmethyl-1-piperazineacetamide (1g). Yellow solid; 28.14 mg (63.1%); IR (film) νmax 3317, 2945, 2834, 1654, 1450, 1414, 1113, 1018 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.25 (s, 1H), 8.83 (s, 1H), 7.63 (s, 1H), 6.74 (s, 1H), 3.61 (s, 3H), 3.37 (s, 2H), 3.02–2.51 (m, 10H), 1.18 (t, J = 7.3 Hz, 1H), 0.60–0.53 (m, 2H), 0.29–0.16 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 183.3, 176.2, 170.7, 162.1, 145.4, 142.3, 138.7, 116.1, 114.4, 109.3, 60.9 (C × 2), 51.9 (C × 2), 45.8 (C × 2), 37.8, 8.7, 4.0 (C × 2); MS (ESI) m/z [M+H]+ 385.1, [M+Na]+ 407.2; HRESIMS m/z 385.1872 [M+H]+ (calcd for C20H25N4O4+, 385.1871).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-acetyl-1-piperazineacetamide (1h). Yellow solid; 37.06 mg (85.8%); IR (film) νmax 3315, 3284, 2949, 2837, 1648, 1450, 1413, 1113, 1014 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.34 (s, 1H), 8.84 (s, 1H), 7.62 (s, 1H), 6.74 (s, 1H), 3.60 (s, 3H), 3.50 (t, J = 5.0 Hz, 4H), 3.32 (s, 2H), 2.58 (t, J = 5.0 Hz, 2H), 2.53–2.51 (m, 2H), 2.02 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 183.6, 176.7, 171.1, 168.8, 162.6, 145.9, 142.7, 139.1, 116.3, 114.8, 109.7, 61.6, 53.2, 52.8, 46.4, 41.5, 38.2, 21.7; MS (ESI) m/z [M+H]+ 373.0, [M+Na]+ 395.1; HRESIMS m/z 373.1520 [M+H]+ (calcd for C18H21N4O5+, 373.1507).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(4-fluorobenzoyl)-1-piperazineacetamide (1i). Yellow solid; 28.44 mg (54.2%); IR (film) νmax 3315, 3286, 2949, 2920, 2838, 1647, 1450, 1414, 1113, 1014 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.35 (s, 1H), 8.37 (s, 1H), 7.90 (s, 1H), 7.45–7.42 (m, 2H), 7.13–7.10 (m, 3H), 4.00–3.79 (m, 2H), 3.69 (s, 3H), 3.27 (s, 2H), 2.73–2.59 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 182.8, 176.8, 169.6, 169.6, 163.6 (d, J = 250.0 Hz), 162.6, 143.3, 141.5, 138.7, 131.3 (d, J = 3.4 Hz), 129.5 (d, J = 8.2 Hz, C × 2), 118.3, 117.1, 115.7 (d, J = 22.3 Hz, C × 2), 110.0, 62.0, 53.4, 53.3, 47.9, 42.5, 38.7; MS (ESI) m/z [M+H]+ 453.1, [M+Na]+ 475.1; HRESIMS m/z 475.1382 [M+Na]+ (calcd for C23H21FN4O5Na+, 475.1389).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(2-chlorobenzoyl)-1-piperazineacetamide (1j). Yellow solid; 33.02 mg (60.7%); IR (film) νmax 3315, 3286, 2947, 2835, 1649, 1450, 1413, 1113, 1015 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.39 (s, 1H), 8.89 (s, 1H), 7.64 (s, 1H), 7.58 (dd, J = 7.6, 1.5 Hz, 1H), 7.52–7.46 (m, 2H), 7.44 (dd, J = 7.2, 2.1 Hz, 1H), 6.77 (s, 1H), 3.83–3.73 (m, 2H), 3.64 (s, 3H), 3.39 (s, 2H), 3.26 (t, J = 5.1 Hz, 2H), 2.71 (t, J = 5.1 Hz, 2H), 2.64–2.57 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 186.3, 179.4, 173.7, 168.8, 165.3, 148.6, 145.4, 141.8, 138.8, 133.8, 132.6, 132.3, 131.2, 130.9, 119.0, 117.6, 112.4, 64.1, 55.7, 55.3, 49.7, 44.5, 40.9; MS (ESI) m/z [M+H]+ 469.0, [M+Na]+ 491.1; HRESIMS m/z 491.1102 [M+Na]+ (calcd for C23H21ClN4O5Na+, 491.1093).