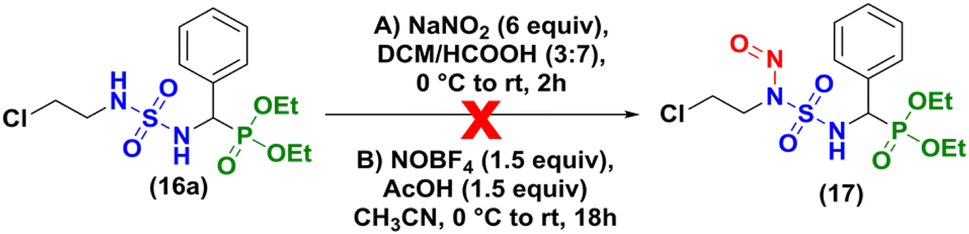DOI:
10.1039/D3RA07001H
(Paper)
RSC Adv., 2023,
13, 35741-35754
An attempt to prepare sulfonyl analogues of fotemustine: unexpected rearrangement to sulfamate during nitrosation step†
Received
14th October 2023
, Accepted 17th November 2023
First published on 11th December 2023
Abstract
This paper describes a flexible strategy to access diethyl ((((N-(2-chloroethyl)-N-nitrososulfamoyl)amino)arylmethyl) phosphonates, as aryl analogues of fotemustine. The new aryl sulfamidophosphonates prepared from 2-chloroethylamine were successfully obtained under eco-environmental conditions using ultrasound irradiation. These compounds did not produce the expected nitroso analogues of fotemustine after the nitrosation reaction but the corresponding sulfamates which were fully characterized. Some attempts to understand this rearrangement reaction were conducted, and particularly the corresponding nitrosoureas analogues could be isolated with good yield. The novel sulfonamidophosphonates as well as their sulfamate derivatives were evaluated for their cytotoxic effect on a panel of tumor cells.
Introduction
Cancer is a significant public health concern in modern society and is the second leading cause of death worldwide, following cerebrovascular diseases. According to recent WHO estimates, there were 19.3 million new cases of cancer diagnosed globally in 2020,1 with 10 million patients expected to die from the disease. The chloroethyl nitrosoureas (CENUs) have shown efficacy in specific types of cancers such as melanoma and glioma. For example, fotemustine a third-generation of nitrosoureas, developed and commercialized in the 90 s by the Servier laboratory as Muphoran®, has proven beneficial effects in the treatment of metastatic melanoma.2
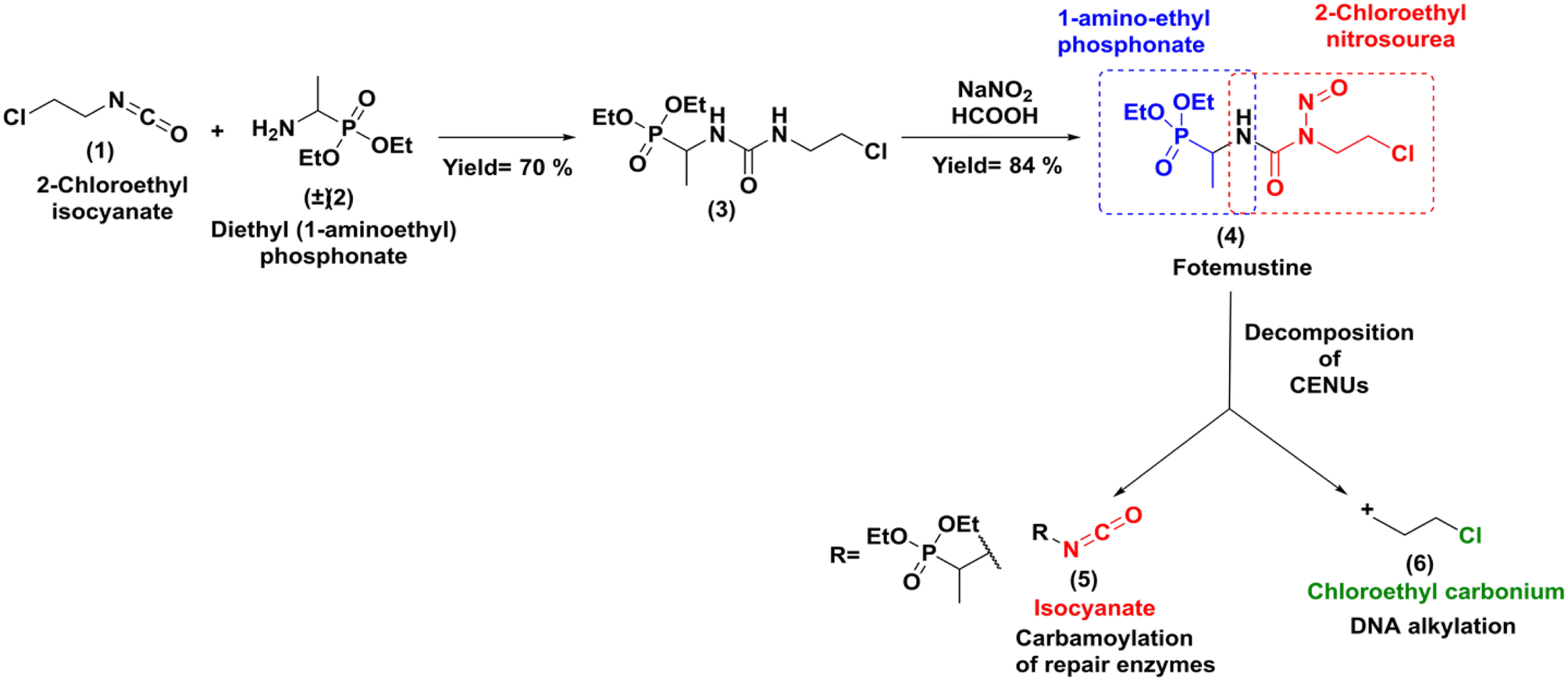
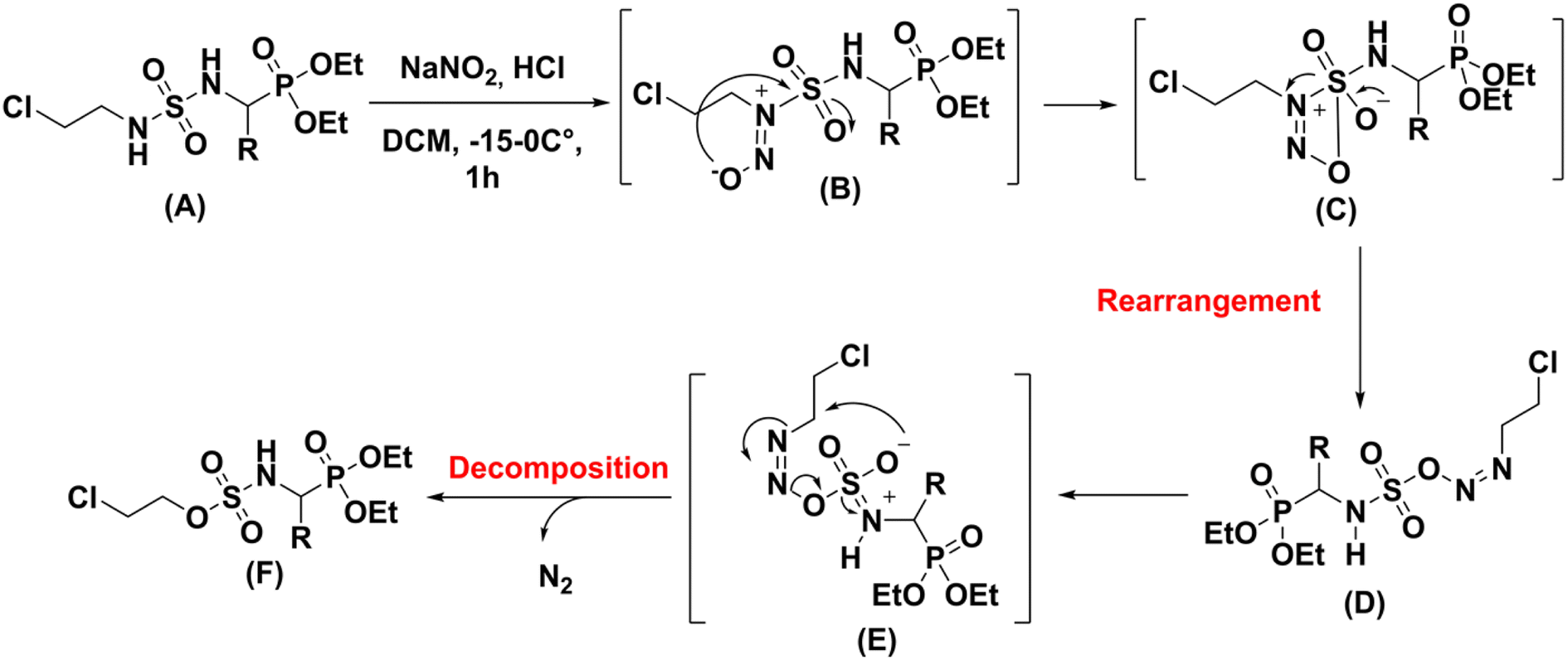
Due to its high lipophilicity facilitating entry in the central nervous system; it was also used as a rescue therapy for recurrent malignant glioma.3 The synthesis of this molecule was accomplished through a classical method that involved addition of the racemic diethylaminoethylphosphonate (2) to 2-chloroethyl isocyanate (1) followed by nitrosation reaction with NaNO2 as shown in Scheme 1.4
 |
| | Scheme 1 Classical synthesis of fotemustine and in cells decomposition. | |
Fotemustine (4), as well as the other members of the CENUs family undergo spontaneous decomposition in aqueous media leading to two electrophilic chemical species, the 2-chloroethyl carbonium ion (6), which contributes to the antitumor activity by DNA alkylation leading to inter/intra-chain bridges,5 and the isocyanate group (5), which acts as acylating agent for proteins (Scheme 1).
Both mechanisms may work synergistically to produce the antitumor effects. While CENUs have shown their usefulness in cancer chemotherapy, their potential contribution is limited by their toxic side effects, such as mutagenicity or myelotoxicity maily due to carbamoylation of repair enzymes.6
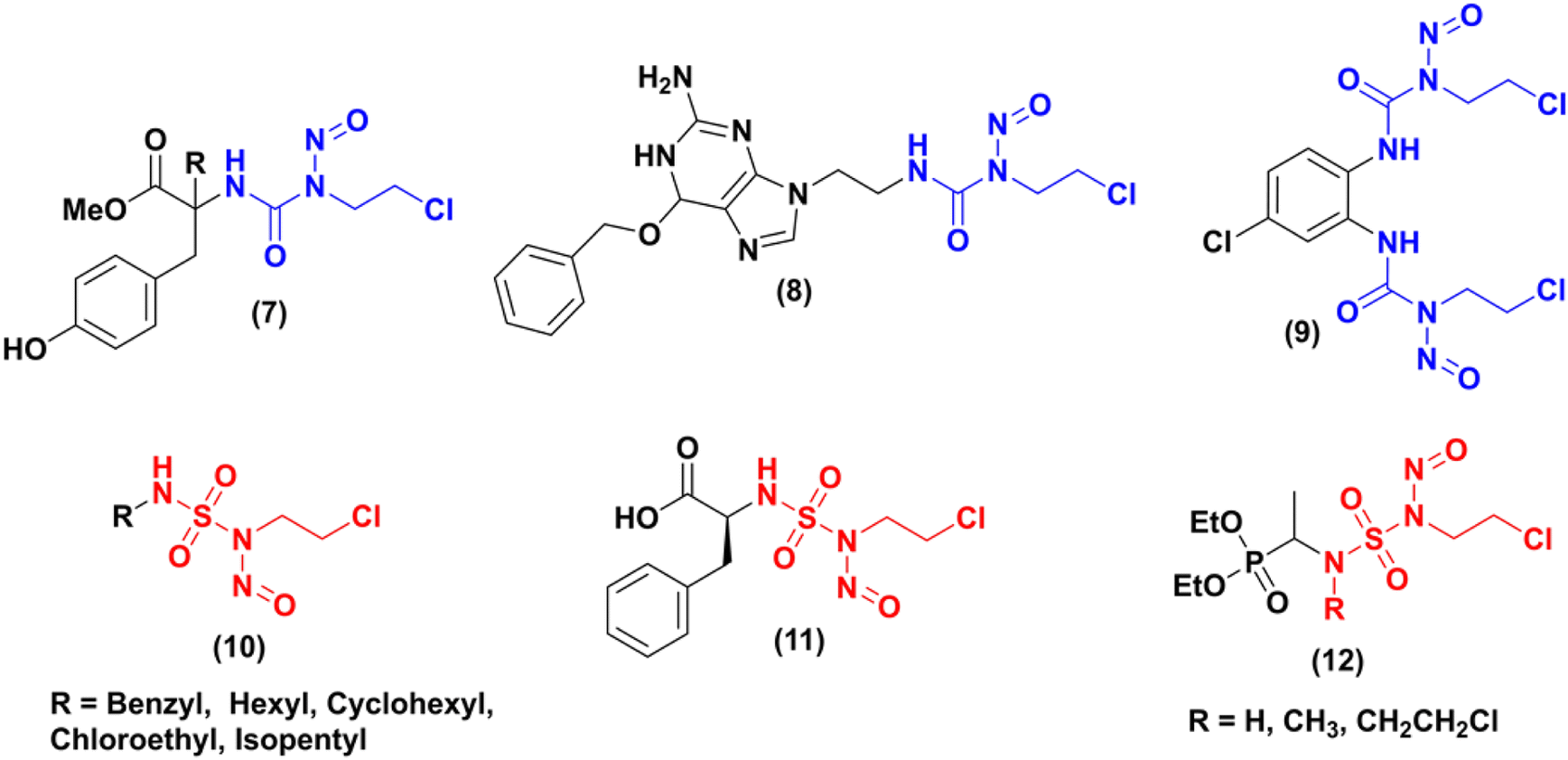
In the literature, numerous studies have been carried out on the synthesis of new nitrosourea analogues (CENUs) by integrating the 2-chloroethyl isocyanate group into various structures. Some examples are shown in Fig. 1. Gadjeva7 synthesized a new nitrosourea (Fig. 1, cpd 7) that incorporates a tyrosine moiety. In vitro evaluations of the biological activity of this product have demonstrated that it has a greater potency, selectivity, and lower toxicity towards melanoma (B16, YAC-1) and lymphocyte (NL) cells compared to lomustine (N-(2-chloroethyl)-N′-cyclohexyl-N-nitrosourea, CCNU), which was used as a reference compound. In 2017, Zhong team8 proposed the synthesis of a new compound (N-2-chloroethyl)-N′-2-(O6-benzyl-9-guanine) ethyl-N-nitrosourea (Fig. 1, cdp 8, BGCNU), containing O6-benzylguanine fragment. This compound has exhibited promising anticancer properties in human glioma cells, demonstrating much higher efficacy than the clinically-used CENU drug carmustine (N,N′-bis(2-chloroethyl)-N-nitrosourea, BCNU). Its mode of action is attributed to its ability to inhibit the activity of AGT (O6-alkylguanine DNA alkyltransferase). Another example9 describes the synthesis of the bis-N-(2-chloroethyl)-N-nitrosourea (9) derived from diamino chloro benzene, its stability during aqueous decomposition, and its cytotoxic effects on two cancer cell lines (human breast (MCF-7) and lung (A549) adenocarcinoma), compared to the clinical compound BCNU.
