
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective and sensitive fluorescent staining of serum albumin in protein gel electrophoresis via sequence-defined oligo-dithiocarbamate†
Anna Josea and
Mintu Porel *ab
*ab
aDepartment of Chemistry, Indian Institute of Technology Palakkad, Indian Institute of Technology, Palakkad, Kerala 678577, India. E-mail: mintu@iitpkd.ac.in
bEnvironmental Sciences and Sustainable Engineering Centre, Indian Institute of Technology, Palakkad, Kerala 678577, India
First published on 8th December 2023
Abstract
This work presents a fluorescent sequence-defined oligo dithiocarbamate platform with a dansyl appendage for interaction studies with a range of proteins including BSA, HSA, proteinase, trypsin, lysozyme, hemoglobin, and amylase. The platform involves six distinct sequence-defined oligomers (SDOs), each offering varied functionalities – dithiocarbamate (DTC), ester, and amide – within the backbone and side chains; different architectures (linear and branched); and introduction of polar or non-polar groups. Fluorescence titration experiments and molecular docking were used to explore the interaction between the synthesized SDOs and the listed proteins. This analysis identified two promising candidates, particularly SDOs 1 and 2, based on higher FRET efficiency, indicating a stronger interaction with serum albumins. SDO 1, demonstrating the highest FRET, was utilized for specific and sensitive staining of serum albumin in native-polyacrylamide gel electrophoresis (Native-PAGE), providing selective fluorescent staining with a 25 times lower concentration of staining agent compared to conventional Coomassie blue staining. This innovative approach serves as an alternative tool for gel staining, especially for selective fluorescent staining of BSA and HSA.
Introduction
Spectroscopic techniques are used as high-throughput tools for clinical diagnosis and detection of various protein components. Among the various proteins, serum albumins, namely human serum albumin (HSA) and bovine serum albumin (BSA), constitute the most abundant and vital protein constituent that performs a plethora of biological functions in humans and animals.1–3 This includes the regulation of physiological activities such as the maintenance of oncotic pressure of plasma, controlling vascular permeability and clotting.2,4–6 Serum proteins also contain copious water-soluble amino acids, which render water solubility and the stability required for the transport of endogenous biomolecules like fatty acids, progesterone, thyroxine, calcium, ferroheme and bilirubin.7–9 In addition to this, exogenous molecules including drugs are also transported to their target sites through serum albumins. This is viable due to the packing of drugs into the serum cavity via hydrophobic and non-covalent interactions. Hence, for the proper functioning of the body, an optimum level of serum albumins is to be maintained and any abnormality is an indication to certain disease states. The optimum concentration of serum albumin in blood ranges from 35–55 g L−1 and less than 20 g L−1 in urine.10 HSA level in urine between the range of 20 to 200 g L−1 is termed as microalbuminuria11 and is observed in patients with cardiovascular disorders and acute kidney issues. The severe stage of kidney damage is indicated as macroalbuminuria, where the serum albumin content in urine goes higher than 200 g L−1. An abnormal serum albumin level is also found in the case of liver failure, cirrhosis, and chronic hepatitis.12 Thus, serum albumin acts as a vital biomarker for the early detection of harmful diseases.Chemosensors based on fluorescence or energy transfer are extensively used for understanding the protein levels in humans. High sensitivity, technical simplicity, cost-effectiveness, rapid response time, non-destructive and non-intrusive nature are the irrefutable traits that lead to the extensive use of fluorescent sensors.13,14 Native-polyacrylamide gel electrophoresis (Native-PAGE) is the most versatile technique used for identifying proteins from a mixture of biological samples based on its molecular weight.15,16 The staining of the gel is traditionally done using Coomassie blue dye. However, Coomassie blue stains all proteins without any specificity. Fluorescent probes that selectively detect and stain serum albumins are hence important owing to their direct concurrence with diseases.
In this work, we report the employment of fluorescent sequence-defined oligo dithiocarbamate platform with dansyl appendage for the interaction studies with several proteins such as BSA, HSA, proteinase, trypsin, lysozyme, hemoglobin, and amylase. The proteins selected here are all involved in specific metabolic activities and can influence the interaction with serum albumins. The modular platform based on sequential design enables the synthesis of macromolecules with tunable functionality and structure via the custom synthesis of monomer units. Using this approach, we have successfully synthesised six distinct sequence-defined oligomers (SDOs). These SDOs exhibit various features such as (1) differing numbers and types of functionalities-dithiocarbamate (DTC), ester, and amide, found in both the main chain and side chains, (2) diverse range of architectures including linear and branched and (3) introduction of polar or non-polar groups to modify the hydrophilic and hydrophobic characteristics. The interaction of the synthesized SDOs with the above listed proteins were studied using fluorescence titration experiments via Förster resonance energy transfer (FRET). The primary requirement for FRET to take place is the spectral overlap between the emission spectra of donor and absorption spectra of acceptor. In this situation, there was a notable alignment between the proteins' emission spectra (acting as the donor) and the absorption spectra of dansyl SDOs (acting as the acceptor). FRET occurred from the tryptophan and tyrosine amino acid residues of serum albumins to the dansyl SDO. Thus, the effect of structure, type and number of functional groups and hydrophobic/hydrophilic character in bringing out selective and efficient FRET behavior towards serum albumins are deduced. From the extent of FRET efficiency, we have selected the SDO with the highest extent of FRET efficiency for selective and sensitive staining of serum albumins in native-PAGE.