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(4-bromobenzoyl)-1-piperazineacetamide (1k). Yellow solid; 48.71 mg (81.8%); IR (film) νmax 3316, 3282, 2948, 2836, 1650, 1450, 1413, 1113, 1015 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.32 (s, 1H), 8.83 (s, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.61 (s, 1H), 7.39 (d, J = 8.4 Hz, 2H), 6.73 (s, 1H), 3.78–3.62 (m, 2H), 3.61 (s, 3H), 3.47–3.37 (m, 2H), 3.34 (s, 2H), 2.67–2.54 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 183.6, 176.7, 171.0, 168.5, 162.5, 145.9, 142.6, 139.1, 135.3, 131.9 (C × 2), 129.7 (C × 2), 123.5, 116.3, 114.9, 109.7, 61.5, 53.0, 52.7, 49.1, 42.4, 38.2; MS (ESI) m/z [M+H]+ 513.0, [M+Na]+ 535.1; HRESIMS m/z 535.0607 [M+Na]+ (calcd for C23H21BrN4O5Na+, 535.0588).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-[2-(2-bromophenyl)acetyl]-1-piperazineacetamide (1l). Yellow solid; 39.12 mg (65.7%); IR (film) νmax 3315, 3284, 2946, 2834, 1654, 1450, 1416, 1113, 1017 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.34 (s, 1H), 8.37 (s, 1H), 7.90 (s, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.38 (t, J = 7.9 Hz, 1H), 7.29–7.26 (m, 2H), 7.11 (s, 1H), 4.05–3.97 (m, 1H), 3.97–3.89 (m, 1H), 3.69 (s, 3H), 3.46–3.41 (m, 1H), 3.37–3.32 (m, 1H), 3.27 (s, 2H), 2.79–2.74 (m, 1H), 2.73–2.67 (m, 2H), 2.56–2.52 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 182.8, 176.9, 169.6, 167.7, 162.6, 143.3, 141.5, 138.7, 137.6, 132.9, 130.5, 127.8, 127.7, 119.1, 118.4, 117.1, 110.0, 62.0, 53.4, 53.0, 46.9, 41.7, 38.7; MS (ESI) m/z [M+H]+ 513.0, [M+Na]+ 535.1; HRESIMS m/z 513.0781 [M+H]+ (calcd for C23H22BrN4O5+, 513.0769).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(2,4-dibromobenzoyl)-1-piperazineacetamide (1m). Yellow solid; 44.52 mg (64.83%); IR (film) νmax 3316, 2949, 2921, 2837, 1648, 1450, 1414, 1113, 1015 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.34 (s, 1H), 8.84 (s, 1H), 7.74 (d, J = 2.0 Hz, 1H), 7.61 (s, 1H), 7.53 (dd, J = 8.3, 2.0 Hz, 1H), 7.46 (d, J = 8.3 Hz, 1H), 6.74 (s, 1H), 3.76–3.69 (m, 2H), 3.60 (s, 3H), 3.36–3.34 (m, 2H), 3.24–3.22 (m, 2H), 2.68–2.65 (m, 2H), 2.56–2.54 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 183.6, 176.7, 171.0, 165.2, 162.5, 145.9, 142.6, 139.1, 135.0, 134.8, 130.8, 129.9, 129.6, 128.4, 116.3, 114.9, 109.7, 61.4, 53.0, 52.5, 46.9, 41.9, 38.2; MS (ESI) m/z [M+H]+ 592.9, [M+Na]+ 614.9; HRESIMS m/z 590.9876 [M+H]+ (calcd for C23H21Br2N4O5+, 590.9874).