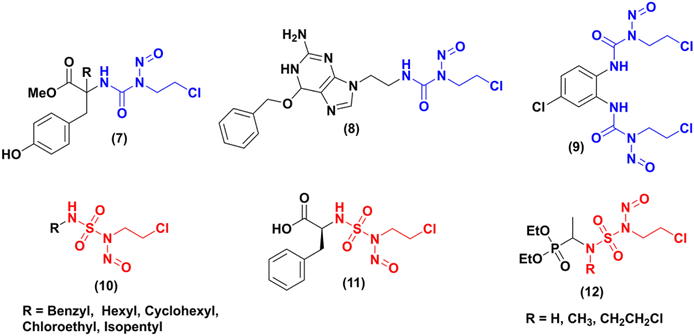 |
| | Fig. 1 Some examples of CENUs and CENSs analogues. | |
Therefore, in this area researchers have focused their interest on the development of new series of compounds that could produce similar alkylating entities in vivo while preventing the formation of isocyanate by products (5), as shown in Scheme 1. The 2-chloroethyl nitroso sulfamides (CENSs) appeared so as bioisosteres of 2-chloroethylnitrosoureas (CENUs) and as novel type of anticancer agents with no carbamoylating potency, avoiding so enzymes alteration.
Dewynter and collaborators,10 proposed the first synthesis of 2-chloroethylnitrososulfamides in 1991. During the last years, few publications have reported on various aspects of CENSs compounds, including their synthesis, biological evaluation, or kinetic study. Montero group11 developed a new series of 2-chloroethylnitrososulfamides (CENSs) in a four-step manner introducing the chloroethyl chain with a Mitsunobu reaction between chloroethanol and the key intermediate N-Boc-sulfamides bearing aliphatic chains. Poor regioselectivity could be observed and the bis-N-chloroethyl byproduct was also obtained. N-Boc deprotection followed by nitrosation gave access to a series of nitrososulfonamides (Fig. 1, cpd 10) in 71–88% yield. In 2000,12 the same group expanded this synthesis to another class of CENSs with amino acid and amino ester terminal moiety such as compound 11 (Fig. 1). The latest example published by Winum and coworkers in 2003 (ref. 13) has particularly caught our attention. They outlined the synthesis of three novel compounds 12 and subsequently evaluated their effects on melanoma cell lines. Compared to fotemustine, these analogues demonstrated significantly greater potency against the A375 cell line, which is known for its expression of the MGMT enzyme associated with tumor cell resistance to chemotherapy.
In continuity of our investigations concerning the preparation of novel bioactive compounds containing organo-phosphorus and sulfur moieties,14 and in order to minimize the toxic effect of conventional nitrosoureas,15 our aim in this paper was oriented towards the synthesis of new analogues of fotemustine. Particularly, we focused our work on the synthesis of analogues bearing a sulfamide function and an aryl substituent in place of the methyl one, on the terminal amino phosphonate moiety of fotemustine.
Results and discussion
Chemistry
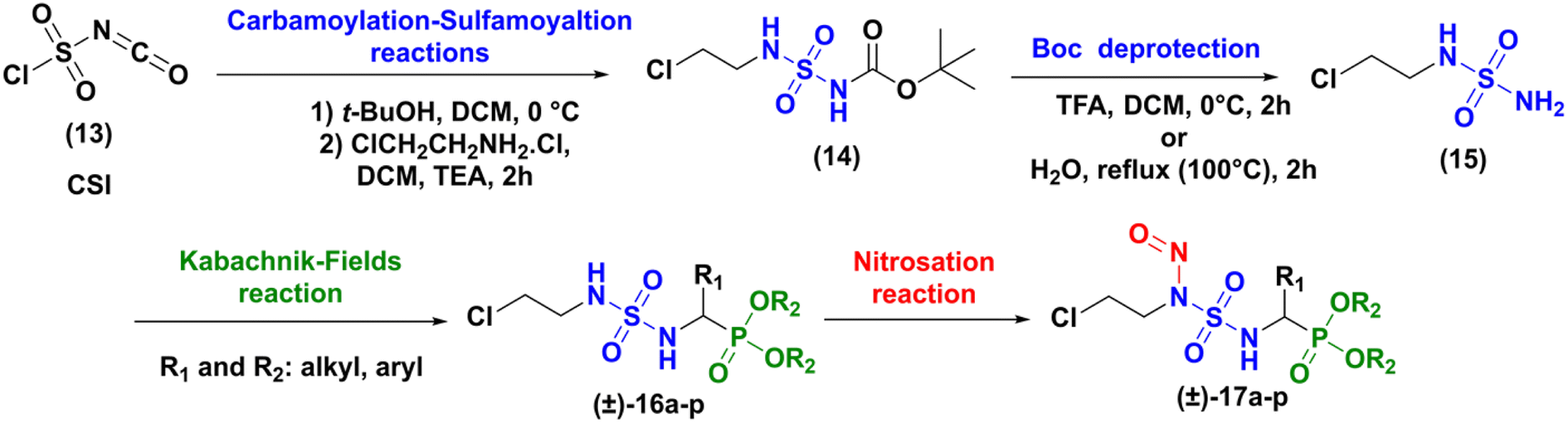
The synthesis of the targeted compounds occurs in four steps (carbamoylation–sulfamoylation reaction, deprotection of Boc group, Kabachnik–Fields reaction and nitrosation), as illustrated in Scheme 2. Compared to Winum strategy,13 the chloroethyl chain was first introduced on the sulfamide structure, in order to open the way to the preparation of a library of analogues, applying then the Kabachnik–Fields multi-component reaction to rapidly introduce a variety of amino phosphonate fragments.
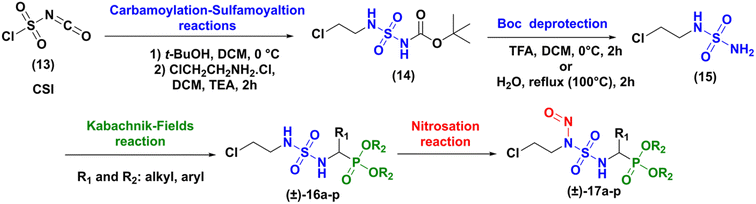 |
| | Scheme 2 Proposed strategy for the synthesis of sulfonyl analogues of fotemustine. | |
Firstly, the synthesis of N-carboxysulfamide derived of 2-chloroethylamine (14) was achieved through the functional one pot two-steps arrangement of chlorosulfonyl isocyanate (CSI) using literature processes.16 The condensation of tert-butyl alcohol with CSI at 0 °C in anhydrous dichloromethane led to the formation of chlorosulfonyl tert-butyl carbamate as an unstable intermediate. The latter product underwent a sulfamoylation reaction with 2-chloroethylamine hydrochloride in a basic medium resulting after 2 h, in the attainment of N-Boc-N-chloroethyl sulfamides (14) in excellent yield (91%).
The deprotection of (14) was achieved through two methods: the first method involved the removal of the Boc group in water under reflux (100 °C) for 2 h, a technique that had been developed in our group.17 In the second method, the Boc protective group was classically removed with trifluoroacetic acid (TFA, 5 equiv.)18 in dichloromethane at 0 °C for two hours. Both methods gave the expected compound (15) with yield around 75%.
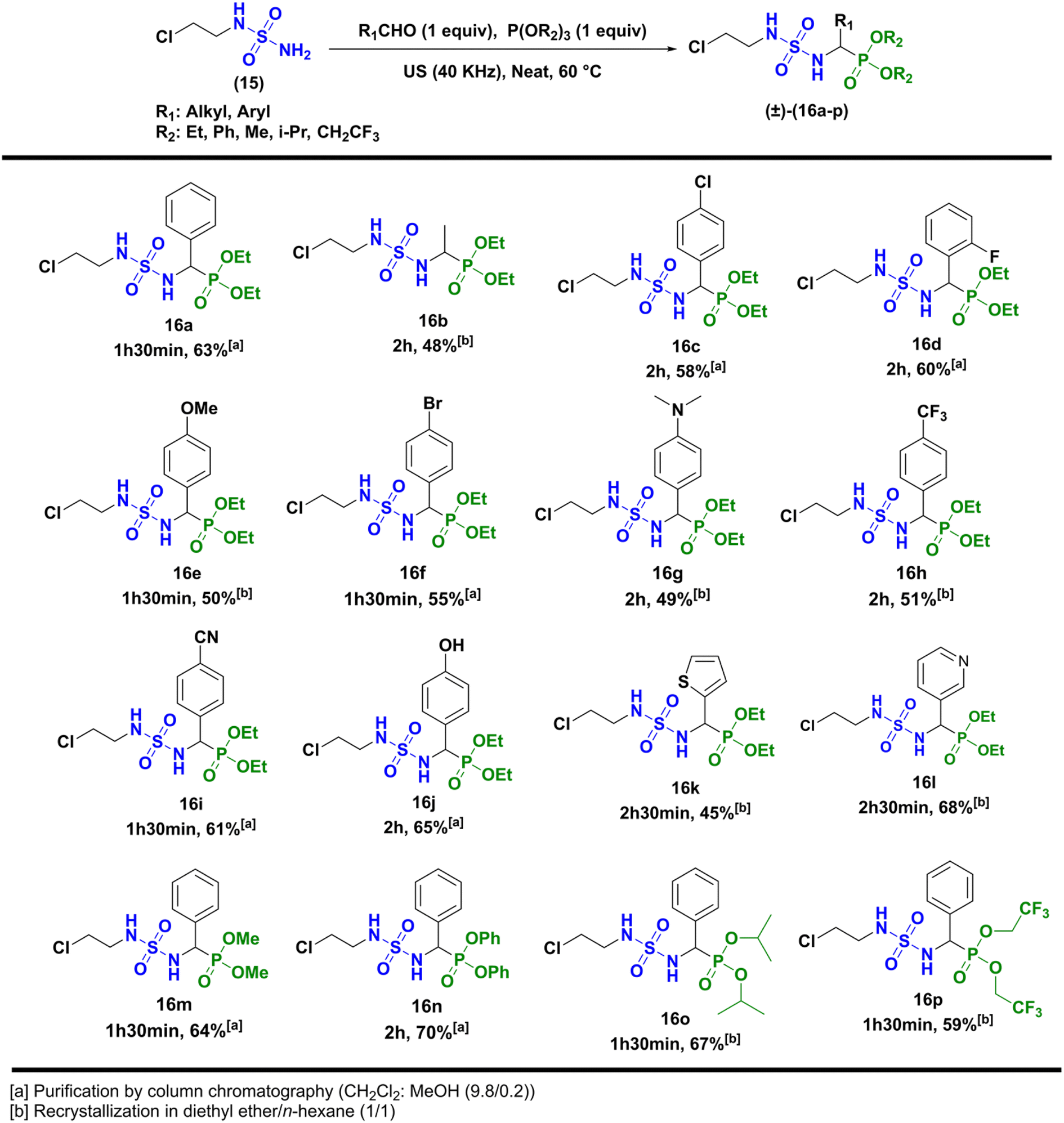
Next, the formation of sulfamidophosphonates was performed via a multi-component Kabachnik–Fields reaction, according to the conditions described by Belhani and collaborators.19 A series of α-sulfamidophosphonates was obtained from the reaction of N-(2-chloroethyl) sulfamide (15), with selected aldehydes and trialkyl-phosphite reagents, without solvent and catalyst, at 60 °C under ultrasonic irradiation (40 kHz). The expected products (16a–p) were obtained with acceptable yield in the range of 48–70% within 1h30-2h30 (Scheme 3).
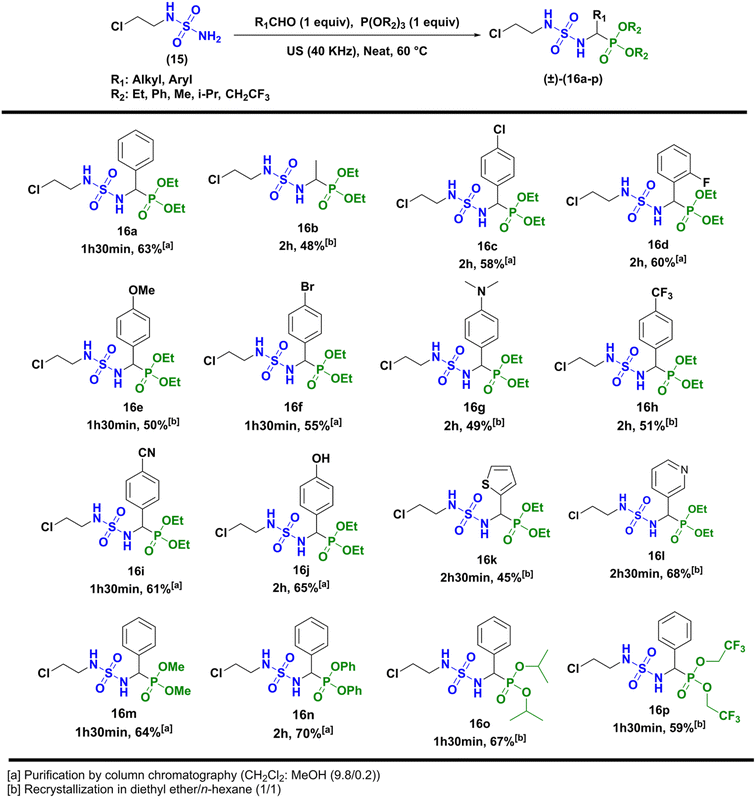 |
| | Scheme 3 Kabachnik–Fields reaction leading to α-sulfamidophosphonates products (±)-16a-p. | |
The compounds (±)-16a-p were fully characterized by NMR analysis. Particularly the chemical shift of the hydrogen of the asymmetric center (*CH) appeared between 3.75 and 5.20 ppm as a doublet split (JH–H ∼9.5 Hz and JH–P ∼23.5 Hz). In addition, the NH hydrogens appeared with specific signals, one as a triplet at 4.50 ppm (JH–H ∼7.0 Hz), and the second as a doublet split (JH–H ∼6.0 Hz, JH–P ∼9.0 Hz) between 6.50 and 6.80 ppm. Those were also identified by infrared analysis in the range of 3050–3300 cm−1, as well as the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group with stretching vibrations at 1210–1245 cm−1, and the symmetric/antisymmetric sulfonyl function SO2 characterized by specific bands at 1305 ± 50 cm−1/1145 ± 50 cm−1. In the last step, N-nitrosation reaction was performed in various reaction conditions described in the literature, as summarized in Scheme 4.
O group with stretching vibrations at 1210–1245 cm−1, and the symmetric/antisymmetric sulfonyl function SO2 characterized by specific bands at 1305 ± 50 cm−1/1145 ± 50 cm−1. In the last step, N-nitrosation reaction was performed in various reaction conditions described in the literature, as summarized in Scheme 4.
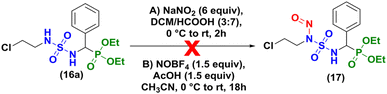 |
| | Scheme 4 Nitrosation assays with compound (±)-16a. | |
Our initial attempt to prepare 2-chloroethylnitroso sulfamido phosphonates was based on the conditions of Winum and coworkers13 in acidic conditions (DCM/HCOOH, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7), at 0 °C during 2 hours (Scheme 4A). Degradation of the starting material was observed without formation of the expected product and the same result was obtained when HCl (35%) was used instead of HCOOH (98%). As proposed by Sun et al.20 for the nitrosation of ureas, nitrosonium tetrafluoroborate was also tested without success (Scheme 4B).
