
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric total synthesis strategies of halichlorine and pinnaic acid
Lu Liu†
a,
Minghua Jiang†a,
Qingkang Zhanga,
Hong Chena,
Yifu Zhang*ab and
Jian Zhang *abc
*abc
aSchool of Pharmacy, Gansu University of Chinese Medicine, Lanzhou 730000, P. R. China. E-mail: zhangjian@gszy.edu.cn; hanyuelongfei@qq.com
bNorthwest Collaborative Innovation Center for Traditional Chinese Medicine Co-Constructed by Gansu Province & MOE of PRC, Lanzhou 730000, P. R. China
cKey Laboratory of Chemistry and Quality for TCM of the College of Gansu Province, Lanzhou 730000, P. R. China
First published on 17th November 2023
Abstract
Halichlorine and pinnaic acid are structurally related natural alkaloids isolated from different marine organisms. These two marine alkaloids bearing a 6-azaspiro[4.5]decane skeleton demonstrate a wide range of biological effects. It is this kind of unique structure and potentially valuable biological activity that have prompted strong synthetic interest, making it a research focus in recent years. Since the first total synthesis of halichlorine and pinnaic acid completed by Danishefsky's group, many groups have reported their outstanding synthesis methods especially the asymmetric synthesis strategies. This review summarizes the asymmetric synthesis strategies of halichlorine and pinnaic acid using a 6-azaspiro[4.5]decane skeleton as the key intermediate, which can provide some guidance for related work.
1 Introduction
Halichlorine (1) and the pinnaic acid (2) are structurally related natural products isolated from different marine organisms (Fig. 1). The marine alkaloid halichlorine (1) was isolated from the black sponge Halichondria okadai Kadota by Uemure and co-workers in 1996.1 It selectively inhibits the expression of the inducible cell surface protein VCAM-1 (vascular cell adhesion molecule-1), and can be used to treat atherosclerosis, coronary artery disease, angina pectoris, and non-cardiovascular inflammatory diseases. Then Uemura's Laboratory isolated pinnaic acid (2) from the Okinawan bishell Pinna muricata in the same year, which was structurally closely related to halichlorine (1).2 Pinnaic acid (2) was found to inhibit cytoplasmic phospholipase A2(cPLA2) at a semi-inhibitory concentration of 0.2 mm in vitro. These two marine alkaloids possess significant biological properties and may have potential for use as biochemical tools or as leads for drug design.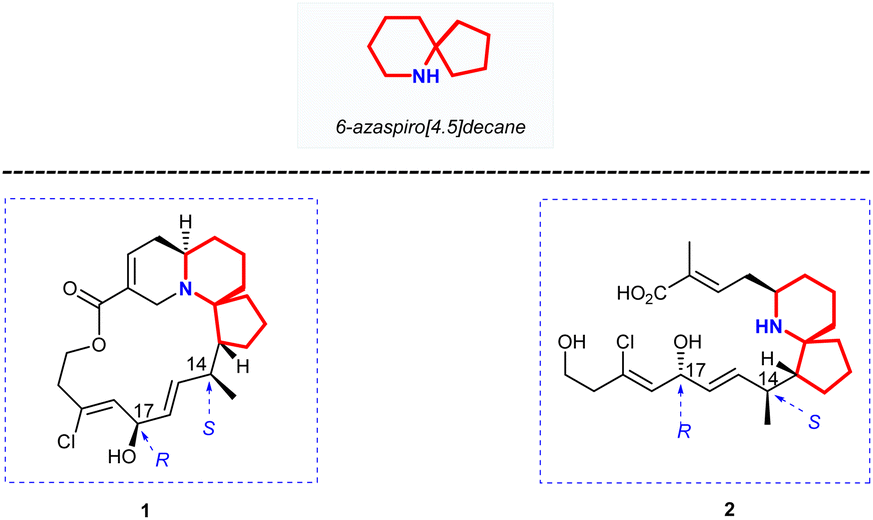 | ||
| Fig. 1 Structure of halichlorine (1) and pinnaic acid (2). | ||
The marine natural products halichlorine (1) and pinnaic acid (2) have attracted a lot of attention because of their good physiological activities and unique structures, and many research groups have carried out outstanding synthesis research on these two alkaloids. In 2005, Clive's group made a systematic and detailed summary on the synthesis of these two alkaloids.3 In recent years, lots of new asymmetric synthesis strategies of halichlorine (1) and pinnaic acid (2) have been reported. This review first summarizes the asymmetric synthesis strategies of halichlorine and pinnaic acid using 6-azaspiro[4.5]decane skeleton as the key intermediate over a span of approximately 20 years (1999–2022), which can provide some guidances for related workers. Furthermore, we also took into account the racemic synthesis work conducted between 2005 and 2022, which was not included in Clive's review. The constructions of 6-azaspiro[4.5]decane framework are the key steps of these asymmetric synthesis methods. In our efforts directed toward the synthesis of these two alkaloids base on the [2,3]-Stevens rearrangement, we have previously disclosed an efficient strategy for construction of the 6-azaspiro[4.5]decane skeleton.4
2 Absolute configuration determination and asymmetric synthesis of halichorine
2.1 Absolute configuration determination of halichorine
In 1998, Uemura's Laboratory verified the absolute configuration of 1 through degradation studies (Scheme 1).5 Methanolysis of 1, followed by ozonolysis with reductive workup and global acetyl protection of the exposed alcohol functionalities yielded 1.1. The degradation product was compared with the sample prepared from a known alcohol 1.2, which it can be obtained from D-(+)-tartaric acid. The hydroxyl group of 1.2 was protected to a THP ether. Then the carbon chain extension product 1.5 was obtained in a high yield with the glycidyl ether 1.4. Then the final product (S)-triacetate 1.1 was obtained by the additional steps, whose NMR spectrum agreed with degradation product of natural halichlorine. It must be noted that the stereochemistry of C(17) is S in 1.1 but R in halichlorine, although there is no difference in the spatial arrangement of the key atoms. The enantiomer of 1.1 was made from L-tartaric acid, and HPLC analysis on a chiral column showed that 1.1 from D-tartaric acid corresponds to the degradation product from halichlorine, thereby establishing the absolute configuration of halichlorine, which was subsequently confirmed by the Danishefsky synthesis.6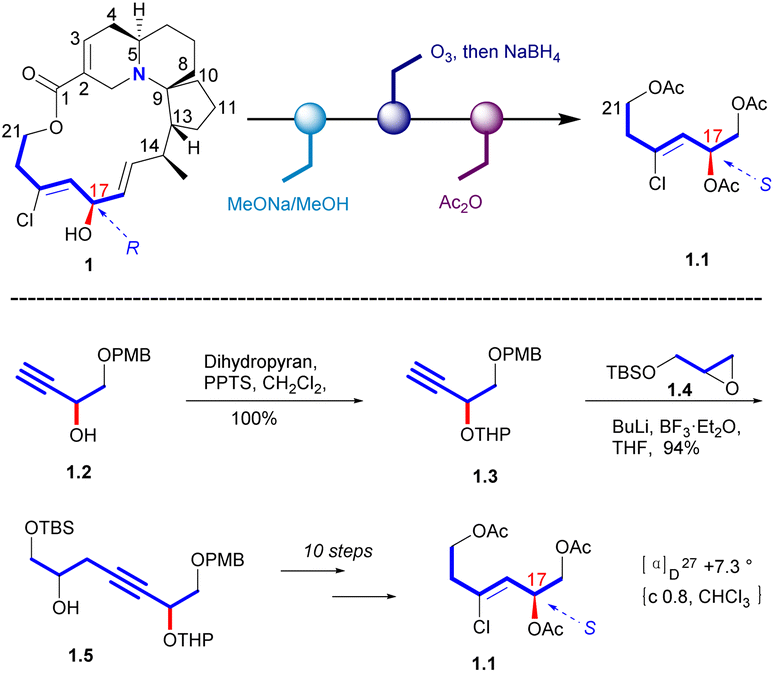 | ||
| Scheme 1 Confirmation of C17 stereochemistry of halichlorine. | ||
2.2 Asymmetric synthesis of halichorine
Since its initial proposal by Danishefsky in 1999, the synthesis strategy for halichlorine has attracted increasing attention from synthetic scholars. Over the years, numerous research groups have successfully completed the synthesis of halichlorine using various strategies. Each group's research work had their own characteristics in the synthesis strategies employed. We summarized the asymmetric synthesis strategies of halichlorine using 6-azaspiro[4.5]decane skeleton as the key intermediate, in which we can see the subtleties of the synthesis strategies of different research groups (Scheme 2).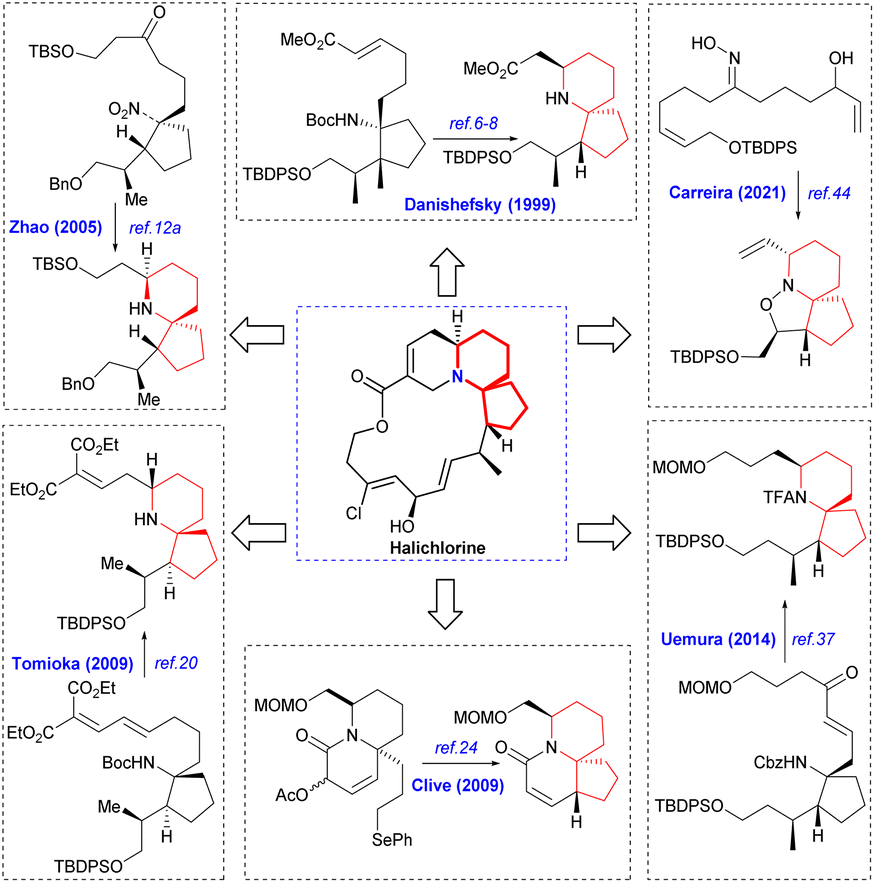 | ||
| Scheme 2 Asymmetric synthesis of halichorine via the 6-azaspiro[4.5]decane skeleton. | ||
Then the compound 3.7 was obtained through multiple steps. Compound 3.7 was borohydrided with 9-BBN to obtain borane, and 3.8 was used as coupling agent to have Suzuki coupling reaction with it to extend the side chain (3.7 → 3.9). The Boc group was removed by TFA and then neutralized with K2CO3, resulting in spontaneous intramolecular Michael addition, providing the 6-azaspiro[4.5]decane skeleton with the desired configuration at C5 (3.9 → 3.10). This method is very effective to construct the 6-azaspiro[4.5]decane skeleton and the stereochemistry of the Michael addition presumably results from reaction via the conformation that with the larger substituent pseudoequatorial (Scheme 3).
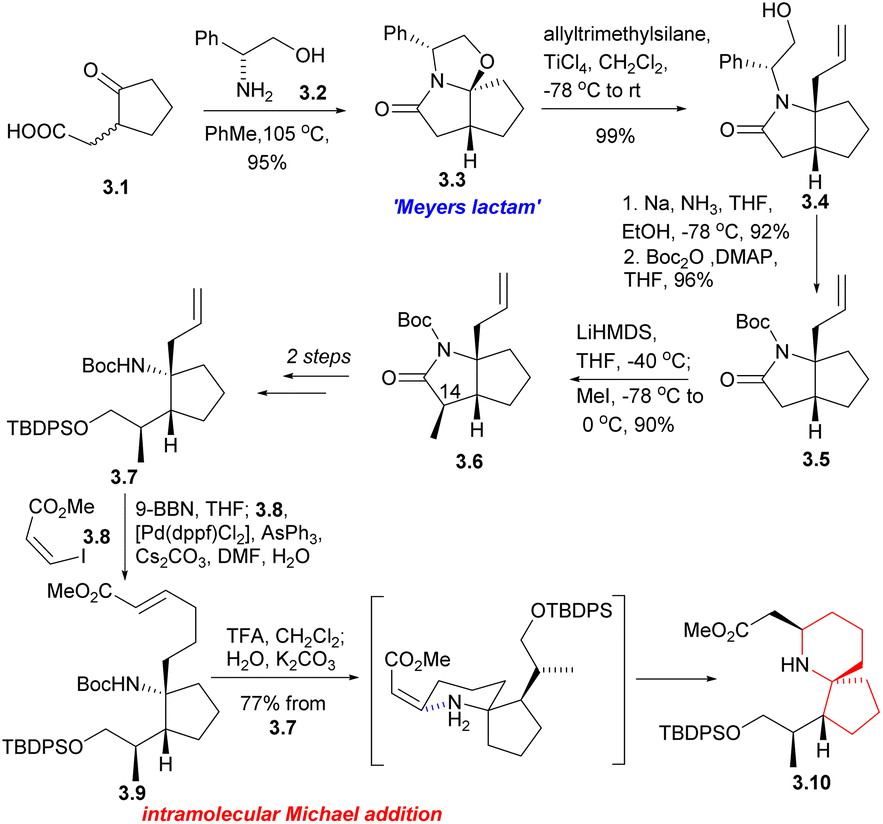 | ||
| Scheme 3 Danishefsky's synthesis of the 6-azaspiro[4.5]decane skeleton of halichlorine. | ||
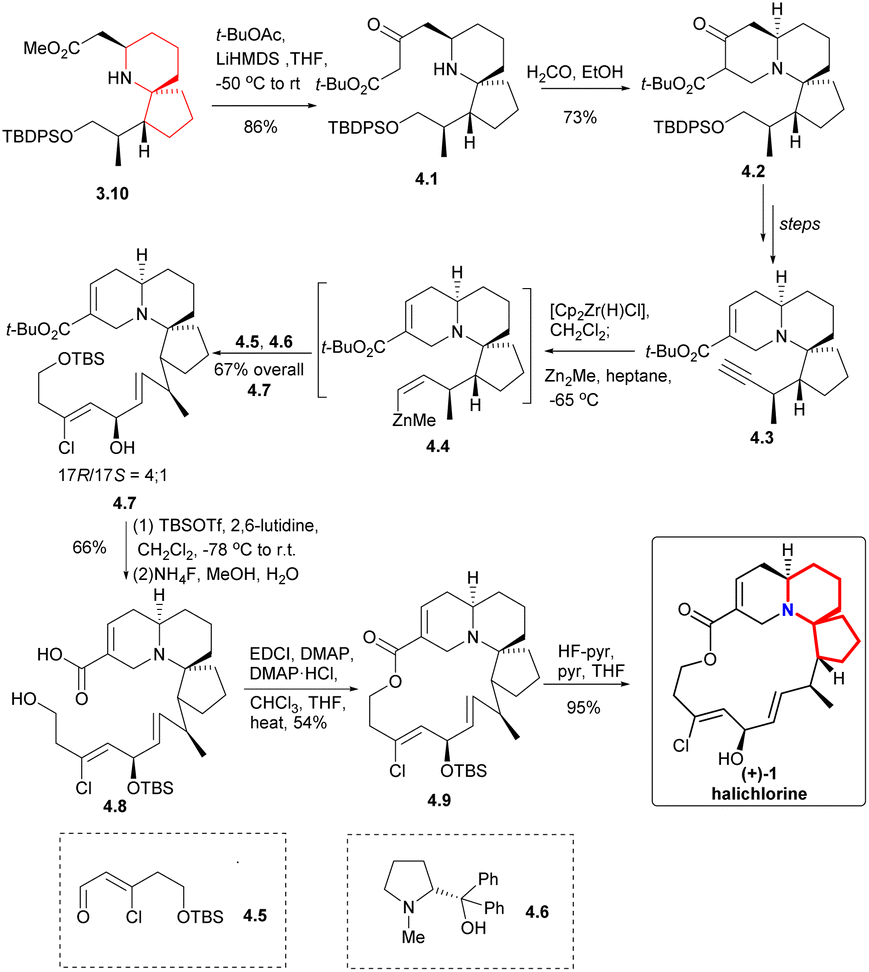
Claisen condensation of 3.10 with t-BuOCOMe produced the β-keto ester 4.1 (Scheme 4), which was then subjected to Mannich reaction with formaldehyde to construct the A ring to obtain the compound 4.2. This process yielded the desired products as a mixture of diastereomer and tautomer. The compound 4.3 was obtained through multiple steps. The terminal alkyne was transformed by zirconation into organozirconium compounds, which were metallized with dimethylzinc to give compounds 4.4. The resulting zinc substance was successfully coupled with aldehyde 4.5, and the reaction was carried out in the presence of optical pure amino alcohol 4.6 (ref. 10) to obtain a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the required 17R epimer 4.7 and the corresponding 17S epimer (17R/17S = 4/1). Conversion of the tert-butyl ester was accomplished by the action of TBSOTf, which also protected the secondary alcohol.11 Then the TBS group was removed by NH4F in aqueous MeOH solution while leaving the protected secondary alcohol intact to obtained 4.8. At the same time, Keck macrocyclic esterification to form 17-OTBS-halichlorine (4.9), at staged the 17R and 17S series (8:1 mixture) which were separated, followed by desilylation to give (+)-halichlorine.
