
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of radical initiator based on o-imino-isourea capable of photo/thermal polymerization †
Eun Jeong Seo ab,
Hyocheol Junga,
Ji-Eun Jeonga,
Sang-Ho Leea,
Jin Chul Kima,
Dong Yeon Kimc,
Seungju Kimb,
Kyu Cheol Lee*ab and
Young Il Park*ad
ab,
Hyocheol Junga,
Ji-Eun Jeonga,
Sang-Ho Leea,
Jin Chul Kima,
Dong Yeon Kimc,
Seungju Kimb,
Kyu Cheol Lee*ab and
Young Il Park*ad
aResearch Center for Advanced Specialty Chemicals, Korea Research Institute of Chemical Technology, Ulsan 44412, Republic of Korea. E-mail: ypark@krict.re.kr
bDepartment of Applied Chemistry·Food Science Technology, Dong-Eui University, 176 Eomgwangro, Busan 47340, South Korea. E-mail: kclee@deu.ac.kr
cDepartment of Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
dAdvanced Materials and Chemical Engineering, University of Science and Technology (UST), Daejeon 34113, Republic of Korea
First published on 14th December 2023
Abstract
Using o-imino isourea, three photo- and thermal dual-responsive radical initiators dicyheDCC, CyheDCC, and BnDCC were systematically developed and synthesized. By adding an aromatic ring to the free radical initiators, the ultraviolet-visible absorption was redshifted, and the absorption coefficient was increased. Compared with other initiators, BnphDCC exhibited an exceptional photoinitiation rate under photo-differential scanning calorimetry (DSC) and a high absorption coefficient (ε = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 420 M−1 cm−1). Therefore, it is an appropriate potential photoinitiator. DicyheDCC, which was composed of a cyclic hydrocarbon, exhibited rapid thermal initiation (Tpeak = 82 °C) during thermal DSC, making it a valuable thermal radical initiator. Because of the low stiffness of the N–O link in radical initiators, density functional theory predicts that the aliphatic ring has a significantly lower enthalpy than the aromatic ring. Moreover, in this study, CyhephDCC and BnphDCC, as dual-responsive radical initiators, indicated the potential for a photo- and heat dual-curing system through the universal free-radical polymerization of acrylates. These significant discoveries may be useful for developing efficient and diversified polymer network systems that require synergistic photo- and thermal effects.
420 M−1 cm−1). Therefore, it is an appropriate potential photoinitiator. DicyheDCC, which was composed of a cyclic hydrocarbon, exhibited rapid thermal initiation (Tpeak = 82 °C) during thermal DSC, making it a valuable thermal radical initiator. Because of the low stiffness of the N–O link in radical initiators, density functional theory predicts that the aliphatic ring has a significantly lower enthalpy than the aromatic ring. Moreover, in this study, CyhephDCC and BnphDCC, as dual-responsive radical initiators, indicated the potential for a photo- and heat dual-curing system through the universal free-radical polymerization of acrylates. These significant discoveries may be useful for developing efficient and diversified polymer network systems that require synergistic photo- and thermal effects.
1. Introduction
Free radical initiators (FRIs) have been widely employed for polymer synthesis in academia and industry because of their high polymer conversion rates, cost-effectiveness, and simple synthesis process.1–3 Recently, they have been used as additives in advanced materials, such as smart coatings, electrolytes, microelectronics, and bioadhesives.4–6 Depending on the structure, FRIs have two distinct radical generating mechanisms, i.e., photo- and thermal, and both are necessary for different applications.7 For instance, owing to its fast curing and low temperature, a photo-radical initiator is required for rapid hardening procedures such as dentistry and surface cracking; however, a thermal radical initiator is needed to build a large-scale, uniform curing system. Recently, the results that improves polymerization effect by adding thermal radical functionality to commercialized photoinitiators (Darocur 2959, Irgacure 184 and Darocur 1173) has been reported.8 In addition, photothermal dual-curing or polymerization can realize a more efficient and convenient curing process using highly crosslinked and high-modulus polymer systems. Despite the considerable interest in curing and polymerization, the study of radical initiators remains stalled, with many researchers focusing solely on polymer methods or physical properties using existing radical initiators such as benzoyl peroxide as a photoinitiator and azobisisobutyronitrile as a thermal initiator.9–11 Thus, it is critical to produce radical initiators with a variety of photothermal properties. Radical initiators with both photo- and thermal effects are uncommon.Our group previously reported stable o-imino-isourea-based thermal radical initiators and established an effective free-radical polymerization system at temperatures below 100 °C.12 In the present study, we implemented a new radical active system based on o-imino-isourea radical initiators, i.e., DicyheDCC, CyhephDCC, and BnphDCC, by systematically introducing an aromatic group for photoactivation, as shown in Fig. 1. In addition, we investigated their intrinsic properties by performing tests such as thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), and ultraviolet-visible (UV-vis) spectroscopy, and we explored their polymerization with n-butyl acrylate by performing thermal and photo-DSC measurements.
 | ||
| Fig. 1 Chemical structures of FRIs based on o-imino-isourea. | ||
Interestingly, among the suggested radical initiators, CyhephDCC exhibited the best photocuring performance, and this phenomenon was addressed in terms of the radical stabilities that arise when radicals are created by light via real-time nuclear magnetic resonance (NMR) monitoring and density functional theory (DFT) calculations. Depending on the functional groups, the proposed o-imino-isourea-based radical initiator can be employed as both a thermal radical initiator and a photo-radical initiator, as determined by experimental and analytical findings. This advantageous result also permits photo- and heat dual polymerization as a new all-in-one system, which will pave the way for a new dual-curing system in the future.