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(2-bromo-5-methoxy-benzoyl)-1-piperazineacetamide (1n). Yellow solid; 38.39 mg (60.9%); IR (film) νmax 3316, 2946, 2834, 1653, 1450, 1414, 1113, 1017 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.35 (s, 1H), 8.84 (s, 1H), 7.60 (s, 1H), 7.55 (d, J = 8.5 Hz, 1H), 6.95 (d, J = 3.0 Hz, 1H), 6.94–6.91 (m, 1H), 6.73 (s, 1H), 3.78–3.76 (m, 5H), 3.60 (s, 3H), 3.34 (s, 2H), 3.22 (t, J = 5.1 Hz, 2H), 2.67 (t, J = 5.1 Hz, 2H), 2.61–2.53 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 183.6, 176.7, 171.0, 166.6, 162.5, 159.3, 145.9, 142.6, 139.1, 134.1, 133.9, 117.1, 116.3, 114.9, 113.8, 109.7, 108.9, 61.4, 56.2 (C × 2), 56.1, 52.9, 52.5, 38.2; MS (ESI) m/z [M+H]+ 543.0, [M+Na]+ 565.1; HRESIMS m/z 565.0709 [M+Na]+ (calcd for C24H23BrN4O6Na+, 565.0694).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(2-bromo-4,5-dimethoxybenzoyl)-1-piperazineacetamide (1o). Yellow solid; 38.25 mg (57.53%); IR (film) νmax 3326, 2948, 2836, 1648, 1450, 1413, 1113, 1015 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.40 (s, 1H), 8.88 (s, 1H), 7.65 (s, 1H), 7.22 (s, 1H), 6.99 (s, 1H), 6.77 (s, 1H), 3.84 (s, 3H), 3.81 (s, 3H), 3.80–3.69 (m, 2H), 3.65 (s, 3H), 3.38 (s, 2H), 3.28 (t, J = 5.1 Hz, 2H), 2.70 (t, J = 5.1 Hz, 2H), 2.66–2.57 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 185.8, 178.9, 173.2, 169.2, 164.8, 152.3, 151.2, 148.1, 144.8, 141.3, 132.2, 118.5, 117.9, 117.1, 113.6, 111.9, 111.3, 63.7, 58.7, 58.6, 55.2, 54.8, 51.3, 44.0, 40.4; MS (ESI) m/z [M+H]+ 573.0, [M−H]− 570.9; HRESIMS m/z 595.0807 [M+Na]+ (calcd for C25H25BrN4O7Na+, 595.0799).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(4-methybenzoyl)-1-piperazineacetamide (1p). Yellow solid; 33.09 mg (63.6%); IR (film) νmax 3321, 2953, 2842, 1643, 1453, 1419, 1305, 1113, 1014 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.35 (s, 1H), 8.36 (s, 1H), 7.87 (s, 1H), 7.30 (d, J = 7.8 Hz, 2H), 7.20 (d, J = 7.8 Hz, 2H), 7.07 (s, 1H), 3.97–3.80 (m, 2H), 3.67 (s, 3H), 3.65–3.53 (m, 2H), 3.24 (s, 2H), 2.68–2.56 (m, 4H), 2.36 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 182.9, 176.8, 170.7, 169.8, 162.6, 143.4, 141.5, 140.2, 138.7, 132.3, 129.2 (C × 2), 127.2 (C × 2), 118.2, 117.0, 110.0, 62.0, 53.5, 53.4, 47.9, 42.4, 38.6, 21.4; MS (ESI) m/z [M−H]− 447.1, [M+Na]+ 471.2; HRESIMS m/z 465.1774 [M+H]+ (calcd for C24H25N4O6+, 465.1769).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-(4-nitrobenzoyl)-1-piperazineacetamide (1q). Yellow solid; 36.53 mg (47.7%); IR (film) νmax 3318, 2945, 2833, 1652, 1450, 1413, 1113, 1018 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.31 (s, 1H), 8.83 (s, 1H), 8.30 (d, J = 8.7 Hz, 2H), 7.70 (d, J = 8.7 Hz, 2H), 7.63 (s, 1H), 6.75 (s, 1H), 3.73 (brs, 2H), 3.61 (s, 3H), 3.36 (s, 2H), 3.30 (brs, 2H), 2.68 (brs, 2H), 2.56 (brs, 2H); 13C NMR (150 MHz, CDCl3) δ 183.2, 176.3, 170.6, 167.2, 162.1, 147.9, 145.5, 142.3, 142.1, 138.7, 128.4 (C × 2), 123.9 (C × 2), 116.0, 114.4, 109.3, 61.1, 52.5, 52.1, 47.3, 41.8, 37.8; MS (ESI) m/z [M+H]+ 480.0, [M+Na]+ 502.1; HRESIMS m/z 502.1335 [M+Na]+ (calcd for C23H21N5O7Na+, 502.1334).