:1 mixture of the required 17R epimer 4.7 and the corresponding 17S epimer (17R/17S = 4/1). Conversion of the tert-butyl ester was accomplished by the action of TBSOTf, which also protected the secondary alcohol.11 Then the TBS group was removed by NH4F in aqueous MeOH solution while leaving the protected secondary alcohol intact to obtained 4.8. At the same time, Keck macrocyclic esterification to form 17-OTBS-halichlorine (4.9), at staged the 17R and 17S series (8:1 mixture) which were separated, followed by desilylation to give (+)-halichlorine.
 | ||
| Scheme 4 Danishefsky's synthesis of (+)-halichlorine. | ||
 | ||
| Scheme 5 Zhao and Ding's formal synthesis of halichlorine. | ||
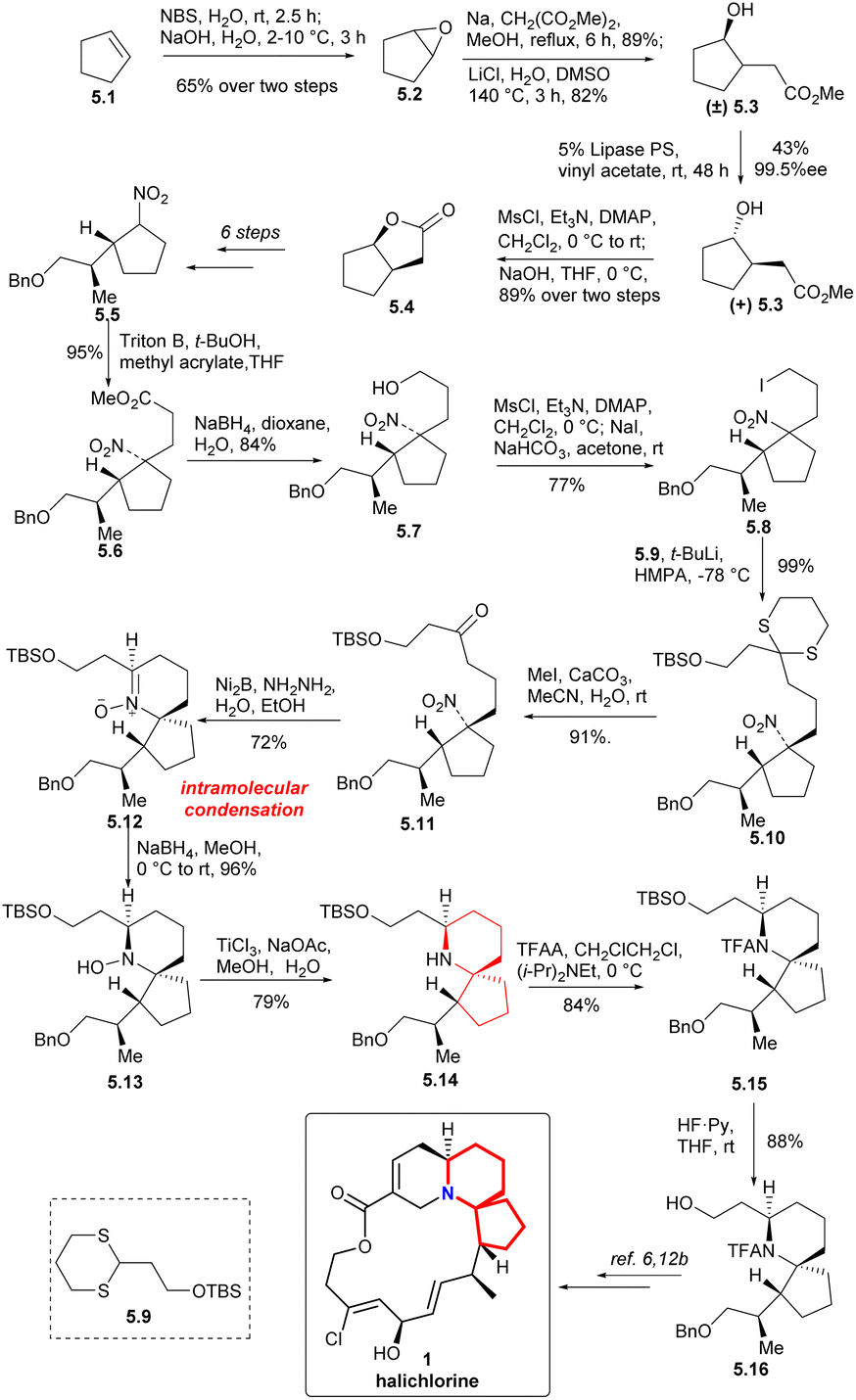
To form the 6-azaspiro[4.5]decane skeleton by the intramolecular condensation of δ-aminones, the tert-nitro in 5.11 must be reduced to an amino group. Ni2B-hydrazine combination was used to reduce the nitro group to obtain N-oxide 5.12.15 Reduction of the N-oxide 5.12 with NaBH4 (ref. 16 and 17b) followed by reduction of the hydroxyl group with TiCl3 (ref. 16c and 17) to obtain the 6-azaspiro[4.5]decane skeleton (5.12 → 5.13 → 5.14). Compound 5.16 was obtained by N-acylation with TFA and deprotection of TBS group (5.14 → 5.15 → 5.16). Since the intermediate 5.16 could be transformed to halichlorine,6,12b so the formal synthesis of halichlorine was achieved.
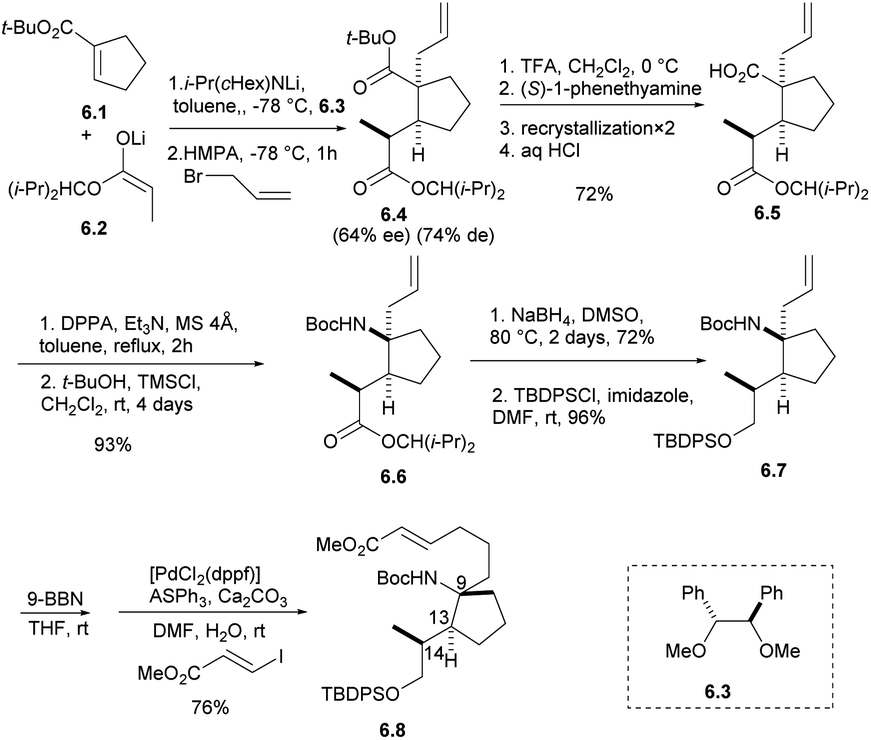
The synthetic strategy started with the addition of the lithium enolate 6.2 of propionate to 6.1 that would proceed by keeping the methyl group of 6.2 away from the cyclopentene moiety of 6.1 to give enolate intermediate, whose allylation was expected to proceed trans to the introduced propionate giving adduct 6.4.21 After the formation of carboxylic acid with TFA, carboxylic acid 6.5 was obtained by optical resolution of (S)-1-phenethylamine, and isocyanate was obtained by Curtius rearrangement with DPPA,22 which was inert to a nucleophilic addition of t-BuOH under refluxing conditions. The addition of TMSCl was effective to give Boc-amide 6.6 in 93% yield from 6.4 in two steps. The ester group was reduced by NaBH4 in DMSO and protected by TBDPSCl to obtain the intermediate 6.7.7,8 The stereochemistry of 6.7 was confirmed by spectroscopic data and the specific rotation of 6.8, which was prepared by hydroboration of 6.7 followed by the Suzuki coupling reaction, to be opposite that of natural halichlorin (Scheme 6).
 | ||
| Scheme 6 Tomioka's formal synthesis of (−)-halichlorine. | ||
Next, from the compound 6.7, the Tomioka's group synthesized the tricyclic core of halichlorine by a new strategy. Compound 6.7 hydroboride underwent a Suzuki coupling reaction with dienyl iodide 7.1, followed by the removal of the Boc group, gave the intramolecular Michael addition product 7.2 in 74% yield in two steps. The next stage was the formation of amides between the secondary amine and the ester moiety. After reduction of the double bond, the monosaponification product was obtained by KOH treatment and subsequent condensation of the amine with the carboxylic acid gave the lactam 7.4 with a tricyclic core of halichlorine (Scheme 7).
 | ||
| Scheme 7 Construction of the tricyclic core of halichlorine by Tomioka. | ||
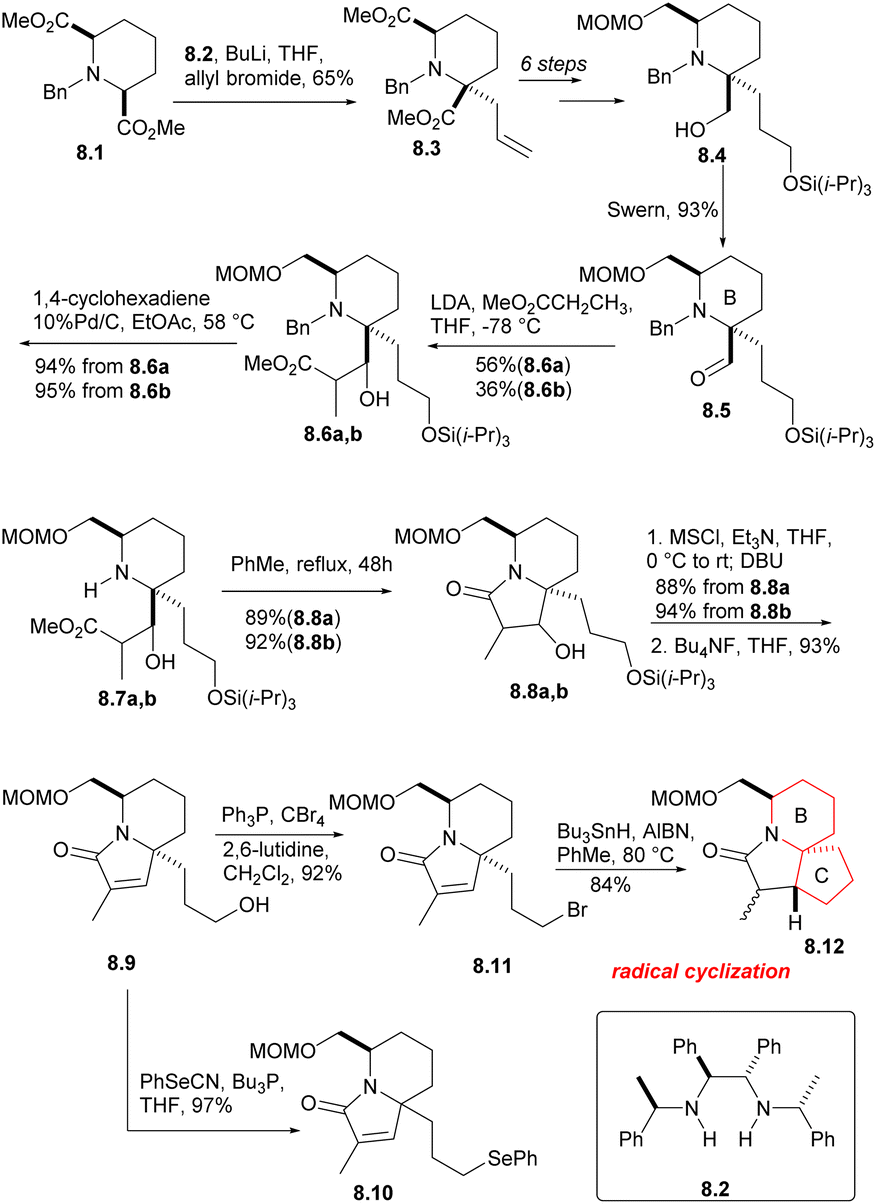
The cis-diester 8.1 was obtained from pyridine 2,6 dicarboxylic acid by esterification, hydrogenation and N-benzylation using the methods reported in the literature (Scheme 8). The symmetrical diester 8.1 was then subjected to asymmetric allylation (8.1 → 8.3), using the chiral base 8.2 (ref. 25 and 26) and allyl bromide. After a few simple steps, they got the compound 8.4, and Swern oxidation produced the expected aldehyde 8.5, which corresponds to the B ring of halichlorine and contains the required stereochemical and structural features. Aldol condensation of aldehyde 8.5 with methyl propionate yielded diastereomer alcohols of 8.6a and 8.6b, which were separable, but both can take subsequent reactions without further separation. The N-benzyl group was removed by heating with 10% Pd/C in the presence of 1,4-cyclohexadiene, and then the isomeric lactams were formed in the PhMe by heating the hydrogenolysis product (8.6a,b → 8.7a,b → 8.8a,b). The two lacamides 8.8a,b afforded the same unsaturated lactam 8.9 via mesylation, prolonged heating with DBU in THF an desilication. The hydroxyl of 8.9 was replaced by bromine under the conditions of Ph3P/CBr4, then radical cyclization produced the desired the tricyclic lactam 8.12. However, the stereochemical result at C (17) was unfavorable, the β-isomer required for the main isomer (4:1) was the secondary component. Although this isomer ratio could be almost reversed by using the t-BuOK/t-BuOH equilibrium, the differential isomers were too difficult to separate. As a result, the team decided to utilize the phenylseleno group 8.10.
 | ||
| Scheme 8 Synthesis of the 6-azaspiro[4.5]decane skeleton of halichlorine. | ||
The phenyl selenide 8.10 was subjected to ozonolysis and in situ reduction at a low temperature to give tricarbonyl selenide 9.1 (Scheme 9).27 After treatment with DBU, it underwent consecutive intramolecular aldol condensation and dehydration of hydroxylaldehydes (9.1 → 9.2), and was reduced to the corresponding α-hydroxy lactam under Luche condition,28 which reduced its reactivity. The hydroxyl group was then protected by acetylation to afford 9.3a,b. In the radical cyclization of 9.3a,b, the main products were the expected acetates 9.4a [AcO and C(17a)H syn] and 9.4b [AcO and C(17a)H anti], but the corresponding rearranged acetates29 9.5a, 9.5b and the enone 9.6 were also isolated. 9.4 were hydrolyzed at a yield of at least 95% to obtain the corresponding alcohol (MeONa, MeOH), and these can be converted to 9.6, either by mesylation and base treatment, or treated with o-(O2N)C6H4SeCN and Bu3P,30 then oxidized with 30% H2O2. 9.5 were hydrolyzed and converted to 9.6, too. At this stage, they needed to introduce a methyl at the final C(17) position, and the reaction yield of conjugated addition31 of Me2CuLi and unsaturated lactam was very high, providing the product saturated lactam 9.9.