Results and discussion
Design of dansyl appended sequence-defined oligomers (SDOs)
In this report, we synthesized six dansyl tagged SDOs for the interaction studies with serum albumins (HSA and BSA) and other proteins like proteinase, trypsin, hemoglobin, lysozyme, and amylase. The synthetic scheme of the SDOs were designed to incorporate several characteristics: (i) a varying count and variety of functionalities, specifically dithiocarbamate (DTC), ester, and amide, present both in the backbone and side chains; (ii) a range of architectures, encompassing linear as well as branched forms; and (iii) the inclusion of distinct hydrophilic (polar) and hydrophobic (non-polar) groups (Fig. 1). The SDO platform offers several advantages including tunability of number and type of functional groups and structure, which in turn will be reflected in the properties of the macromolecule when compared with conventional polymers. The SDOs were synthesized by the sequential addition of required monomers and dansyl chloride was clipped at the beginning to serve as the fluorophore. The choice of dansyl chloride was made to effectively enable energy transfer from serum albumins through FRET, which was facilitated by the favorable spectral overlap between the emission spectra of serum albumins and the absorption spectra of dansyl SDOs. The comprehensive procedure for synthesis is outlined in Scheme S1–S8.† The synthesis process for linear SDOs primarily encompasses chloroacetylation of dansyl ethanolamine, followed by a three-component reaction involving carbon disulfide (CS2), a suitable monomer, and the chloro terminal from the previous reaction. In the case of branched systems, the approach involves reacting dansyl diethanolamine with chloroacetyl chloride, followed by the three-component reaction. The specific structures of the synthesized SDOs are depicted in Fig. 1, with 1, 2, 3, and 4 representing linear SDOs, while 5 and 6 depict the branched variants. Characterization was carried out using techniques such as 1H-NMR (Fig. S8–S18†), high-performance liquid chromatography (HPLC, Fig. S1–S7†), and liquid chromatography-mass spectrometry (LC-MS, Fig. 1).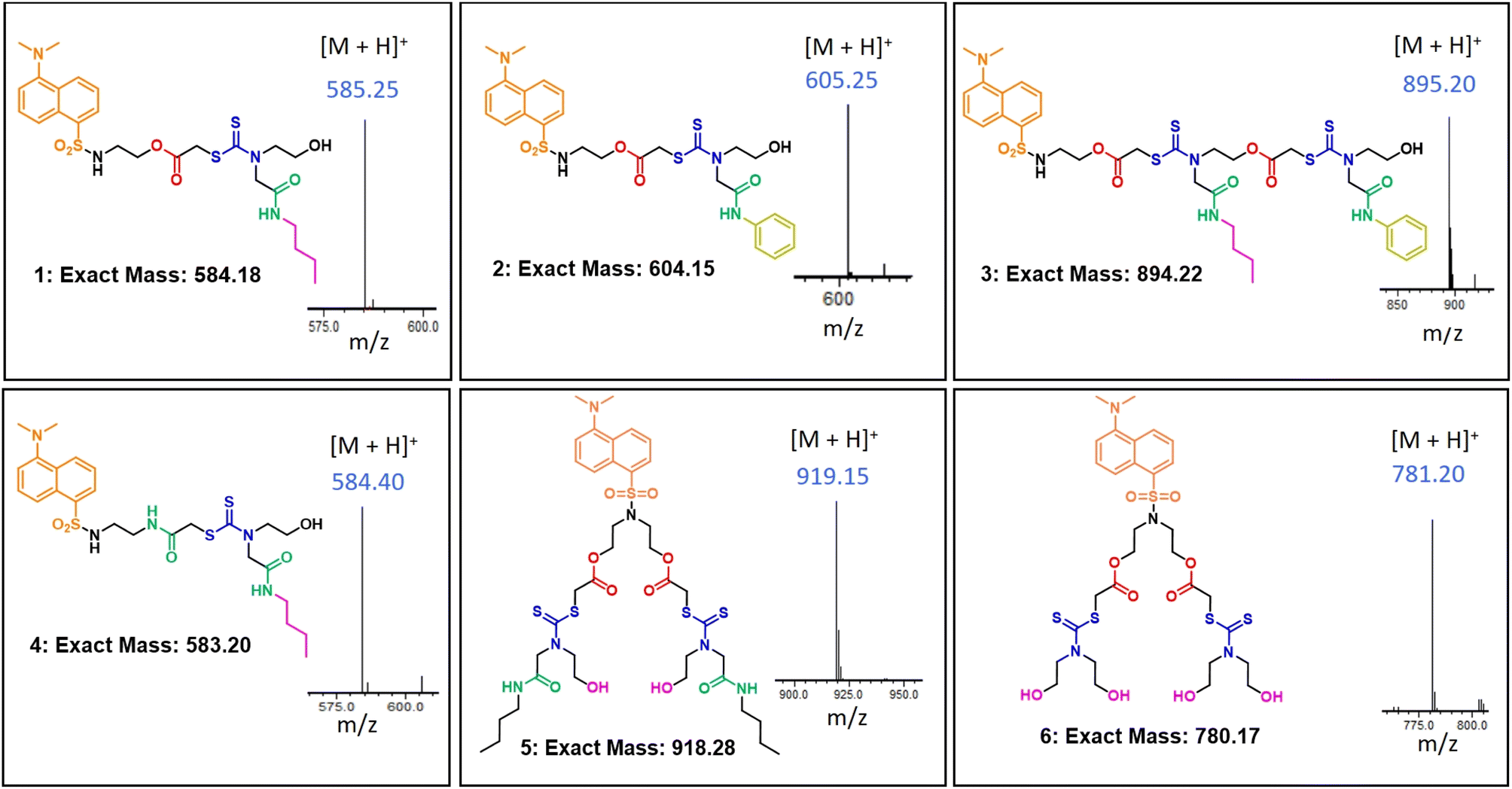 | ||
| Fig. 1 The structure of the synthesised macromolecules along with their exact mass and acquired [M + H]+ from mass spectrometry. | ||
Interaction studies of the SDOs with different proteins
The sequence-defined modular platform bestows several advantages of tuning the functional groups and structure of the synthesized macromolecules, which will in turn result in modulating the properties of the SDO for a desired application. In the present study, the interactions of the SDOs with different proteins were studied with the aim to comprehend the effect of the afore-mentioned parameters (functionality, chain length, architecture, and hydrophobicity) in bringing about an effective and selective interaction with serum albumins in presence of other proteins. In silico studies were also performed to calculate the binding energy for each interaction and to further support the experimental findings.The fluorescence titrations for the interaction of the six SDOs with different proteins (BSA, HSA, proteinase, trypsin, lysozyme, hemoglobin, and amylase) were performed. Initially SDO 1 was selected as the model compound for this study and its absorption and emission spectra were recorded along with HSA (Fig. S19†). The normalized emission and absorption spectra of these entities reveal an overlapping range between the emission spectrum of HSA and the absorption spectrum of 1. This implies the feasibility of energy transfer from HSA (as the donor) to dansyl-SDOs (acting as acceptors), suggesting their potential as a FRET pair. To investigate further, incremental additions of 1 were made to a solution containing 10 μM HSA, and subsequent emission spectra were recorded. The emission peak of HSA was observed at 350 nm upon being excited at 280 nm. Upon the introduction of 1, the intensity of the emission peak at 350 nm decreased while a new peak emerged at 500 nm, corresponding to the emission of 1 (Fig. 2a). An isoemissive point at 430 nm was also noted, indicating the formation of a complex between the donor (HSA) and the acceptor (dansyl SDO, denoted as 1).