2. Results and discussions
2.1. Synthesis of FRIs
The syntheses of radical initiators 3a–c are shown in Fig. 2. According to the literature, general oximes (1a–c) were prepared using hydroxylamine and sodium acetate.13–15 All the oximes were in the form of white crystal-like solids after recrystallization via ethyl acetate and hexane, affording a yield of 65–80%. Benzophenone oxime was stored in a glovebox in the dark owing to its light- and air-sensitive properties. The final products 3a–c were synthesized from oximes 1a–c and N,N′-dicyclohexylcarbodiimide (DCC). These were mixed with sodium hydroxide as the base reagent and tetrahydrofuran (THF) as the solvent to produce white solids in yields of 45–60%. The purified compounds were analyzed using NMR spectroscopy, elemental analysis (EA), and mass spectrometry (MS) using a liquid chromatography-mass spectrometry (LC-MS) system. | ||
| Fig. 2 General synthesis route for FRIs. | ||
2.2. Photo- and thermal properties of FRIs
Fig. 3 and Table 1 present the absorption coefficients of all the FRIs, as well as their normalized absorption peaks from the UV-vis spectrum. DicyheDCC lacked an absorption peak owing to the absence of a conjugated building block, whereas CyhephDCC and BnphDCC exhibited absorption peaks between 240 and 300 nm in the UV region. In addition, depending on the number of aromatic groups, the absorption coefficients varied between 8570 and 15420 M−1 cm−1. This indicated that the optical absorption increased with the number of aromatic groups.
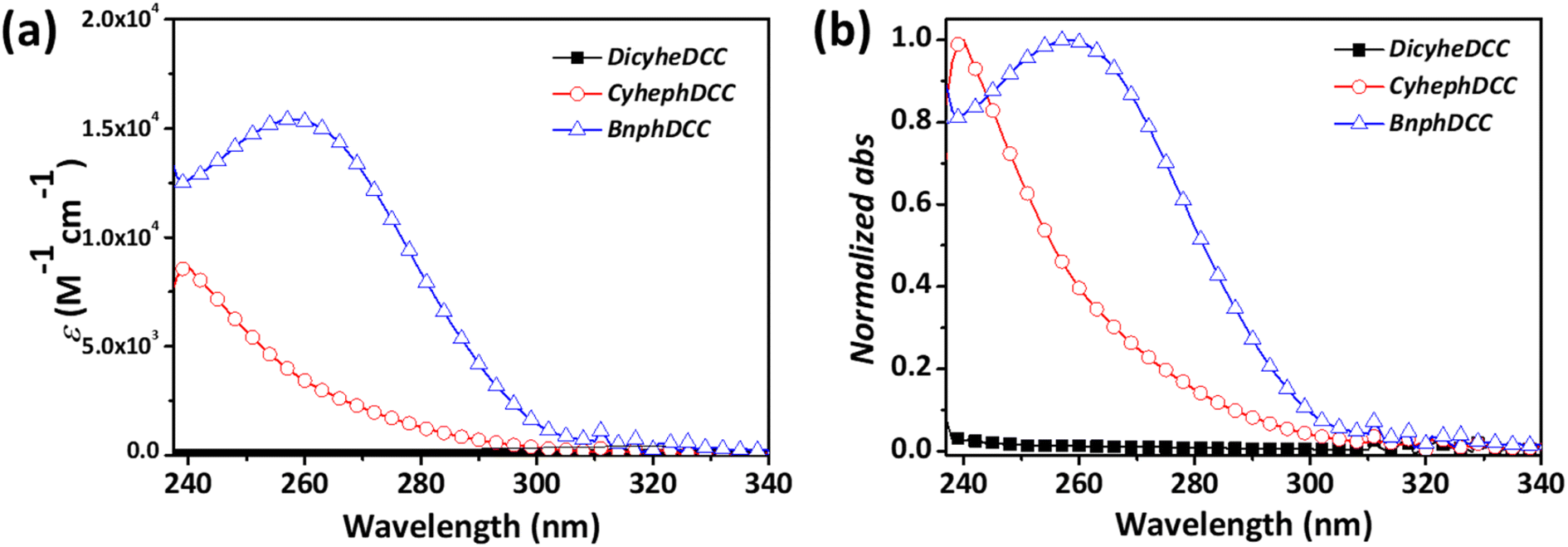 | ||
| Fig. 3 (a) Absorption coefficient and (b) normalized UV-vis absorption curves of FRIs. | ||
| Sample | Abs. coefficiency [M−1 cm−1] | λonseta [nm] | λmaxa [nm] | Tdb [°C] | Tonsetc [°C] |
|---|---|---|---|---|---|
| a Absorption spectra in the dichloromethane solution.b Initiation of the decomposition of only FRIs in the TGA curves.c Cleavage temperatures of the N–O bonds in only FRIs obtained using DSC curves. | |||||
| DicyheDCC | — | 117 | 112 | ||
| CyhephDCC | 8.57 | 291 | 239 | 122 | 116 |
| BnphDCC | 15.42 | 300 | 259 | 129 | 122 |
Fig. 4 shows the TGA and DSC results for the intrinsic thermal properties of the FRIs. The detailed experimental data are presented in Table 1. In the case of TGA, the overall thermal properties were measured, and the initial decomposition temperatures were 117, 122, and 129 °C for DicyheDCC, CyhephDCC, and BnphDCC, respectively. For DSC, DicyheDCC, CyhephDCC, and BnphDCC were measured at 112, 116, and 122 °C, respectively, and the data indicated the cleavage temperature of N–O bonds in the FRIs. According to the thermal properties of the FRIs, we predicted that cyclic aliphatic groups could reduce the N–O bond cleavage temperature under thermal conditions and lead to polymerization at low temperatures.16,17