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-[2-(4-methylphenyl)acetyl]-1-piperazineacetamide (1r). Yellow solid; 26.23 mg (48.93%); IR (film) νmax 3326, 2945, 2834, 1644, 1450, 1415, 1113, 1018 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.30 (s, 1H), 8.35 (s, 1H), 7.89 (s, 1H), 7.14–7.12 (m, 4H), 7.10 (s, 1H), 3.78 (t, J = 5.1 Hz, 2H), 3.72 (s, 2H), 3.68 (s, 3H), 3.57 (t, J = 4.8 Hz, 2H), 3.18 (s, 2H), 2.57 (t, J = 5.1 Hz, 2H), 2.41 (t, J = 4.8 Hz, 2H), 2.33 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 182.8, 176.8, 169.8, 169.7, 162.6, 143.3, 141.5, 138.7, 136.6, 131.6, 129.5 (C × 2), 128.4 (C × 2), 118.3, 117.0, 110.0, 62.0, 53.3, 53.1, 46.1, 41.8, 40.7, 38.7, 21.1; MS (ESI) m/z [M+H]+ 463.1, [M+Na]+ 485.2; HRESIMS m/z 463.1975 [M+H]+ (calcd for C25H27N4O5+, 463.1976).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-[2-(4-methoxyphenyl)acetyl]-1-piperazineacetamide (1s). Yellow solid; 27.64 mg (49.83%); IR (film) νmax 3332, 2946, 2834, 1651, 1450, 1412, 1113, 1017 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.37 (s, 1H), 8.88 (s, 1H), 7.64 (s, 1H), 7.19 (d, J = 8.5 Hz, 2H), 6.92 (d, J = 8.5 Hz, 2H), 6.77 (s, 1H), 3.77 (s, 3H), 3.70 (s, 2H), 3.64 (s, 3H), 3.58 (t, J = 4.9 Hz, 4H), 3.33 (s, 2H), 2.58–2.55 (m, 2H), 2.53–2.50 (dm, 2H); 13C NMR (150 MHz, CDCl3) δ 183.2, 176.3, 170.6, 169.3, 162.1, 157.9, 145.5, 142.2, 138.7, 129.9 (C × 2), 127.6, 115.8, 114.4, 113.8 (C × 2), 109.3, 61.1, 55.1, 52.7, 52.3, 45.7, 41.5, 38.7, 37.8; MS (ESI) m/z [M+H]+ 479.1, [M+Na]+ 501.2; HRESIMS m/z 501.1745 [M+Na]+ (calcd for C25H26N4O6Na+, 501.1745).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-[2-(3,4-dichloro-phenyl)acetyl]-1-piperazineacetamide (1t). Yellow solid; 27.13 mg (46.0%); IR (film) νmax 3334, 2947, 2920, 2835, 1648, 1450, 1415, 1113, 1017 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.30 (s, 1H), 8.36 (s, 1H), 7.89 (s, 1H), 7.10 (s, 1H), 6.82 (d, J = 8.1 Hz, 1H), 6.80 (d, J = 2.0 Hz, 1H), 6.75 (dd, J = 8.1, 12.0 Hz, 1H), 3.88 (s, 3H), 3.87 (s, 3H), 3.78 (t, J = 5.1 Hz, 2H), 3.70 (s, 2H), 3.68 (s, 3H), 3.59 (t, J = 4.8 Hz, 2H), 3.19 (s, 2H), 2.58 (t, J = 5.1 Hz, 2H), 2.44 (t, J = 4.8 Hz, 2H); 13C NMR (150 MHz, CDCl3) δ 182.8, 176.8, 169.7, 169.7, 162.6, 149.2, 148.0, 143.3, 141.5, 138.7, 127.2, 120.6, 118.3, 117.0, 111.6, 111.3, 110.0, 61.9, 56.0, 55.9, 53.3, 53.1, 46.1, 41.9, 40.6, 38.6; MS (ESI) m/z [M+H]+ 509.1, [M+Na]+ 531.2; HRESIMS m/z 509.2033 [M+H]+ (calcd for C26H29N4O7+, 509.2031).