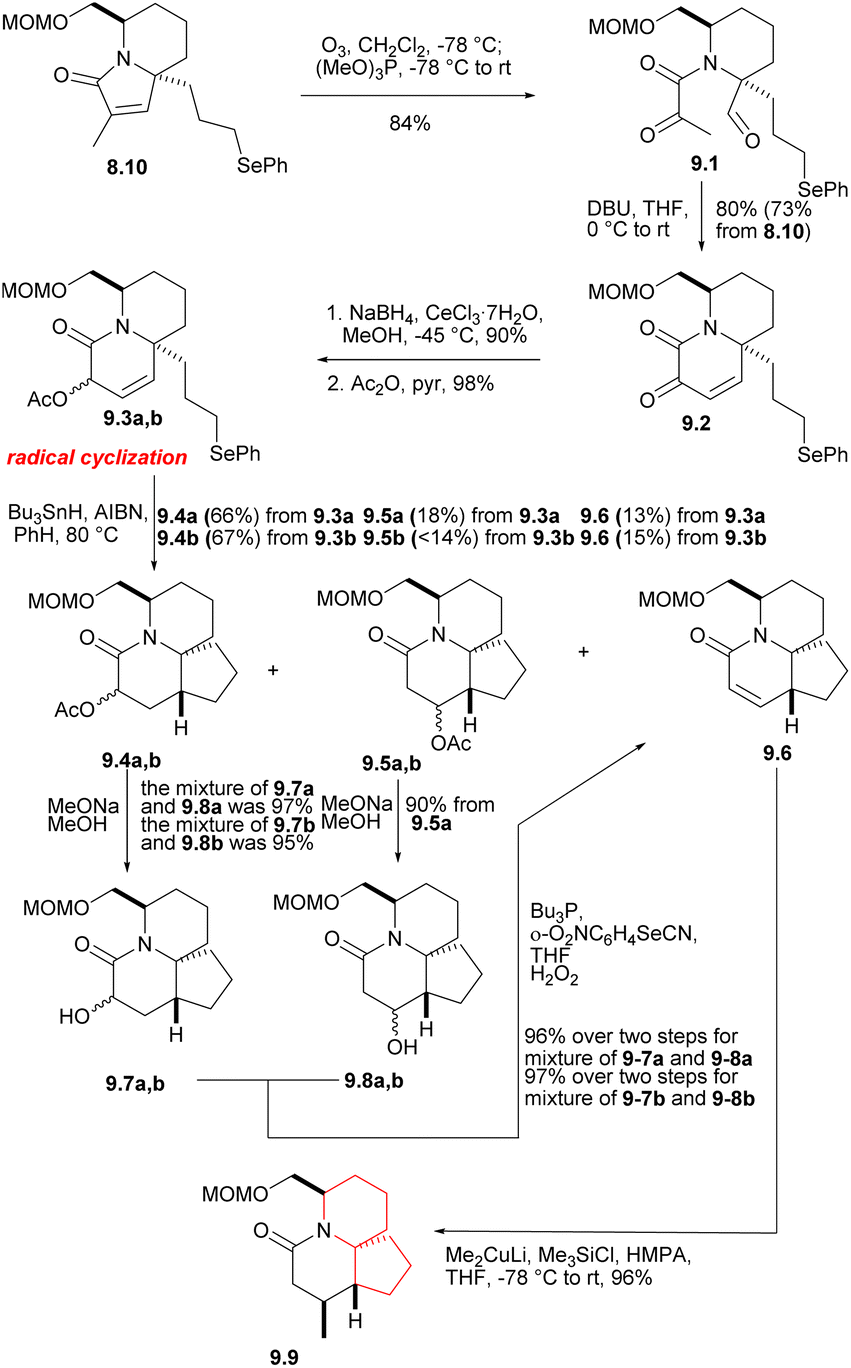 | ||
| Scheme 9 The exploration of ring C and introduction of the eventual C(17) methyl group. | ||
Removing the MOM group of 9.9 via Me3SiBr, n-PR4NRuO4, oxidizesing the primary alcohol to provide the corresponding aldehyde, Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(OMe) for Wittig olefination(9.9 → 10.1 → 10.2 → 10.3), and followed by acid hydrolysis afforded compound 10.4 (aldehyde 10.2 and the formation of enol ethers 10.3 did not cause any epimerization at C5). For the sake of constructing ring A, aldehyde 10.4 underwent a Baylis–Hillman reaction with acrylonitrile to provide the required alcohols at a high yield, and AcCl acetylation instantly converted these alcohols into the corresponding acetate 10.5a,b. With opening the lactam ring with Meerwein salt Me3O+BF4−, the resulting amines were subjected to spontaneous intramolecular conjugate displacement32 to unsaturated nitrile 10.7. At this point, the A, B, and C rings were constructed (Scheme 10).
CH(OMe) for Wittig olefination(9.9 → 10.1 → 10.2 → 10.3), and followed by acid hydrolysis afforded compound 10.4 (aldehyde 10.2 and the formation of enol ethers 10.3 did not cause any epimerization at C5). For the sake of constructing ring A, aldehyde 10.4 underwent a Baylis–Hillman reaction with acrylonitrile to provide the required alcohols at a high yield, and AcCl acetylation instantly converted these alcohols into the corresponding acetate 10.5a,b. With opening the lactam ring with Meerwein salt Me3O+BF4−, the resulting amines were subjected to spontaneous intramolecular conjugate displacement32 to unsaturated nitrile 10.7. At this point, the A, B, and C rings were constructed (Scheme 10).
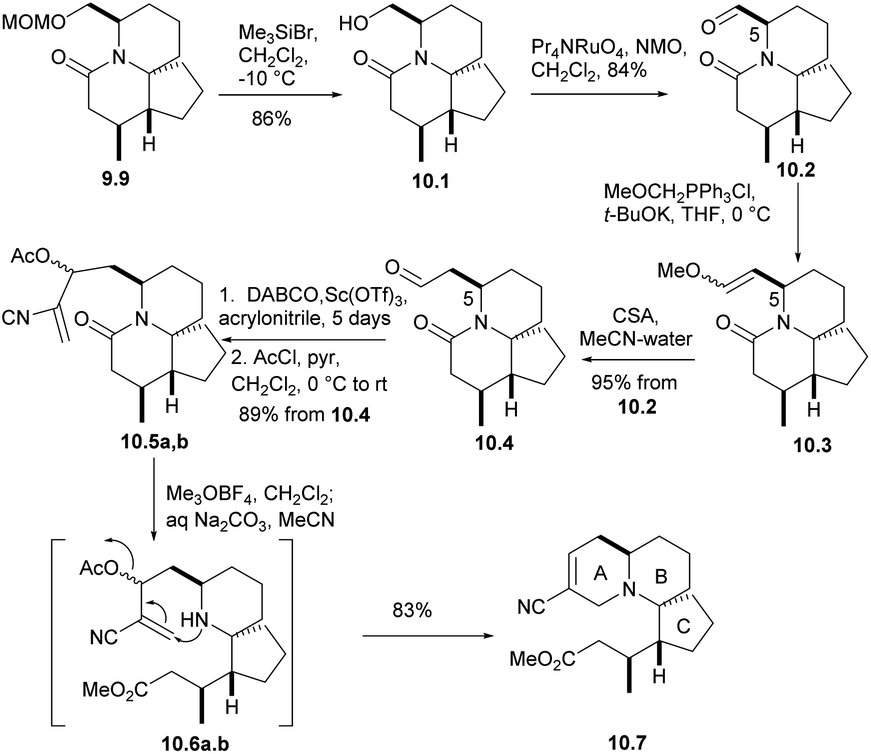 | ||
| Scheme 10 Construction of ring A. | ||
In the presence of DIBAL-H, nitrile can be reduced to imine, which was then hydrolyzed and reduced to eventually yield alcohol 11.1 (Scheme 11). The processes of multiple steps could afford the compound aldehydes 11.2. By now the research group hoped to form a carboanion on C15 of halichlorine, which needed to produce selenium-stabilized carbanion and use selenium unit to make C15–C16 double bond. So as to, the stannyl alcohols mixture obtained by treating aldehydes with Bu3SnLi was immediately converted to the corresponding selenide 11.3. When the compound was treated with BuLi (to produce the desired selenium-stabilized carbanion33 by preferential C–Sn heterolysis) and then treated with the known compound 11.4,6,34 a mixture of β-hydroxyselenides 11.5 was obtained. The selenium was removed to liberate the double bond to give 11.6 during oxidation. Finally, the hydroxyl group was protected by silylation to obtain 11.7. By multi-step simple reactions, the hydroxyl and carboxyl groups of the compound 11.8 was cyclized by Keck macrocyclization and deprotected to form (±) halichlorine.
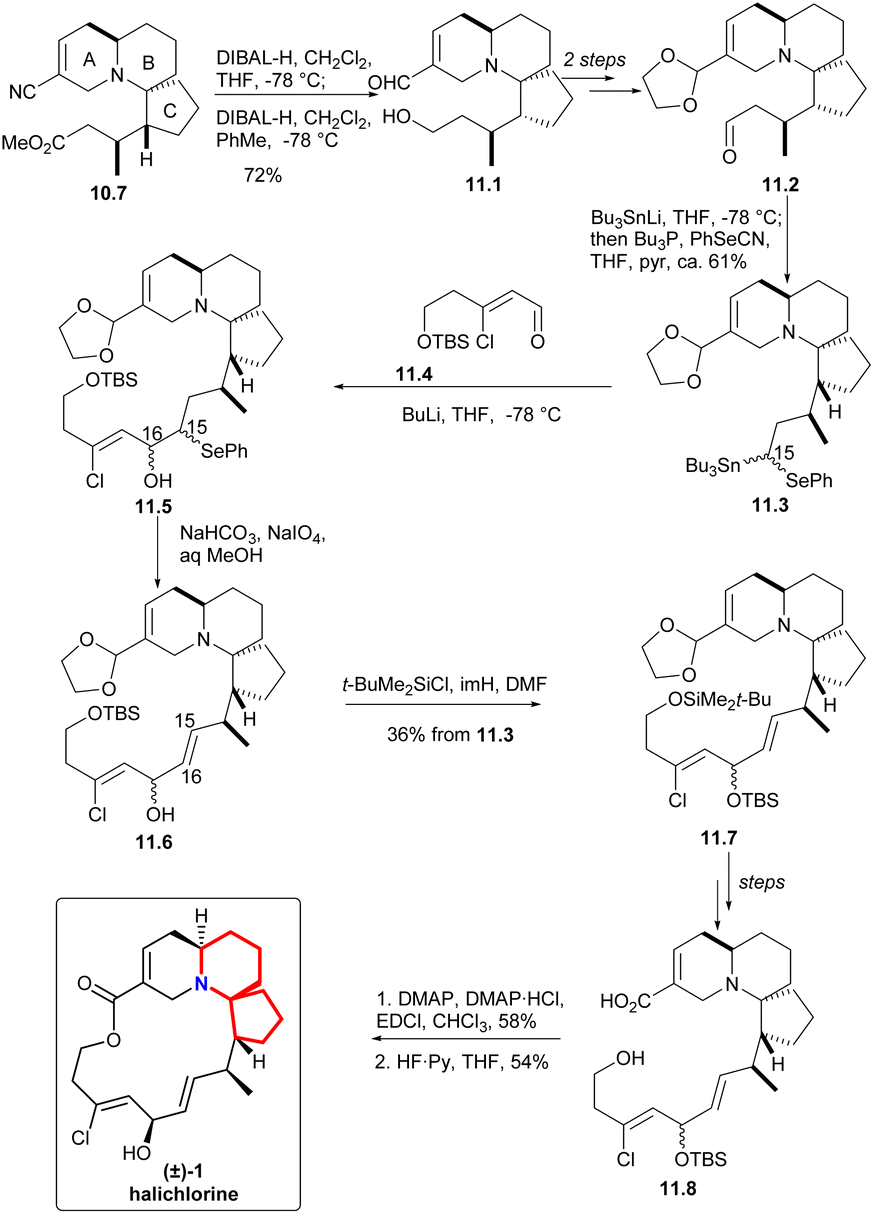 | ||
| Scheme 11 Clive's total synthesis of (±)-halichlorine. | ||
Iodide 12.1 was treated with lithium divinyl cuprate to produce 12.2 (Scheme 12). The terminal double bond of 12.2 performed ozone REDOX and reacted with compound 12.4 (ref. 35) under alkaline conditions to obtain the corresponding alcohols 12.5, which were oxidized to ketone 12.6. After treatment with Cs2CO3 in MeOH, ketone was converted into dihydropyridinone 12.7 at 80% yield. Each of the steps from L-serine to 12.7 occurred without loss of stereochemical integrity. After several reactions, alcohol 12.9 was obtained.
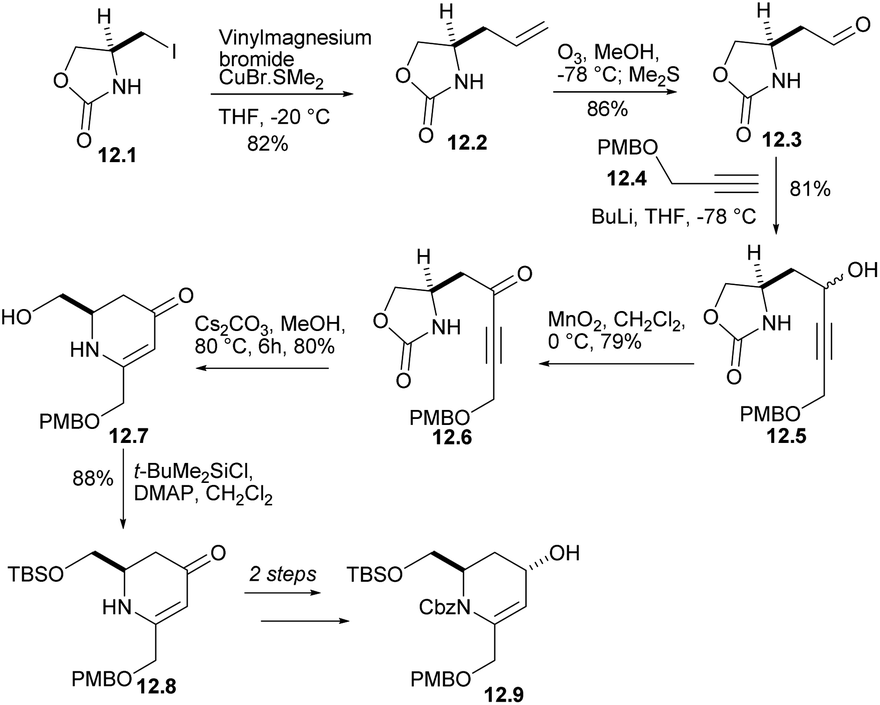 | ||
| Scheme 12 Synthesis of the precursor to the rearrangement substrate 12.9. | ||
The crucial claisen rearrangement reaction occurred when alcohol 12.9 was heated in butyl vinyl ether in the presence of Hg(OAc)2 and Et3N, which was finally converted to 13.1 with 79% yield (Scheme 13), and then Wittig enylation reaction occurred, and the enol ether 13.2 was converted into aldehyde 13.3. This was reduced and silylated (13.3 → 13.4 →13.5). The removal and oxidation of the PMB group gave aldehyde 13.6 and converted it to 13.7. Condensation of the enolate derived from EtCO2Me then gave conpound 13.8 and then reduced double bond. When these were heated for 35 h in boiling toluene, the lactam 13.10 were formed in good yield. Mesylation and heating with DBU in THF then converted compound 13.11. Next, the silicon masking the primary hydroxyl was removed with AcCl and the resulting alcohol 13.12 was protected as its MOM ether 13.13. The formation of optically pure 13.13 constitutes a formal synthesis, based on racemic route, of (+)-halichlorine.