 | ||
| Fig. 4 (a) TGA and (b) DSC curves of synthesized FRIs. | ||
2.3. Photo- and thermal polymerization of acrylate monomer
To evaluate the polymerization efficiency of the synthesized FRIs in the acrylate monomer, photo- and thermal-curing processes were performed using DSC, as shown in Fig. 5 and summarized in Table 2. In the thermal-DSC experiment, all initiators exhibited curing peaks between 85 and 100 °C, with DicyheDCC generating radicals most effectively at the lowest temperature. For photo-DSC, curing curves were acquired in the range of 254–365 nm at a light intensity of 2 mW cm−2. With the exception of DicyheDCC, the FRIs exhibited a considerable increase in heat flow within 1–2 min of UV irradiation, indicating that they are useful initiators for photopolymerization. | ||
| Fig. 5 Polymerization of n-butyl acrylate with new radical initiatiors: (a) thermal/photo-radical polymerization mechanism (b) thermal polymerization; (c) photopolymerization. | ||
| Sample | DSCexp | Simulation | ||
|---|---|---|---|---|
| Tonset [°C] | Tpeak [°C] | BDE [kcal mol−1] | N–O bond length (Å) | |
| DicyheDCC | 82.5 | 86.0 | 27.7 | 1.339 |
| CyhephDCC | 84.2 | 88.1 | 28.1 | 1.393 |
| BnphDCC | 93.8 | 96.1 | 29.6 | 1.388 |
According to the photo-DSC results, we conducted tests to determine the polymer conversion rate for the FRIs, as shown in Fig. 6. Thermal radical polymerization of a mixture of butyl acrylates (200 mmol) and FRIs (1 mmol) in toluene (1.5 mL) under N2 stirred for 1–5 h at different temperatures (90, 100, 120 °C) was conducted. The molecular weight and conversion (%) were monitored during polymerization. The conversion (%) was calculated from the integration ratio of the methylene unit next to the vinyl group in acrylate in the NMR spectra. As shown in Fig. 6, 80% of the vinyl group may be converted by thermal radical polymerization of DicyheDCC and CyhephDCC in less than 5 h at ≤100 °C. At 120 °C, the BnphDCC conversion rate was 80%, which corresponded to the radical's initiation temperature. The photo-radical polymerization of n-butyl acrylates using FRIs (2.5 wt%) was performed under irradiation at 254–365 nm without a solvent. The conversion (%) was calculated by integrating the ratio of the typical double bond peaks of acrylate (1619 and 1636 cm−1) in the Fourier transform infrared (FTIR) spectra, as shown in Fig. S4.† As indicated by Fig. 6d, CyhephDCC and BnphDCC had a high conversion rate of 90%; however, DicyheDCC did not respond to light, because it lacked a structure that reacted with light. The photo/light DSC values of the synthesized FRIs and the polymer conversion rate exhibited a strong connection, indicating that the conversion rate can be predicted via DSC analysis.