:7), at 0 °C during 2 hours (Scheme 4A). Degradation of the starting material was observed without formation of the expected product and the same result was obtained when HCl (35%) was used instead of HCOOH (98%). As proposed by Sun et al.20 for the nitrosation of ureas, nitrosonium tetrafluoroborate was also tested without success (Scheme 4B).
Finally, we attempted the last assay concerning the nitrosation of sulfamidophosphonate (±)-16a based on a different study published in 2000,12 which involved 3 equivalents of sodium nitrite (NaNO2) and 3 equivalents of concentrated HCl or HCOOH at −15 °C to 0 °C in DCM.
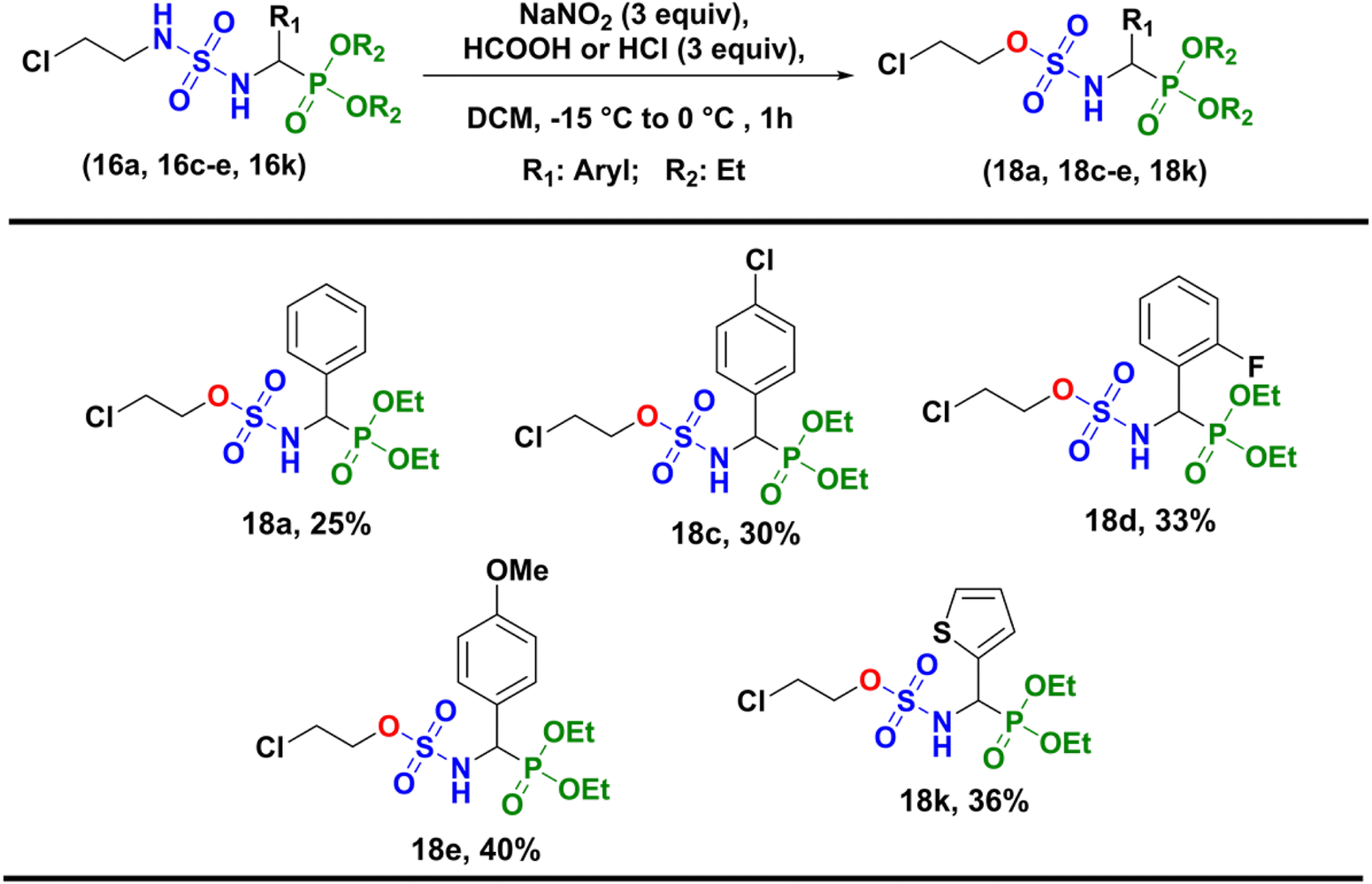
Unfortunately, the nitrosation of α-sulfamidophosphonates under these conditions only resulted in the formation of new rearranged products (±)-18 (2-chloroethyl (diethoxyphosphoryl)methyl sulfamates) instead of 2-chloroethylnitroso sulfamido phosphonate compounds (±)-17, as summarized in Scheme 5.
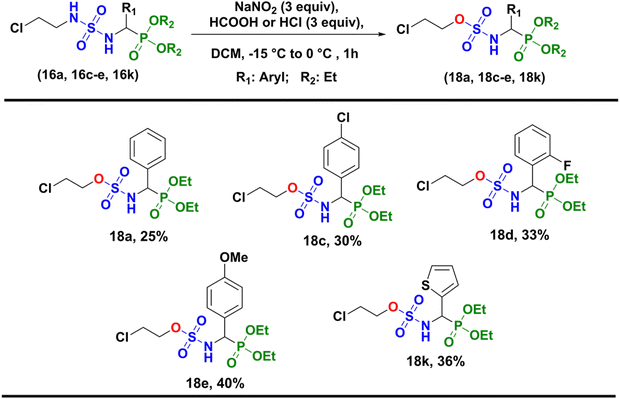 |
| | Scheme 5 Synthesis of 2-chloroethyl (diethoxyphosphoryl) methylsulfamate derivatives (±)-18. | |
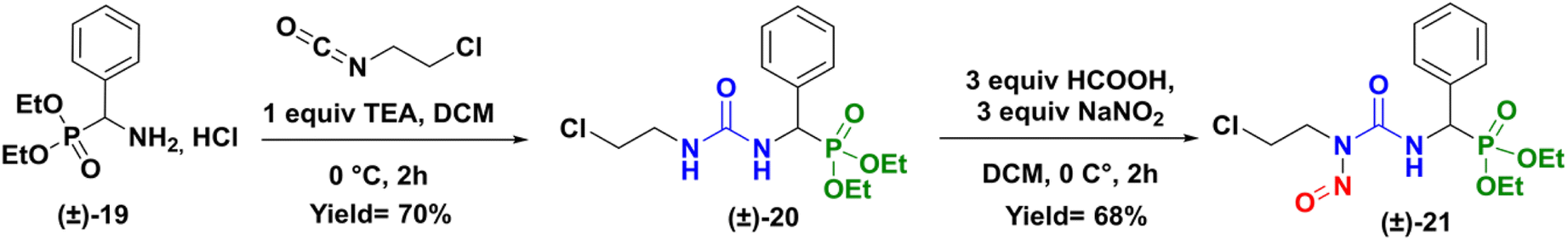
To gain a better understanding of the rearrangement phenomenon observed with N-chloroethyl sulfamido phosphonates compounds, we first compared the reactivity of urea compound versus sulfamide. We so prepared the urea analogue (CENU) of compound (±)-16a, which was subjected to nitrosation reaction, to give the phenyl analogue of fotemustine (Scheme 6). The 2-chloroethylureidophosphonate (±)-20 was achieved with a 70% yield via nucleophilic addition of diethylamino(phenyl)methylphosphonate hydrochloride (±)-19 with 2-chloroethyl isocyanate in basic conditions at 0 °C.21 It was then treated with sodium nitrite (3 equiv.) using formic acid at low 0 °C in DCM for 2 hours. The desired novel nitroso-2-chloroethylureidophosphonate (cpd 21) was obtained as a yellow powder with good yield (68%) after purification by column chromatography. This result proved that substitution of the methyl group in fotemustine with an aromatic one is not a limiting parameter in the nitrosation reaction.
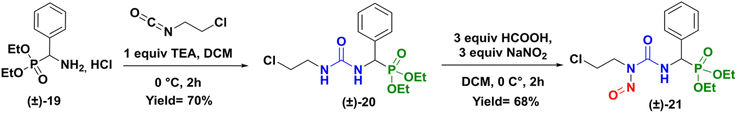 |
| | Scheme 6 Synthesis of nitroso-2-chloroetylureidophosphonate (±)-21. | |
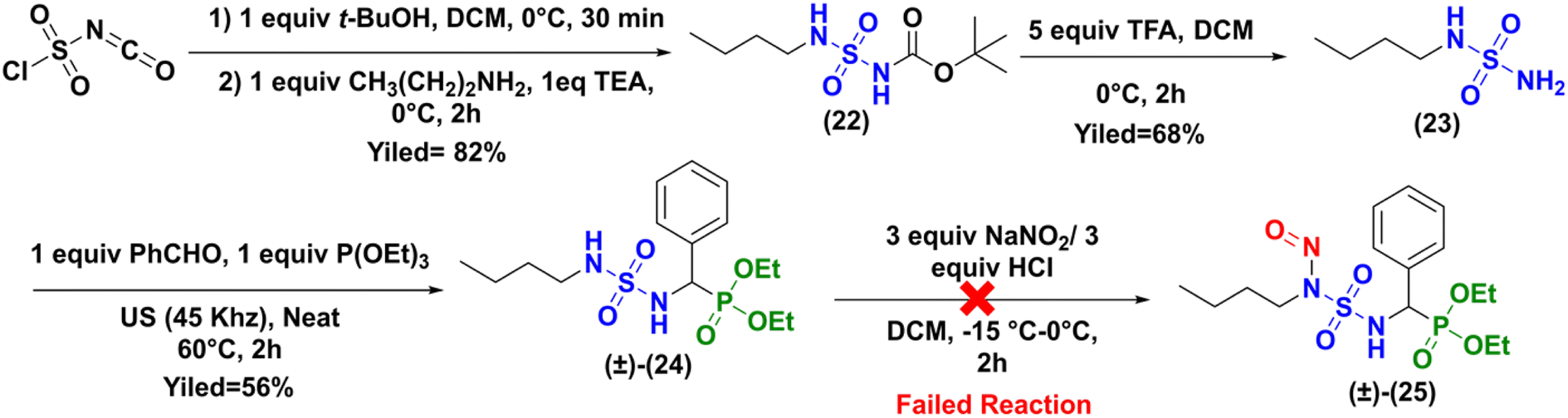
Because a number of CENSs without terminal phosphonate function are described in the literature (Fig. 1), we hypothesized that the obtention of the expected nitroso compound failed due to this particular sulfamido fragment of compound (±)-16a. Therefore, we prepared the N-butyl analogue of compound (±)-16a, which was subjected to nitrosation, as depicted in Scheme 7.
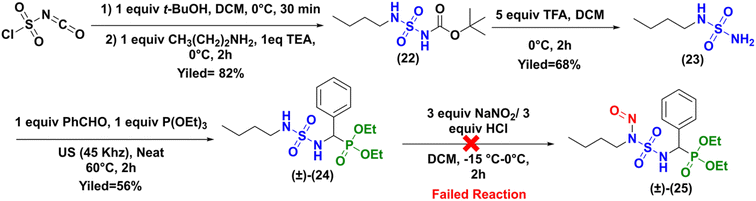 |
| | Scheme 7 Synthesis of nitroso-sulfamidophosphonate derivative of N-butylamine (±)-25. | |
The synthesis to access to the N-butyl analogue (±)-24 was identical to that previously described for the synthesis of the sulfonyl analogue (±)-16a (Scheme 2). The intermediates 23, 24, and 25 were prepared with good yields and full characterized by NMR and HRMS analysis (see ESI†).
Here too, attempts to perform the nitrosation reaction on the sulfamidophosphonate derivative of N-butylamine (±)-24 with the classical protocol were unsuccessful.
To our knowledge, the nitrosation of chloroethylsulfamido phosphonate bearing an aryl substituent in place of an alkyl one has never been studied in the literature. Moreover, Winum and collaborators attempted to prepare the strict sulfonamide analogue of fotemustine (Fig. 1, cpd 12 (RH)), but the authors specified that the compound observed by TLC was strongly unstable and could'nt be obtained pure and fully characterized.13 In our study, we also observed complete consumption of starting material (16a) or the butyl analogue (24), but we never isolated the corresponding nitroso derivatives.
The most significant changes observed in the IR spectra data of compounds (±)-18 were the absence of the NH stretching band vibration and the lack of a characteristic band for the NO bond around 1555–1570 cm−1.
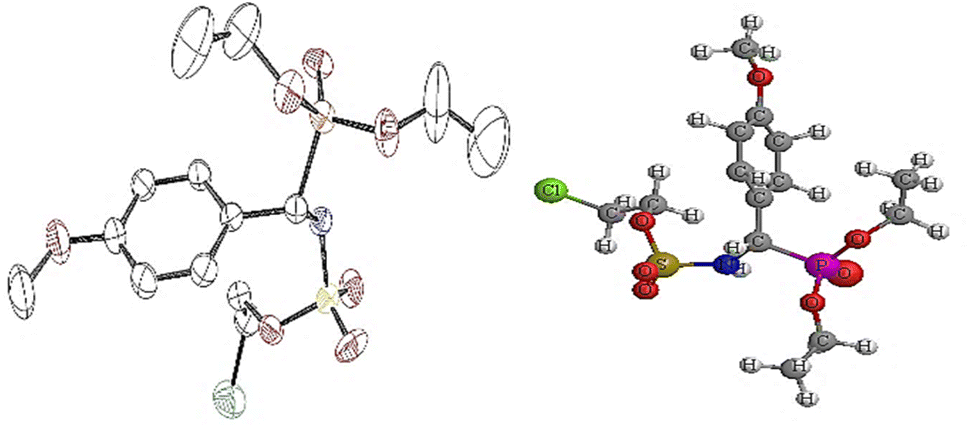
1H NMR spectrum also pointed out the disappearance of the signal around 3.75–5.20 ppm, which corresponds to the NH proton of the chloroethylamine moiety. It should also be noted that the characteristic chemical shift of the NH tethered to the phosphonate fragment undergoes downfield shift to 9.20 ppm. The 31P NMR spectra also confirmed the presence of the phosphonate functional group in the range of 13.0–23.0 ppm for α-sulfamidophosphonates (±)-16 and sulfamate analogues (±)-18, which were fully characterized by mass spectrometry analysis. Finally, we could obtain a crystal structure of the molecule (±)-18e which unambiguously confirmed us the structure of this novel family of compounds 18 (Fig. 2). The crystallographic study revealed that the molecule contained an oxygen atom bound to the SO2 group instead of the N–NO moiety, as shown in Fig. 2.
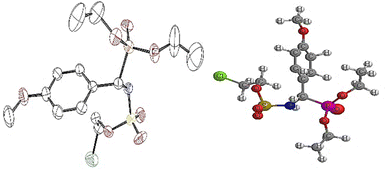 |
| | Fig. 2 ORTEP of 2-chloroethyl ((diethoxyphosphoryl)(4-methoxyphenyl)methyl) sulfamate 18e. | |
Thermal rearrangement of N-nitroso amides to the corresponding esters22 as well as thermal decomposition of nitroso sulfonamides have been earlier described in the literature.23 Based on these results we propose the following reaction mechanism the nitrosation of N-chloroethysulfamidophosphonate derivatives (A) with NaNO2 in acidic conditions at low temperature allows to the formation of the unstable cyclic intermediate (C), which rearranged to azo (D). A second concerted six centers rearrangement (E) turned to decomposition with N2 elimination to give the sulfamate (F) (Scheme 8).