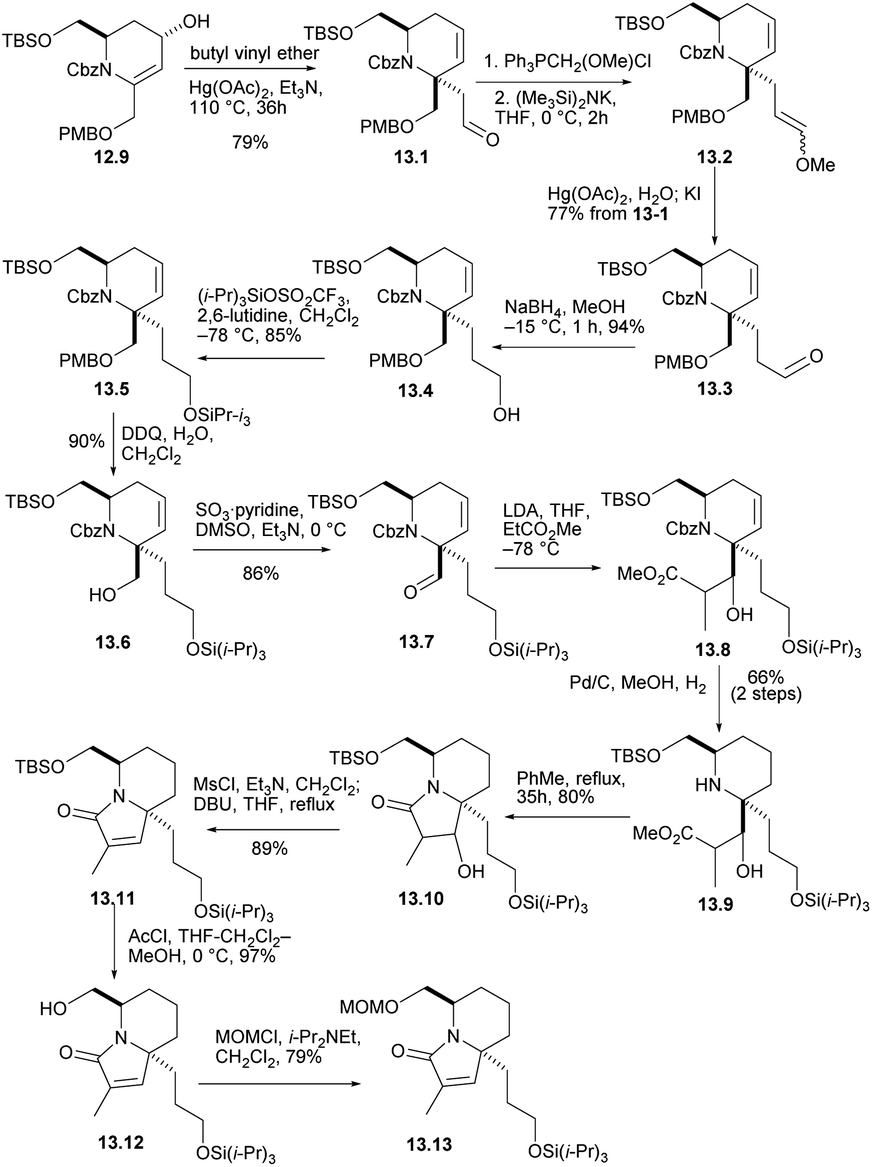 | ||
| Scheme 13 Preparation of optically pure intermediate. | ||
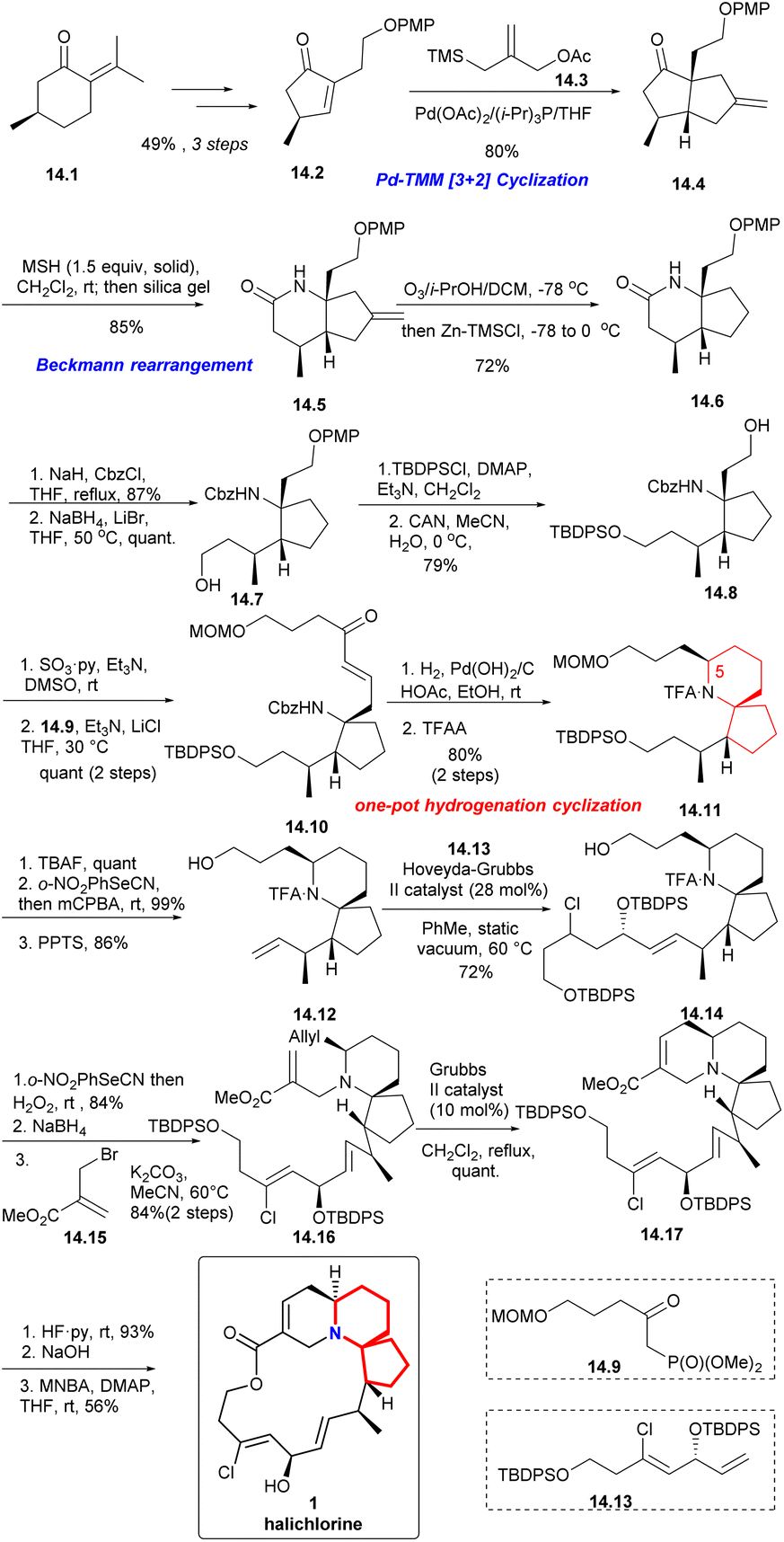 | ||
| Scheme 14 Uemura's enantioselective total synthesis of halichlorine. | ||
With (R)-pulegone 14.1 as the starting material, enone 14.2 obtained through 3 steps in 49% yield. Compound 14.2 reacted with the TMM38 precursor 14.3 highly selectively and only the diastereomer 14.4 was obtained in 80% yield. After Beckmann rearrangement39 and a one-pot ozonolysis-Clemmensen reduction process,40 14.6 could be obtained in a high yield. Protection of 14.6 with Cbz and reduction with LiBH4 led to compound 14.7. Then, protection with TBDPS, removal of PMP via CAN, Parikh–Doering oxidation and the H–W–E reaction with 14.9 afforded spiro-cyclization precursor 14.10 as the single trans isomer (14.7 → 14.8 → 14.10). Next, using a one-pot hydrogenation-cyclization41 efficiently constructed the 6-azaspiro[4.5]decane skeleton with the desired C5 configuration and then TFA protected the amino group to obtained 14.11 through 2 steps in 80% yield. Selective removal of TBS, Grieco elimination30 and removal of MOM led to terminal olefin (14.11 → 14.12). The cross metathesis of 14.12 then proceeded smoothly with the lower-chain unit 14.13 under static vacuum conditions to afford compound 14.14 in 72% yield. Then, 14.14 via 3 steps including Grieco elimination,30 deprotection of TFA and alkylation with bromo-substituted upper-chain unit 14.15 gave compound 14.16 in 84% yield.42 Six-membered RCM of 14.16 under the conditions reported by Kibayashi et al.12b afforded the tricyclic compound 14.17 quantitative. Deprotection of the TBDPS groups, basic hydrolysis of the methyl ester and Shiina macrolactonization in overall 56% yield completed the total synthesis of (+)-halichlorine (14.17 → 1).
Synthesis began with the Grignard addition reaction of 15.2 (Scheme 15) and racemic lactone 15.1.45 After treatment with hydroxylamine hydrochloride, oxime 15.3 was obtained by two-step separation. Iridium-catalyzed chemoselective N-alkylation and thermal 1,3-dipolar cycloaddition reactions provided compound 15.4 with four stereogenic centers with a yield of 42%, 98% ee and 18:1 d.r. on 2.5 mmol scale. The compound 15.4 was hydroborated and oxidized to the corresponding primary alcohol 15.5. The sequence of esterification by Ley oxidation, Pinnick oxidation, and Steglich provided a pathway for ester 15.6 to act as a single diastereomer. Finally, the silyl ether was cracked to obtain alcohol 15.7 with 91% yield,46 completing the synthesis of (+)-halichlorine.
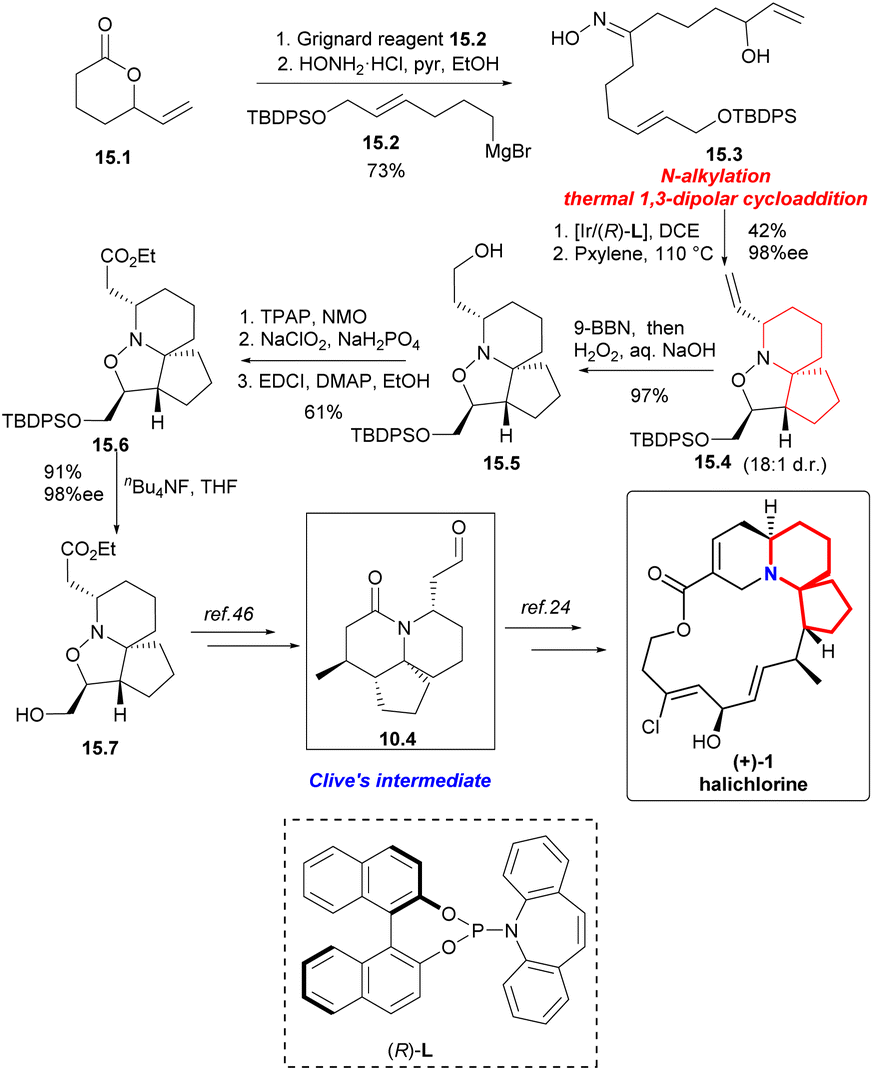 | ||
| Scheme 15 Carreira's formal synthesis of (+)-halichlorine. | ||
3. Absolute configuration determination and asymmetric synthesis of pinnaic acid
3.1 Absolute configuration determination of pinnaic acid
In 1996, Uemura's Laboratory isolated pinnaic acid from the Okinawan bishell Pinna muricata, which was structurally closely related to halichlorine. Since only 1 mg of pinnaic acid was isolated from 3000 individual specimens of Okinawa bishell (Pinna muricata), it was not possible to correctly establish the stereochemistry of its structure in this case. Until 2001, Danishefsky's Laboratory determined the absolute configuration of pinnaic acid through synthetic method and clarified the stereochemistry of C14 and C17 chiral centers.47,48 The absolute configuration of the chiral center at C17 was established by degradation studies (Scheme 16). After acetylation of the key intermediate 16.1, the desired fragment was extracted through ozonolysis, resulting in aldehyde 16.2. Reduction and final acetylation of 16.2 gave the known compound 16.3 with the absolute configuration determined at C17. This correlation confirmed the absolute configuration of pinnaic acid and thus established the 17R configuration, as depicted in (and 2). The confirmation of the C14 configuration would be seen in subsequent Danishefsky's route.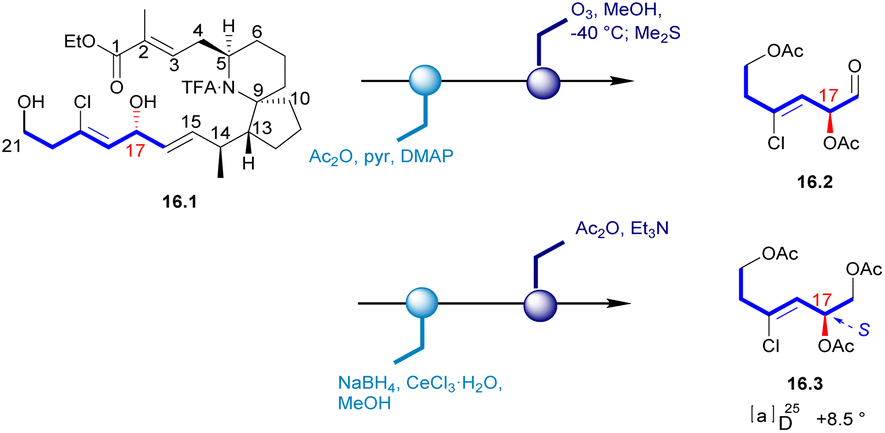 | ||
| Scheme 16 Confirmation of C17 stereochemistry of pinnaic acid. | ||
3.2 Asymmetric synthesis of pinnaic acid
The synthesis of pinnaic acid was initially accomplished by Danishefsky in 2001, and this achievement held reference significance for subsequent researchers working on synthesis. Other research groups have also successfully synthesized the pinnaic acid by constructing the 6-azaspiro[4.5]decane skeleton as the key steps through different strategies (Scheme 17). | ||
| Scheme 17 Asymmetric synthesis of pinnaic acid via the 6-azaspiro[4.5]decane skeleton. | ||
Compound 3.7 was obtained by a multi-step reaction starting with ‘Meyers lactam’ 3.3, which was then borohydrided again, followed by coupling with iodide 18.1 in the presence of palladium(II) catalyst to obtain 18.2 (Scheme 18). Removal of the Boc group and treatment with DBU led to clean and stereoselective cyclization while retaining the E geometry of the resulting olefin 18.3. After three steps, the compound 18.4 C(14) isomer was synthesized. The aldehyde 18.4 underwent a H–W–E reaction using known phosphonate 18.5 (ref. 34) to generate α, β -unsaturated ketone 18.6. The reaction was incomplete and it was challenging to separate the desired product from the initial aldehyde 18.4. When the mixture of 18.4 and 18.6 was reduced by Alpine hydride,49 the resulting alcohol 18.7 exhibited the desired C17 configuration, irrespective of whether chiral R or S-hydride was used. Desilylation (18.7 → 18.8) and few steps reaction led to natural pinnaic acid.
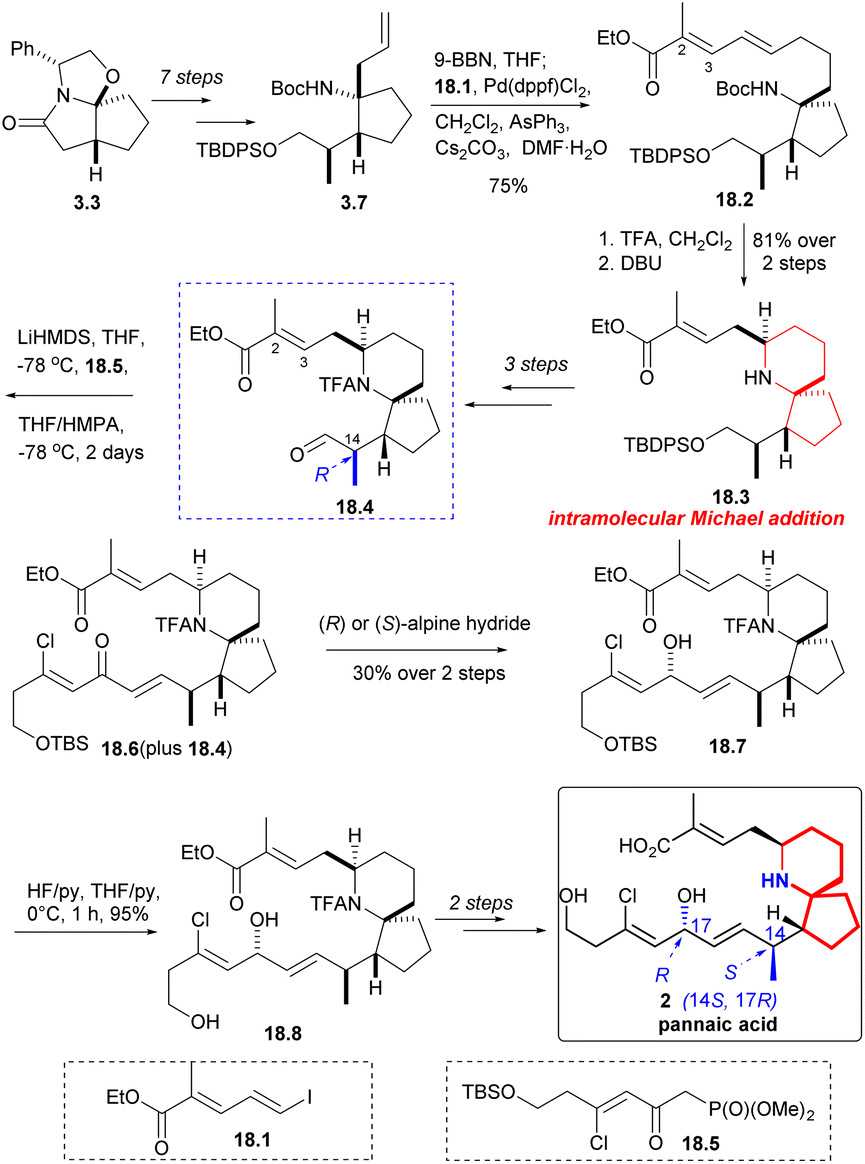 | ||
| Scheme 18 Danishefsky's synthesis of pinnaic acid and confirmation of C14 stereochemistry. | ||
During the course of the study, it was discovered that ketone 18.6 predominantly yielded alcohol 19.1 (Scheme 19) and its C17 epimers, alcohol 19.2, when subjected to Luche reduction conditions. Desilication, N-acetyl cleavage, and final ester hydrolysis yielded the non-natural pinnaic acid (19.2 → 19.3 → 19.4 → 19.5), whose 1HNMR spectrum was different from that of the natural compound.