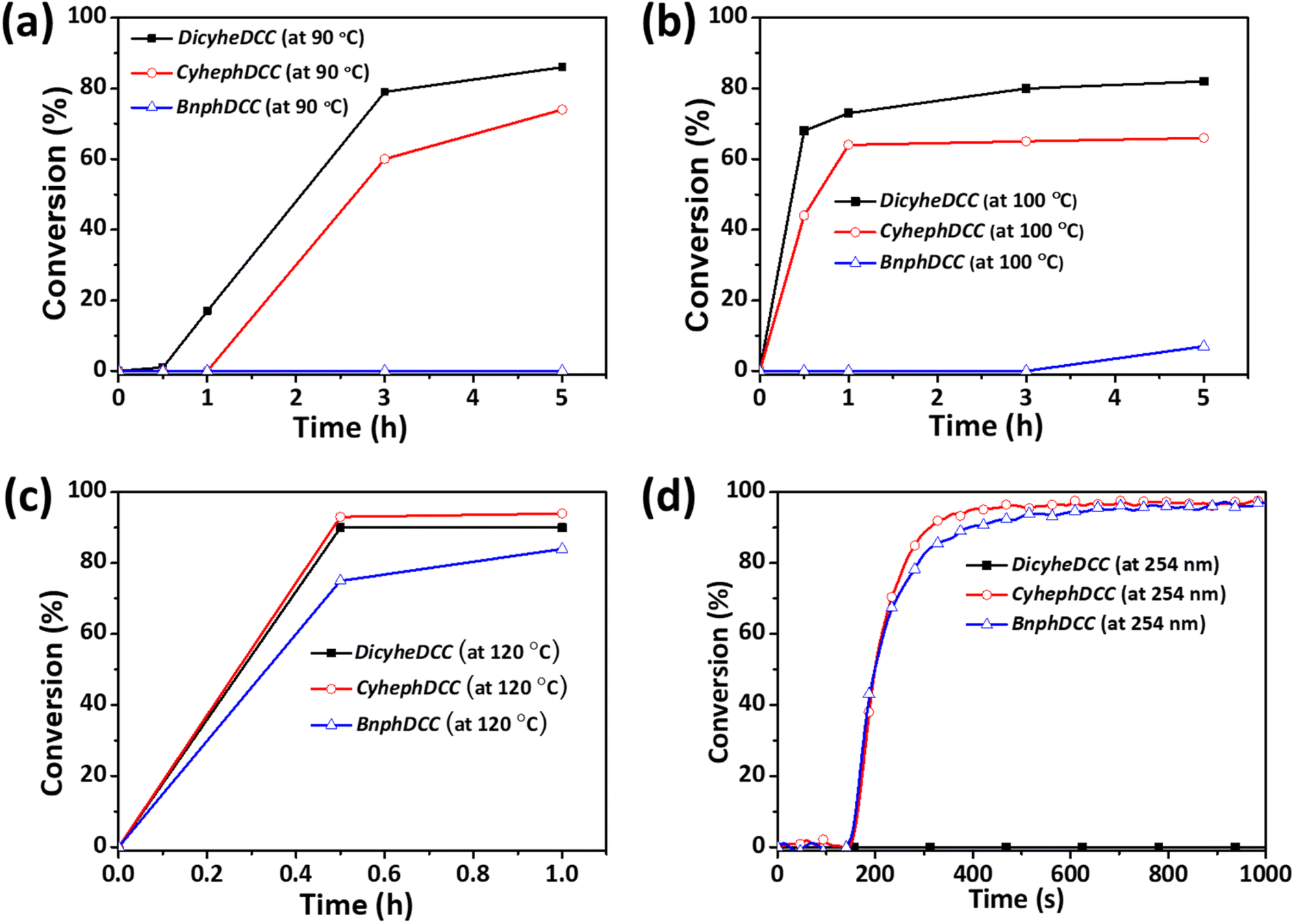 | ||
| Fig. 6 Conversion rates of thermal radical polymerization at different temperatures: (a) 90 °C; (b) 100 °C; (c) 110 °C. (d) Conversion rates of photo-radical polymerization under 254 nm UV irradiation. | ||
To assess the polymerization characteristics of the synthesized FRIs, polymerization reactions were conducted using n-butyl acrylate. For CyhephDCC and BnphDCC, polymerization occurs only under photo. Therefore, to compare the properties of FRIs, polymerization was carried out at 120 °C, and the molecular weights were measured using GPC. The results have been summarized in Table 3. Molecular weights (Mn, Mw) of polymerized n-butyl acrylate for example, CyhephDCC, BnphDCC, and DicyheDCC, were found to be (3400, 7600), (3900, 9900), and (5100, 12700) g mol−1, respectively (see Table 3). The conversion (%) was calculated by integration of each peak at 5.8–6.5 ppm (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) and 4.23 ppm (–C(O)–O–CH2–) in the 1H-NMR spectra. The conversion (%) of polymerized n-butyl acrylate with CyhephDCC, BnphDCC, and DicyheDCC were found to 88, 90 and 94% (see Fig. S5†). These results suggest that substituted FRIs with an aliphatic group showed significantly higher conversion and exhibited higher molecular weights compared to the FRIs substituted with an aromatic group.
CH–) and 4.23 ppm (–C(O)–O–CH2–) in the 1H-NMR spectra. The conversion (%) of polymerized n-butyl acrylate with CyhephDCC, BnphDCC, and DicyheDCC were found to 88, 90 and 94% (see Fig. S5†). These results suggest that substituted FRIs with an aliphatic group showed significantly higher conversion and exhibited higher molecular weights compared to the FRIs substituted with an aromatic group.
| Sample | Conversion (%) | Mn (g mol−1) | Mw (g mol−1) | D |
|---|---|---|---|---|
| DicyheDCC | 88 | 3400 | 7600 | 2.22 |
| CyhephDCC | 90 | 3900 | 9900 | 2.52 |
| BnphDCC | 94 | 5100 | 12700 |
2.47 |
2.4. Real-time NMR measurement and DFT calculations of FRIs