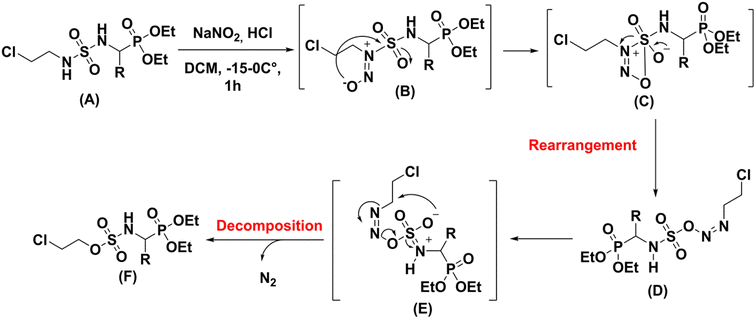 |
| | Scheme 8 Proposed mechanism for the rearrangement process. | |
In vitro anti-proliferative activity of sulfamide (16) and sulfamates (18). Sulfamides as well as sulfamates have received great attention as therapeutic agents.24 In order to evaluate the cytotoxic effect of some compounds synthesized in this work; we selected some α-sulfamidophosphonates (16a, 16c–e, 16k) and their sulfamates derivatives (18a, 18c–e, 18k).In vitro cytotoxicity of these compounds was evaluated on seven tumor cell lines: Huh7 and NCI–H727 (liver), Caco2 and HCT116 (lung), PC3 (prostate), MCF7 and MDA-MB-231 (breast). They were compared to known anticancer agents such as Doxorubicin and Taxol (Table 1). Both series of compounds, lacking the nitroso function, have not shown anti-proliferative effect at a unique dose of 25 μM.
Table 1 Anti-proliferative activity of α-sulfamidophosphonates (16a, 16c–e, 16k) and sulfamates derivatives (18a, 18c–e, 18k) against tumor cell lines
| Compounds |
% of survivala of tumor cells with the selected compounds at a dose of 25 μM |
| Huh7 D12 |
CaCo2 |
MDA |
HCT 116 |
PC3 |
NCI–H727 |
MCF7 |
| Percentage of survival was determined at a single dose of 25 μM, in triplicate measurements taken after 48 hours. |
| DMSO |
100 |
100 |
100 |
100 |
100 |
100 |
100 |
| Doxorubicine |
54 |
60 |
35 |
18 |
36 |
63 |
45 |
| Taxol |
47 |
58 |
44 |
10 |
37 |
71 |
33 |
| 16a |
104 |
125 |
112 |
97 |
96 |
110 |
126 |
| 16c |
103 |
111 |
120 |
101 |
90 |
115 |
119 |
| 16d |
96 |
115 |
111 |
97 |
92 |
111 |
118 |
| 16e |
86 |
122 |
117 |
110 |
118 |
102 |
106 |
| 16k |
95 |
111 |
96 |
82 |
89 |
89 |
92 |
| 18a |
109 |
110 |
116 |
107 |
91 |
101 |
120 |
| 18c |
97 |
112 |
105 |
100 |
98 |
98 |
110 |
| 18d |
97 |
126 |
106 |
91 |
85 |
88 |
118 |
| 18e |
105 |
124 |
118 |
102 |
90 |
112 |
132 |
| 18k |
109 |
126 |
112 |
101 |
96 |
113 |
127 |
It is to be noted that more recently, our laboratory have published anticancer activity of some α-sulfamidophosphonates containing nitrogen mustard fragment and so analogs of our compounds 16.
The authors also found very modest IC50 values (from 52 to 182 μM) for the best compounds, on selected cancer cell lines (PRI, K562 and JURKAT).25
Experimental
General information
Name of the compounds adheres to the international IUPAC convention; the reagents were acquired from commercial suppliers (Merck, Alfa Aesar or Fluka) and employed without undergoing additional purification. Melting points were determined on Electrothermal IA9100 apparatus in open capillary tubes. Thin layer chromatography (TLC) was conducted on aluminum plates coated with silica gel (Kieselgel 60F254, Merck) and visualized using ultraviolet (UV) radiation with a wavelength of 254 nm and a solution containing ninhydrine or vanillin. Purification by flash column chromatography was conducted on silica gel 60 (ACC 40–63 μm 5SDS-CarloErba). A FUNGILAB ultrasonic bath with a frequency of 40 kHz and nominal power of 250 W was used for ultrasound-assisted reactions. All analysis were performed in CEISAM laboratory (Nantes University). The infrared spectra were measured on an IRTF Bruker Tensor spectrophotometer with a specac Quest ATK setup, with the samples being in a neat state. NMR spectra were recorded on an Avance I 300 MHz Bruker, or an Avance III 400 MHz Bruker at room temperature, on CEISAM-NMR platform. The samples were dissolved in a suitable deuterated solvent prior to analysis. Tetramethylsilane (TMS) and the deuterated solvent (CDCl3, DMSO-d6) signals were chosen as the reference standards for 1H and 13C, respectively. Chemical displacement values (δ) are reported in parts per million (ppm), while coupling constants (J) are reported in Hertz (Hz). Signals are described by their multiplicity, which can be indicated by terms such as singlet (s), doublet (d), triplet (t), quadruplet (q), multiplet (m), or broad singlet (brs). Low and high-resolution mass spectrometry (MS and HRMS in Da unit) analyses were recorded on a Xevo-Q-Tof waters, on CEISAM AMaCC platform. Ionization sources were performed with the available methods among electrospray ionization (ESI+) or (ESI−).
Preparation of tert-butyl (N-(2-chloroethyl)sulfamoyl) carbamate (14)
A solution of tert-butanol (1 equiv., 546.50 μL, 5.76 mmol) in anhydrous CH2Cl2 (10 mL) was added dropwise to an equimolar solution of chlorosulfonyl isocyanate (CSI) (500.00 μL, 5.76 mmol) dissolved in the same solvent at 0 °C for 30 min. The resulting solution was added to a mixture of 2-chloroethylamine hydrochloride (1 equiv., 662.11 mg, 5.76 mmol) dissolved in anhydrous CH2Cl2 (10 mL) in the presence of triethylamine (2 equiv., 1.61 mL, 11.52 mmol) at 0 °C, then stirred at room temperature during 2 h. The reaction mixture was diluted with CH2Cl2 (50 mL) and a solution of HCl (25 mL) (0.1 N), the resulting mixture was extracted with CH2Cl2 (3 × 50 mL), the organic phase was washed with water to pH = 7 and extracted with CH2Cl2 (3 × 50 mL). The organic layer was combined and dried over anhydrous magnesium sulfate and concentrated. The pure products were crystallized in a mixture of diethyl ether/n-hexane (1:1) at 5 °C overnight to give the carboxyl sulfonamide correspondent as a white powder (1.36 g, 91%); mp 82–84 °C; Rf = 0.70 (EtOAc/EP 95: 5); IR (cm−1): 3294 (NH), 3188 (NH), 1696 (CO), 1370 and 1133 (SO2); 1H NMR (300 MHz, DMSO-d6): δ = 1.42 (s, 9H, 3CH3), 3.18–3.25 (q, J = 6.3 Hz, 2H, CH2–NH), 3.61 (t, J = 6.3 Hz, 2H, CH2–Cl), 7.90 (t, J = 6.0 Hz, 1H, NH–CH2), 10.93 (s, 1H, NH–CO) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 27.7 (3CH3), 42.7 (CH2–NH), 44.7 (CH2–Cl), 81.3 (C–CH3), 150.5 (CO) ppm; MS (ESI+) m/z = 281 [M + Na]+; HRMS (ESI+): calcd for C7H15ClN2O4NaS [M + Na]+ m/z 281.0333, found 281.0330.
Preparation of N-(2-chloroethyl)sulfamide (15)
Deprotection with TFA. A solution of trifluoroacetic acid 5 equiv. (744 μL, 9.66 mmol) was dropwise added into a stirred solution of tert-butyl (N-(2-chloroethyl)sulfamoyl)carbamate 1 equiv. (500 mg, 1.93 mmol) in dried dichloromethane (15 mL) at 0 °C. The reaction medium was stirred during two hours, concentrated under reduced pressure and co-evaporated with diethyl ether. The residue was recrystallized in a mixture of diethyl ether/n-hexane (1:1) to give the N-(2-chloroethyl)sulfamide as a white powder (0.24 g, mmol, 78%); mp 75–77 °C; Rf = 0.55 (EtOAc/EP 95: 5).
Deprotection in water. Water (10 mL) was added to tert-butyl (N-(2-chloroethyl)sulfamoyl) carbamate, 1 equiv. (500 mg, 1.93 mmol) and the reaction mixture was heated to reflux, until TLC indicated no remaining starting material. The aqueous layer was extracted with AcOEt (3 × 50 mL) then the organic layers was dried over MgSO4 and the solvent was removed in vacuum.The crude product was obtained after recrystallized in diethyl ether with the same yield (78%); IR (cm−1): 3344 (NH2), 3289 (NH), 1331 and 1141 (SO2); 1H NMR (300 MHz, DMSO-d6): δ = 3.14–3.20 (q, J = 6.6 Hz, 2H, CH2–NH), 3.64 (t, J = 6.9 Hz, 2H, CH2–Cl), 6.64 (s, 1H, NH2), 6.85 (t, J = 6.3 Hz, 1H, NH–CH2) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 43.1 (CH2–NH), 44.3 (CH2–Cl) ppm; MS (ESI+) m/z = 181 [M + Na]+; HRMS (ESI+): calcd for C2H7O2N2ClNaS [M + Na]+ m/z 181.9809, found 181.9809.
Preparation of N-(2-chloroethyl) sulfamidophosphonates (16a–16p)
General procedure 1. N-(2-Chloroethyl) sulfamide (1 equiv.), the selected aldehyde (1 equiv.) and trialkylphosphite (1 equiv.), were placed in a bottom flask of 10 mL under ultrasound irradiations (45 kHz) at 60 °C for the appropriate times (Scheme 3). The mixture was stirred until TLC indicated the appearance of a new less polar product and total consumption of starting material.Sulfamidophosphonates 16 were obtained as white powder after recrystallization of crude reaction in a mixture of diethyl ether/n-hexane or purification by flash column chromatography on silica gel column eluted with ethyl acetate/petroleum ether (95/5).
Diethyl (((N-(2-chloroethyl)sulfamoyl)amino)(phenyl)methyl)phosphonate (16a). Following general procedure 1, compound (16a) was obtained after recrystallization as a white powder, 63% yield; Rf: 0.50 (EtOAc/EP 95: 5); mp: 125–127 °C; IR (cm−1): 3273 (NH), 3132 (NH), 1325 and 1151 (SO2), 1228 and 1014 (PO); 1H NMR (300 MHz, CDCl3): δ = 1.06 (t, JH–H = 7.2 Hz, 3H, CH3), 1.36 (t, JH–H = 7.2 Hz, 3H, CH3), 2.81–2.92 (m, 1H, CH2–N), 3.14–3.19 (m, 1H, CH2–N), 3.21–3.26 (m, 1H, CH2–Cl), 3.33–3.41 (m, 1H, CH2–Cl), 3.66–3.69 (m, 1H, CH2–PO), 3.86–3.92 (m, 1H, CH2–PO), 4.18–4.26 (m, 2H, CH2–PO), 4.63 (t, JH–H = 6.0 Hz, 1H, NH–CH2), 4.68–4.79 (dd, JH–H = 9.2 Hz, 2JH–P = 23.5 Hz, 1H, CH*), 6.56–6.59 (dd, JH–H = 5.8 Hz, 3JH–P = 9.0 Hz, 1H, NH–CH), 7.35–7.37 (m, 3H, HAr), 7.49–7.52 (m, 2H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.1 (d, 4JC–P = 5.25 Hz, CH3), 16.4 (d, 4JC–P = 6.0 Hz, CH3), 43.2 (CH2–Cl), 44.4 (CH2–N), 54.2 (d, 2JC–P = 154.5 Hz, CH*), 63.8 (t, 3JC–P = 6.8 Hz, CH2–PO), 128.3 (CHAr), 128.4 (CHAr), 128.8 (CHAr), 128.9 (CHAr), 128.8 (CHAr), 134.9 (CAr) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 19.78 ppm; MS (ESI+) m/z = 385 [M + H]+; HRMS (ESI+): calcd for C13H23O5N2ClPS [M + H]+ m/z 385.0748, found 385.0741.
Diethyl (1-((N-(2-chloroethyl)sulfamoyl)amino)ethyl)phosphonate (16b). Following general procedure 1, compound (16b) was obtained after purification by column chromatography as a yellow oil; 56% yield; Rf: 0.36 (EtOAc/EP 95: 5); IR (cm−1): 3139 (NH), 1330 and 1154 (SO2), 1223 and 1017 (PO); 1H NMR (300 MHz, CDCl3): δ = 1.35 (t, JH–H = 7.2 Hz, 6H, 2CH3), 1.42–1.51 (dd, JH–H = 7.2 Hz, 3JH–P = 16.5 Hz, 3H, CH3–C), 3.38–3.44 (m, 2H, CH2–N), 3.68–3.72 (m, 2H, CH2–Cl), 3.73–3.79 (m, 1H, CH*), 4.11–4.22 (m, 4H, CH2–PO), 4.66–4.71 (dd, JH–H = 4.5 Hz, 3JH–P = 9.6 Hz, 1H, NH–CH), 5.18 (t, JH–H = 6.3 Hz, 1H, NH–CH2) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.5 (CH3–CH2), 16.6 (CH3–CH), 43.5 (CH2–Cl), 44.9 (CH2–N), 45.5 (d, 2JC–P = 159.0 Hz, CH*), 62.7 (d, 3JC–P = 6.7 Hz, CH2–PO), 62.8 (d, 3JC–P = 7.2 Hz, CH2–PO) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 24.19 ppm; MS (ESI+) m/z = 345 [M + Na]+; HRMS (ESI+): calcd for C7H15ClN2O4NaS [M + Na]+ m/z 345.0567, found 345.0567.
Diethyl (((N-(2-chloroethyl)sulfamoyl)amino)(4-chlorophenyl)methyl)phosphonate (16c). Following general procedure 1, compound (16c) was obtained after recrystallization as a white powder, 58% yield; Rf: 0.64 (EtOAc/EP 95: 5); mp: 128–130 °C; IR (cm−1): 3269 (NH), 3102 (NH), 1337 and 1151 (SO2), 1232 and 1013 (PO); 1H NMR (300 MHz, CDCl3): δ = 1.06 (t, JH–H = 7.0 Hz, 3H, CH3), 1.29 (t, JH–H = 7.0 Hz, 3H, CH3), 2.91–2.97 (m, 1H, CH2–N), 3.11–3.20 (m, 1H, CH2–N), 3.31–3.35 (m, 1H, CH2–Cl), 3.37–3.41 (m, 1H, CH2–Cl), 3.68–3.71 (m, 1H, CH2–PO), 3.84–3.90 (m, 1H, CH2–PO), 4.09–4.19 (m, 2H, CH2–PO), 4.63 (t, JH–H = 8.5 Hz, 1H, NH–CH2), 4.66 (d, JH–H = 8.4 Hz, 1H, CH*), 6.21 (dd, JH–H = 6.4 Hz, 3JH–P = 8.5 Hz, 1H, NH–CH), 7.27 (s, 1H, HAr), 7.30 (s, 1H, HAr), 7.34 (d, J = 2.1 Hz, 1H, HAr), 7.37 (d, J = 1.8 Hz, 1H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.1 (d, 4JC–P = 6.0 Hz, CH3), 16.4 (d, 4JC–P = 6.0 Hz, CH3), 43.2 (CH2–Cl), 44.4 (CH2–N), 53.4 (d, 2JC–P = 154.5 Hz, CH*), 63.8 (d, 3JC–P = 7.1 Hz, CH2–PO), 63.9 (d, 3JC–P = 6.9 Hz, CH2–PO), 128.9 (CHAr), 129.5 (d, JC–P = 6.0 Hz, CAr), 133.4 (CHAr), 134.6 (d, JC–P = 3.8 Hz, CAr) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 19.31 ppm; MS (ESI+) m/z = 419 [M + H]+; HRMS (ESI+): calcd for C13H22N2O5PSCl2 [M + H]+ m/z 419.0364, found 419.0368.