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-[2-(4-chlorophenyl)acetyl]-1-piperazineacetamide (1u). Yellow solid; 22.80 mg (40.7%); IR (film) νmax 3334, 3284, 2952, 2844, 1644, 1015 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.33 (s, 1H), 8.84 (s, 1H), 7.61 (s, 1H), 7.38–7.36 (m, 2H), 7.27–7.25 (m, 2H), 6.74 (s, 1H), 3.75 (s, 2H), 3.60 (s, 3H), 3.58–3.53 (m, 4H), 3.31 (s, 2H), 2.52 (s, 4H); 13C NMR (150 MHz, CDCl3) δ 183.6, 176.7, 171.0, 169.1, 162.5, 145.9, 142.6, 139.1, 135.4, 131.5, 131.5 (C × 2), 128.7 (C × 2), 116.3, 114.8, 109.7, 61.5, 53.1, 52.7, 46.1, 42.0, 39.0, 38.2; MS (ESI) m/z [M+H]+ 483.0, [M+Na]+ 505.1; HRESIMS m/z 483.1432 [M+H]+ (calcd for C24H24ClN4O5+, 483.1430).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-[2-(4-bromophenyl)acetyl]-1-piperazineacetamide (1v). Yellow solid; 26.37 mg (43.1%); IR (film) νmax 3330, 2947, 2835, 1651, 1450, 1411, 1113, 1016 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.33 (s, 1H), 8.83 (s, 1H), 7.60 (s, 1H), 7.50 (d, J = 8.3 Hz, 2H), 7.20 (d, J = 8.3 Hz, 2H), 6.73 (s, 1H), 3.74 (s, 2H), 3.60 (s, 3H), 3.57–3.54 (m, 4H), 3.31 (s, 2H), 2.53 (s, 4H); 13C NMR (150 MHz, CDCl3) δ 183.2, 176.3, 170.6, 168.6, 162.1, 145.5, 142.2, 138.7, 135.4, 131.5 (C × 2), 131.2 (C × 2), 119.6, 115.9, 114.4, 109.3, 61.1, 52.7, 52.3, 45.7, 41.6, 38.7, 37.8; MS (ESI) m/z [M+H]+ 527.0, [M−H]− 525.0; HRESIMS m/z 527.0923 [M+H]+ (calcd for C24H24BrN4O5+, 527.0925).
N-(2-methyl-3,5,8-trioxo-2,3,5,8-tetrahydroisoquinolin-7-yl)-4-[2-(4-nitrophenyl)acetyl]-1-piperazineacetamide (1w). Yellow solid; 19.01 mg (33.2%); IR (film) νmax 3314, 2949, 2837, 1648, 1450, 1411, 1113, 1014 cm−1; 1H NMR (600 MHz, CDCl3) δ 10.33 (s, 1H), 8.83 (s, 1H), 8.19 (d, J = 8.6 Hz, 2H), 7.61 (s, 1H), 7.52 (d, J = 8.6 Hz, 2H), 6.74 (s, 1H), 3.94 (s, 2H), 3.60 (s, 3H), 3.60–3.59 (m, 2H), 3.58–3.55 (m, 2H), 3.32 (s, 2H), 2.58–2.54 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 183.6, 176.6, 171.0, 168.5, 162.5, 146.7, 145.8, 144.7, 142.6, 139.1, 131.2 (C × 2), 123.7 (C × 2), 116.3, 114.8, 109.6, 61.4, 53.1, 52.7, 46.0, 42.0, 39.4, 38.2; MS (ESI) m/z [M+H]+ 494.1, [M+Na]+ 516.1; HRESIMS m/z 494.1674 [M+H]+ (calcd for C24H24N5O7+, 494.1671).
Biological evaluation
Cytotoxicity assay. The cytotoxic activity of compounds 1–4 and 1g was tested on six human cancer cell lines, including MDA-MB-231, HCT-116, BGC-823, A-549, HepG2, and Ishikawa cells. And the cytotoxicity of compounds 1a–1f and 1h–1w was detected on MDA-MB-231 cell. The cells were cultivated in DMEM culture medium including 10% FBS, 1% penicillin and 1% streptomycin for 12 h under incubation conditions of 5% CO2 and 37 °C. The cells were treated with different doses (200–1.56 μM) of 1–4 and 1g for 48 h. The results of cytotoxic were measured by the MTT assay, while adriamycin was selected as a positive control drug. The experiment was replicated three times in each group.