Through real-time 1H NMR and DFT computations, we analyzed the general stability of the synthesized FRIs by tracking their radical production capabilities. For 1H NMR, the generation of radical species was monitored every 15 min after exposure to 254 nm UV irradiation. As shown in Fig. 7, DicyheDCC without an aromatic group had no effect on the NMR signal due to light, indicating that no radical species were produced. In contrast, for CyhephDCC and BnphDCC, the formation of radicals was confirmed by the measured 8.0 ppm N–H peak caused by the breakdown of the N–O bond after 15 min. In addition, the CyhephDCC radicals remained relatively stable under UV irradiation, in contrast to the BnphDCC radicals, which were rapidly destroyed after 60 min. Thus, these results led to an associated polymer conversion rate. Furthermore, we optimized all the FRIs and calculated the thermal enthalpy via computational simulations, as shown in Fig. 8 and Table 2. Fig. 8a shows the optimized N–O bond lengths of various FRIs at the most sterically stable level calculated using the M06/6-31g(d) method. Because they represent the strength of the N–O bond, our calculated data can provide theoretical insights into the thermal properties of FRIs, as radicals are generated by N–O bond cleavage.12 The simulated N–O bond lengths of the synthesized FRIs DicyheDCC, CyhephDCC, and BnphDCC were 1.399, 1.393, and 1.388 Å, respectively. The calculated N–O bond dissociation energies (BDEs) were 27.73, 28.15, and 29.62 kcal mol−1, respectively. Thus, the orders of the simulated N–O bond lengths and BDEs well matched the FRIs. With an increase in the degree of conjugation of the imine radical, the BDE increased and the bond length decreased, in the order of Bnph–Cyheph–Dicyhe. When the simulation results were interpreted according to the chemical structure, the degree of conjugation from the aromatic ring increased from DicyheDCC to BnphDCC. Using DSC data, we obtained thermal curing initiators that were active at high temperatures. These results supported our expectations regarding the relationship between the stiffness effects of substitution. It may be that the higher stiffness of the aromatic group compared with the cyclohexyl group suppressed the steric effect between the cyclohexyl groups on the DCC side because there was less rotation or vibration. This reduced steric effect between N–O bonds reduced the BDE and increased the bond length. All the calculation results agreed well with the experimental thermal analysis and other curing processes.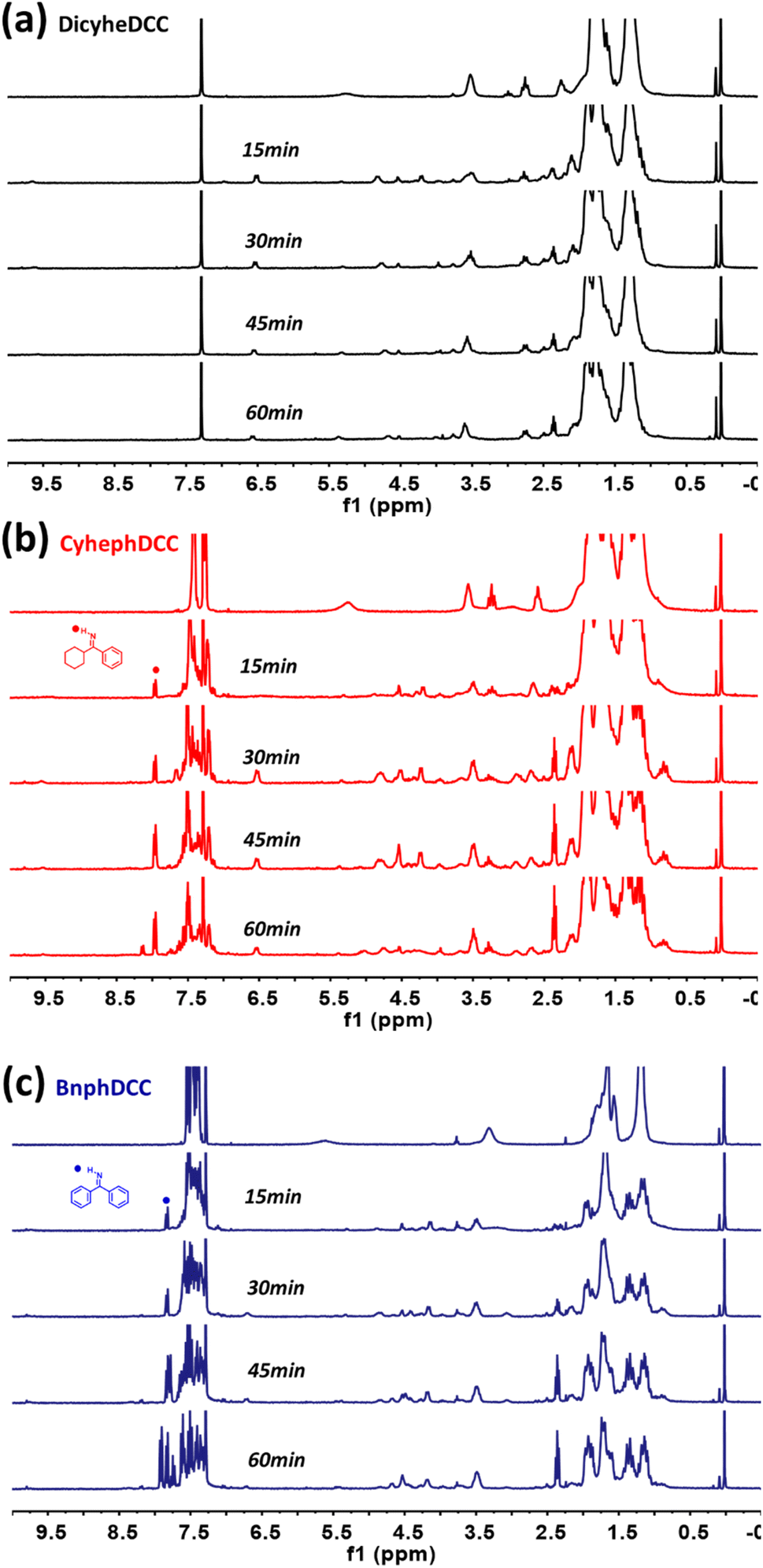 | ||
| Fig. 7 1H NMR screening to analyze the decomposition of the synthesized radical initiators ((a) DicyheDCC, (b) CyhephDCC, (c) BnphDCC) under 254 nm irradiation in CDCl3. | ||
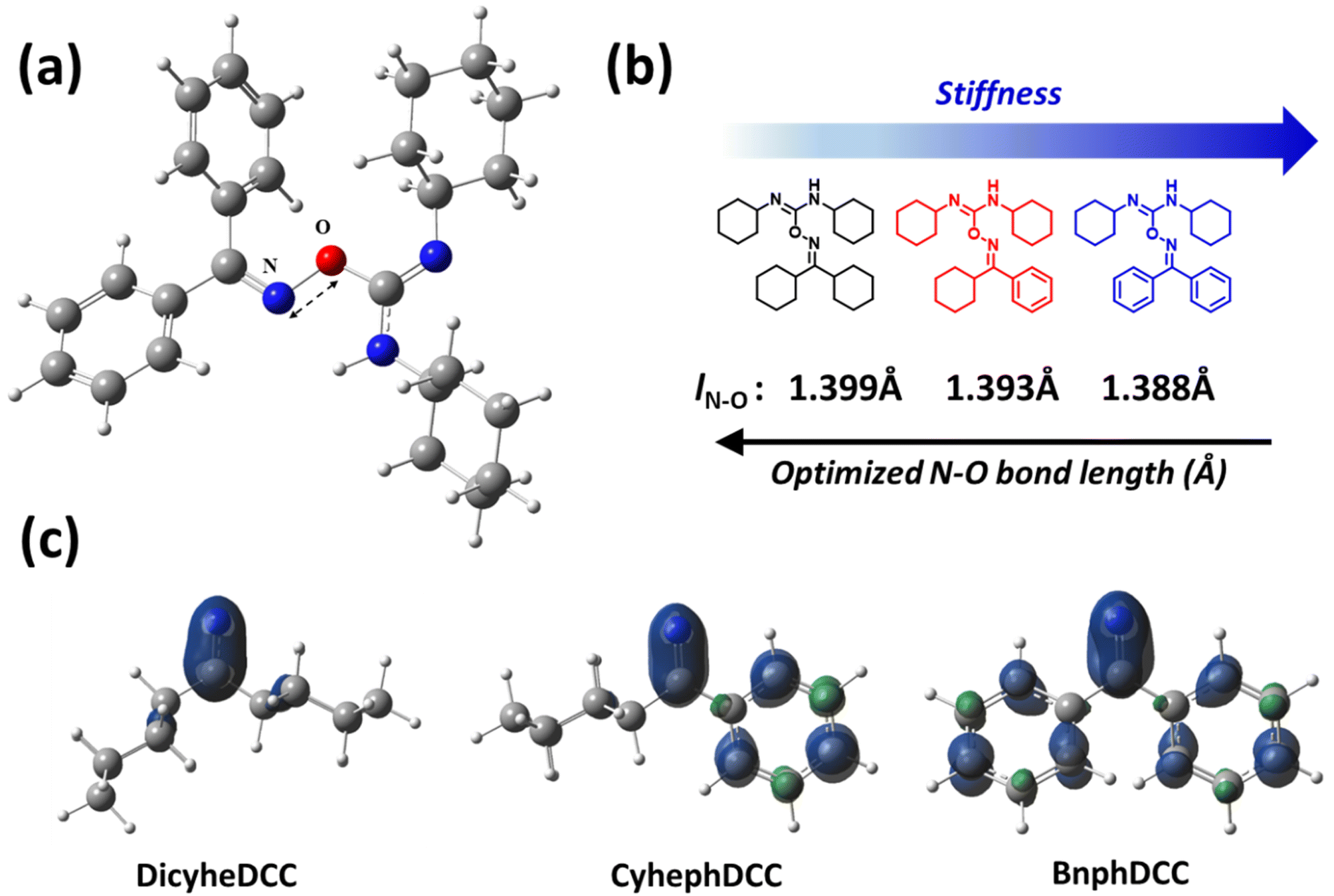 | ||
| Fig. 8 (a) Optimized structure of Bnph-DCC (H: white, C: dark gray, N: blue, O: red). (b) Optimized N–O bond lengths of the various FRIs. All calculations were performed at the M06/6-31g(d) level of theory. (c) Electron distributions of the FRIs. | ||