Diethyl (((N-(2-chloroethyl)sulfamoyl)amino)(2-fluorophenyl)methyl)phosphonate (16d). Following general procedure 1, compound (16d) was obtained after recrystallization as a white powder, 60% yield; Rf: 0.55 (EtOAc/EP 95: 5); mp: 126–128 °C; IR (cm−1): 3265 (NH), 3120 (NH), 1316 et 1151 (SO2), 1235 and 1010 (PO); 1H NMR (300 MHz, CDCl3): δ = 1.10 (t, JH–H = 7.0 Hz, 3H, CH3), 1.38 (t, JH–H = 7.0 Hz, 3H, CH3), 2.90–3.01 (m, 1H, CH2–N), 3.17–3.28 (m, 1H, CH2–N), 3.33–3.37 (m, 1H, CH2–Cl), 3.41–3.45 (m, 1H, CH2–Cl), 3.79–3.82 (m, 1H, CH2–PO), 3.92–3.98 (m, 1H, CH2–PO), 4.21–4.28 (q, J = 7.2 Hz, 2H, CH2–PO), 4.82 (t, JH–H = 6.3 Hz, 1H, NH–CH2), 5.08–5.19 (dd, JH–H = 9.5 Hz, 2JH–P = 23.9 Hz, 1H, CH*), 6.45–6.51 (dd, JH–H = 5.3 Hz, 3JH–P = 9.7 Hz, 1H, NH–CH), 7.09 (t, JH–H = 9.1 Hz, 1H, HAr), 7.18 (t, JH–H = 7.5 Hz, 1H, HAr), 7.32–7.39 (m, 1H, HAr), 7.62–7.69 (tt, JH–H = 2.2 Hz, JH–P = 9.1 Hz, 1H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.0 (d, 4JC–P = 5.2 Hz, CH3), 16.3 (d, 4JC–P = 6.0 Hz, CH3), 43.2 (CH2–Cl), 44.3 (CH2–N), 46.8 (d, 2JC–P = 157.5 Hz, CH*), 63.7 (t, 3JC–P = 7.0 Hz, CH2–PO), 64.0 (d, 3JC–P = 7.2 Hz, CH2–PO), 115.4 (d, JC–P = 21.9 Hz, CHAr), 122.5 (d, JC–P = 14.2 Hz, CHAr), 124.6 (CHAr), 129.5 (CAr), 130.2 (d, JC–P = 8.2 Hz, CAr-F) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 19.00 (d, JP–F = 9.1 Hz) ppm; MS (ESI+) m/z = 403 [M + H]+; HRMS (ESI+): calcd for C13H22N2O5PSClF [M + H]+ m/z 403.0660, found 403.0674.
Diethyl (((N-(2-chloroethyl)sulfamoyl)amino)(4-methoxyphenyl)methyl)phosphonate (16e). Following general procedure 1, compound (16e) was obtained after purification by column chromatography as white powder, 50% yield; Rf: 0.52 (EtOAc/EP 95: 5); mp: 121–123 °C; IR (cm−1): 3284 (NH), 3133 (NH), 1321 and 1147 (SO2), 1243 and 1014 (PO); 1H NMR (300 MHz, CDCl3): δ = 1.09 (t, JH–H = 7.1 Hz, 3H, CH3), 1.36 (t, JH–H = 7.1 Hz, 3H, CH3), 2.80–2.95 (m, 1H, CH2–N), 3.16–3.24 (m, 1H, CH2–N), 3.30–3.37 (m, 1H, CH2–Cl), 3.38–3.42 (m, 1H, CH2–Cl), 3.67–3.70 (m, 1H, CH2–PO), 3.80 (s, 3H, CH3–O), 3.88–3.93 (m, 1H, CH2–PO), 4.16–4.26 (q, JH–H = 7.2 Hz, 2H, CH2–PO), 4.60 (br, 1H, NH–CH2), 4.63–4.73 (dd, JH–H = 9.0 Hz, 2JH–P = 23.1 Hz, 1H, CH*), 6.35–6.40 (dd, JH–H = 5.8 Hz, 3JH–P = 8.9 Hz, 1H, NH–CH), 6.88 (d, JH–H = 8.6 Hz, 2H, HAr), 7.41 (d, JH–H = 2.0 Hz, 1H, HAr), 7.43 (d, JH–H = 2.0 Hz, 1H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.1 (d, 4JC–P = 5.5 Hz, CH3), 16.4 (d, 4JC–P = 5.8 Hz, CH3), 43.2 (CH2–Cl), 44.3 (CH2–N), 53.4 (d, 2JC–P = 156.1 Hz, CH*), 55.3 (CH3–O), 63.7 (d, 3JC–P = 6.9 Hz, CH2–PO), 114.2 (CHAr), 126.6 (CHAr), 129.5 (d, JC–P = 6.0 Hz, CAr), 159.8 (d, JC–P = 2.25 Hz, CAr-OMe) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 19.99 ppm; MS (ESI+) m/z = 437 [M + Na]+; HRMS (ESI+): calcd for C14H24N2O6PSClNa [M + Na]+ m/z 437.0679, found 437.0674.
Diethyl ((4-bromophenyl)((N-(2-chloroethyl)sulfamoyl)amino)methyl)phosphonate (16f). Following general procedure 1, compound (16f) was obtained after recrystallization as a white powder, 55% yield; Rf: 0.81 (EtOAc/EP 95: 5); mp: 130–132 °C; IR (cm−1): 3296 (NH), 3168 (NH), 1340 and 1156 (SO2), 1232 and 1015 (PO); 1H NMR (300 MHz, CDCl3): δ = 1.12 (t, JH–H = 7.0 Hz, 3H, CH3), 1.35 (t, JH–H = 6.9 Hz, 3H, CH3), 2.91–3.04 (m, 1H, CH2–N), 3,19–3.28 (m, 1H, CH2–N), 3.37–3.47 (m, 2H, CH2–Cl), 3.75–3.80 (m, 1H, CH2–PO), 3.91–3.96 (m, 1H, CH2–PO), 4.16–4.25 (m, 2H, CH2–PO), 4.64 (t, JH–H = 7.5 Hz, 1H, NH–CH2), 4.67–4.75 (dd, JH–H = 9.0 Hz, 2JH–P = 22.8 Hz, 1H, CH*), 6.36–6.41 (dd, JH–H = 6.3 Hz, 3JH–P = 8.7 Hz, 1H, NH–CH), 7.35 (d, JH–H = 2.1 Hz, 1H, HAr), 7.38 (d, JH–H = 2.1 Hz, 1H, HAr), 7.49 (s, 1H, HAr), 7.51 (s, 1H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.1 (d, 4JC–P = 5.2 Hz, CH3), 16.4 (d, 4JC–P = 5.2 Hz, CH3), 43.2 (2CH2–N), 44.4 (2CH2–Cl), 53.5 (d, 2JC–P = 156.0 Hz, CH*), 63.8 (d, 3JC–P = 4.5 Hz, CH2–PO), 63.9 (d, 3JC–P = 4.5 Hz, CH2–PO), 122.8 (CHAr), 129.8 (d, JC–P = 6.0 Hz, CAr), 131.9 (CAr), 133.9 (CAr-Br) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 19.15 ppm; MS (ESI+) m/z = 463 [M + H]+; HRMS (ESI+): calcd for C13H22N2O5PSClBr [M + H]+ m/z 462.9859, found 462.9858.
Diethyl (((N-(2-chloroethyl)sulfamoyl)amino)(4-(dimethylamino)phenyl)methyl)phosphonate (16g). Following general procedure 1, compound (16g) was obtained after purification by column chromatography as an orange powder; 49% yield; Rf: 0.61 (EtOAc/EP 95: 5); mp: 120–122 °C; IR (cm−1): 3261 (NH), 3127 (NH), 1321 and 1150 (SO2), 1230 and 1011 (PO); 1H NMR (300 MHz, CDCl3): δ = 1.10 (t, JH–H = 7.0 Hz, 3H, CH3), 1.35 (t, JH–H = 6.9 Hz, 3H, CH3), 2.80–290 (m, 1H, CH2–N), 2.92 (s, 6H, 2CH3–N), 3.13–3.20 (m, 1H, CH2–N), 3.28–3.40 (m, 2H, CH2–Cl), 3.67–3.70 (m, 1H, CH2–PO), 3.87–3.93 (m, 1H, CH2–PO), 4.16–4.23 (m, 2H, CH2–PO), 4.54 (t, JH–H = 7.5 Hz, 1H, NH–CH2), 4.57–4.67 (dd, JH–H = 9.0 Hz, 2JH–P = 22.8 Hz, 1H, CH*), 6.24 (brs, 1H, NH–CH), 6.66 (d, JH–H = 8.7 Hz, 2H, HAr), 7.32 (d, JH–H = 2.1 Hz, 1H, HAr), 7.35 (d, JH–H = 2.1 Hz, 1H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.2 (d, 4JC–P = 5.2 Hz, CH3), 16.4 (d, 4JC–P = 5.2 Hz, CH3), 40.4 (2CH3–N), 43.2 (2CH2–N), 44.4 (2CH2–Cl), 53.6 (d, 2JC–P = 156.0 Hz, CH*), 63.5 (d, 3JC–P = 4.5 Hz, CH2–PO), 63.5 (d, 3JC–P = 4.5 Hz, CH2–PO), 112.3 (CHAr), 122.4 (CHAr), 129.1 (CAr), 129.2 (CAr-N) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 20.34 ppm; MS (ESI+) m/z = 450 [M + Na]+; HRMS (ESI+): calcd for C15H27N3O5NaPSCl [M + Na]+ m/z 450.0995, found 450.0992.
Diethyl (((N-(2-chloroethyl)sulfamoyl)amino)(4-(trifluoromethyl)phenyl)methyl)phosphonate (16h). Following general procedure 1, compound (16h) was obtained after purification by column chromatography as a white powder; 51% yield; Rf: 0.63 (EtOAc/EP 95: 5); mp: 131–133 °C; IR (cm−1): 3264 (NH), 3124 (NH), 1325 and 1152 (SO2), 1229 and 1024 (PO); 1H NMR (300 MHz, CDCl3): δ = 1.13 (t, JH–H = 7.2 Hz, 3H, CH3), 1.36 (t, JH–H = 7.2 Hz, 3H, CH3), 2.93–3.05 (m, 1H, CH2–N), 3.18–3.29 (m, 1H, CH2–N), 3.37–3.47 (m, 2H, CH2–Cl), 3.78–3.84 (m, 1H, CH2–PO), 3.93–3.99 (m, 1H, CH2–PO), 4.18–4.27 (m, 2H, CH2–PO), 4.74 (t, JH–H = 6.3 Hz, 1H, NH–CH2), 4.76–4.87 (dd, JH–H = 9.0 Hz, 2JH–P = 24.0 Hz, 1H, CH*), 6.51–6.56 (dd, JH–H = 6.6 Hz, 3JH–P = 9.0 Hz, 1H, NH–CH), 7.64 (s, 4H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.1 (d, 4JC–P = 5.2 Hz, CH3), 16.4 (d, 4JC–P = 5.2 Hz, CH3), 43.1 (CH2–Cl), 44.4 (CH2–N), 53.7 (d, 2JC–P = 153.0 Hz, CH*), 63.8 (d, 3JC–P = 6.7 Hz, CH2–PO), 64.1 (d, 3JC–P = 6.7 Hz, CH2–PO), 125.6 (CHAr), 128.5 (d, JC–P = 5.2 Hz, CAr), 131.0 (CAr), 139.0 (CAr-CF3) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 18.96 (d, JF–P = 34.5) ppm; MS (ESI+) m/z = 453 [M + H]+; HRMS (ESI+): calcd for C14H22N2O5F3PSCl [M + H]+ m/z 453.0628, found 453.0623.
Diethyl (((N-(2-chloroethyl)sulfamoyl)amino)(4-cyanophenyl)methyl)phosphonate (16i). Following general procedure 1, compound (16i) was obtained after recrystallization as a white powder, 61% yield; Rf: 0.45 (EtOAc/EP 95: 5); mp: 118–120 °C; IR (cm−1): 3295 (NH), 3153 (NH), 1340 and 1153 (SO2), 1230 and 1006 (PO); 1H NMR (400 MHz, DMSO-d6): δ = 1.09 (t, JH–H = 6.8 Hz, 3H, CH3), 1.22 (t, JH–H = 7.2 Hz, 3H, CH3), 2.78–2.81 (m, 1H, CH2–N), 2.97–3.07 (m, 1H, CH2–N), 3.38–4.47 (m, 2H, CH2–Cl), 3.80–3.88 (m, 1H, CH2–PO), 3.90–3.96 (m, 1H, CH2–PO), 4.02–4.07 (m, 2H, CH2–PO), 4.70–4.79 (dd, JH–H = 10.4 Hz, 2JH–P = 25.2 Hz, 1H, CH*), 7.26 (t, JH–H = 6.0 Hz, 1H, NH–CH2), 7.67 (d, JH–H = 2.0 Hz, 1H, HAr), 7.69 (d, JH–H = 2.0 Hz, 1H, HAr), 7.82 (s, 1H, HAr), 7.84 (s, 1H, HAr), 8.34–8.37 (dd, JH–H = 3.2 Hz, 3JH–P = 10.0, 1H, NH–CH) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 15.9 (d, 4JC–P = 6.0 Hz, CH3), 16.1 (d, 4JC–P = 6.0 Hz, CH3), 42.8 (2CH2–Cl), 43.8 (2CH2–N), 53.1 (d, 2JC–P = 153.4 Hz, CH*), 62.6 (d, 3JC–P = 7.0 Hz, 2CH2–PO), 62.9 (d, 3JC–P = 7.0 Hz, 2CH2–PO), 110.4 (CAr-CN), 118.6 (CN), 129.3 (d, JC–P = 6.0 Hz, CHAr), 131.8 (CHAr), 141.8 (CAr) ppm; 31P NMR (161.97 MHz, DMSO-d6): δ = 19.30 ppm; MS (ESI+) m/z = 410.1 [M + H]+; HRMS (ESI+): calcd for C14H22N2O5F3PSCl [M + H]+ m/z 410.0706, found 410.0693.