Cell cycle assay. MDA-MB-231 were inoculated in a 6-well plate with an amount of 5 × 105 cells in each well and treated with 1g (0 or 5.0 μM) for 24 h. The cells were harvested and fixed in −20 °C pre-cooled 75% ethanol for 12 h. The cells were laved with 4 °C PBS and resuspended in 0.5 mL of propidium iodide (PI) solution (Beijing Dingguo Changsheng Biotechnology Co., Ltd, China), and then incubated at room temperature in darkness for 15 min. Cell cycle experiments were conducted on LSR Fortessa flow cytometer and each experiment was repeated three times. The content of cells at different stages was calculated using ModFit LT 4.1 software (Verity Software House, Topsham, ME). The experiment was replicated three times in each group.
Cell apoptosis assay. MDA-MB-231 were plated at 5 × 105 cells per well in a 6-well plate and exposed to 1g (0 or 5.0 μM) for 24 h. Afterward, cells were washed with cold PBS and then the collected cells were resuspended in a solution consisting of 490 μL buffer, 5 μL PI (BD, Franklin Lakes, NJ, USA) and 5 μL of fluorescein isothiocyanate (FITC)-conjugated annexin-V reagent. After 15 min incubation at ordinary temperature under dark condition, the apoptosis analysis was carried out on flow cytometry using the Annexin V-FITC/PI cell apoptosis method. Cytographs were drawn by BD FACSDiva software (BD, Franklin Lakes, NJ). Repeat each experiment three times. The experiment was replicated three times in each group.
Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 342 staining. Morphological changes of nucleus were widely evaluated by Hoechst 33342 staining. MDA-MB-231 cells (1 × 105 cells per well) were vegetated in a 24-well plate and encountered different amounts of 1g (0 and 5.0 μM) for 24 h. Subsequently, the cells, which washed three times using PBS, were fixed in 4% paraformaldehyde for 30 min. Following three washes with PBS, Hoechst 33258 dye was added for nuclear staining under darkness at room temperature for 10 min. The morphological changes of nucleus were observed by fluorescence microscope and photographed in time. The experiment was replicated three times in each group.
342 staining. Morphological changes of nucleus were widely evaluated by Hoechst 33342 staining. MDA-MB-231 cells (1 × 105 cells per well) were vegetated in a 24-well plate and encountered different amounts of 1g (0 and 5.0 μM) for 24 h. Subsequently, the cells, which washed three times using PBS, were fixed in 4% paraformaldehyde for 30 min. Following three washes with PBS, Hoechst 33258 dye was added for nuclear staining under darkness at room temperature for 10 min. The morphological changes of nucleus were observed by fluorescence microscope and photographed in time. The experiment was replicated three times in each group.
Western blot analysis. At a density 5 × 105 cells per well, MDA-MB-231 were added to per well of the six-well plate for 12 h and then treated with multiple concentrations of compound 1g (0, 0.56, 1.67 and 5.0 μM) for 24 h. After the cells were lysed in RIPA buffer, proteins were extracted with the extraction kits (Beyotime, Shanghai, China). The protein concentration was measured using the BCA protein assay kit (Beyotime, Shanghai, China). The samples were separated by 12% SDS-PAGE gel, and the loading amount was 30–50 μg. After transferring the sample to a 0.45 μm PVDF membrane, seal it in 5% skimmed milk for one hour. The PVDF membranes were incubated overnight with the primary antibodies at 4 °C, followed by one hour of incubation with the secondary antibodies at room temperature. The details of primary antibodies were reported in the Table S1.† The grayscale of the bands was quantitatively analyzed using GIS Gel Image System (Tanon, Shanghai, China). The experiment was replicated three times in each group.
Statistical analysis. The results were expressed as mean ± SEM and generated from at least three independent experiments. Statistical analysis was carried out by GraphPad Prism 5.0 software and the significant differences were assumed at p < 0.05.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Science and Technology Major Project of Guangxi, P. R. China (Grant No. AA18242026), National Natural Science Foundation of China, (Grant No. 32060001), the Fundamental Research Funds for the Central Universities, China (No. N2220003) and the Construction Project of Liaoning Provincial Key Laboratory, China (2022JH13/10200026).
Notes and references
- J. L. Bolton and T. Dunlap, Chem. Res. Toxicol., 2017, 30, 13–37 Search PubMed.
- V. Jaime, D. Virginia, S. Sandra, B. Julio, T. Cristina, B. C. Pedro and M. Giulio, Molecules, 2016, 21, 1199 Search PubMed.