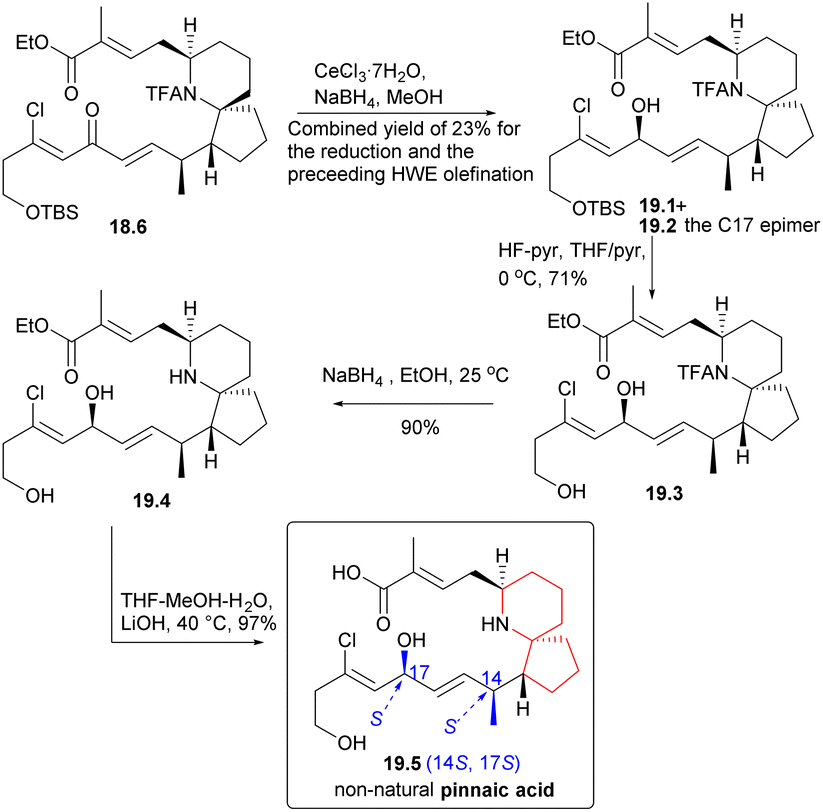 | ||
| Scheme 19 Danishefsky's synthesis of non-natural pinnaic acid. | ||
Next, Danishefsky intended to prepare the C14 epimer of aldehyde 18.4. The route began with intermediate 3.6, which was deprotonated using LiHMDS and then re-deprotonated using BHT. This process led to a significant inversion of stereochemistry at the final C(14) position (3.6 → 20.1). The subsequent reactions followed the same procedure as described earlier, and the precursor compound 20.2 of the azabicyclic core was synthesized through a series of straightforward reactions. When treated with DBU, Micheal addition occurred to obtain the 6-azaspiro[4.5]decane skeleton 20.3, and aldehyde 20.4 was obtained after several steps reactions. The aldehyde 20.4 and phosphonate 18.5 underwent H–W–E reaction in the presence of lithium anion (20.4 → 20.5). Hydride reduction was employed once more, but this time without selectivity, resulting in a mixture of 20.6 and exomer alcohol in a ratio of 1.7:1. The diastereomers were subsequently isolated and transformed into non-native pinnaic acids 20.7 and 20.8 (Scheme 20).
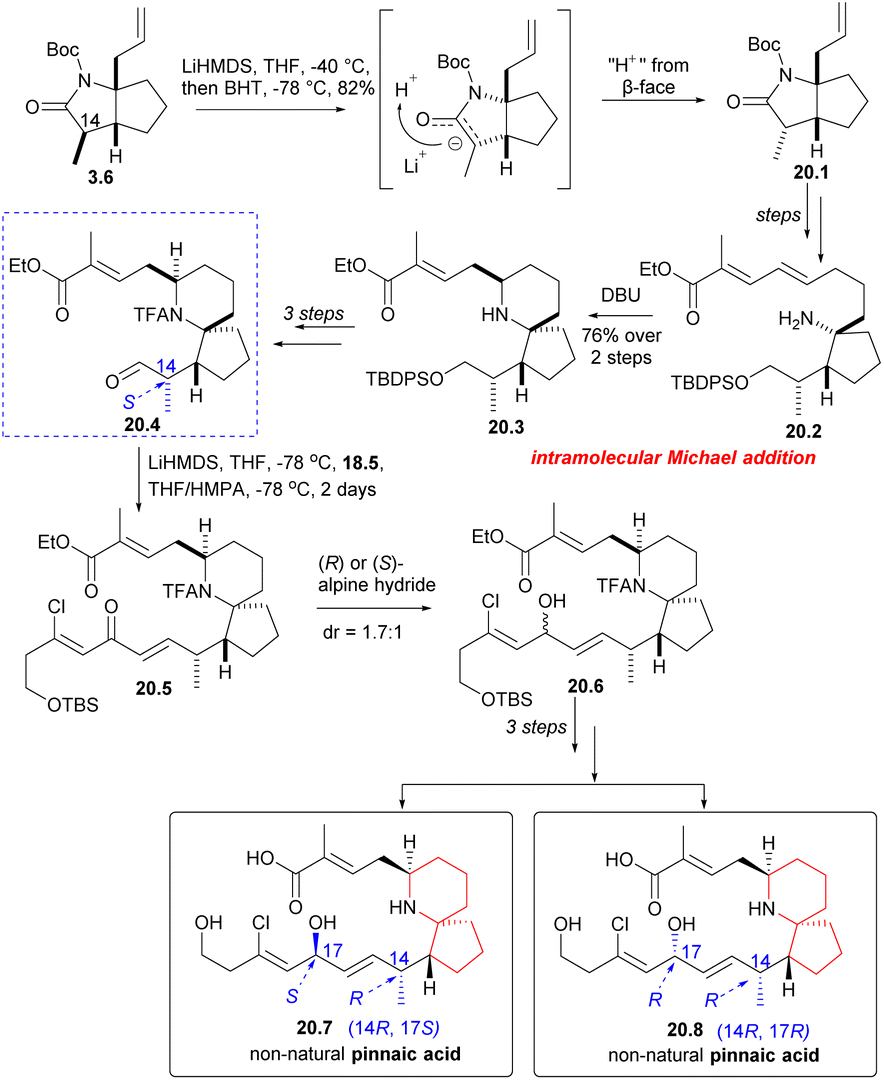 | ||
| Scheme 20 Danishefsky's synthesis of non-natural pinnaic acids. | ||
After obtaining four diastereomers (2, 19.5, 20.7, 20.8), only 2 exhibited spectra that matched the spectral data of native pinnaic acid. The final task was to confirm the absolute configuration of C17, which was accomplished through degradation studies (see: Scheme 16).
Compound 21.1 was obtained oxidation of PCC (Scheme 21). Based on 21.1, they extended its side chain by alkenization. After H–W–E enylation, they constructed (E)-C2C3, which was labelled as 21.2. and then alcohols were obtained by debenzoylation. The key intermediate 21.3 in Danishefsky route was obtained successfully.
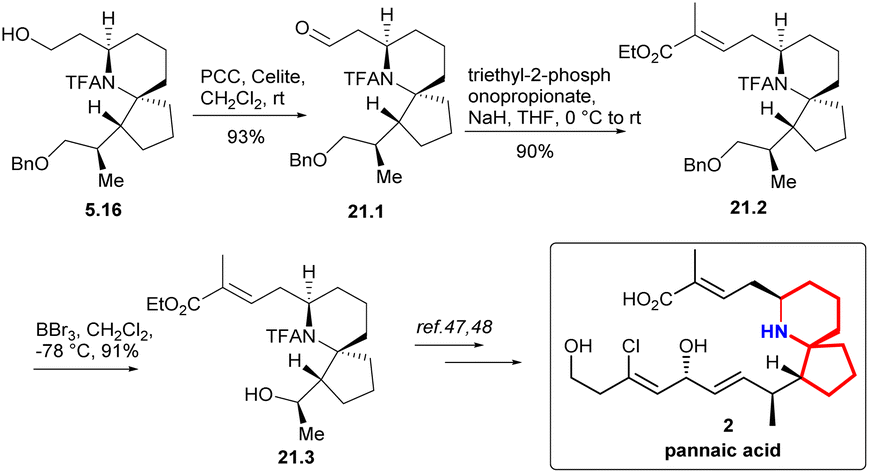 | ||
| Scheme 21 Zhao and Ding's formal synthesis of pinnaic acid. | ||
The work of Zhao’ s group further enriches the research on the synthesis of halichlorine and pinnaic acid. They presented a novel and efficient method for the highly stereoselective synthesis of azospira rings using mild conditions.
The BINAP–Ru complex has been found to be an efficient catalyst for the asymmetric hydrogenation of β-keto ester.51 In this synthesis, the catalyst was utilized to hydrogenate γ-ketone ester 22.1 (Scheme 22), resulting in the successful production of bicyclolactone 22.2 with satisfactory results. The asymmetric hydrogenation of racemic γ-keto ester 22.1 was conducted using [(R)-BINAP-RuCl2](DMF)n] as a catalyst. The desired (1R,5R)-lactone 22.2 was obtained in gram scale with a yield of 61% and an enantiomeric excess (ee) of 90%. The yield of the required lactone 22.2 was found to be higher than 50%, suggesting some deracemization of the α-position of the carbonyl group occurred under the reaction conditions. Asymmetric methylation of dicyclolactone 22.2 was carried out at −78 °C using LDA as a non-nucleophilic base, resulting in the formation of 22.3. LAH was used for reduction, followed by position-selective protection of primary alcohols with benzyl bromide. The secondary alcohols were then oxidized in CH2Cl2 using PCC to obtain the desired cyclopentanone 22.5. Hydroxylamine was condensed with cyclopentanone and then oxidized using m-CPBA to give nitrocyclopentane 22.6 with a yield of 64% in two steps.52 Compound 22.6 was a pair of diastereoisomers without further separation. The Michael addition reaction of nitrocyclopentane with methyl acrylate provided the foundation for the construction of the spirocenter. The desired nitroester 22.7 was obtained with 97% yield as a single diastereoisomer,12a which can be explained by assuming that the acrylate approximated the less hindered face of the nitrocyclopentane. The nitroester was reduced with LiBH4, followed by mesylation and iodination to obtaine iodide 22.8. Subsequently, dithiane 22.9 was subjected to metallization using t-BuLi,14 and then alkylated with iodide 22.8.12a,53 Dithiane was then deprotected in the presence of I2 and NaHCO3 in acetone to obtaine ketone 22.10, which served as a crucial precursor for the cyclization of the piperidine ring. The nitro group in 22.10 was reduced to an amino group using RANEY® Ni and H2 at 1 atmosphere pressure, and then the cycloimine compound 22.11 was formed through a simple nucleophilic reaction involving its carbonyl group. Nitrogen spiral ring 22.12 was successfully constructed by reducing cycloimine compound with NaBH4 in mixed solvent.
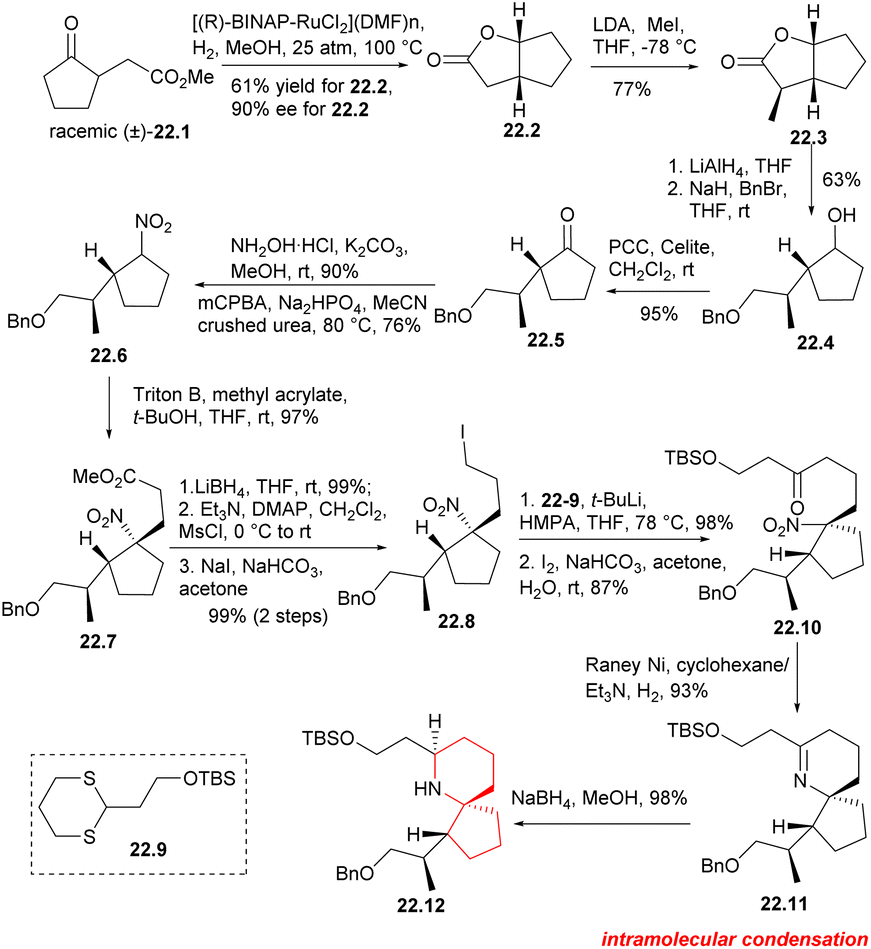 | ||
| Scheme 22 Construction of the azaspirocyclic core. | ||
The secondary amino group was protected by TFAA,47,48 the TBS group was deprotected to obtain 23.1 (Scheme 23), and the aldehyde was oxidized with DMP and converted into compound 23.2 through H–W–E reaction. Subsequently, benzyl was removed using BBr3 at −78 °C, the resulting compound was then subjected to DMP oxidation, resulting in the desired aldehyde 23.4.and DMP oxidation yielded the desired aldehyde 23.4. A H–W–E reaction was performed between aldehyde 23.4 and Weinreb's phosphonate34 23.5 to produce trace products. The desired dienone 23.7 was obtained at a moderate yield (60%) by heating aldehyde 23.4 and phosphorane 23.6 in benzene. Under the Luche reduction condition,28 the dienone was converted in a 3:1 mixture favoring the desired diastereomer 23.8. The TBS group was deprotected by HF-Py complex to produce the corresponding alcohol. To obtain lithium carboxylate salt of pinnaic acid, the trifluoroacetamide underwent reductive cleavage, followed by hydrolysis of the ethyl ester in the presence of LiOH. Pinnaic acid was synthesized enantioselectively by dissolving salt in an aqueous buffer solution of pH = 7, extracting it with 1-butanol and purifying it by liquid chromatography.