Furthermore, we calculated the electron density distributions of various FRIs at the TD-M06/6-31g(d) level of theory using the M06/6-31g(d) optimized geometry, as shown in Fig. 8c. The electrodensity distribution is related to the degree of conjugation in the systems and the range of absorption.18,19 Therefore, absorption occurs in FRIs containing aromatic groups and is activated by photons. The simulation results, including the BDEs, bond lengths, and absorption spectra, agreed well our experimental results.
3. Experimental section
Reagents and starting materials were purchased from Aldrich Co., Alfa Aesar, and TCI Co. and were used without further purification. All solvents were of ACS grade, unless otherwise noted. THF was used as the solvent for purification. 1H and 13C NMR spectra were recorded using a UI-trashied 300 MHz spectrophotometer (Bruker, Germany). FTIR spectra were recorded using a Nicolet 6700/Nicolet Continuum spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). EA was performed using FLASH EA-2000 and Finnigan FLASH EA-1112 (Thermo Fisher Scientific). LC-MS was performed using a Quadrupole LC/MS6130 system (HPLC 1260, Agilent, USA). The thermal stability was evaluated via TGA (TA Instruments, USA) in the range of 25–400 °C at a heating rate of 5 °C min−1, and thermal DSC curves were recorded using a differential scanning calorimeter (TA Instruments, USA) at a heating rate of 1 °C min−1 between 25 and 130 °C. For photo-DSC (TA Instruments, DSC-Q100, USA), the polymerization curves were recorded at a light intensity of 2 mW cm−2 in the range of 254–365 nm after mixing the monomer and initiators (2.5 wt%). The BDE simulations and geometry optimization of the FRIs were performed via DFT calculations using Gaussian09, and details are presented below. The molecular weight and molecular weight distribution of the polymers were analyzed by GPC (Agilent Tech 1260) using a liquid chromatograph equipped with a series of Agilent PLgel 5 μm MIXED-D columns, and a 1260 Isocratic pump at room temperature. THF was used as an eluent at 23 °C, the flow rate was set to 1 mL min−1, and the pump pressure was maintained at 80 MPa. All of the GPC data were calibrated with polystyrene standard in the range of molecular weight from 650 to 6375000.
Polymerization of n-butyl acrylate: Molecular weights (Mn, Mw) of polymerized n-butyl acrylate at 120 °C with the FRIs in toluene, the FRIs and n-butyl acrylate were reacted in a ratio of 1:200 for overnight, following which the molecular weight was measured using GPC.