- R. D. Kruschel, A. Buzid, U. B. R. Khandavilli, S. E. Lawrence, J. D. Glennon and F. O. McCarthy, Org. Biomol. Chem., 2020, 18, 557–568 RSC.
- I. Klopčič and M. S. Dolenc, Chem. Res. Toxicol., 2019, 32, 1–34 Search PubMed.
- A. Tadashi, Y. Katsukiyo, M. Yuzuru, K. Akinori and T. Katsuhiro, J. Antibiot., 1976, 4, 398–407 Search PubMed.
- J. M. Frincke and D. J. Faulkner, J. Am. Chem. Soc., 1982, 104, 265–269 CrossRef CAS.
- D. J. Milanowski, K. R. Gustafson, J. A. Kelley and J. B. Mcmahon, J. Nat. Prod., 2004, 67, 70–73 CrossRef CAS.
- R. Y. Huang, W. T. Chen, T. Kurtán, A. Mándi and Y. W. Guo, Future Med. Chem., 2016, 8, 17–27 CrossRef CAS PubMed.
- U. W. Hawas, M. Shaaban, K. A. Shaaban, M. Speitling and H. Laatsch, J. Nat. Prod., 2009, 72, 2120–2124 CrossRef CAS.
- M. Shaaban, K. A. Shaaban, G. Kelter, H. H. Fiebig and H. Laatsch, Mar. Drugs, 2021, 19, 715 CrossRef CAS PubMed.
- A. Plubrukarn, S. Yuenyongsawad, T. Thammasaroj and A. Jitsue, Pharm. Biol., 2003, 41, 439–442 CrossRef CAS.
- E. D. Jesus, J. S. Hur, K. I. Notarte, K. Santiago and T. D. Cruz, CREAM, 2016, 6, 173–183 CrossRef.
- G. Shrestha, A. M. El-Naggar, L. L. S. Clair and K. L. O'Neill, Phytother. Res., 2015, 29, 100–107 CrossRef.
- E. Z. Studzinska-Sroka, D. Szwajgier, A. Majchrzak-Celinska, P. Zalewski, D. Szwajgier, E. Baranowska-Wójcik, B. Kapron, T. Plech, M. Zarowski and J. Cielecka-Piontek, Pharmaceuticals, 2021, 14, 1293 CrossRef CAS PubMed.
- S. Nakahara and A. Kubo, Heterocycles, 2004, 63, 1849 CrossRef CAS.
- S. N. Goodman and E. N. Jacobsen, Adv. Synth. Catal., 2002, 344, 953–956 CrossRef CAS.
- P. Loupias, I. Dechamps-Olivier, L. Dupont, P. Vanlemmens, C. Mullié, N. Taudon, A. Bouchut, A. Dassonville-Klimpt and P. Sonnet, Pharmaceuticals (Basel), 2019, 12, 160 CrossRef CAS PubMed.
- Z. Ye, S. Adhikari, Y. Xia and M. Dai, Nat. Commun., 2018, 9, 721 CrossRef PubMed.
- E. Pitta, M. K. Rogacki, O. Balabon, S. Huss, F. Cunningham, E. M. Lopez-Roman, J. Joossens, K. Augustyns, L. Ballell and R. H. Bates, J. Med. Chem., 2016, 59, 6709–6728 CrossRef CAS PubMed.
- K. Vermeulen, D. Bockstaele and Z. N. Berneman, Cell Proliferation, 2010, 36, 131–149 CrossRef PubMed.
- S. Tadesse, E. C. Caldon, W. Tilley and S. Wang, J. Med. Chem., 2018, 62, 4233–4251 CrossRef PubMed.
- R. Agami and R. Bernards, Cell, 2000, 102, 55–66 CrossRef CAS PubMed.
- J. Neuzil, X. F. Wang, L. F. Dong, P. Low and S. J. Ralph, FEBS Lett., 2006, 580, 5125–5129 CrossRef CAS PubMed.
- P. Huang, D. Wang, Y. Su, W. Huang and Y. Zhou, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef CAS.
- P. Guan, X. Wang, Y. Jiang, N. Dou, X. Qu, J. Liu, B. Lin, L. Han, X. Huang and C. Jiang, Bioorg. Chem., 2020, 95, 103057 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab and
Xueshi Huang
ab and
Xueshi Huang