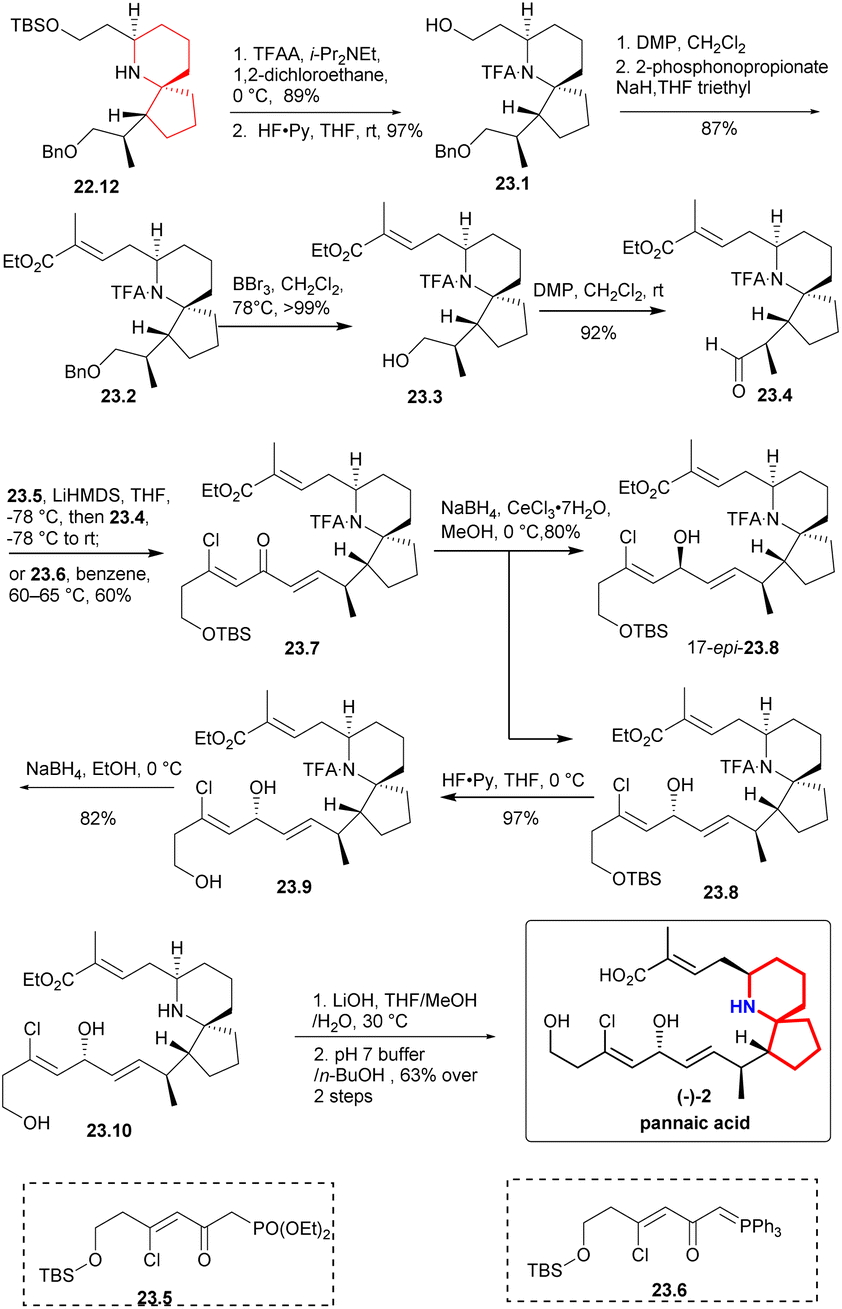 | ||
| Scheme 23 Zhao's total synthesis of pinnaic acid. | ||
The highlight of this study was the use of a Ru complex as a catalyst for the asymmetric hydrogenation reaction, and facilitated the formation of the nitrogen azaspirocyclic core through multiple key reactions, leading to a shorter and more efficient synthesis route for pinnaic acid.
Under the condition of (COCl)2, the known acid 24.1 was converted into the corresponding acyl halide, and then ammonolysis occurred under basic conditions to obtain chiral amides 24.3 (Scheme 24). The asymmetric [2 + 2] cycloaddition of compound 24.3 using Ghosez's protocol,55 and the obtained intermediate iminium ions were alkaline hydrolyzed to cyclobutanone 24.5, which continued to undergo cross-metathesis,56 NaN3 displacement gave an interconverting mixture of allylic azides 24.6a–d. The Schmidt reaction of isomeric allyl azides, which was treated with TiCl4, was the key reaction, and the mixture of lactam 24.7a and 24.7b can be separated in a ca. 10:1 ratio. The stereochemistry of the reaction was controlled only by the position of the vinyl in the product. Finally, hydroboration/oxidation gave Kibayashi's pinnaic acid intermediate 24.8 in 83% yield.
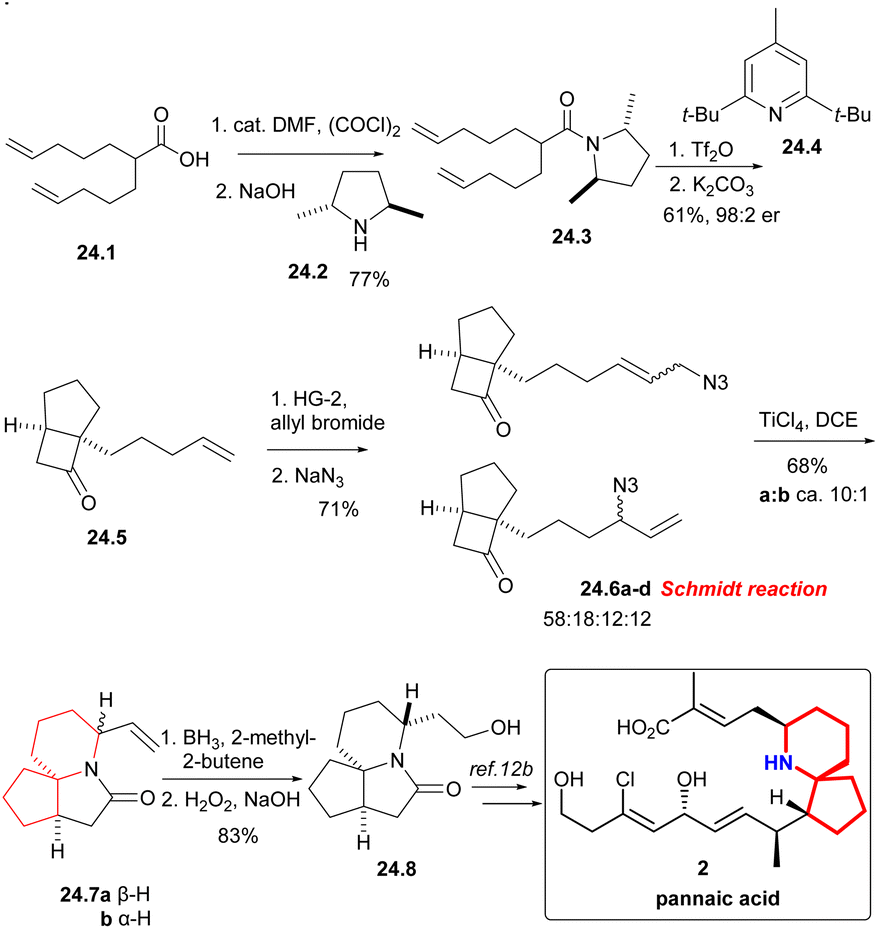 | ||
| Scheme 24 Aubé’s formal synthesis of pinnaic acid. | ||
Aldehydes were formed by Parkin–Doering oxidation (SO3·Py, DMSO) from the key intermediate 14.8 and reacted with phosphonate 25.1 (Horner–Wadsworth–Emmons reaction). The product 25.2 was dominated by E isomer. This was followed by a series of hydrogenation–cyclization to construct a piperidine ring 25.3 with the desired C5 configuration.41 It consisted of four successive one-pot transformations: reduction of alkene double bonds; Cbz protective group was removed; formation of intramolecular cyclic imine; stereoselective reduction of imine/enamine intermediates. After multiple steps of reaction, 25.4 was obtained. By combining compound 25.5 (Scheme 25) with 10 mol% Hoveyda–Grubbs second-generation catalyst 25.6 (ref. 57) under reflux conditions, they obtained compound 25.7 with a yield of 74% during the above metathesis. TBDPS deprotection and Grieco elimination30 were employed to produce terminal olefin 25.8. This strategy utilized olefin cross-metathesis to establish the C17 center (90% ee) of segment 25.9 before it was incorporated into the spirocyclic core 25.8. Compound 25.9, which underwent a six-step transformation, was synthesized from but-3-yn-1-ol. The next step involved repeating the olefin cross-metathesis process. However, in the two precursors 25.8 and 25.9, there were four different types of carbon–carbon double bonds. It was observed that the two terminal double bonds were more reactive, possibly due to spatial factors. This reactivity led to the formation of a single trans isomer 25.11. The next step performed the olefin cross-metathesis again.58 However, in the two precursors 25.8 and 25.9, there were four types of carbon–carbon double bonds. It was observed that the two terminal double bonds were more reactive, possibly due to spatial factors. This reactivity led to the formation of a single trans isomer 25.11. Compound 25.11 was successively deprotected by the two silyl protecting groups, the TFA amide, and the ethyl ester using common methods48 to obtain chiral pinnaic acid (25.12) in the form of sodium salt.
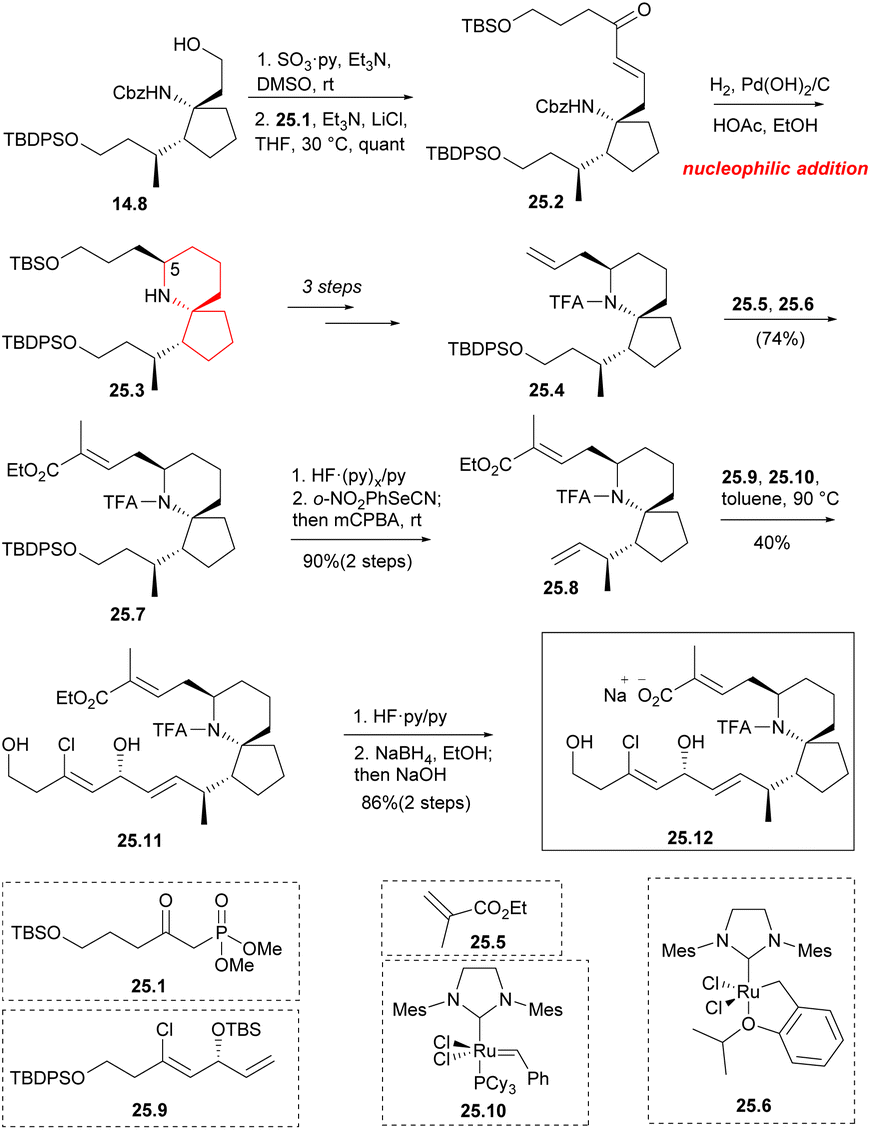 | ||
| Scheme 25 Uemura's total synthesis of (−)-pinnaic acid. | ||
4. Racemic synthesis of halichlorine and pinnaic acid
In addition to Clive group's review,3 we also summarized the racemization work of halichlorine and pinnaic acid that was not included.4.1 Martin's formal synthesis of pinnaic acid and halichlorine
Martin's group synthesized the known intermediates 26.9 and 27.5 to achieve the formal synthesis of halichlorine and pinnaic acid.59 This route effectively installed the upper chain through the key reaction: cross-olefin-metathesis,60 which illustrated the utility of selective olefin cross metathesis methodologies for the elaboration of advanced synthetic intermediates in complex molecule synthesis.The synthesis commenced with the compound 26.1 (Scheme 26), which was converted to alcohol 26.2 by literature.61 The resulting primary alcohol 26.2 was protected as its tert-butyl-diphenylsilyl derivative 26.3. Oxidation with Jones' reagent then generated carboxylic acid 26.4 as expected, which was subjected to a Curtius rearrangement with DPPA22 and t-BuOH to afford 26.5. At this point cross metathesis of 26.5 with the ester 26.6, using the Grubbs II catalyst 25.10,58 produced the E-olefin 26.7, which upon deprotection of the amino function with TFA and cyclization of the intermediate amino dienoate via intramolecular 1,6-conjugate addition to afford piperidine 26.8 in 34% yield for the three steps. The nitrogen was protected as its trifluoroacetate 26.9, an intermediate in the Danishefsky's synthesis of pinnaic acid.48
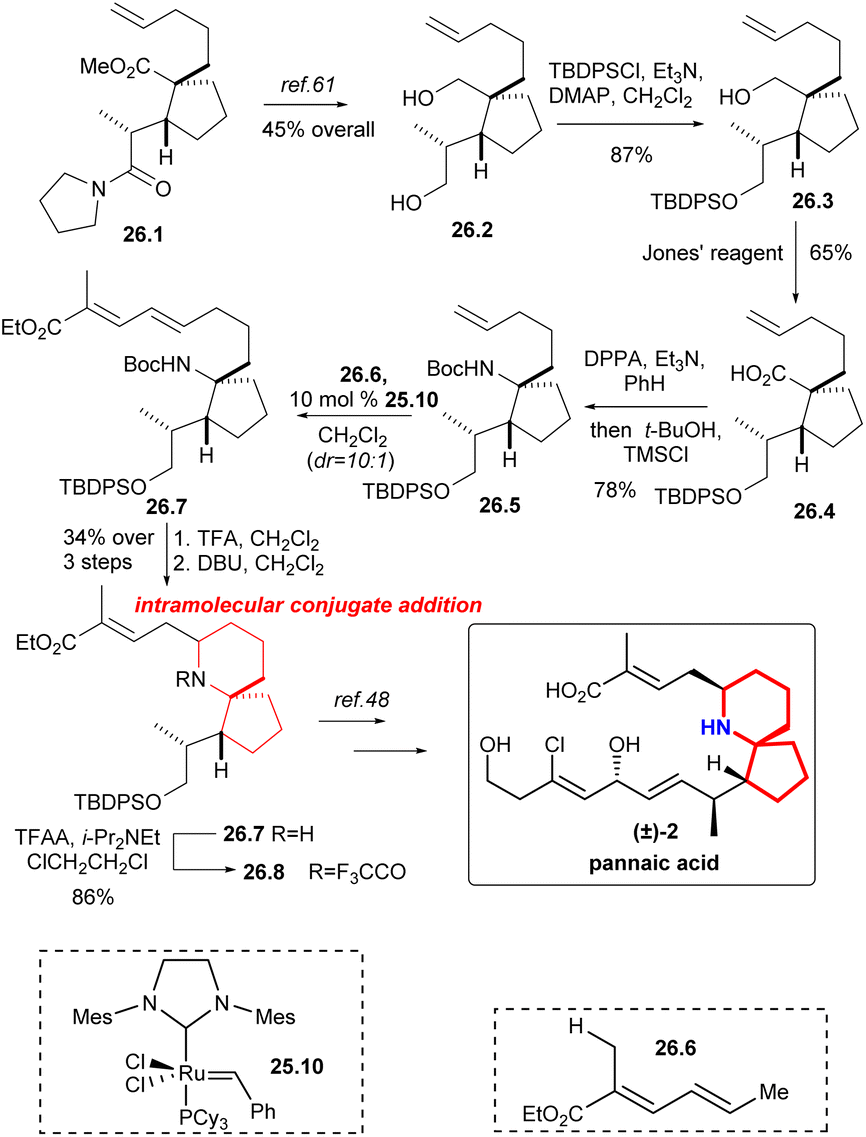 | ||
| Scheme 26 Martin's formal synthesis of (±)-pinnaic acid. | ||
Compound 27.1 (Scheme 27) was obtained in 89% yield with excellent diastereoselectivity by compound 26.5 and 10 mol% Grubbs II catalyst 25.10.58 Carbamate cleavage facilitated spontaneous Michael addition affording spirocycle 27.2. The reaction of ethyl propiolate in THF with DIBAL-H/NMO complex provided the vinylaluminum reagent.62 Aldehyde 27.2 was added to the vinylaluminum reagent to gain compound 27.3 as a mixture of diastereomers (dr = 2:1). Acetylation of this mixture under standard conditions led to a facile cyclization that provided the known tricycle 27.4. Final desilylation provided ester 27.5, a substance synthesized previously by Kibayashi.12b
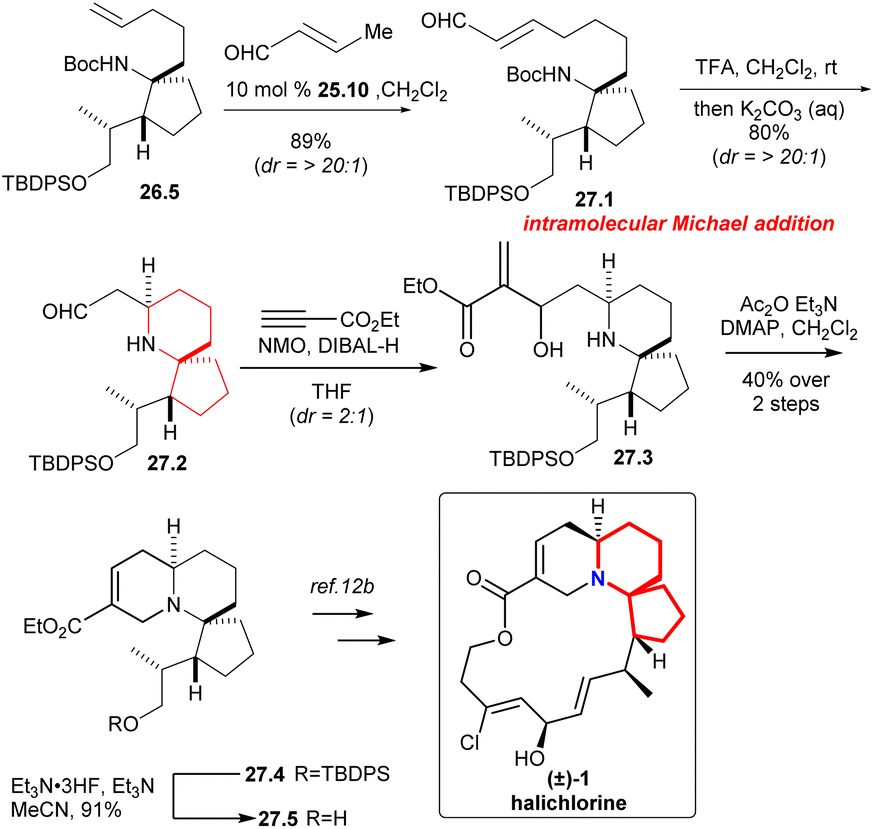 | ||
| Scheme 27 Martin's formal synthesis of (±)-halichlorine. | ||
4.2 Clive's total synthesis of (±) halichlorine
(See 2.2.4).4.3 Stockman's formal synthesis of halichlorine
In 2004, Stockman's group reported a concise method for preparing the azaspirocyclic core structure of halichlorine and pinnaic acid with a tandem cascade strategy.63 In 2012, they improved this strategy and completed the formal synthesis of halichlorine.46 The resulting synthesis of azaspirocyclic aldehyde 10.4 was an intermediate for the total synthesis of halichlorine by Clive's laboratory.24By utilizing an electrophilic centrepiece for the two-directional approach, symmetrical alcohol 28.1 (Scheme 28) was successfully synthesized through double addition of pent-5-enyl magnesium bromide on ethyl formate.64 The alcohol function of 28.1 was then oxidized, resulting in the formation of dialkene 28.2 with a high overall yield. Ketodiester 28.3 could through either the two-directional cross-metathesis method or a two-step approach involving oxidative addition of the alkenes of 28.2 followed by two-directional Wittig homologation. When performed on a 10 g scale, the two-step approach gave the ketodiester 28.3 in 47–53% yield after purification, and was found to be more cost-effective compared to the cross-metathesis route. Upon treatment with hydroxylamine hydrochloride in the presence of sodium acetate, symmetrical ketodiester 28.3 underwent a transformation the [6,5,5]tricycle 28.4 in 88% yield, this transformation was achieved through a tandem oxime formation/Michael addition/1,4-prototopic shift/[3 + 2]-cycloaddition.65
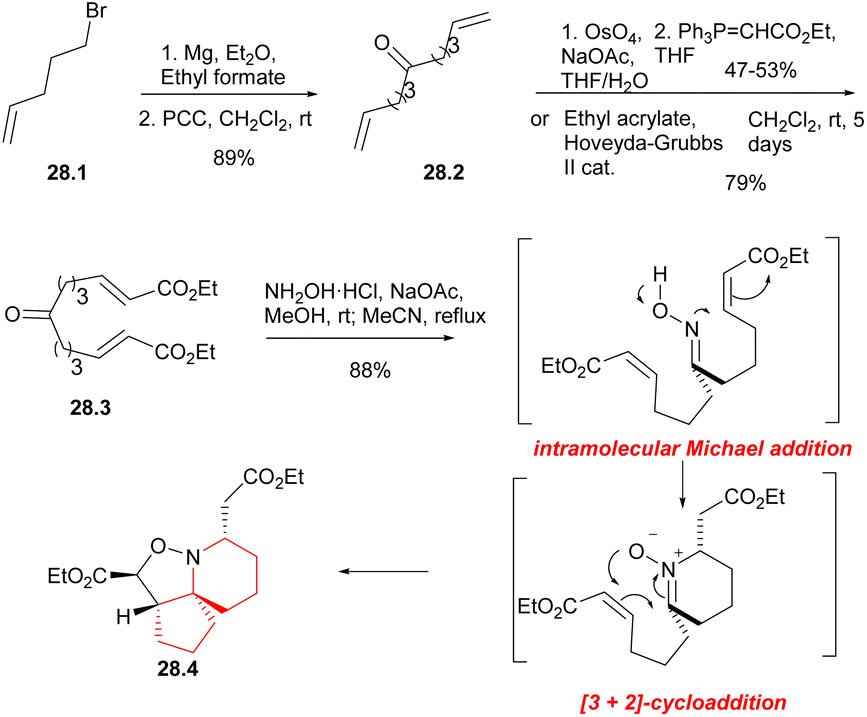 | ||
| Scheme 28 Synthesis of the 6-azaspiro[4.5]decane skeleton of halichlorine. | ||