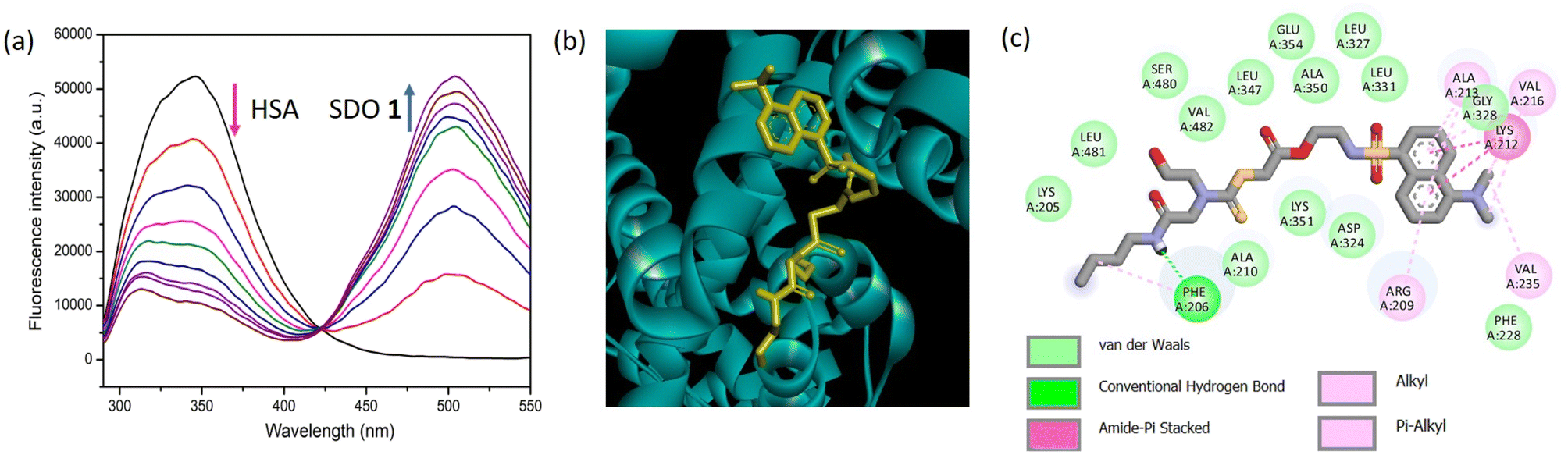 | ||
| Fig. 2 The fluorescence intensity of HSA (10 μM) recorded for the incremental addition of 1 (4.25 μM to 51 μM) in phosphate buffer solution at a temperature of 25 °C with an excitation wavelength of 280 nm, (b) molecular docking of 1 with HSA and (c) the two-dimensional depiction illustrating the interaction between 1 and the amino acid residues of HSA. | ||
Molecular docking was conducted as an additional step to corroborate the experimental findings. The interaction between HSA and 1 through docking produced a binding energy of −7.1 kcal mol−1. The visual representation of this docking is presented in Fig. 2b, illustrating the binding of 1 to HSA. It is known that for HSA, the amino acid residues from 1–195 constitute domain I, 196–383 constitute domain II and 384–585, domain III.17 To attain a more comprehensive view of these interactions, a 2-D depiction of the docking interaction between 1 and HSA was generated. The image highlights the engagement of amino acid residues from domain I and II with 1. This interaction involves substantial forces like van der Waals interactions, hydrogen bonds, and Pi-sulfur interactions, as revealed in Fig. 2c. Similarly, the docking study of 1 with BSA is presented in Fig. S27,† and it also demonstrates a notable binding affinity.
The fluorescence emission spectra obtained for the titration of 1 with BSA is shown in Fig. 3a. Here also, the intensity of the peak at 350 nm is decreasing and that at 500 nm is increasing, indicating the FRET phenomenon. For the addition of 1 to proteinase (Fig. 3b), the peak intensity at 350 nm decreased to some extent, however, it was not accompanied by a significant increase in the emission intensity at 500 nm when compared to that obtained for BSA/HSA. Similar was the case for the titrations with other proteins like trypsin, lysozyme, amylase, and hemoglobin (Fig. 3c–f). Unlike BSA and HSA, the other proteins did not exhibit FRET behavior and this indicates a much weaker interaction than BSA/HSA. Therefore, 1 exhibit excellent FRET behavior only with BSA and HSA indicating stronger interaction with BSA/HSA than the other proteins. Moreover, the docking studies of 1 with other proteins were performed (Fig. S27, S33, S39, S45, S51, S57 and S63†). The binding energy obtained for 1 with BSA, proteinase, trypsin, amylase, lysozyme, and hemoglobin were −7.1, −6.5, −6.6, −6.0, −5.1 and −6.4 kcal mol−1 respectively. This corroborates the higher interaction of 1 with serum albumins.
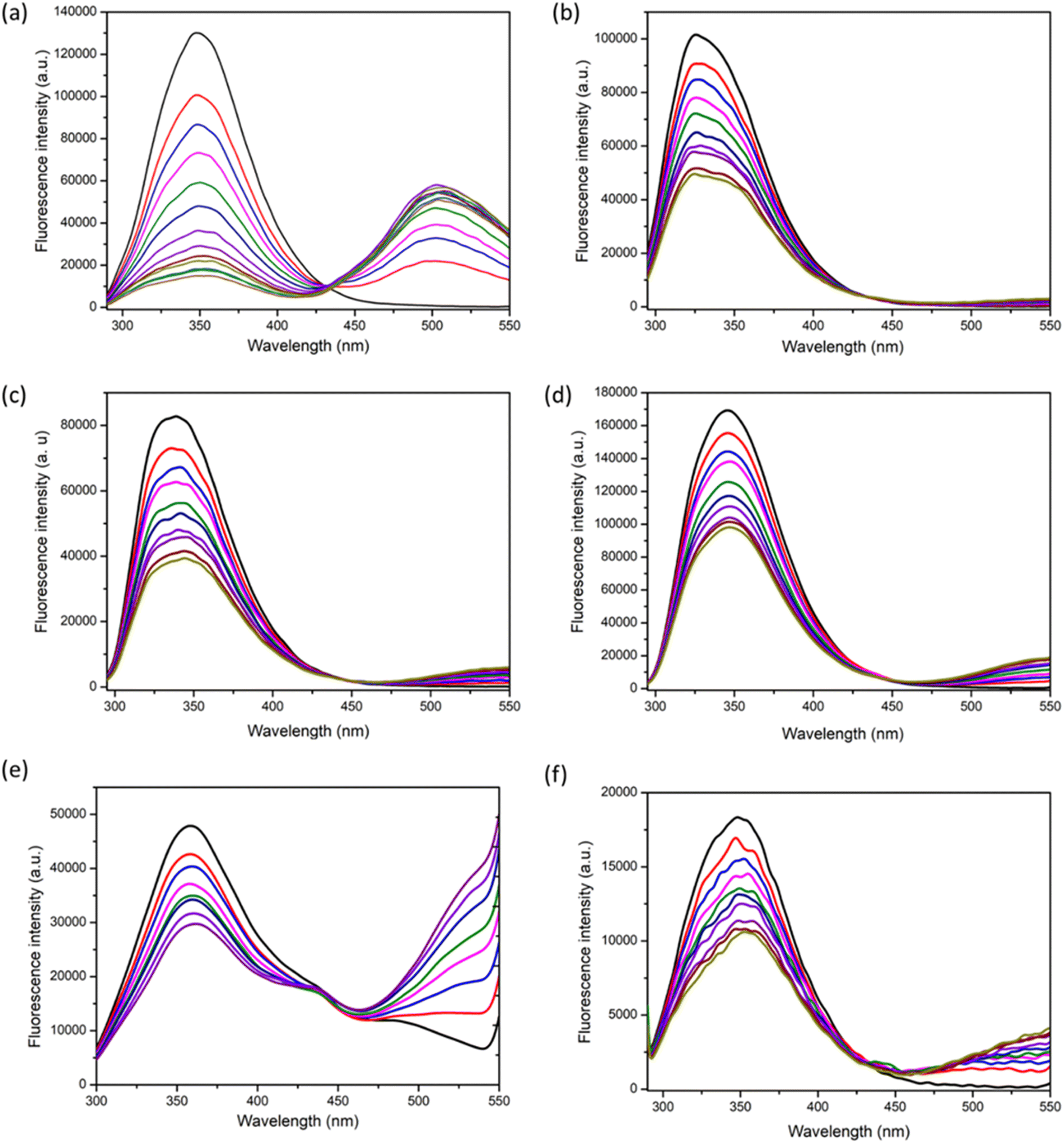 | ||
| Fig. 3 The fluorescence emission spectra for the titration of 1 (4.25 μM to 51 μM) with (a) BSA (10 μM) (b) proteinase (10 μM), (c) trypsin (10 μM), (d) lysozyme (10 μM), (e) haemoglobin (10 μM), and (f) amylase (10 μM) in phosphate buffer at 25 °C with an excitation wavelength of 280 nm. | ||
Next, we studied the interaction of 2 with various proteins of interest. This was done with the aim to understand the effect of the side-chain functionality in determining the interaction pattern with proteins. The only structural difference that 1 and 2 have is in their side-chain functionality. The SDO 1 possess a butyl entity and 2 possess phenyl entity. The fluorescence titrations of 2 with HSA, BSA, proteinase, trypsin, lysozyme, hemoglobin, and amylase are shown in Fig. S20.† The FRET behavior was significant only for the addition of serum albumins and not with other proteins. The hydrophobicity of 2 is higher than that of 1 and that is reflected in the slightly higher extent of binding of 2 with serum albumins. This indicates that the structure and hydrophobicity of the molecule greatly influences the binding with proteins.
The other factors that were considered apart from side-chain functional group (case of 2), for its influence on the interaction with proteins were (i) backbone functional group (case of 4), (ii) chain length (case of 3) and (iii) architecture (5 and 6). When the tuning of the functional group in the backbone was considered, 4 was titrated to different protein solutions. 4 was different from other linear counterparts in the fact that it does not contain ester linkage in the backbone. The interactions of 4 with the serum albumins did not produce a significant FRET behavior as compared with 1 and 2 (Fig. S22†). The interactions with other proteins were also weak. This signifies the importance of functional groups in bringing about efficient protein interactions. We also studied the interactions with 3, having an increased number of functional groups. The fluorescence titration of 3 with all the proteins are shown in Fig. S21.† It was found that 3 interacted well with hemoglobin and lysozyme in addition to mild interactions with BSA and HSA. Thus, selectivity was compromised in this case. The hydrophobicity of 1, 2, and 3 varies as 3 > 2 > 1. Therefore, an increase in hydrophobicity should theoretically enhance the interaction with serum albumins. However, in this case, the binding affinity observed with both BSA and HSA was lower in comparison to that with hemoglobin. This implies that, aside from hydrophobicity, the chain length of the SDO also plays a role in influencing the binding with serum albumins. Additionally, to explore the impact of architecture, a comparison between linear and branched SDOs was conducted. The branched SDOs, represented by 5 and 6, similarly did not display substantial interactions as FRET pairs (as shown in Fig. S23 and S24†). This might be attributed to the presence of polar hydroxy groups – four in the case of 6 and two in the case of 5. This underscores the significance of the SDO platform in tailoring both structure and functionality to attain the desired property or function as required.
For better comparison and to understand which compound binds well with each protein, we have plotted the fluorescence spectra of each protein with all the six SDOs. A typical example is shown for HSA in Fig. S25.† It was found that SDOs 1 and 2 were interacting with HSA to a greater extent. We observed similar observations with BSA as well (Fig. S26a†). Here also SDOs 1 and 2 were showing the best extent of interaction. Similar plots for the other proteins are shown in Fig. S26b–f.†
The similar experiments with other proteins indicated the relevance of structure–activity relationship. The interaction of SDOs with BSA was similar to that of HSA. However, in the case of proteinase, trypsin, lysozyme, hemoglobin, and amylase the interaction was weak, which is indicated by the weak emission maxima of the peak at 500 nm. A bar diagram for the ratio of fluorescence intensities (F500/F350) of BSA and HSA after addition of the SDO is plotted in Fig. 4a. This indicates the enhanced selectivity of SDOs 1 and 2 towards serum albumin. Further, the F500/F350 for the addition of 1 and 2 towards different proteins (Fig. 4b) also shows the selectivity of these probes towards BSA and HSA over other proteins.
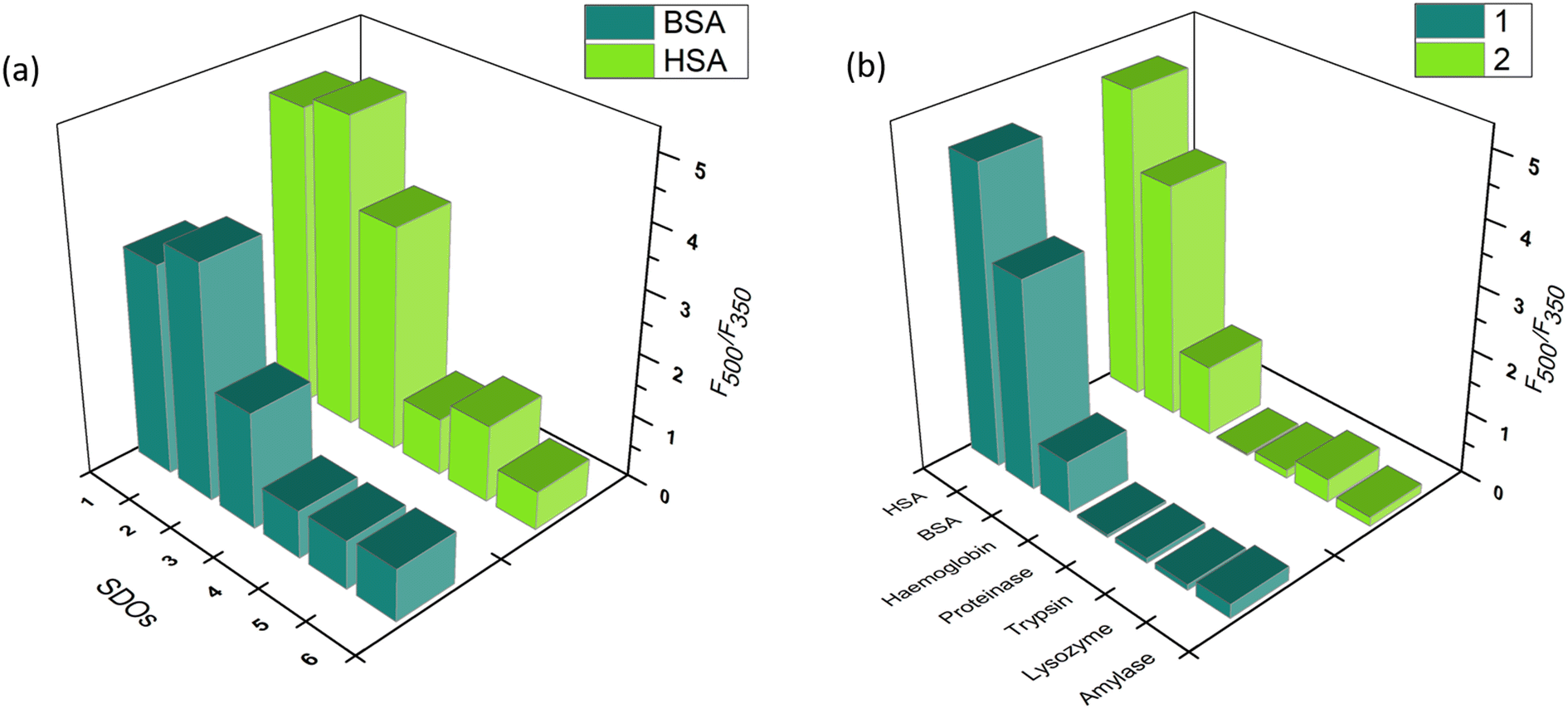 | ||
| Fig. 4 The bar diagram for the ratio of fluorescence intensities (F500/F350) of (a) BSA and HSA after addition of the different SDOs and (b) that for the addition of 1 and 2 to different proteins. | ||
The experimental data was also correlated with the in silico studies (Fig. S27–S68†). The interactions of the synthesized SDOs with the amino acid residues of the proteins were identified. Strong interactions like conventional hydrogen bond were identified along with van der Waals, Pi alkyl and Pi sulphur interactions. The binding energy obtained for the interaction of the SDOs with each protein is tabulated in Table 1. From the binding energy, dissociation constant was also calculated and the obtained values are given in Table S1 in the ESI.† The binding energy obtained was highest for the docking of SDOs with serum albumins especially in the case of 1 and 2. This reiterates the high binding affinity obtained from the experimental results for the linear SDOs 1 and 2.
| Protein | Binding energy obtained for SDOs in kcal mol−1 | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| HSA | −7.1 | −8.0 | −8.1 | −6.7 | −6.6 | −5.9 |
| BSA | −7.1 | −7.2 | −7.4 | −6.8 | −6.6 | −6.6 |
| Proteinase | −6.5 | −6.1 | −5.6 | −5.8 | −6.0 | −5.7 |
| Trypsin | −6.6 | −6.7 | −6.5 | −6.7 | −5.9 | −5.6 |
| Amylase | −6.0 | −6.9 | −5.3 | −6.4 | −5.5 | −5.5 |
| Lysozyme | −5.1 | −6.8 | −6.4 | −5.9 | −5.4 | −5.3 |
| Haemoglobin | −6.4 | −6.4 | −5.8 | −5.7 | −5.7 | −6.3 |
Staining of native-polyacrylamide gel electrophoresis (PAGE) with 1
The selectivity towards serum albumins is vital considering its role as a biomarker for the early detection of certain diseases. The FRET based spectroscopic results indicate that out of the six SDOs, 1 and 2 shows significant selectivity towards serum albumins over the others. This prompted us to utilize the synthesized SDOs for real-end application as serum albumin selective fluorescent staining agent in native-PAGE. From the two SDOs (1 and 2) screened from the basket, 1 was selected for this application. A 10% gel was prepared according to procedure given in SI and the staining reagent was prepared with 0.01% w/v of 1. HSA and BSA were run separately and also as a mixture with other proteins like lysozyme, hemoglobin, and amylase. This was used to understand the staining capacity of the SDO in presence of other competitive proteins (Fig. 5). Trypsin and proteinase were not included due to their ability to cleave other proteins. When the proteins were run in native-PAGE, and post staining with 1, it was found that the serum albumin were identified efficiently and selectively (Fig. 5). From this gel image the intensity of the protein bands (molecular weight 66 kDa) was obtained using ImageJ software and the results are shown in Fig. S69.† The intensity is proportional and in accordance with the concentration of the protein loaded in each well. Also, the selectivity of the synthesized probe was further tested by running a gel with fetal serum, a real serum sample. The obtained gel image with the intensity of the protein bands is shown in Fig. S70† and this further reinstates the selective nature of the SDO. To compare the staining efficiency of SDO 1 with the conventional staining agent-Coomassie blue, another gel was prepared and stained with Coomassie blue (0.25% w/v). However, unlike 1, Coomassie blue stained all the proteins (Fig. S71†) and required 25 times more compound than 1. Also, lower concentrations of BSA/HSA, even up to 0.3 μg was also stained by 1 (Fig. S72†). Taken together, this work puts forth the advantages of an SDO platform to tune the functionality and structure of a macromolecule to modify the properties, so as to attain a specified application. The fluorescence and molecular docking studies aided the selection of the most suitable candidate for probing serum albumins. In this way, a selective probe for the fluorescent labelling of BSA/HSA was identified from a basket of six SDOs and was proven to be an efficient substitute for conventional staining process. Taken together, this sequence-defined oligomeric system with dansyl appendage has significant potential to move this field ahead.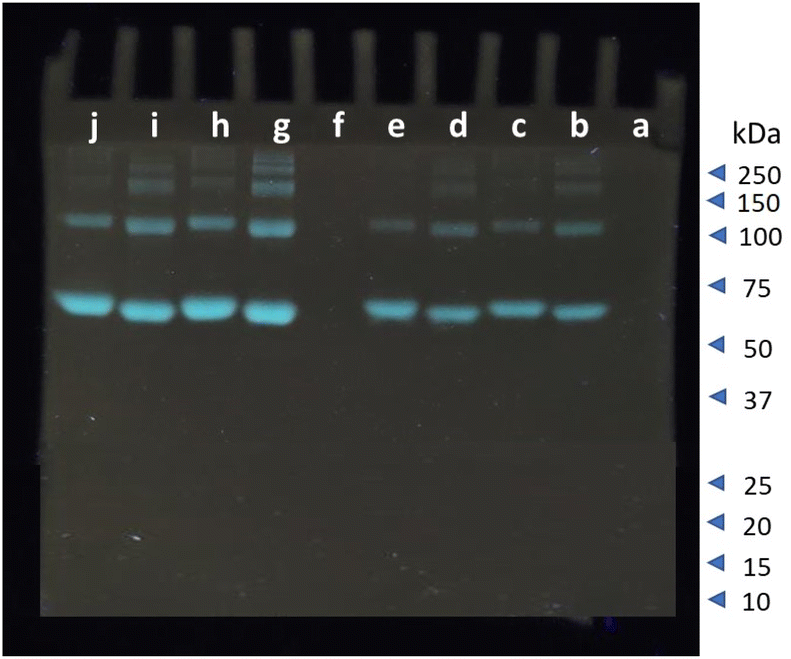 | ||
| Fig. 5 Native-PAGE of a mixture of BSA, HSA, amylase, lysozyme, and haemoglobin. The proteins loaded in each well are; (a) ladder, (b) BSA (20 μg), (c) HSA (20 μg), (d) BSA (20 μg) + mixture (2.5 μg each), (e) HSA (20 μg) + mixture (2.5 μg each), (f) mixture (2.5 μg each) of proteins without BSA/HSA, (g) BSA (40 μg), (h) HSA (40 μg), (i) BSA (40 μg) + mixture (5 μg each), (j) HSA (40 μg) + mixture (5 μg each) stained with SDO 1. | ||
Conclusion
We have employed six different fluorescent labelled sequence-defined oligomers (SDOs) with (i) a varying count and variety of functionalities, specifically dithiocarbamate (DTC), ester, and amide, present both in the backbone and side chains; (ii) a range of architectures, encompassing linear as well as branched forms; and (iii) the inclusion of distinct hydrophilic (polar) and hydrophobic (non-polar) groups for the interaction studies with several proteins. The proteins chosen in addition to serum albumins (BSA and HSA) were proteinase, trypsin, lysozyme, haemoglobin, and amylase. Experimental studies, especially by fluorescence analysis, revealed the occurrence of FRET (Förster resonance energy transfer) between serum albumins and SDOs 1 and 2 unlike in the case of SDOs 3, 4, 5 and 6. This revealed the importance of structure, hydrophobic/hydrophilic character, and chain length in bringing out selective and efficient FRET behaviour towards serum albumins. This data further prompted us to utilize the SDO 1 as a staining agent in native-PAGE. We attained selective staining of serum albumins in native-PAGE with 25 times lower concentration of the staining agent (here SDO 1) than the conventional Coomassie staining. In this way, a selective probe for staining serum albumins was identified from a basket of six SDOs which works as a substitute for conventional staining process. Therefore, this work provides an insight in to the importance of structure and functional group tunability to aid any given application and proves to be significant advancement in the field to move this forward.Experimental section
Materials and method
The chemicals utilized in this study were obtained from standard commercial suppliers. Bovine serum albumin (BSA) was acquired from spectrochem, while human serum albumin (HSA) was obtained from HiMedia. Proteinase, trypsin, lysozyme, hemoglobin, and amylase were sourced from SRL chemicals. The solvents employed were of analytical quality and were used without further purification.Instrumentation
The LC-MS (Liquid chromatography-mass spectrometry) measurements were conducted using a Shimadzu LC-MS-8045 system equipped with a Sprite TARGA C18 column measuring 40 × 2.1 mm and having a particle size of 5 μm. The UV detector on this instrument operated in positive ionization mode, detecting at both 254 nm and 210 nm. The solvent system comprised LC-MS grade water with 0.1% formic acid as solvent A, and LC-MS grade acetonitrile as solvent B. For High Performance Liquid Chromatography (HPLC), a Shimadzu Nexera X2 instrument was employed. This setup utilized a Shim-pack C-18 column sized 20 × 250 mm with a particle size of 5 μm. The detection was accomplished using a diode array detector. HPLC-grade water and acetonitrile were used as the solvents in this context. Fluorescence studies were carried out employing a PerkinElmer FL 6500 instrument.Synthetic strategy for the macromolecules
The sequence-defined oligomers (SDOs) employed in this investigation were synthesised using an iterative process involving the gradual inclusion of reactive substrates or individual monomeric units. To introduce fluorescence, dansyl chloride was utilized during the synthesis. The synthetic approach begins with modifying dansyl chloride and involves two primary steps: (1) the introduction of chloroacetyl chloride in the presence of triethylamine (acting as base) and dichloromethane (acting as solvent), and (2) the incorporation of carbon disulfide and the monomer through a three-component reaction route utilizing polyethylene glycol. These two reactions were performed consecutively to craft the necessary macromolecules.General procedure for fluorescence analysis
To conduct fluorescence experiments, initial solutions of SDOs were formulated using DMSO. The stock solutions of proteins, including BSA, HSA, proteinase, trypsin, lysozyme, hemoglobin, and amylase, were created using phosphate buffer with a pH of 7.4 at a concentration of 10 μM. Fluorescence analysis was conducted by taking a 10 μM solution of distinct proteins in a quartz cuvette and progressively adding the prepared SDOs (ranging from 4.25 μM to 51 μM) in phosphate buffer. The experiments were conducted at 25 °C at an excitation wavelength of 280 nm. The emission and excitation slit widths were both set at 5 nm.General procedure for docking analysis
Molecular docking investigations were carried out using Autodock Vina18 with the integration of executables facilitated by a Perl Script. On 23 December 2022, the protein data bank was accessed to retrieve the PDB structures of HSA (1BM0, Chain A), BSA (4F5V, Chain A), proteinase (4NKK), trypsin (2STB), lysozyme (5LVK), hemoglobin (1GZX), and amylase (3VX0). To prepare the structures, hydrogen atoms were added and Gasteiger charges were assigned using Autodock tools 4.19 For the docking simulations, grid sizes were selected as follows: HSA with dimensions (60 × 60 × 70) and center at (x29.61, y31.78, z23.48), BSA with dimensions (62 × 80 × 74) and center at (x34.16, y24.80, z41.47), proteinase with dimensions (64 × 104 × 126) and center at (x2.45, y8.34, z25.63), trypsin with dimensions (42 × 36 × 52) and center at (x2.49, y7.53, z21.19), lysozyme with dimensions (100 × 90 × 110) and center at (x24.21, y31.71, z10.18), hemoglobin with dimensions (100 × 90 × 90) and center at (x14.42, y67.71, z4.18), and amylase with dimensions (54 × 54 × 53) and center at (x21.79, y15.32, z40.94). Prior to docking, the ligands underwent energy minimization utilizing Argus lab 4.0.1.20 Subsequently, the docked structures were visualized using Pymol, and interactions with amino acid residues were explored through Discovery studio.21General procedure for running native gel
The resolving gel and stacking gel were prepared according to reported procedure. Since the gel ran is native gel, SDS or 2-mercaptoethanol was not added anywhere. In place of that distilled water was added. The detailed procedure is available in ESI.† After the separation of the proteins in the gel, the staining was done using 1. The similar composition of a duplicate gel was stained using Coomassie blue for comparison. After staining, the gel stained with 1 was viewed under a transilluminator and the one stained in Coomassie blue was viewed under normal light.Author contributions
M. P. conceived the concept for Dansyl-appended sequence-defined oligo-dithiocarbamate as a sensitive and serum albumin-selective fluorescent staining agent for protein gel electrophoresis. The conceptualization of molecular design and synthetic methodologies was undertaken by M. P. and A. J. The synthesis, characterizations, fluorescence analyses, and electrophoresis was executed by A. J. The data analysis and paper writing were jointly carried out by A. J. and M. P.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Indian Institute of Technology Palakkad, India; Ramanujan Fellowship (SB/S2/RJN-145/2017) and Core Research Grant (CRG/2019/002495), Science and Engineering Research Board-Department of Science and Technology, India; Scheme for Transformational and Advanced Research in Sciences (MoE/STARS1/293), Ministry of Education, India; The Central Instrumentation Facility (CIF) at the Indian Institute of Technology Palakkad and Indian Institute of Technology Madras are acknowledged for the analytical instrumentation support. We thank Dr Sushabhan Sadhukhan, Revathy S and Dr Anupama Binoy for their guidance in running native-PAGE.Notes and references
- G. J. Quinlan, G. S. Martin and T. W. Evans, Hepatology, 2005, 41, 1211–1219 CrossRef CAS PubMed.
- T. Peters Jr, All about Albumin: Biochemistry, Genetics, and Medical Applications, Academic press, 1995 Search PubMed.
- B. T. Doumas and T. Peters Jr, Clin. Chim. Acta, 1997, 258, 3–20 CrossRef CAS PubMed.
- G. Fanali, A. Di Masi, V. Trezza, M. Marino, M. Fasano and P. Ascenzi, Mol. Aspects Med., 2012, 33, 209–290 CrossRef CAS PubMed.
- C.-E. Ha and N. V Bhagavan, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5486–5493 CrossRef CAS PubMed.
- J. Reichenwallner and D. Hinderberger, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5382–5393 CrossRef CAS PubMed.
- B. R. Patel and K. Kerman, Analyst, 2018, 143, 3890–3899 RSC.
- G. P. Szekeres and J. Kneipp, Analyst, 2018, 143, 6061–6068 RSC.
- C. Wang, Q. Wang and R. Tan, Analyst, 2018, 143, 4118–4127 RSC.
- J.-F. Xu, Y.-S. Yang, A.-Q. Jiang and H.-L. Zhu, Crit. Rev. Anal. Chem., 2022, 52, 72–92 CrossRef CAS PubMed.
- D. de Zeeuw, H.-H. Parving and R. H. Henning, J. Am. Soc. Nephrol., 2006, 17, 2100–2105 CrossRef CAS PubMed.
- K. Ohnishi, A. Kawaguchi, S. Nakajima, H. Mori and T. Ueshima, J. Clin. Pharmacol., 2008, 48, 203–208 CrossRef CAS PubMed.
- J. Li and N. Wu, Biosensors Based on Nanomaterials and Nanodevices, CRC Press, 2013 Search PubMed.
- I. L. Medintz, A. R. Clapp, H. Mattoussi, E. R. Goldman, B. Fisher and J. M. Mauro, Nat. Mater., 2003, 2, 630–638 CrossRef CAS PubMed.
- A. Chrambach and D. Rodbard, Science, 1971, 172, 440–451 CrossRef CAS PubMed.
- J. V Maizel Jr, in Methods in Virology, Elsevier, 1971, Vol. 5, pp. 179–246 Search PubMed.
- F. Dangkoob, M. R. Housaindokht, A. Asoodeh, O. Rajabi, Z. R. Zaeri and A. V. Doghaei, Spectrochim. Acta, Part A, 2015, 137, 1106–1119 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- D. Ranjith, J. Pharm. Innov., 2019, 8, 481–487 Search PubMed.
- D. S. Biovia, 2019. Discovery Studio Modeling Environment; Release 2017, Dessault Systemes, San Diego, CA, USA, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06985k |
| This journal is © The Royal Society of Chemistry 2023 |