Computational details: For reliable results, we used DFT. Geometry optimization and BDE calculations were performed at the M06/6-31g(d) level of theory.20,21 The BDE was defined by the equation BDE (kcal mol−1) = HTRI − Himine − Hisourea, where HTRI, Himine, and Hisourea represent the thermal enthalpies of the FRIs, the imine radical, and the isourea radical, respectively, at 298.15 K. It was used as an indicator of the strength of the N–O bonds in the FRIs. Because a lower BDE indicates a lower bond strength and cleavage at lower temperatures, our calculated BDE values can provide theoretical insights into the thermally active temperatures for various FRIs. Furthermore, we calculated the excitation to obtain the electron density distributions for various FRIs at the TD-M06/6-31g(d) level of theory using the M06/6-31g(d) optimized geometry. All the calculations in this study were performed using the Gaussian09 package.
3.1. Synthesis method for oximes and FRIs
:5) was refluxed. After TLC monitoring, the solvent was removed using a rotary evaporator and extracted with ethyl acetate and water. The organic layers were dried over sodium sulfate and recrystallized from ethyl acetate and hexane to obtain a pure white crystal-like solid. (3.25 g, 60%) 1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.66 (s, OH), 7.44–7.40 (m, 3H), 7.39–7.36 (m, 2H), 2.49–2.41 (m, 1H), 1.90–1.60 (m, 4H), 1.30–1.19 (m, 6H).1H NMR (CDCl3, 300 MHz): δ (ppm) = 7.47–7.32 (m, 4H), 7.25–7.21 (m, 1H), 5.20 (brs, 1H–NH), 3.54 (brs, 2H), 2.64–2.50 (m, 1H), 2.15–1.57 (m, 13H), 1.44–0.70 (m, 17H). 13C NMR (CDCl3, 75 MHz): δ (ppm) = 166.07, 164.93, 148.29, 148.03, 135.34, 133.62, 129.08, 128.61, 128.22, 127.96, 127.92, 127.16, 44.19, 40.82, 30.25, 29.26, 26.44, 26.03, 25.96, 25.09. Element anal. calc. for C26H39N3O: C, 76.24; H, 9.66; N, 10.26; O. Found: C, 76.11; H, 9.63; N, 10.32. LC-MS calc. for C26H39N3O: 409.31. Found: 410.5.
FTIR = 3416, 2927, 2848, 1671, 1627, 1488, 1444, 1365, 1347, 1328, 1310, 1249, 1223, 1183, 1152, 1124, 1103, 1086, 1028, 1003, 987, 971, 942, 919, 891, 771, 715, 702.
13C NMR (CDCl3, 75 MHz): δ (ppm) = 159.77, 148.33, 135.77, 133.11, 130.84, 129.85, 129.49, 129.24, 128.95, 128.58, 128.18, 51.89, 36.64, 26.47, 25.65. FTIR = 3419, 2922, 2850, 1669, 1477, 1444, 1371, 1334, 1313, 1301, 1249, 1221, 1152, 1117, 1099, 1029, 1007, 966, 950, 931, 889, 844, 796, 783, 776, 740, 694, 668. Element anal. calc. for C26H33N3O: C, 77.38; H, 8.24; N, 10.41. Found: C, 77.21; H, 8.46; N, 10.53. LC-MS calc. for C26H33N3O: 403.57. Found: 404.4.
4. Conclusions
To realize o-imino isourea-based radical initiators with photo- and thermal dual effects, three radical initiators (DicyheDCC, CyhephDCC, and BnphDCC) were systematically designed and synthesized. DicyheDCC, which consisted of cyclic hydrocarbons, exhibited rapid thermal initiation (Tpeak = 82 °C) under thermal DSC, whereas CyhephDCC and BnphDCC with aromatic groups exhibited excellent photoinduced polymerization under photo-DSC (254 nm). This indicates that the active source for the generation of radical species varies with an increasing amount of aliphatic or aromatic groups in the radical initiator. The conversion rates matched the trends of photo- and thermal DSC, and the conventional radical polymerization of n-butyl acrylate utilizing all the radical initiators was performed satisfactorily. Interestingly, compared with the rapid radical generation in the photoreaction exhibited by BnphDCC, CyhephDCC contained steady radical species, resulting in a comparatively high rate of polymerization conversion. In particular, CyhephDCC and BnphDCC can be used in efficient and diversified polymer networks that require a synergistic effect system and a novel all-in-one dual-curing system because of their ability to generate photo- and heat radicals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Technology Innovation Program funded by the Ministry of Trade, Industry, and Energy (MOTIE, Korea) [RS-2023-00269383] and the Ministry of Trade, Industry & Energy of Korea Industrial Technology Innovation Program [grant number 20017536]. This research was supported by the Traditional Culture Innovative Convergence Research Program through the National Research Foundation of Korea (NRF) funded by Ministry of Science and ICT and funded by Ministry of Culture, Sports and Tourism (RS-2023-00301733).References
- M. Ouchi, T. Terashima and M. Sawamoto, Transition metal-catalyzed living radical polymerization: toward perfection in catalysis and precision polymer synthesis, Chem. Rev., 2009, 109, 4963–5050, DOI:10.1021/cr900234b.
- Y. Yagci, S. Jockusch and N. J. Turro, Photoinitiated Polymerization: Advances, Challenges, and Opportunities, Macromolecules, 2010, 43, 6245–6260, DOI:10.1021/ma1007545.
- S. Liu, D. Brunel, K. Sun, Y. Xu, F. Morlet-Savary, B. Graff, P. Xiao, F. Dumur and J. Lalevée, Polym. Chem., 2020, 11, 3551–3556 RSC.
- J. Lopez, D. G. Mackanic, Y. Cui and Z. Bao, Designing polymers for advanced battery chemistries, Nat. Rev. Mater., 2019, 4, 312–330, DOI:10.1038/s41578-019-0103-6.
- Y. Zhang, Y. Xu, A. Simon-Masseron and J. Lalevée, Radical photoinitiation with LEDs and applications in the 3D printing of composites, Chem. Soc. Rev., 2021, 50, 3824–3841, 10.1039/d0cs01411g.
- R. E. Richard, M. Schwarz, S. Ranade, A. K. Chan, K. Matyjaszewski and B. Sumerlin, Evaluation of acrylate-based block copolymers prepared by atom transfer radical polymerization as matrices for paclitaxel delivery from coronary stents, Biomacromolecules, 2005, 6, 3410–3418, DOI:10.1021/bm050464v.
- Q. Ma, S. Liu, M. Le Dot, H. Mokbel, Y. Zhang, B. Graff and J. Lalevée, Imidazole based dual photo/thermal initiators for highly efficient radical polymerization under air with a metal-free approach, Polym. Chem., 2021, 12, 6386–6391, 10.1039/D1PY01079D.
- F. Li, Y. Song, M. Yao, J. Nie and Y. He, Design and properties of novel photothermal initiators for photoinduced thermal frontal polymerization, Polymer, Chemistry, 2020, 11, 3980 RSC.
- A. Mariani, D. Nuvoli, V. Alzari and M. Pini, Phosphonium-Based Ionic Liquids as a New Class of Radical Initiators and Their Use in Gas-Free Frontal Polymerization, Macromolecules, 2008, 41, 5191–5196, DOI:10.1021/ma800610g.
- J. Ueda, H. Yamaguchi, K. Shirai, T. Yamauchi and N. Tsubokawa, Radical polymerization of vinyl monomers in the presence of carbon black initiated by 2,2’-azobisisobutyronitrile and benzoyl peroxide in ionic liquid, J. Appl. Polym. Sci., 2008, 107, 3300–3305, DOI:10.1002/app.27448.
- J. C. Bevington, B. J. Hunt and J. Warburton, Effects of stabilized radicals upon polymerizations initiated by benzoyl peroxide, Polymer, 2003, 44, 3469–3475, DOI:10.1016/S0032-3861(03)00292-1.
- B. Kim, D. G. Lee, D. Y. Kim, H. J. Kim, N. S. Kong, J. C. Kim, S. M. Noh, H. W. Jung and Y. I. Park, Thermal radical initiator derivatives based on O -imino-isourea: Synthesis, polymerization, and characterization, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3593–3600, DOI:10.1002/pola.28244.
- R. X. Ren and W. Ou, Preparation of cyclic ketoximes using aqueous hydroxylamine in ionic liquids, Tetrahedron Lett., 2001, 42, 8445–8446, DOI:10.1016/S0040-4039(01)01851-2.
- X. Mo, T. D. R. Morgan, H. T. Ang and D. G. Hall, Scope and Mechanism of a True Organocatalytic Beckmann Rearrangement with a Boronic Acid/Perfluoropinacol System under Ambient Conditions, J. Am. Chem. Soc., 2018, 140, 5264–5271, DOI:10.1021/jacs.8b01618.
- S. Morales, J. L. Aceña, J. L. García Ruano and M. B. Cid, Sustainable Synthesis of Oximes, Hydrazones, and Thiosemicarbazones under Mild Organocatalyzed Reaction Conditions, J. Org. Chem., 2016, 81, 10016–10022, DOI:10.1021/acs.joc.6b01912.
- C. A. Knoop and A. Studer, Hydroxy- and silyloxy-substituted TEMPO derivatives for the living free-radical polymerization of styrene and n-butyl acrylate: synthesis, kinetics, and mechanistic studies, J. Am. Chem. Soc., 2003, 125, 16327–16333, DOI:10.1021/ja037948o.
- W. G. Skene, S. T. Belt, T. J. Connolly, P. Hahn and J. C. Scaiano, Decomposition Kinetics, Arrhenius Parameters, and Bond Dissociation Energies for Alkoxyamines of Relevance in “Living” Free Radical Polymerization, Macromolecules, 1998, 31, 9103–9105, DOI:10.1021/ma9812229.
- U. Warde, L. Rhyman, P. Ramasami and N. Sekar, DFT studies of the photophysical properties of fluorescent and semiconductor polycyclic benzimidazole derivatives, J. Fluoresc., 2015, 25, 685–694, DOI:10.1007/s10895-015-1554-9.
- M. E. Alberto, B. C. De Simone, G. Mazzone, T. Marino and N. Russo, Photophysical properties of free and metallated meso-substituted tetrabenzotriazaporphyrin from density functional theory investigation, Dyes Pigm., 2015, 120, 335–339, DOI:10.1016/j.dyepig.2015.04.032.
- T. Delgado-Montiel, J. Baldenebro-López, R. Soto-Rojo and D. Glossman-Mitnik, Theoretical Study of the Effect of π-Bridge on Optical and Electronic Properties of Carbazole-Based Sensitizers for DSSCs, Molecules, 2020, 25, 3670, DOI:10.3390/molecules25163670.
- G. Arhin, A. H. Adams, E. Opoku, R. Tia and E. Adei, 1, 3-Dipolar cycloaddition reactions of selected 1,3-dipoles with 7-isopropylidenenorbornadiene and follow-up thermolytic cleavage: A computational study, J. Mol. Graphics Modell., 2019, 92, 267–279, DOI:10.1016/j.jmgm.2019.08.004.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07296g |
| This journal is © The Royal Society of Chemistry 2023 |