The isoxazolidine 28.4 was selectively reduced to 29.1 in ethanol using sodium borohydride64 and subsequent hydrogenation cleaved the N–O bond, resulting in the formation of diol 29.2 with a quantitative yield (Scheme 29). Ester 29.4 with Z-conformation was produced by oxidative cleavage of diol followed immediately by an Ando homologation reaction.66 It was observed that the reaction yielded the best results when conducted in refluxing toluene with stoichiometric acetic acid, resulting in a high yield of lactam 29.5. Addition of the Gilman reagent67 to 29.5 afforded the lactam 29.6 in excellent yield. They found that this reaction required both TMSCl and triethylamine to proceed. With full stereoscopic control of C14 methyl, the ester functional group was converted to the target aldehyde 10.4 using DIBAL-H reduction at low temperature. So they had completed a short and efficient synthesis of Clive's aldehyde 10.4 in 12 steps and 13.2% overall yield from ethyl formate. This study represents the formal synthesis of halichlorine through the rapid and efficient synthesis of Clive aldehyde 10.4.24
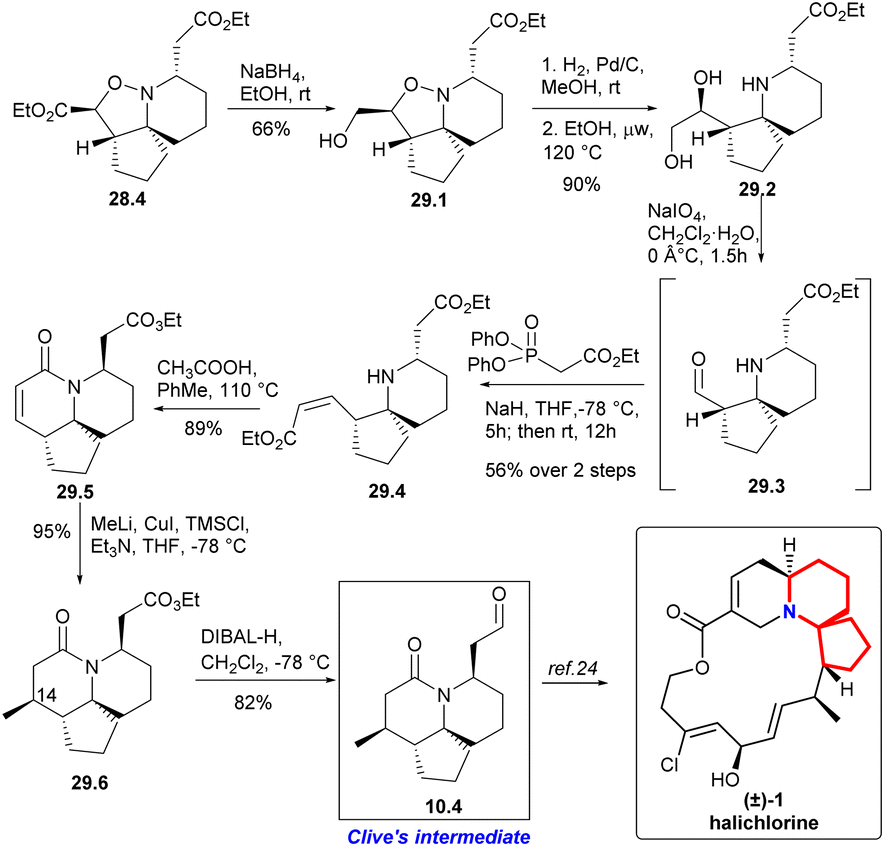 | ||
| Scheme 29 Stockman's formal synthesis of halichlorine. | ||
4.4 Zhu's formal synthesis of halichlorine
In 2015, a AuI/CuII-cocatalyzed tandem cyclization/semipinacol reaction68 had been developed by Zhu's group to provide an effective method for the construction of the 6-aza/oxa-spiro[4.5]decane skeletons. The synthetic utility of the approach was demonstrated by the efficient, formal synthesis of the marine natural product (±)-halichlorine.69The AuI/CuII-cocatalyzed tandem cyclization/semipinacol reaction was conducted with substrate 30.1a as the model substrate. After trying many conditions, [(BINAP)(AuCl)2]/AgBF4/Cu(OTf)2(1:2:2) was selected as the catalyst to construct the 6-aza/oxa-spiro[4.5]decane skeletons 30.2a,b (Scheme 30).70 The aza-spiro-ketone 30.2b was subjected to Wittig olefination with Ph3PBrCH3 in order to protect the carbonyl group (because the carbonyl group on the piperidine ring would cause side reactions in the subsequent nucleophilic steps), followed by removing the Ts group and amidation with 2-bromoacetyl bromide (30.2b → 30.4). For the construction of tricyclic intermediate 30.5, the bromine substitute compound 30.4 was subjected to intramolecular Reformatsky reaction with iodine as the initiator in a 60% yield. Ozonolysis and reductive workup gave 30.6, and dehydration led to amide 30.7,71 which gave 30.8 by catalytic hydrogenation. Introduction of the desired carbon–carbon double bond into 30.9 was achieved using copper bromide and DBU, which can get α-bromination compound, and then followed by the elimination reaction. At the same time, the configuration reversion of the C-14 methyl, was also successfully achieved. Subsequently, intermediate 30.10 was obtained by a 1,4-addition of amide 30.9 with allylstannane. Reaction of 4-piperidonone 30.10 with 1,3-propane thiol gave in a 90% yield the expected dithiane 30.11, they had completed an advanced intermediate reported by Padwa's group,72 representing a formal synthesis of (±)-halichlorine.
 | ||
| Scheme 30 Zhu's formal synthesis of (±)-halichlorine. | ||
5 Conclusions
In summary, we mainly focus on the construction of the 6-azaspiro[4.5]decane skeleton and provide many brief account of the asymmetric synthesis routes of two marine alkaloids: halichlorine and pinnaic acid. The unique structures and interesting bioactivity of pinnaic acid and halichlorine have motivated the exploration of a number of total syntheses. The synthesis of halichlorine and pinnaic acid was first accomplished by Danishefsky's research group in 1999 and 2001. This groundbreaking work holds great historic significance as it fills a gap in the total synthesis history of these compounds. In this review, we can see from the research works of various groups that the construction of C5, C9 and C13 three stereogenic carbons in the 6-azaspiro[4.5]decane skeleton is somewhat the challenging and key step. And different construction strategies of 6-azaspiro[4.5]decane frameworks of halichlorine and pinnaic acid have been also developed.73 In addition to the asymmetric synthesis methods summarized in this review, many research groups have also successfully completed the racemic synthesis of the halichlorine and pinnaic acid.3,24,46,59,69 These outstanding synthetic works make the two alkaloids more attractive. We hope that more relevant researchers will have the courage to challenge and synthesize halichlorine and pinnaic acid in a more concise and efficient way.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21562001), the Natural Science Foundation of Gansu Province (22JR5RA586) and the Educational Science and Technology Innovation Program of Gansu Province (2021CYZC-13).Notes and references
- M. Kuramoto, C. Tong, K. Yamada, T. Chiba, Y. Hayashi and D. Uemura, Tetrahedron Lett., 1996, 37, 3867–3870 CrossRef CAS.
- T. Chou, M. Kuramoto, Y. Otani, M. Shikano, K. Yazawa and D. Uemura, Tetrahedron Lett., 1996, 37, 3871–3874 CrossRef CAS.
- D. L. J. Clive, M. L. Yu, J. Wang, V. S. C. Yeh and S. Z. Kang, Chem. Rev., 2005, 105, 4483–4514 CrossRef CAS PubMed.
- (a) J. Y. Liu, J. Zhang, Y. X. Fan, H. L. Ding, T. F. Liu, S. S. Li, M. H. Jiang and L. Liu, Org. Biomol. Chem., 2022, 20, 1879–1882 RSC; (b) J. Zhang, H. L. Ding, Y. X. Fan, H. Chen, H. X. Dai and M. Jing, Heterocycles, 2023, 106, 166–173 CrossRef CAS; (c) J. Zhang, Y. Q. Wang, X. W. Wang and W. D. Z. Li, J. Org. Chem., 2013, 78, 6154–6162 CrossRef CAS PubMed.
- H. Arimoto, I. Hayakawa, M. Kuramoto and D. Uemura, Tetrahedron Lett., 1998, 39, 861–862 CrossRef CAS.
- D. Trauner, J. B. Schwarz and S. J. Danishefsky, Angew. Chem., Int. Ed., 1999, 38, 3542–3545 CrossRef CAS PubMed.
- D. Trauner and S. J. Danishefsky, Tetrahedron Lett., 1999, 40, 6513–6516 CrossRef CAS.
- D. Trauner, D. G. Churchill and S. J. Danishefsky, Helv. Chim. Acta, 2000, 83, 2344–2351 CrossRef CAS.
- Cf. L. E. Burgess and A. I. Meyers, J. Am. Chem. Soc., 1991, 113, 9858–9859 CrossRef.
- (a) K. Soai and S. Niwa, Chem. Rev., 1992, 92, 833–856 CrossRef CAS; (b) K. Soai and K. Takahashi, J. Chem. Soc., Perkin Trans. 1 (1972–1999), 1994, 1257–1258 RSC.
- (a) D. Meng, P. Bertinato, A. Balog, D. S. Su, T. Kamenecka, E. J. Sorensen and S. J. Danishefsky, J. Am. Chem. Soc., 1997, 119, 10073–10092 CrossRef CAS; (b) A. B. Jones, A. Villalobos, R. G. Linde and S. J. Danishefsky, J. Org. Chem., 1990, 55, 2786–2797 CrossRef CAS.
- (a) H. L. Zhang, G. Zhao, Y. Ding and B. Wu, J. Org. Chem., 2005, 70, 4954–4961 CrossRef CAS PubMed; (b) Y. Matsumura, S. Aoyagi and C. Kibayashi, Org. Lett., 2004, 6, 965–968 CrossRef CAS PubMed.
- S. Hashimoto, Y. Miyazaki and S. Ikegami, Synth. Commun., 1992, 22, 2717–2722 CrossRef CAS.
- (a) B. Tarnchompoo and Y. Thebtaranonth, Tetrahedron Lett., 1984, 25, 5567–5570 CrossRef CAS; (b) A. Braun, I. H. Cho, S. Ciblat, D. Clyne, P. Forgione, A. C. Hart, G. X. Huang, J. Kim, I. Modolo, L. A. Paquette, X. W. Peng, S. Pichlmair, C. A. Stewart, J. Z. Wang and D. Zuev, Collect. Czech. Chem. Commun., 2009, 74, 651–769 CrossRef CAS.
- (a) D. H. Lloyd and D. E. Nichols, J. Org. Chem., 1986, 51, 4294–4295 CrossRef CAS; (b) B. Ganem and J. O. Osby, Chem. Rev., 1986, 86, 763–780 CrossRef CAS.
- (a) G. Stork, D. Niu, A. Fujimoto, E. R. Koft, J. M. Balkovec and J. R. Tata, J. Am. Chem. Soc., 2001, 123, 3239–3242 CrossRef CAS PubMed; (b) L. E. Overman, D. Lesuisse and M. Hashimoto, J. Am. Chem. Soc., 1983, 105, 5373–5379 CrossRef CAS; (c) S. H. Yang and V. Caprio, Synlett, 2007, 8, 1219–1222 Search PubMed.
- (a) K. Yamdada, K. Kishikawa and M. Yamamoto, J. Org. Chem., 1987, 52, 2327–2330 CrossRef; (b) S. H. Yang, G. R. Clark and V. Caprio, Org. Biomol. Chem., 2009, 7, 2981–2990 RSC.
- (a) D. P. Barre, Z. Y. Zhang, A. Cador, T. Vives, T. Roisnel, O. Basle, L. Jarrige, L. Cavallo, L. Falivene and M. Mauduit, Chem. Sci., 2022, 13, 8773–8780 RSC; (b) H. C. Guo and J. A. Ma, Angew. Chem., Int. Ed., 2006, 45, 354–366 CrossRef CAS PubMed.