Diethyl (((N-(2-chloroethyl)sulfamoyl)amino)(4-hydroxyphenyl)methyl)phosphonate (16j). Following general procedure 1, compound (16j) was obtained after recrystallization as a white powder, 65% yield; Rf: 0.33 (EtOAc/EP 95: 5); mp: 123–125 °C; IR (cm−1): 3345 (OH), 3278 (NH), 3105 (NH), 1324 and 1143 (SO2), 1218 and 1015 (PO); 1H NMR (300 MHz, DMSO-d6): δ = 1.04 (t, JH–H = 7.0 Hz, 3H, CH3), 1.21 (t, JH–H = 7.0 Hz, 3H, CH3), 2.63–2.68 (m, 1H, CH2–N), 2.86–2.91 (m, 1H, CH2–N), 3.32–3.42 (m, 2H, CH2–Cl), 3.68–3.80 (m, 1H, CH2–PO), 3.82–3.86 (m, 1H, CH2–PO), 3.97–4.05 (m, 2H, CH2–PO), 4.36–4.48 (dd, JH–H = 10.2 Hz, 2JH–P = 24.0 Hz, 1H, CH*), 6.68 (d, JH–H = 8.4 Hz, 2H, HAr), 7.14 (t, JH–H = 6.0 Hz, 1H, NH–CH2), 7.27 (d, JH–H = 1.5 Hz, 1H, HAr), 7.29 (d, JH–H = 1.5 Hz, 1H, HAr), 8.03 (d, JH–H = 9.6 Hz, 1H, NH–CH), 9.43 (s, 1H, OH) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 15.9 (d, 4JC–P = 5.2 Hz, CH3), 16.2 (d, 4JC–P = 5.2 Hz, CH3), 42.7 (CH2–Cl), 43.6 (CH2–N), 52.4 (d, 2JC–P = 83.2 Hz, CH*), 62.2 (d, 3JC–P = 6.7 Hz, CH2–PO), 62.4 (d, 3JC–P = 6.7 Hz, CH2–PO), 114.6 (CHAr), 125.8 (CHAr), 129.7 (d, JC–P = 6.0 Hz, CAr), 156.8 (CAr-OH) ppm; 31P NMR (125.5 MHz, DMSO-d6): δ = 20.98 ppm; MS (ESI+) m/z = 401.1 [M + H]+; HRMS (ESI+): calcd for C13H23N2O6SClP [M + H]+ m/z 401.0703, found 401.0692.
Diethyl (((N-(2-chloroethyl)sulfamoyl)amino)(thiophen-2-yl)methyl)phosphonate (16k). Following general procedure 1, compound (16k) was obtained after purification by column chromatography as a white powder; 50% yield, Rf: 0.50 (EtOAc/EP 95: 5); mp: 119–121 °C; IR (cm−1): 3165 (NH), 3138 (NH), 1326 and 1151 (SO2), 1230 and 1014 (PO); 1H NMR (300 MHz, CDCl3): δ = 1.16 (t, JH–H = 7.2 Hz, 3H, CH3), 1.36 (t, JH–H = 7.2 Hz, 3H, CH3), 2.93–3.18 (m, 1H, CH2–N), 3.21–3.32 (m, 1H, CH2–N), 3.39–3.52 (m, 2H, CH2–Cl), 3.85–3.88 (m, 1H, CH2–PO), 4.00–4.18 (m, 1H, CH2–PO), 4.20–4.26 (m, 2H, CH2–PO), 4.71 (t, JH–H = 7.5 Hz, 1H, NH–CH2), 4.95–5.06 (dd, JH–H = 9.0 Hz, 2JH–P = 23.1 Hz, 1H, CH*), 6.02 (dd, JH–H = 6.6 Hz, 3JH–P = 10.2 Hz, 1H, NH–CH), 7.00–7.03 (dd, JH–H = 3.6 Hz, JH–P = 5.2 Hz, 1H, HAr), 7.26 (s, 1H, HAr), 7.27 (d, JH–H = 9.6 Hz, 1H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.1 (d, 4JC–P = 6.0 Hz, CH3), 16.4 (d, 4JC–P = 5.2 Hz, CH3), 43.3 (CH2–Cl), 44.5 (CH2–N), 49.5 (d, 2JC–P = 161.2 Hz, CH*), 64.0 (d, 3JC–P = 3.0 Hz, CH2–PO), 64.1 (d, 3JC–P = 3.0 Hz, CH2–PO), 126.5 (CHAr), 127.3 (CHAr), 127.8 (d, JC–P = 6.7 Hz, CHAr), 136.8 (CAr) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 18.15 ppm; MS (ESI+) m/z = 413 [M + Na]+; HRMS (ESI+): calcd for C11H21N2O5PS2Cl [M + H]+ m/z 391.0318, found 391.0317.
Diethyl (((N-(2-chloroethyl)sulfamoyl)amino)(pyridin-3-yl)methyl)phosphonate (16l). Following general procedure 1, compound (16l) was obtained after purification by column chromatography as a white powder; 68% yield; Rf: 0.42 (EtOAc/EP 95: 5); mp: 122–124 °C; IR (cm−1): 3150 (NH), 3077 (NH), 1337 and 1149 (SO2), 1222 and 1017 (PO); 1H NMR (300 MHz, CDCl3): δ = 1.13 (t, JH–H = 7.2 Hz, 3H, CH3), 1.36 (t, JH–H = 7.2 Hz, 3H, CH3), 2.97–3.07 (m, 1H, CH2–N), 3.20–3.25 (m, 1H, CH2–N), 3.40–3.50 (m, 2H, CH2–Cl), 3.82–3.85 (m, 1H, CH2–PO), 3.92–4.01 (m, 1H, CH2–PO), 4.20–4.27 (m, 2H, CH2–PO), 4.72–4.83 (dd, JH–H = 8.6 Hz, 2JH–P = 23.6 Hz, 1H, CH*), 5.15 (t, JH–H = 6.1 Hz, 1H, NH–CH2), 6.47 (brs, 1H, NH–CH), 7.31–7.35 (dd, JH–H = 4.8 Hz, JH–P = 7.8 Hz, 1H, HAr), 7.88–7.91 (dd, JH–H = 1.8 Hz, JH–P = 8.1 Hz, 1H, HAr), 8.58–8.61 (2 t, JH–H = 1.8 Hz, JH–P = 4.5 Hz, 1H, HAr), 8.65 (brs, 1H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.1 (d, 4JC–P = 5.2 Hz, CH3), 16.4 (d, 4JC–P = 6.0 Hz, CH3), 43.2 (CH2–Cl), 44.4 (CH2–N), 51.3 (d, 2JC–P = 154.5 Hz, CH*), 63.8 (d, 3JC–P = 6.7 Hz, CH2–PO), 64.1 (d, 3JC–P = 6.7 Hz, CH2–PO), 123.5 (CHAr), 131.1 (CHAr), 135.7 (d, JC–P = 4.5 Hz, CHAr), 149.3 (d, JC–P = 6.7 Hz, CHAr), 149.6 (d, JC–P = 2.5 Hz, CAr) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 18.99 ppm; MS (ESI+) m/z = 386 [M + H]+; HRMS (ESI+): calcd for C12H22N3O5PSCl [M + H]+ m/z 386.0706, found 386.0710.
Dimethyl (((N-(2-chloroethyl)sulfamoyl)amino)(phenyl) methyl)phosphonate (16m). Following general procedure 1, compound (16m) was obtained after recrystallization as a white powder, 65% yield; Rf: 0.49 (EtOAc/EP 95: 5); mp: 142–144 °C; IR (cm−1): 3304 (NH), 3180 (NH), 1336 and 1155 (SO2), 1237 and 1026 (PO); 1H NMR (300 MHz, DMSO-d6): δ = 2.64–2.71 (m, 1H, CH2–N), 2.89–2.97 (m, 1H, CH2–N), 3.30–3.37 (m, 2H, CH2–Cl), 3.47 (d, 3JH–P = 10.8 Hz, 3H, CH3–PO), 3.67 (d, 3JH–P = 10.8 Hz, 3H, CH3–PO), 4.58–4.70 (dd, JH–H = 10.2 Hz, 2JH–P = 24.3 Hz, 1H, CH*), 7.26 (t, JH–H = 6.0 Hz, 1H, NH–CH2), 7.32–7.39 (m, 3H, HAr), 7.49–7.55 (m, 2H, HAr), 8.29–8.33 (dd, JH–H = 2.1 Hz, 3JH–P = 10.2, 1H, NH–CH*) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 42.7 (CH2–Cl), 44.6 (CH2–N), 52.4 (d, 2JC–P = 155.2 Hz, CH*), 53.2 (d, 3JC–P = 7.5 Hz, CH3–PO), 53.6 (d, 3JC–P = 6.7 Hz, CH3–PO), 127.7 (CHAr), 127.9 (CHAr), 128.3 (d, JC–P = 6.0 Hz, CHAr), 135.6 (CAr) ppm; 31P NMR (125.5 MHz, DMSO-d6): δ = 22.78 ppm; MS (ESI+) m/z = 357 [M + H]+; HRMS (ESI+): calcd for C11H19N2O5PSCl [M + H]+ m/z 357.0441, found 357.0437.
Diphenyl (((N-(2-chloroethyl)sulfamoyl)amino)(phenyl)methyl)phosphonate (16n). Following general procedure 1, compound (16n) was obtained after recrystallization as a white powder, 72% yield; Rf: 0.8 (EtOAc/EP 95: 5); mp: 143–145 °C; IR (cm−1): 3291 (NH), 3164 (NH), 1329 and 1150 (SO2), 1212 and 1096 (PO); 1H NMR (300 MHz, DMSO-d6): δ = 2.60–2.72 (m, 1H, CH2–N), 2.86–3.00 (m, 1H, CH2–N), 3.28–3.33 (m, 2H, CH2–Cl), 5.01–5.14 (dd, JH–H = 10.5 Hz, 2JH–P = 24.5 Hz, 1H, CH*), 6.87–6.90 (m, 2H, CHAr), 7.10–7.22 (m, 4H, CHAr), 7.30 (t, JH–H = 8.3 Hz, 1H, NH–CH2), 7.34–7.41 (m, 7H, CHAr), 7.63–7.66 (m, 2H, CHAr), 8.68–8.72 (dd, JH–H = 1.5 Hz, 3JH–P = 10.3, 1H, NH–CH) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 42.6 (CH2–Cl), 43.6 (CH2–N), 53.6 (d, 2JC–P = 161.2 Hz, CH*), 120.2 (d, JC–P = 4.0 Hz, CHAr), 120.4 (d, JC–P = 4.1 Hz, CHAr), 125.5 (CHAr), 128.2 (CHAr), 128.8 (d, JC–P = 6.4 Hz, CHAr), 129.7 (CHAr), 134.5 (CAr), 149.7 (d, JC–P = 10.0 Hz, CAr), 150.0 (d, JC–P = 9.9 Hz, CAr) ppm; 31P NMR (125.5 MHz, DMSO-d6): δ = 14.13 ppm; MS (ESI+) m/z = 481 [M + H]+; HRMS (ESI+): calcd for C21H23N2O5PSCl [M + H]+ m/z 481.0754, found 481.0758.
Diisopropyl (((N-(2-chloroethyl)sulfamoyl)amino)(phenyl)methyl)phosphonate (16o). Following general procedure 1, compound (16o) was obtained after purification by column chromatography as a white powder, 68% yield; Rf: 0.65 (EtOAc/EP 95: 5); mp: 141–143 °C; IR (cm−1): 3290 (NH), 3156 (NH), 1334 and 1149 (SO2), 1229 and 1017 (PO); 1H NMR (400 MHz, CDCl3): δ = 0.84 (d, JH–H = 6.1 Hz, 3H, CH3), 1.20 (d, JH–H = 6.1 Hz, 3H, CH3), 1.37 (t, JH–H = 6.7 Hz, 6H, CH3), 2.82–2.86 (m, 1H, CH2–N), 3.12–3.17 (m, 1H, CH2–N), 3.20–3.26 (m, 1H, CH2–Cl), 3.31–3.38 (m, 1H, CH2–Cl), 4.39–4.41 (m, 1H, CH-i-pro), 4.47 (t, JH–H = 6.3 Hz, 1H, NH–CH2), 4.64–4.71 (dd, JH–H = 8.7 Hz, 2JH–P = 23.7 Hz, 1H, CH*), 4.75–4.83 (m, 1H, CH-i-pro), 6.32 (t, JH–H = 7.0 Hz, 1H, NH–CH), 7.32–7.35 (m, 3H, HAr), 7.49 (d, JH–H = 6.7 Hz, 2H, HAr) ppm; 13C NMR (100 MHz, CDCl3): δ = 22.9 (d, 4JC–P = 5.3 Hz, CH3), 23.8 (d, 4JC–P = 5.0 Hz, CH3), 24.1 (2CH3), 43.1 (CH2–Cl), 44.3 (CH2–N), 55.0 (d, 2JC–P = 160.5 Hz, CH*), 72.6 (d, 3JC–P = 7.0 Hz, CH-i-pro), 72.7 (d, 3JC–P = 6.9 Hz, CH-i-pro), 128.4 (CHAr), 128.5 (CHAr), 128.6 (CHAr), 128.7 (CHAr), 135.2 (CAr) ppm; 31P NMR (161.97 MHz, CDCl3): δ = 18.06 ppm; MS (ESI+) m/z = 413 [M + H]+; HRMS (ESI+): calcd for C15H27N2O5SClP [M + H]+ m/z 413.1067, found 413.1065.
Bis(2,2,2-trifluoroethyl)(((N-(2-chloroethyl)sulfamoyl)amino)(phenyl)methyl)phosphonate (16p). Following general procedure 1, compound (16p) was obtained after purification by column chromatography as a white powder; 60% yield; Rf: 0.70 (EtOAc/EP 95: 5); mp: 138–140 °C; IR (cm−1): 3235 (NH), 3151 (NH), 1305 and 1153 (SO2), 1245 and 1098 (PO); 1H NMR (300 MHz, CDCl3): δ = 2.83–2.93 (m, 1H, CH2–N), 3.08–3.20 (m, 1H, CH2–N), 3.24–3.28 (m, 1H, CH2–Cl), 3.32–3.43 (m, 1H, CH2–Cl), 3.63–3.73 (m, 1H, CH2–PO), 4.08–4.15 (m, 1H, CH2–PO), 4.40–4.49 (m, 2H, CH2–PO), 4.65 (t, JH–H = 6.4 Hz, 1H, NH–CH2), 4.81–4.92 (dd, JH–H = 9.9 Hz, 2JH–P = 24.3 Hz, 1H, CH*), 6.27–6.30 (dd, JH–H = 4.5 Hz, 3JH–P = 9.9 Hz, 1H, NH–CH), 7.39–7.44 (m, 5H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 43.1 (CH2–Cl), 44.3 (CH2–N), 53.9 (d, 2JC–P = 164.2 Hz, CH*), 62.9 (t, 3JC–P = 7.0 Hz, 2CH2–PO), 127.9 (d, JC–P = 6.9 Hz, CHAr), 129.3 (d, JC–P = 1.8 Hz, CHAr), 129.6 (d, JC–P = 3.0 Hz, CHAr), 137.7 (CAr) ppm. 31P NMR (125.5 MHz, CDCl3): δ = 22.53 ppm; MS (ESI+) m/z = 493 [M + H]+; HRMS (ESI+): calcd for C13H17N2O5SClPF6 [M + H]+ m/z 493.0189, found 493.0192.
Nitrosation reaction
General procedure 2. To a stirred solution of N-(2-chloroethyl) sulfamidophosphonates (1 equiv.) in a minimum of dichloromethane (10 mL) and 3 equiv. of hydrochloric acid/or formic acid was added in fractions dried sodium nitrite (3 equiv.) at −15 °C to 0 °C during 1 h (Scheme 5). The insoluble by-product (NaCl) was removed by filtration and the organic phase was washed with distilled water, dried over anhydrous sodium sulfate, and the solvent evaporated under reduced pressure. The 2-chloroethyl((diethoxyphosphoryl)(phenyl)methyl)sulfamates (18a, 18c–e, 18k) were obtained after purification by column chromatography on silica gel, with DCM as the eluent.
2-Chloroethyl ((diethoxyphosphoryl)(phenyl)methyl)sulfamate (18a). Following general procedure 2, compound (18a) was obtained after purification by column chromatography as a white powder; 25% yield; Rf: 0,48 (EtOAc/EP 95: 5); mp: 102–104 °C; IR (cm−1): 3074 (NH), 1375 et 1173 (SO2), 1248 et 1023 (PO); 1H NMR (300 MHz, DMSO-d6): δ = 1.04 (t, JH–H = 7.1 Hz, 3H, CH3), 1.24 (t, JH–H = 7.1 Hz, 3H, CH3), 3.26–3.32 (m, 1H, CH2–O), 3.42–3.51 (m, 1H, CH2–O), 3.70–3.81 (m, 2H, CH2–Cl), 3.84–3.92 (m, 1H, CH2–PO), 4.02–4.10 (m, 3H, CH2–PO), 4.65–4.76 (dd, JH–H = 10.1 Hz, 2JH–P = 24.0 Hz, 1H, CH*), 7.35–7.38 (m, 3H, HAr), 7.50–7.54 (m, 2H, HAr), 9.31–9.35 (dd, JH–H = 2.1 Hz, 3JH–P = 10.1 Hz, 1H, NH–CH*) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 15.9 (d, 4JC–P = 5.1 Hz, CH3), 16.2 (d, 4JC–P = 5.5 Hz, CH3), 41.3 (CH2–Cl), 53.5 (d, 2JC–P = 153.5 Hz, CH*), 62.2 (CH2–PO), 62.9 (CH2–PO), 68.8 (CH2–O), 128.1 (CHAr), 128.2 (CHAr), 128.3 (CHAr), 128.4 (CHAr), 135.1 (CAr) ppm; 31P NMR (121.5 MHz, DMSO-d6): δ = 19.46 ppm; MS (ESI+) m/z = 386.1 [M + H]+.
2-Chloroethyl((4-chlorophenyl)(diethoxyphosphoryl)methyl)sulfamate (18c). Following general procedure 2, compound (18c) was obtained after purification by column chromatography as a white powder; 30% yield; Rf: 0.60 (EtOAc/EP 95: 5); mp: 106–108 °C; IR (cm−1): 3078 (NH), 1370 et 1177 (SO2), 1225 et 1009 (PO); 1H NMR (300 MHz, DMSO-d6): δ = 1.06 (t, JH–H = 7.1 Hz, 3H, CH3), 1.23 (t, JH–H = 7.1 Hz, 3H, CH3), 3.43–3.49 (m, 1H, CH2–O), 3.50–3.60 (m, 1H, CH2–O), 3.80–3.85 (m, 2H, CH2–Cl), 3.86–3.92 (m, 1H, CH2–PO), 4.04–4.12 (m, 3H, CH2–PO), 4.65–4.76 (dd, JH–H = 10.0 Hz, 2JH–P = 23.9 Hz, 1H, CH*), 7.43 (s, 1H, HAr), 7.45 (s, 1H, HAr), 7.52 (d, JH–H = 1.9 Hz, 1H, HAr), 7.55 (d, JH–H = 1.9 Hz, 1H, HAr), 9.32–9.39 (dd, JH–H = 2.3 Hz, 3JH–P = 10.1 Hz, 1H, NH–CH*) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 15.9 (d, 4JC–P = 5.7 Hz, CH3), 16.1 (d, 4JC–P = 5.7 Hz, CH3), 41.5 (CH2–Cl), 52.6 (d, 2JC–P = 153.3 Hz, CH*), 62.7 (d, 3JC–P = 6.8 Hz, CH2–PO), 62.9 (d, 3JC–P = 6.9 Hz, CH2–PO), 69.1 (CH2–O), 128.2 (CHAr), 130.1 (d, JC–P = 5.4 Hz, CHAr), 132.8 (CAr), 134.2 (CAr-Cl) ppm; 31P NMR (121.5 MHz, DMSO-d6): δ = 19.06 ppm; MS (ESI+) m/z = 420 [M + H]+.
2-Chloroethyl((diethoxyphosphoryl)(2-fluorophenyl)methyl)sulfamate (18d). Following general procedure 2, compound (18d) was obtained after purification by column chromatography as a brown powder; 33% yield; Rf: 0,53 (EtOAc/EP 95: 5); mp: 105–107 °C; IR (cm−1): 3093 (NH), 1363 et 1173 (SO2), 1222 et 1014 (PO); 1H NMR (300 MHz, DMSO-d6): δ = 1.04 (t, JH–H = 7.1 Hz, 3H, CH3), 1.27 (t, JH–H = 7.1 Hz, 3H, CH3), 3.37–3.42 (m, 1H, CH2–O), 3.51–3.55 (m, 1H, CH2–O), 3.78–3.85 (m, 2H, CH2–Cl), 3.88–3.93 (m, 1H, CH2–PO), 4.06–4.14 (m, 3H, CH2–PO), 4.91–5.02 (dd, JH–H = 10.1 Hz, 2JH–P = 24.3 Hz, 1H, CH*), 7.21–7.30 (m, 2H, HAr), 7.42–7.54 (s, 1H, HAr), 7.72 (t, JH–H = 2.0 Hz, 1H, HAr), 9.48–9.54 (dd, JH–H = 2.2 Hz, 3JH–P = 10.0 Hz, 1H, NH–CH*) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 15.8 (d, 4JC–P = 5.4 Hz, CH3), 16.1 (d, 4JC–P = 5.4 Hz, CH3), 41.3 (CH2–Cl), 46.6 (d, 2JC–P = 154.7 Hz, CH*), 62.8 (d, 3JC–P = 7.0 Hz, CH2–PO), 63.1 (d, 3JC–P = 6.9 Hz, CH2–PO), 69.1 (CH2–O), 115.0 (d, JC–P = 24.4 Hz, CHAr), 122.3 (d, JC–P = 16.2 Hz, CHAr), 124.5 (CHAr), 129.6 (CAr), 130.3 (d, JC–P = 8.0 Hz, CAr) ppm; 31P NMR (121.5 MHz, DMSO-d6): δ = 18.47 (d, JP–F = 4.8 Hz) ppm; MS (ESI+) m/z = 404.1 [M + H]+.
2-Chloroethyl((diethoxyphosphoryl)(4-methoxyphenyl)methyl)sulfamate (18e). Following general procedure 2, compound (18e) was obtained after purification by column chromatography as a white crystal; 40% yield; Rf: 0,50 (EtOAc/EP 95: 5); mp: 94–96 °C; IR (cm−1): 3114 (NH), 1356 et 1170 (SO2), 1226 et 1051 (PO); 1H NMR (300 MHz, DMSO-d6): δ = 1.05 (t, JH–H = 7.2 Hz, 3H, CH3), 1.25 (t, JH–H = 6.9 Hz, 3H, CH3), 3.31–3.33 (m, 1H, CH2–O), 3.46–3.48 (m, 1H, CH2–O), 3.75 (s, 3H, OCH3), 3.81–3.84 (m, 2H, CH2–Cl), 3.86–3.91 (m, 1H, CH2–PO), 4.00–4.10 (m, 3H, CH2–PO), 4.58–4.70 (dd, JH–H = 10.3 Hz, 2JH–P = 23.7 Hz, 1H, CH*), 6.92–6.94 (m, 2H, HAr), 7.42–7.46 (m, 2H, HAr), 9.22–9.26 (dd, JH–H = 1.9 Hz, 3JH–P = 10.3 Hz, 1H, NH–CH*) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 15.9 (d, 4JC–P = 5.4 Hz, CH3), 16.1 (d, 4JC–P = 5.4 Hz, CH3), 41.3 (CH2–Cl), 52.8 (d, 2JC–P = 159.3 Hz, CH*), 54.9 (CH3–O), 62.5 (d, 3JC–P = 6.7 Hz, CH2–PO), 62.7 (d, 3JC–P = 6.9 Hz, CH2–PO), 68.8 (CH2–O), 113.6 (CHAr), 126.8 (CAr), 129.7 (d, JC–P = 5.9 Hz, CHAr), 150.0 (CAr-OMe) ppm; 31P NMR (121.5 MHz, DMSO-d6): δ = 19.76 ppm; MS (ESI+) m/z = 416.1 [M + H]+.
2-Chloroethyl((diethoxyphosphoryl)(thiophen-2-yl)methyl)sulfamate (18k). Following general procedure 2, compound (18k) was obtained after purification by column chromatography as a brown powder; 36% yield; Rf: 0,45 (EtOAc/EP 95: 5); mp: 98–100 °C; IR (cm−1): 3087 (NH), 1358 et 1169 (SO2), 1229 et 1006 (PO); 1H NMR (300 MHz, DMSO-d6): δ = 1.10 (t, JH–H = 7.0 Hz, 3H, CH3), 1.26 (t, JH–H = 7.0 Hz, 3H, CH3), 3.33–3.44 (m, 1H, CH2–O), 3.55–3.61 (m, 1H, CH2–O), 3.83–3.89 (m, 2H, CH2–Cl), 3.91–3.99 (m, 1H, CH2–PO), 4.06–4.16 (m, 3H, CH2–PO), 4.93–5.05 (dd, JH–H = 10.0 Hz, 2JH–P = 23.7 Hz, 1H, CH*), 7.04 (t, JH–H = 4.2 Hz, 1H, HAryl), 7.29 (brs, 1H, HAryl), 7.54 (d, JH–H = 4.7 Hz, 1H, HAryl), 9.37 (d, 3JH–P = 9.8 Hz, 1H, NH–CH) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 15.9 (d, 4JC–P = 5.2 Hz, CH3), 16.1 (d, 4JC–P = 5.2 Hz, CH3), 41.4 (CH2–Cl), 48.7 (d, 2JC–P = 162.7 Hz, CH*), 62.9 (d, 3JC–P = 6.7 Hz, CH2–PO), 63.1 (d, 3JC–P = 6.7 Hz, CH2–PO), 69.0 (CH2–O), 126.9 (CHAr), 128.0 (CHAr), 128.1 (CHAr), 136.5 (CAr) ppm; 31P NMR (121.5 MHz, DMSO-d6): δ = 18.08 ppm; MS (ESI+) m/z = 392.0 [M + H]+.
Synthesis of carbonyl analogue of fotemustine
Preparation of 2-chloroethylureidophosphonate (20). To 1 equiv. of diethyl(amino(phenyl)methyl)phosphonate hydrochloride (400 mg, 1.4 mmol 10 mL of DCM), cooled at 0 °C is added 1 equiv. of TEA (1.4 μL, 1.4 mmol) and 1.2 equiv. of 2-chloroethyl isocyanate (147.2 μL, 1.72 mmol) successively. The progress of the reaction was followed by TLC (EtOAc/EP 95:5), the organic middle was washed with HCl (1 M) and with water. The ureidophosphonate derived of 2-chloroethylamine was obtained after purification by column chromatography on silica gel eluted with (EtOAc/EP 95:5) to give (20) as a white powder; 50% yield; Rf: 0.58 (EtOAc/EP 95: 5); mp: 60–62 °C; IR (cm−1): 3389 (NH), 3152 (NH), 1666 (CO), 1246 and 1015 (PO); 1H NMR (300 MHz, CDCl3): δ = 1.06 (t, JH–H = 7.0 Hz, 3H, CH3), 1.35 (t, JH–H = 7.1 Hz, 3H, CH3), 3.45–3.51 (m, 4H, 2CH2–Cl), 3.64–3.70 (m, 1H, CH2–PO), 3.84–3.90 (m, 1H, CH2–PO), 4.18–4.24 (m, 2H, CH2–PO), 5.34–5.45 (dd, JH–H = 10.0 Hz, 2JH–P = 22.0 Hz, 1H, CH*), 6.15 (brs, 1H, NH–CH2), 7.04 (brs, 1H, NH–CH*), 7.27–7.35 (m, 3H, HAr), 7.43–7.47 (m, 2H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.0 (d, 4JC–P = 5.8 Hz, CH3), 16.4 (d, 4JC–P = 6.0 Hz, CH3), 42.0 (CH2–Cl), 45.1 (CH2–N), 49.6 (d, 2JC–P = 156.4 Hz, CH*), 63.4 (d, 3JC–P = 7.6 Hz, CH2–PO), 63.6 (d, 3JC–P = 7.3 Hz, CH2–PO), 127.9 (d, JC–P = 2.2 Hz, CHAr), 128.0 (CHAr), 128.1 (CHAr), 128.4 (CHAr), 136.0 (CAr), 157.4 (d, JC–P = 9.6 Hz, CO) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 23.16 ppm; MS (ESI+) m/z = 371 [M + Na]+; HRMS (ESI+): calcd for C14H22O4N2NaClP [M + Na]+ m/z 371.0903, found 371.0905.
Synthesis of diethyl ((3-(2-chloroethyl)-3-nitrosoureido) (phenyl)methyl) phosphonate (21). To a solution of 2-chloroethylamineuridophosphonate (20) (300 mg, 860.2 μmol, 1 equiv.) in DCM (10 mL) at 0 °C, 3 equiv. (97.4 μL, 2.6 mmol) of acetic acid HCOOH and 3 equiv. (178.1 mg, 2.6 mmol) of NaNO2 are successively added drop-wise over 2 hours. After disappearance of the starting material, the reaction mixture is poured into 100 mL of cold water, followed by the addition of 20 mL of a sodium bicarbonate solution (5%) to adjust the pH. The aqueous phase is extracted twice with ethyl acetate (2 × 15 mL), and the organic phase is washed with brine (15 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting 2-chloroethylnitrosoureidophosphonate is obtained as yellow powder after crystallization of the crude with diethyl ether; 68% yield; Rf: 0.50 (EtOAc/EP 95: 5); mp: 86–88 °C; IR (cm−1): 3173 (NH), 1714 (CO), 1242 and 1021 (PO), 1534 (NO); 1H NMR (300 MHz, CDCl3): δ = 1.14 (t, JH–H = 7.1 Hz, 3H, CH3), 1.29 (t, JH–H = 7.1 Hz, 3H, CH3), 3.47 (t, 2H, JH–H = 6.8 Hz, CH2–Cl), 3.74–3.82 (m, 1H, CH2–N), 3.95–3.98 (m, 1H, CH2–N), 4.09–4.18 (m, 4H, 2CH2–PO), 5.45–5.55 (dd, JH–H = 9.3 Hz, 2JH–P = 21.2 Hz, 1H, CH*), 7.32–7.43 (m, 3H, HAr), 7.46–7.49 (m, 2H, HAr), 7.73–7.80 (dd, JH–H = 5.6 Hz, 3JH–P = 9.6 Hz, 1H, NH–CH*) ppm; 13C NMR (75 MHz, CDCl3): δ = 16.2 (d, 4JC–P = 5.5 Hz, CH3), 16.4 (d, 4JC–P = 5.6 Hz, CH3), 38.7 (CH2–Cl), 40.3 (CH2–N), 50.9 (d, 2JC–P = 154.6 Hz, CH*), 63.2 (CH2–PO), 63.6 (CH2–PO), 127.8 (CHAr), 127.9 (CHAr), 128.5 (CHAr), 128.8 (CHAr), 134.3 (CAr), 158.4 (CO) ppm; 31P NMR (125.5 MHz, CDCl3): δ = 20.34 ppm; MS (ESI+) m/z = 400 [M + Na]+; HRMS (ESI+): calcd for C14H21O5N3ClP [M + Na]+ m/z 400.0748, found 400.0741.
Preparation of α-sulfonamidophosphonate with butylamine moiety
The preparation of α-sulfamidophosphonate with butylamine moiety is carried out according to the same procedure described for the preparation of α-sulfamidophosphonates derived from 2-chloroethylamine (16).
Tert-butyl N-butylsulfamoylcarbamate (22). Prepared in the same conditions than (14) starting with butylamine and CSI, to give a white powder; 82%; mp 102–104 °C; Rf = 0.62 (EtOAc/EP 95: 5); IR (cm−1): 3291 (NH), 3203 (NH), 1693 (CO), 1341 et 1132 (SO2); 1H NMR (300 MHz, CDCl3): δ = 0.92 (t, J = 7.4 Hz, 3H, CH3), 1.33–1.43 (m, 2H, CH2–CH3), 1.49 (s, 9H, 3CH3), 1.50–1.60 (m, 2H, CH2–CH2), 3.03 (q, J = 7.0 Hz, 2H, CH2–NH), 5.09 (t, J = 6.0 Hz, 1H, NH–CH2), 7.25 (s, 1H, NH–CO) ppm; 13C NMR (75 MHz, CDCl3): δ = 13.5 (CH3), 19.7 (CH2–CH3), 28.0 (3CH3), 31.0 (CH2–CH2), 43.6 (CH2–NH), 83.7 (C–3CH3), 150.2 (CO) ppm; MS (ESI+) m/z = 527.2 [2M + Na]+; HRMS (ESI+): calcd for C9H20N2O4NaS [M + Na]+ m/z 275.1041, found 275.1041.
N-Butylsulfamide (23). Prepared in the same conditions than (15), starting from (22) to give white powder; 68%; mp 125–127 °C; Rf = 0.50 (EtOAc/EP 95: 5); IR (cm−1): 3330 (NH), 3280 (NH), 1325 et 1130 (SO2); 1H NMR (300 MHz, DMSO-d6): δ = 0.92 (t, J = 7.4 Hz, 3H, CH3), 1.30–1.42 (m, 2H, CH2–CH3), 1.49–1.59 (m, 2H, CH2–CH2), 3.10–3.19 (m, 2H, CH2–NH), 3.90 (brs, 3H, NH, NH2) ppm; 13C NMR (75 MHz, DMSO-d6): δ = 13.6 (CH3), 19.8 (CH2–CH3), 31.4 (CH2–CH2), 43.3 (CH2–NH) ppm; MS (ESI−) m/z = 151.1 [M − H]−; HRMS (ESI−): calcd for C4H11N2O2S [M − H]− m/z 151.0541, found 151.0546.
Diethyl (((N-butylsulfamoyl)amino)(phenyl)methyl)phosphonate (24). Following general procedure 1, compound (16) was obtained after purification by column chromatography as a white powder; 56% yield; Rf: 0.48 (EtOAc/EP 95: 5); mp: 134–136 °C; IR (cm−1): 3291 (NH), 3124 (NH), 1330 and 1157 (SO2), 1235 and 1013 (PO); 1H NMR (300 MHz, CDCl3): δ = 0.78 (t, J = 7.2 Hz, 3H, CH3), 1.08 (t, JH–H = 7.0 Hz, 3H, CH3–CH2OP), 1.10–1.20 (m, 4H, 2CH2), 1.34 (t, JH–H = 7.1 Hz, 3H, CH3–CH2OP), 2.54–2.62 (m, 1H, CH2–N), 2.81–2.91 (m, 1H, CH2–N), 3.65–3.73 (m, 1H, CH2–PO), 3.82 (brs, 1H, NH–CH2), 3.88–3.94 (m, 1H, CH2–PO), 4.13–4.21 (m, 2H, CH2–PO), 4.64–4.75 (dd, JH–H = 8.2 Hz, 2JH–P = 22.9 Hz, 1H, CH*), 5.48 (t, JH–H = 8.0 Hz, 1H, NH–CH*), 7.32–7.36 (m, 3H, HAr), 7.43–7.46 (m, 2H, HAr) ppm; 13C NMR (75 MHz, CDCl3): δ = 13.5 (CH3), 16.0 (d, 4JC–P = 5.6 Hz, CH3–CH2OP), 16.4 (d, 4JC–P = 5.6 Hz, CH3–CH2OP), 19.7 (CH2–CH3), 31.0 (CH2–CH2), 42.8 (CH2–NH), 54.0 (d, 2JC–P = 156.9 Hz, CH*), 63.7 (t, 3JC–P = 6.8 Hz, 2CH2–PO), 128.1 (d, JC–P = 6.0 Hz, CHAr), 128.6 (CHAr), 128.7 (CHAr), 134.8 (CAr) ppm; 31P NMR (121.5 MHz, CDCl3): δ = 19.95 ppm; MS (ESI+) m/z = 401.1 [M + Na]+; HRMS (ESI+): calcd for C15H27N2O5NaPS [M + Na]+ m/z 401.1276, found 401.1279.
X-ray crystallography
A crystal was mounted at the tip of Lindemann capillary by means of a solvent free glue. The intensity measurement was carried out at 293 K on a Bruker-Nonius Kappa CCD diffractometer, using graphite-monochromatized MoK-L2,3 radiation (λ = 0.71073 Å). Data were corrected for Lorentz-polarization effects and absorption corrections applied using a Gaussian integration. Friedel pairs have been merged. The structure was solved with the Sir 2004 (direct methods and subsequent calculations were carried out with the (Jana 2006)26 program package. All non-hydrogen atoms were refined anisotropically and hydrogen atoms were introduced with geometrical constraints and riding atomic displacement parameters.
Crystal data of compound (18e): C14H23Cl1N1O7P1S1, Mr = 415.8, monoclinic, P21/c, a = 10.1328(6), b = 11.6196(8), c = 16.8769(12) Å, β = 96.810(6)°, V = 1973.1(2) Å3, Z = 4, ρcalcd = 1.3998 g·cm−3, μ = 0.414 mm−1, F(000) = 872, colorless prism, 0.27 × 0.23 × 0.22 mm3, 2θmax = 54.6°, T = 293 K, 39181 reflections, 4356 unique (99% completeness), Rint = 0.1770, 230 parameters, GOF = 1.06, wR2 = 0.1367, R = 0.053 for 1634 reflections with I > 2σ(I).
In vitro cytotoxicity study
ImPACell protocol for cell culture. Skin normal fibroblastic cells were purchased from Lonza (Basel, Switzerland), HuH7, Caco-2, MDA-MB-231, HCT116, PC3, MCF7 and NCI–H727 cancer cell lines were obtained from the ECACC collection (Porton Down, UK). Cells were grown at 37 °C, 5% CO2 in ECACC recommended media: DMEM for HuH7, MDA-MB-231 and fibroblast, EMEM for MCF7 and CaCo-2, McCoy's for HCT116 and RPMI for PC3 and NCI–H727. All culture media were supplemented by 10% of FBS, 1% of penicillin–streptomycin and 2 mM glutamine.
ImPACell protocol for cytotoxic assay. The compounds have been tested at a unique concentration of 25 μM. Cells were plated in 96 wells plates (4000 cells per well). Twenty-four hours after seeding, cells were exposed to chemicals. After 48 h of treatment, cells were washed in PBS and fixed in cooled 90% ethanol/5% acetic acid for 20 minutes and the nuclei were stained with Hoechst 33342 (B2261 Sigma). Image acquisition and analysis were performed using a Cellomics ArrayScan VTI/HCS Reader (ThermoScientific). The survival percentages were calculated as the percentage of cell number after compound treatment over cell number after DMSO treatment.
Conclusions
To summarize, this paper describes the development of a novel series of α-sulfamidophosphonates derived from 2-chloroethylamine, which were synthesized through an efficient flexible three-step process. The formation of chloroethyl sulfamide moiety was achieved in the first step with CSI and chloroethylamine. Then the phosphonate moiety was introduced through a Kabachnik–Fields reaction with a variety of commercial aromatic aldehydes and phosphites with good yields. The nitrosation reaction of these novel α-sulfamidophosphonate compounds derived from 2-chloroethylamine led to the formation of the (2-chloroethyl(diethoxyphosphoryl)methyl)sulfamates instead of the desired nitroso-sulfamidophosphonates products, due to an intramolecular rearrangement. It appeared that the N-nitrosation process of these novel sulfonyl compounds is very challenging compared to the corresponding carbonyl analogues, which gave the expected nitroso ureidophosphonate compounds with good yield. No cytotoxic effect was observed with both series of phosphonosulfamido or phosphonosulfamate compounds.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support for this study was provided by: Applied Organic Laboratory (FNR 2000), General Directorate for Scientific Research and Technological Development (DG-RSDT) and Algerian Ministry of Scientific Research. The study was conducted as part of the PRFU project (2021; ref B00L01UN230120200006). We thank AMaCC platform (CEISAM laboratory) for analyses contribution and Planchat A. (CEISAM laboratory) for X-ray study.
References
-
(a) J. Ferlay, M. Ervik, F. Lam, M. Colombet, L. Mery and M. Piñeros, Global Cancer Observatory: Cancer Today, International Agency for Research on Cancer, Lyon, 2020 Search PubMed;
(b) World Health Organization (WHO), Global Health Estimates 2020: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2019, 2020, https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates Search PubMed.
- G. Quéreux and B. Dréno, Expert OpinPharmacother., 2011, 12, 2891 CrossRef.
- P. Beauchesne, Cancers, 2012, 4, 77 CrossRef CAS.
- Traité de Chimie Thérapeutique, Médicaments Anti-Tumoraux et Perspectives Dans le Traitement Des Cancers, Association française des enseignants de chimie thérapeutique, 2003, vol. 6, p. 879 Search PubMed.
- M. T. Hayes, J. Bartley, P. G. Parsons, G. K. Eaglesham and A. S. Prakash, Biochem, 1997, 36, 10646 CrossRef CAS PubMed.
- T. Nikolova, W. P. Roos, O. H. Krämer, H. M. Strik and B. Kaina, Biochim. Biophys. Acta, 2017, 1868, 29 CAS.
- V. Gadjeva, Eur. J. Med. Chem., 2002, 37, 295 CrossRef CAS.
- Y. Wang, T. Ren, X. Lai, G. Sun, L. Zhao, N. Zhang and R. Zhong, ACS Med. Chem. Lett., 2017, 8, 174 CrossRef CAS PubMed.
- A. Sarkar, S. Karmakar, S. Bhattacharyya, K. Purkait and A. Mukherjee, RSC Adv., 2015, 5, 2137 RSC.
- G. Dewynter, J. L. Montero, B. Agoh, A. Leydet and G. Doukhan, Phosphorus, Sulfur Silicon, 1991, 61, 223 CrossRef CAS.
- M. Abdaoui, G. Dewynter, N. E. Aouf, G. Favre, A. Morère and J. L. Montero, Bioorg. Med. Chem., 1996, 4, 1227 CrossRef CAS PubMed.
- M. Abdaoui, G. Dewynter, L. Toupet and J. L. Montero, Tetrahedron, 2000, 56, 2427 CrossRef CAS.
- J. Y. Winum, J. L Bouissière, I. Passagne, A. Evrard, V. Montero, P. Cuq and J. L. Montero, Eur. J. Med. Chem., 2003, 38, 319 CrossRef CAS PubMed.
-
(a) A. Amira, Z. Aouf, H. K'tir, Y. Chemam, R. Ghodbane, R. Zerrouki and N. E. Aouf, ChemistrySelect, 2021, 6, 6137 CrossRef CAS;
(b) S. Boughaba, S. Bouacida, Z. Aouf, O. Bechiri and N. E. Aouf, Curr. Org. Chem., 2018, 22, 1335 CrossRef CAS;
(c) Z. Aouf, S. Bouacida, C. Benzaid, A. Amira, H. K'tir, M. Mathé -Allainmat, J. Lebreton and N. E. Aouf, ChemistrySelect, 2021, 6, 9722 CrossRef CAS;
(d) H. K'tir, A. Amira, C. Benzaid, Z. Aouf, S. Benharoun, Y. Chemam, R. Zerrouki and N. E. Aouf, J. Mol. Struct., 2022, 1250, 131635 CrossRef;
(e) Y. Chemam, Z. Aouf, A. Amira, H. K’tir, H. Bentoumi, R. Ghodbane, R. Zerrouki and N. E Aouf, Phosphorus, Sulfur, Silicon Relat Elem., 2022, 197, 777 CrossRef CAS;
(f) R. Ghodbane, H. K'Tir, S. Bouacida, M. Ibrahim-Ouali, Z. Aouf and N.-E. Aouf, ChemistrySelect, 2023, 8, e202300157 CrossRef CAS.
-
(a) S. Carter, M. T. Bakowski and K. Hellmann, Chemotherapy of Cancer, John Wiley & Sons: New York, 3rd edn, 1987, p. 55 Search PubMed;
(b) S. M. Bronstein, J. E. Cochrane, T. R. Craft, J. A. Swenberg and T. R. Skopek, Cancer Res., 1991, 51, 5188 CAS;
(c) Nitrosoureas: Current Status and New Developments, ed. A. W. Prestayko, S. T. Crooke, L. H. Baker, S. K. Carter and P. S. Schein, New York, Academic Press, 1981, p. 85 Search PubMed.
- D. N. Dhar and K. S. K. Murthy, Synthesis, 1986, 6, 437 CrossRef.
- Z. Cheraiet, S. Ouarna, Z. Jamel, M. Berredjem and N. E. Aouf, Int. J. Chem., 2012, 4, 73 Search PubMed.
- N. Srinivasan, A. Yurek-George and A. Ganesan, Mol. Diversity, 2005, 9, 291 CrossRef CAS.
- B. Belhani, M. Berredjem, M. Le Borgne, Z. Bouaziz, J. Lebreton and N. E. Aouf, RSC Adv., 2015, 5, 39324 RSC.
- G. Sun, N. Zhang, L. Zhao, T. Fan, S. Zhang and R. Zhong, Bioorg. Med. Chem., 2016, 24, 2097 CrossRef CAS.
- P. P. Onys’ko, K. A. Zamulko, O. I. Kyselyova, I. P. Yelenich and Y. V. Rassukana, J. Fluorine Chem., 2016, 185, 191 CrossRef.
- R. W. Darbeau, R. S. Pease and R. E. Gibble, J. Org. Chem., 2001, 66, 5027 CrossRef CAS PubMed.
- R. Schirrmacher, B. Mathiasch, E. Schirrmacher, D. Radnic and F. Rosch, J. Labelled Compd. Radiopharm., 2003, 46, 959 CrossRef CAS.
-
(a) J. Y. Winum, A. Scozzafava, J. L. Montero and C. T. Supuran, Med. Res. Rev., 2005, 25, 186 CrossRef CAS;
(b) J. J. Jun and X. Q. Xie, ChemistrySelect, 2021, 6, 430 CrossRef CAS;
(c) S. O. Zaraei, A. R Abduelkarem, H. S. Anbar, S. Kobeissi, M. Mohammad, A. Ossama and M. El-Gamal, Eur. J. Med. Chem., 2019, 179, 257 CrossRef CAS PubMed.
- M. Guerfi, M. Berredjem, A. Dekir, R. Bahadi, S. E. Djouad, T. O. Sothea, R. Redjemia, B. Belhani and M. Boussaker, Mol. Diversisty, 2023 DOI:10.1007/s11030-023-10630-w.
- V. Petricek, M. Dusek and L. Palatinus, Jana 2006 – the Crystallographic Computing System, Institute of Physics, Praha, Czech Republic, 2006 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Sara Boughabaab,
Rayene Sayada,
Jacques Lebretonc,
Monique Mathe-Allainmat
*a,
Sara Boughabaab,
Rayene Sayada,
Jacques Lebretonc,
Monique Mathe-Allainmat