- (a) K. Yamada, M. Yamashita, T. Sumiyoshi, K. Nishimura and K. Tomioka, Org. Lett., 2009, 11, 1631–1633 CrossRef CAS PubMed; (b) M. Suzuki, Y. Kawamoto, T. Sakai, Y. Yamamoto and K. Tomioka, Org. Lett., 2009, 11, 653–655 CrossRef CAS PubMed; (c) T. Sakai, Y. Kawamoto and K. Tomioka, J. Org. Chem., 2006, 71, 4706–4709 CrossRef CAS PubMed.
- Y. Yamamoto, Y. Yasuda, H. Nasu and K. Tomioka, Org. Lett., 2009, 11, 2007–2009 CrossRef CAS PubMed.
- Y. Yamamoto, H. Suzuki, Y. Yasuda, A. Iida and K. Tomioka, Tetrahedron Lett., 2008, 49, 4582–4584 CrossRef CAS.
- K. Ninomiya, T. Shioiri and S. Yamada, Tetrahedron, 1974, 30, 2151–2157 CrossRef CAS.
- (a) M. L. Yu, D. L. J. Clive, V. S. C. Yeh, S. Z. Kang and J. Wang, Tetrahedron Lett., 2004, 45, 2879–2881 CrossRef CAS; (b) D. L. J. Clive, M. L. Yu and Z. Y. Li, J. Chem. Soc., Chem. Commun., 2005, 906–908 RSC.
- D. Z. Liu, H. P. Acharya, M. L. Yu, J. Wang, V. S. C. Yeh, S. Z. Kang, C. Chiruta, S. M. Jachak and D. L. J. Clive, J. Org. Chem., 2009, 74, 7417–7428 CrossRef CAS PubMed.
- (a) N. J. Goldspink, N. S. Simpkins and M. Beckmann, Synlett, 1999, 1292–1294 CrossRef CAS; (b) T. Huxford and N. S. Simpkins, Synlett, 2004, 2295–2298 CAS.
- K. Bambridge, M. J. Begley and N. S. Simpkins, Tetrahedron Lett., 1994, 35, 3391–3394 CrossRef CAS.
- (a) H. J. Reich, S. K. Shah and F. Chow, J. Am. Chem. Soc., 1979, 101, 6648–6656 CrossRef CAS; (b) D. L. J. Clive and M. H. D. Postema, J. Chem. Soc., Chem. Commun., 1994, 235–236 RSC.
- A. L. Gemal and J. L. Luche, J. Am. Chem. Soc., 1981, 103, 5454–5459 CrossRef CAS.
- A. L. J. Beckwith, D. Crich, P. J. Duggan and Q. Yao, Chem. Rev., 1997, 97, 3273–3312 CrossRef CAS PubMed.
- P. A. Grieco, S. Gilman and M. Nishizawa, J. Org. Chem., 1976, 41, 1485–1486 CrossRef CAS.
- (a) M. Amat, M. Prez, N. Llor, J. Bosch, E. Lago and E. Molins, Org. Lett., 2001, 3, 611–614 CrossRef CAS PubMed; (b) C. Herdeis, C. Kaschinski, R. Karla and H. Lotter, Tetrahedron: Asymmetry, 1996, 7, 867–884 CrossRef CAS.
- D. L. J. Clive, Z. Y. Li and M. L. Yu, J. Org. Chem., 2007, 72, 5608–5617 CrossRef CAS PubMed.
- S. C. Fernandopulle, D. L. J. Clive and M. L. Yu, J. Org. Chem., 2008, 73, 6018–6021 CrossRef CAS PubMed.
- S. P. Keen and S. M. Weinreb, J. Org. Chem., 1998, 63, 6739–6741 CrossRef CAS.
- D. L. J. Clive, W. Yang, A. C. MacDonald, Z. Wang and M. Cantin, J. Org. Chem., 2001, 66, 1966–1983 CrossRef CAS PubMed.
- S. Xu, H. Arimoto and D. Uemura, Angew. Chem.,Int. Ed., 2007, 119, 5848–5851 (Angew. Chem., Int. Ed., 2007, 46, 5746–5749) CrossRef.
- S. Xu, D. Unabara, D. Uemura and H. Arimoto, Chem.–Asian J., 2014, 9, 367–375 CrossRef CAS PubMed.
- (a) B. M. Trost, B. R. Taft, J. S. Tracy and C. E. Stivala, Org. Lett., 2021, 23, 4981–4985 CrossRef CAS PubMed; (b) S. Yamago and E. Nakamura, Org. React., 2002, 61, 1–217 CAS; (c) B. M. Trost, Pure Appl. Chem., 1988, 60, 1615–1626 CrossRef CAS; (d) R. Baker and R. B. Keen, J. Organomet. Chem., 1985, 285, 419–427 CrossRef CAS.
- (a) R. T. Conley and M. C. Annis, J. Org. Chem., 1962, 27, 1961–1964 CrossRef CAS; (b) Y. Tamura, J. Minamikawa and M. Ikeda, Synthesis, 1977, 1–17 CrossRef CAS; (c) Y. Tamura, H. Fujiwara, K. Sumoto, M. Ikeda and Y. Kita, Synthesis, 1973, 215–216 CAS.
- S. Xu, T. Toyama, J. Nakamura and H. Arimoto, Tetrahedron Lett., 2010, 51, 4534–4537 CrossRef CAS.
- (a) I. Hayakawa, H. Arimoto and D. Uemura, Heterocycles, 2003, 59, 441–444 CrossRef CAS; (b) H. Arimoto, S. Asano and D. Uemura, Tetrahedron Lett., 1999, 40, 3583–3586 CrossRef CAS; (c) I. Hayakawa, H. Arimoto and D. Uemura, Chem. Commun., 2004, 1222–1223 RSC.
- (a) H. E. Blackwell, D. J. OLeary, A. K. Chatterjee, R. A. Washen-felder, D. A. Bussmann and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 58–71 CrossRef CAS; (b) S. J. Meek, R. V. O'Brien, J. Llaveria, R. R. Schrock and A. H. Hoveyda, Nature, 2011, 471, 461–466 CrossRef CAS PubMed; (c) B. K. Keitz, K. Endo, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 9686–9688 CrossRef CAS PubMed.
- T. Y. Zhang, L. Y. Zhang, X. Liang, K. Wei and Y. R. Yang, Org. Lett., 2022, 24, 2905–2909 CrossRef CAS PubMed.
- T. Sandmeier and E. M. Carreira, Angew. Chem., Int. Ed., 2021, 60, 9913–9918 CrossRef CAS PubMed.
- M. Bischöp and J. Pietruszka, Synlett, 2011, 2689–2692 Search PubMed.
- C. Gignoux, A. F. Newton, A. Barthelme, W. Lewis, M. L. Alcaraz and R. A. Stockman, Org. Biomol. Chem., 2012, 10, 67–69 RSC.
- M. W. Carson, G. Kim, M. F. Hentemann, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4450–4452 CrossRef CAS PubMed.
- M. W. Carson, G. Kim and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4453–4456 CrossRef CAS PubMed.
- (a) S. Krishnamurthy, F. Vogel and H. C. Brown, J. Org. Chem., 1977, 42, 2534–2536 CrossRef CAS; (b) L. M. Mikkelsen, C. M. Jensen, B. Høj, P. Blakskjær and T. Skrydstrup, Tetrahedron, 2003, 59, 10541–10549 CrossRef CAS; (c) H. C. Zhang, B. D. Harris, M. J. Costanzo, E. C. Lawson and C. A. Maryanoff, J. Org. Chem., 1998, 63, 7964–7981 CrossRef CAS; (d) H. C. Zhang, B. D. Harris, C. A. Maryanoff and B. E. Maryanoff, Tetrahedron Lett., 1996, 37, 7897–7900 CrossRef CAS; (e) H. C. Zhang, M. J. Costanzo and B. E. Maryanoff, Tetrahedron Lett., 1994, 35, 4891–4894 CrossRef CAS.
- H. Wu, H. L. Zhang and G. Zhao, Tetrahedron Lett., 2007, 63, 6454–6461 CrossRef CAS.
- (a) M. Kitamura, M. Tokunaga, T. Ohkuma and R. Noyori, Org. Synth., 1993, 71, 1–9 CrossRef CAS; (b) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, NY, 1994 Search PubMed.
- (a) D. C. Iffland and G. X. Criner, J. Am. Chem. Soc., 1953, 75, 4047–4048 CrossRef CAS; (b) M. W. Barnes, J. Org. Chem., 1976, 41, 733–735 CrossRef CAS; (c) E. J. Corey and H. Estreicher, Tetrahedron Lett., 1980, 21, 1117–1120 CrossRef CAS; (d) T. R. Walters, W. W. Zajac and J. M. Woods, J. Org. Chem., 1991, 56, 316–321 CrossRef CAS; (e) A. P. Marchand, R. Sharma, U. R. Zope, W. H. Watson and R. P. Kashyap, J. Org. Chem., 1993, 58, 759–762 CrossRef CAS.
- A. B. Smith, S. M. Condon, J. A. McCauley, J. L. Leazer, J. W. Leahy and R. E. Maleczka, J. Am. Chem. Soc., 1997, 119, 947–961 CrossRef CAS.
- R. Z. Liu, O. Gutierrez, D. J. Tantillo and J. Aubé, J. Am. Chem. Soc., 2012, 134, 6528–6531 CrossRef CAS PubMed.
- (a) D. Depré, L. Y. Chen and L. Ghosez, Tetrahedron, 2003, 59, 6797–6812 CrossRef; (b) C. Houge, A. M. Frisque-Hesbain, A. Mockel, L. Ghosez, J. P. Declercq, G. Germain and M. Van Meerssche, J. Am. Chem. Soc., 1982, 104, 2920–2921 CrossRef CAS.
- M. Bandini, P. G. Cozzi, S. Licciulli and A. Umani-Ronchi, Synthesis, 2004, 409–414 CAS.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–965 CrossRef CAS PubMed.
- R. B. Andrade and S. F. Martin, Org. Lett., 2005, 7, 5733–5735 CrossRef CAS PubMed.
- For some selected examples, see: (a) S. F. Martin, J. M. Humphrey, Y. Liao, A. Ali, T. Rein, Y. L. Wong, H. J. Chen and A. K. Courtney, J. Am. Chem. Soc., 2002, 124, 8584–8592 CrossRef PubMed; (b) A. Deiters and S. F. Martin, Org. Lett., 2002, 4, 3243–3245 CrossRef CAS PubMed; (c) S. F. Martin and C. E. Neipp, J. Org. Chem., 2003, 68, 8867–8878 CrossRef PubMed; (d) D. G. Washburn, R. W. Heidebrecht and S. F. Martin, Org. Lett., 2003, 5, 3523–3525 CrossRef CAS PubMed; (e) J. B. Brenneman, R. Machauer and S. F. Martin, Tetrahedron, 2004, 60, 7301–7314 CrossRef CAS.
- G. A. Wallace and C. H. Heathcock, J. Org. Chem., 2001, 66, 450–454 CrossRef CAS PubMed.
- P. V. Ramachandran, M. T. Rudd, T. E. Burghardt and M. V. R. Reddy, J. Org. Chem., 2003, 68, 9310–9316 CrossRef CAS PubMed.
- L. G. Arini, P. Szeto, D. L. Hughes and R. A. Stockman, Tetrahedron Lett., 2004, 45, 8371–8374 CrossRef CAS.
- A. F. Newton, S. J. Roe, J. C. Legeay, P. Aggarwal, C. Gignoux, N. J. Birch, R. Nixon, M. L. Alcaraz and R. A. Stockman, Org. Biomol. Chem., 2009, 7, 2274–2277 RSC.
- (a) R. Grigg, J. Markandu, S. Surendrakumar, M. Thornton-Pett and W. J. Warnock, Tetrahedron, 1992, 48, 10399–10422 CrossRef CAS; (b) R. Grigg, J. Markandu, T. Perrior, S. Surendrakumar and W. J. Warnock, Tetrahedron, 1992, 48, 6929–6952 CrossRef CAS; (c) R. Grigg, M. J. Dorrity, F. Heaney, J. F. Malone, S. Rajviroongit, V. Sridharan and S. Surendrakumar, Tetrahedron, 1991, 47, 8297–8322 CrossRef CAS; (d) R. Grigg, F. Heaney, J. Markandu, S. Surendrakumar, M. Thornton-Pett and W. J. Warnock, Tetrahedron, 1991, 47, 4007–4030 CrossRef CAS; (e) P. Armstrong, R. Grigg, F. Heaney, S. Surerndrakumar and W. J. Warnock, Tetrahedron, 1991, 47, 4495–4518 CrossRef CAS; (f) R. Grigg, F. Heaney, S. Surendrakumar and W. J. Warnock, Tetrahedron, 1991, 47, 4477–4494 CrossRef CAS.
- K. Ando, Tetrahedron Lett., 1995, 36, 4105–4108 CrossRef CAS.
- H. Gilman, R. G. Jones and L. A. Woods, J. Org. Chem., 1952, 17, 1630–1634 CrossRef CAS.
- (a) T. C. Coombs, Y. Q. Zhang, E. C. Garnier-Amblard and L. S. Liebeskind, J. Am. Chem. Soc., 2009, 131, 876–877 CrossRef CAS PubMed; (b) Z. H. Chen, Z. M. Chen, Y. Q. Zhang, Y. Q. Tu and F. M. Zhang, J. Org. Chem., 2011, 76, 10173–10186 CrossRef CAS PubMed; (c) H. Y. Lee and C. K. Sha, J. Org. Chem., 2012, 77, 598–605 CrossRef CAS PubMed; (d) D. J. Canham and L. E. Overman, J. Org. Chem., 2013, 78, 9–34 CrossRef PubMed; (e) M. C. P. Yeh, M. N. Lin, C. H. Hsu and C. J. Liang, J. Org. Chem., 2013, 78, 12381–12396 CrossRef CAS PubMed.
- D. Y. Zhu, Z. Zhang, X. Q. Mou, Y. Q. Tu, F. M. Zhang, J. B. Peng, S. H. Wan and S. Y. Zhang, Adv. Synth. Catal., 2015, 357, 747–752 CrossRef CAS.
- (a) C. Zhu and S. M. Ma, Org. Lett., 2014, 16, 1542–1545 CrossRef CAS PubMed; (b) D. G. Yu, M. Suri and F. Glorius, J. Am. Chem. Soc., 2013, 135, 8802–8805 CrossRef CAS PubMed; (c) Y. P. Yan, P. Feng, Q. Z. Zheng, Y. F. Liang, J. F. Lu, Y. X. Cui and N. Jiao, Angew. Chem., 2013, 125, 5939–5943 (Angew. Chem., Int. Ed., 2013, 52, 5827–5831) CrossRef.
- (a) K. F. Hebenbrock, Adv. Cycloaddit., 1978, 320–336 CAS; (b) J. B. Peng, X. J. Hou, S. Y. Zhang and Y. Q. Tu, Acta Chim. Sin., 2012, 70, 2232–2235 CrossRef CAS.
- A. C. Flick, M. J. A. Caballero, H. I. Lee and A. Padwa, J. Org. Chem., 2010, 75, 1992–1996 CrossRef CAS PubMed.
- For selective synthesis examples, see: (a) D. L. J. Clive and V. S. C. Yeh, Tetrahedron Lett., 1999, 40, 8503–8507 CrossRef CAS; (b) J. L. Koviach and C. J. Forsyth, Tetrahedron Lett., 1999, 40, 8529–8532 CrossRef CAS; (c) S. K. Lee and Z. C. Zhao, Org. Lett., 1999, 1, 681–683 CrossRef CAS; (d) S. Lee and Z. Zhao, Tetrahedron Lett., 1999, 40, 7921–7924 CrossRef CAS; (e) M. Shindo, Y. Fukuda and K. Shishido, Tetrahedron Lett., 2000, 41, 929–932 CrossRef CAS; (f) D. L. Wright, J. P. Schulte II and M. A. Page, Org. Lett., 2000, 2, 1847–1850 CrossRef CAS PubMed; (g) J. D. White, P. R. Blakemore, E. A. Korf and A. F. T. Yokochi, Org. Lett., 2001, 3, 413–415 CrossRef CAS PubMed; (h) W. Yokota, M. Shindo and K. Shishido, Heterocycles, 2001, 54, 871–885 CrossRef CAS PubMed; (i) K. Takasu, H. Ohsato and M. Ihara, Org. Lett., 2003, 5, 3017–3020 CrossRef CAS PubMed; (j) Y. Matsumura, S. Aoyagi and C. Kibayashi, Org. Lett., 2003, 5, 3249–3252 CrossRef CAS PubMed; (k) K. S. Feldman, A. L. Perkins and K. M. Masters, J. Org. Chem., 2004, 69, 7928–7932 CrossRef CAS PubMed; (l) D. L. J. Clive, J. Wang and M. L. Yu, Tetrahedron Lett., 2005, 46, 2853–2855 CrossRef CAS; (m) A. L. Sousa and R. A. Pilli, Org. Lett., 2005, 7, 1617–1619 CrossRef PubMed; (n) G. E. Keck and S. A. Heumann, Org. Lett., 2008, 10, 4783–4786 CrossRef CAS PubMed; (o) F. D. Ferrari, A. J. Ledgard and R. Marquez, Tetrahedron, 2011, 67, 4988–4994 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |