
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePt nanoparticles-decorated molybdenum nitrides for efficient hydrogen evolution reaction†
Jie Wang a and
Xiaorong Zhangb
a and
Xiaorong Zhangb
aCollege of Chemical Engineering, Shanxi Institute of Science and Technology, Jincheng, Shanxi 048000, China. E-mail: wangjie2@sxist.edu.cn
bNational Engineering Laboratory for Reducing Emissions from Coal Combustion, Engineering Research Center of Environmental Thermal Technology of Ministry of Education, Shandong Key Laboratory of Energy Carbon Reduction and Resource Utilization, School of Energy and Power Engineering, Shandong University, Jinan, Shandong 250061, China. E-mail: zhangxr1216@126.com
First published on 21st November 2023
Abstract
Exploring low cost and high efficiency catalysts for hydrogen production from electrochemical water splitting is preferable and remains a significant challenge. As an alternative to Pt-based catalysts, molybdenum nitrides have attracted more attention for their hydrogen evolution reaction (HER). However, their performance is restricted due to the strong bonding of Mo–H. Herein, molybdenum nitrides with Pt-doping are fabricated to enhance the catalytic activity for HER in acidic solution. As expected, Pt (5 wt%)–MoNx delivers a low overpotential of 47 mV at a current density of 10 mA cm−2 with a high exchange current density (j0 = 0.98 mA cm−2). The superior performance is attributed to the modified electronic structure of Mo with Pt incorporation.
Introduction
Hydrogen energy possessing the virtues of high-energy density and being pollution-free has been regarded as an alternative to traditional fossil fuels for energy conversion, storage, and utilization.1–3 Currently, industrial hydrogen production is mainly produced from steam reforming of hydrocarbons and coal gasification, which inevitably releases a large amount of CO2 and causes environmental pollution and global warming.4–6 Hydrogen from electrocatalytic water splitting powered by intermittent electricity generated from renewable energy sources (e.g., wind, solar, and tidal) is recognized as a greener sustainable approach. To date, platinum-based materials having low hydrogen absorption Gibbs free energies are still the state-of-the-art catalysts for the hydrogen evolution reaction (HER).7–9 However, its low abundance and high cost have hindered the large-scale practical application for HER.10 Therefore, it is highly desirable to design and fabricate robust catalysts with low Pt loadings or even Pt-free toward HER.To effectively reduce the Pt-loadings, many efforts have been devoted to develop highly efficient non-noble metal-based materials, such as transition metal carbides,11 nitrides,12 phosphides,13 oxides,14 sulfides,15 and carbon-materials.16 Among these, molybdenum-based carbides or nitrides have been intensively pursued as promising catalysts due to the similarity of their d-band electronic structure to that of platinum-based materials.17–19 Additionally, molybdenum carbides and nitrides possess superior corrosion resistance, high electronic conductivity, and excellent mechanical properties.20,21 Furthermore, Molybdenum carbides have been widely used in various catalytic reactions including hydrogen evolution,3 oxygen reduction,22 hydrogenation reactions,23 the water gas shift,24 methanol reforming,25 and so on. Molybdenum nitrides, as an interstitial compound with similar structure and electronic properties of molybdenum carbide have high unoccupied d-orbitals, which result in a stronger Mo–H bonding and poorer H2 desorption.26–28 Therefore, enhance the catalytic activity of molybdenum nitrides is still a great challenge. Both experimental and theoretical researches have revealed there are strong interactions between molybdenum nitrides and surface metal species.28,29 Hence, it is expected molybdenum nitrides can not only act as an excellent support for the catalysts preparation, but also modulate their electronic structure.30
Traditionally, the synthesis methods of molybdenum nitride (Mo2N) including thermal treatment of metal salts or metal oxides in an inert or NH3 atmosphere is difficult to control the morphologies and dispersion.31 In addition, it was reported that the synthesis of molybdenum nitrides (MoN and Mo5N6) is used by salt-templated methods, which involved using large amounts of NaCl.32,33 Hence, synthesis of molybdenum nitrides with a large amount of catalytic active sites still remains highly challenging to optimize the nanostructure for achieving higher HER activity.
During HER processes, the adsorption and desorption of hydrogen on the catalysts' surfaces play pivotal roles in their electrocatalytic activities.21 Thus, a favourable affinity between catalyst and hydrogen prefers the formation of Hads intermediates and release of H2, which boosts HER activity.17,26 It has been confirmed the introduction of metal vacancies and the group VIII elements are promising strategies for adjusting d-band of Mo.26,34,35 For example, Co-doped Mo2C could effectively improve the HER performance of Mo2C, which is derived from the modifiable electronic structure around the Femi level, leading to an increase of electronic density of around Mo sites.36 Meanwhile, the synergistic effect of Co incorporation and Mo vacancies could tune electronic features of Mo and optimize the strength of Mo–H bond, thereby enhancing HER performance of Mo2C.26
Inspired by the above achievements, we herein developed Pt/MoNx catalysts as active and stable HER electrocatalysts in acidic media. Pt (5 wt%)–MoNx showed excellent catalytic performance with η10 of 47 mV, close to commercial Pt/C. Meanwhile, molybdenum nitrides doped with other precious metals (Pd and Ru) were also fabricated, which can also boost HER performance compared with bare MoNx.
Results and discussion
MoO3 nanobelts were prepared by the hydrothermal method using molybdenum powder as the precursor. In brief, molybdenum powers are oxidized by hydrogen peroxide (H2O2, 30%) followed by solvothermal treatment at 180 °C for 48 h. The crystalline structure and 1D morphology of MoO3 are shown in Fig. S1 and S2.† The characteristic diffraction peaks in its XRD patterns could be assigned to the orthorhombic phase of MoO3 (PDF # 05-0508). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) results display that the MoO3 nanobelts have widths in the range of 120 to 350 nm and their length can reach several micrometers (Fig. S2†).MoNx was fabricated by ammoniation of as-synthesized MoO3 at 750 °C for 5 h. The crystaline structure of the catalysts was firstly investigated by X-ray diffraction (XRD) pattern. XRD result of bare MoNx demonstrates the coexistence of cubic Mo2N and hexagonal MoN phases. The peaks located at about 37.1°, 43.1°, 62.7°, and 75.2° correspond to (111), (200), (220), and (311) planes of Mo2N (PDF # 25-1366), accordingly. Whereas two weak diffraction peaks at 36.1° and 48.8° belong to the (200) and (202) planes of MoN (PDF # 25-1367), respectively (Fig. 1a). When the loading of Pt is about 3 wt%, the characteristic diffraction peaks of Pt tend to appear. Further increasing its loading to 5% (Pt-5), diffraction peaks at 39.8° and 46.3°, corresponding to (111) and (200) crystal planes of Pt, become discernible (Fig. 1b). Specifically, the peaks at about 37.1°, corresponding to the (111) crystal plane of Mo2N, is slightly shifted to a low angle after loading Pt, confirming that Pt-doping changes the lattice parameters of Mo2N. Pt-5 displays the best electrocatalytic performance among as-synthesized samples, therefore, it is used for the following characterizations unless specified.
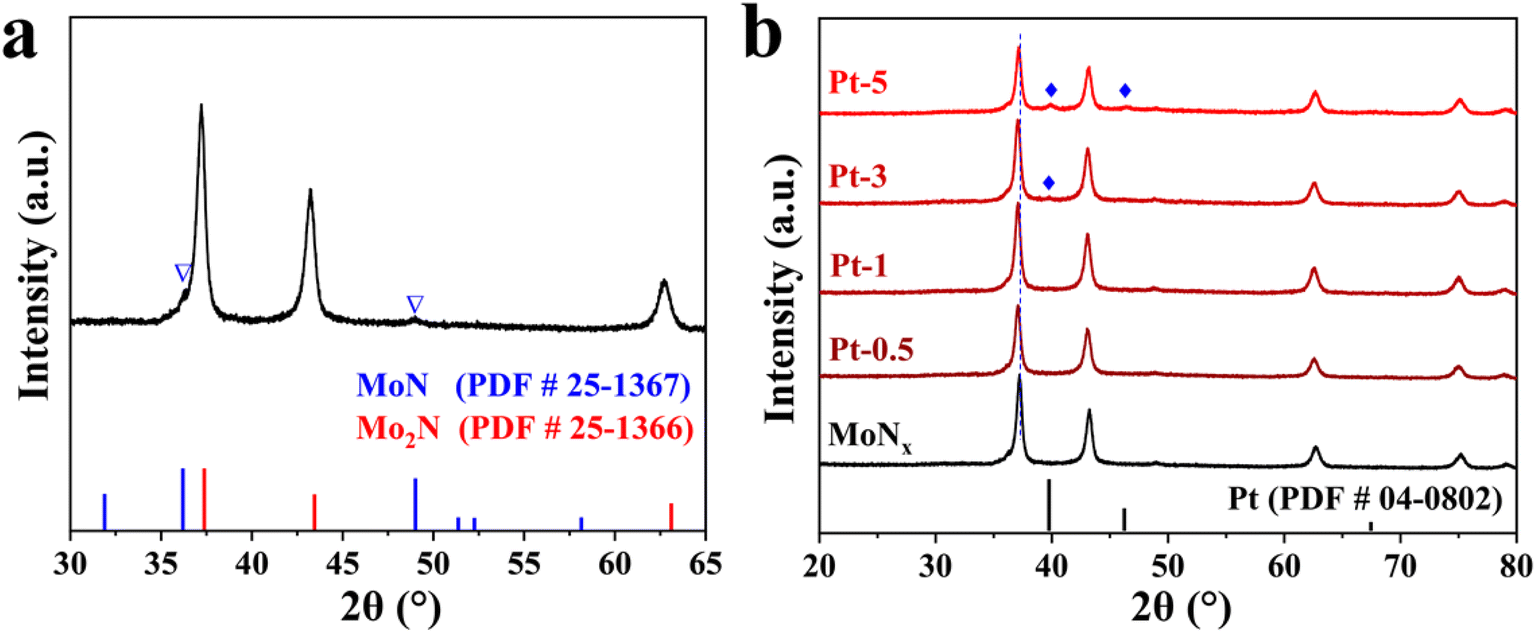 | ||
| Fig. 1 The XRD patterns of the bare MoNx (a) and Pt–MoNx (b). | ||
The morphology of MoNx is not inherited the 1D morphology of MoO3 nanobelts from SEM results (Fig. 2a, S2, S3a and b†). TEM result shows that MoNx is composed of small nanoparticles (≈20 nm). High-resolution TEM (HRTEM) image reveals the lattice fringes with d-spacing of 0.241 nm and 0.248 nm, corresponding to the (111) and (200) planes of the Mo2N and MoN, respectively (Fig. S3c†). SEM and TEM images indicate the synthesized Pt-5 retains the initial morphology of MoNx with a rough and porous surface (Fig. 2a and b). The porous structures are verified by N2 adsorption/desorption isotherms curves (Fig. S4a†). The isotherms exhibit the III-type porous structure, indicating the coexistence of mesopores and macropores for Pt-5.37,38 Pore size distribution curves are multimodal, with mesopores approximately centered at 8 nm and a wide distribution up to 75 nm (Fig. S4b†). The HRTEM image shows, new lattice fringes with interplanar spacings of 0.222 nm are clearly observed and could be assigned to the (111) plane of Pt (Fig. 2c). Selected area electron diffraction (SAED) pattern reveals the characteristic (111) and (220) facets of Mo2N and (111) facets of Pt (111). High-angle annular dark field scanning TEM (HAADF-STEM) image and corresponding elemental mapping images indicate the uniform distribution of N, Mo, and Pt in the selected area (Fig. 2e–h and S3d†).
 | ||
| Fig. 2 SEM (a), TEM (b), HRTEM (c), SAED pattern (d) and TEM elemental mapping (e–h) of Pt-5. | ||
The HER performance of as-synthesized catalysts and commercial Pt/C catalyst was investigated in an acidic electrolyte using a three-electrode system. The loading of each catalyst is fixed at 0.4 mg cm−2 on a carbon paper electrode. Fig. 3a shows the linear sweep voltammetry (LSV) polarization curves of the obtained-catalysts in 0.5 M H2SO4 and 20% Pt/C as reference. The overpotentials (η10) of bare MoNx, Pt-1, Pt-3 and Pt-5 are 349, 65, 52 and 47 mV @10 mA cm−2 (Fig. 3b), respectively. The corresponding overpotentials of catalysts at η50 are depicted in Fig. 3b. Therefore, the activities of these catalysts are significantly improved with the increase of the loading of Pt. However, further increasing the loading of Pt on MoNx to 7% could not lead to a higher catalytic activity as reflected from its overpotential, e.g. 48 mV @10 mA cm−2. Since commercial 20% Pt/C catalyst has the lowest overpotential of 33 mV for achieving the current density of 10 mA cm−2, implying that the loading amount of Pt on MoNx substrate is not the solely factor determining the catalytic activity. Furthermore, the overpotential (η10) of Pt-5 is comparable to those of the reported Mo-based catalysts (Table S1†). Meanwhile, MoNx doped with other noble metallic nanoparticles were also synthesized using the same protocol as Pt-5 and investigated their electrocatalytic HER performance (Fig. S6†). Ru/MoNx, Pd/MoNx, and Au/MoNx require the overpotential of 120, 238, and 320 mV to deliver a current density of 10 mA cm−2, respectively, but inferior to that of Pt-5.
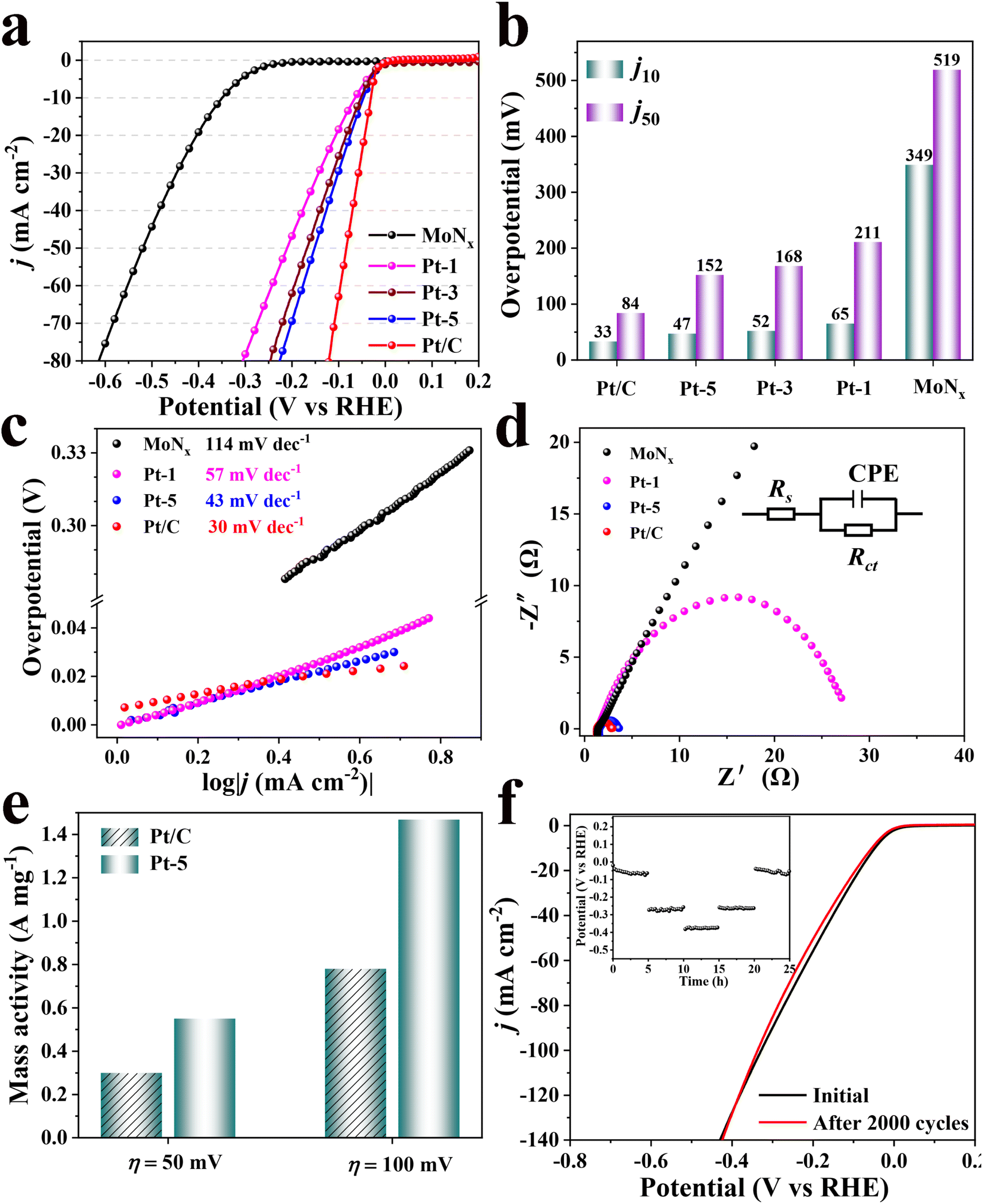 | ||
| Fig. 3 Polarization curves of MoNx, Pt-1, Pt-3, Pt-5, and Pt/C (a); corresponding overpotentials at the current densities of 10 and 50 mA cm−2 (b); Tafel plots of Pt/C, Pt-5, Pt-1, and MoNx (c); Nyquist plots and equivalent circuit of MoNx, Pt-1, Pt-5, and Pt/C (d); mass activities of Pt-5 and Pt/C at an overpotential of 50 and 100 mV, respectively (e); polarization curves of Pt-5 with an initial and after 2000 cycles, and the inset image shows long-time stability of Pt-5 obtained at 10, 50 and 100 mA cm−2 (f). | ||
Tafel slope was used to evaluate reaction kinetics and the rate-determining step by the Tafel equation (η = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logj, where a, b, and j represent intercept, Tafel slope and current density, respectively).39–41 The Tafel slope for Pt-5 is 43 mV dec−1, which is lower than those of Pt-1 (57 mV dec−1) and MoNx (114 mV dec−1). The smaller Tafel slope demonstrates there is a fast increase of the hydrogen generation rate for Pt-5.21,41 It is accepted that HER in acidic condition goes through two processes, including electrochemical reduction step (Volmer step) with a Tafel slope of 120 mV dec−1 and the desorption of adsorbed hydrogen. The latter is either electrochemical desorption (Heyrovsky step) with a Tafel slope of 40 mV dec−1 or recombination step (Tafel step) with a Tafel slope of 30 mV dec−1.42 The Tafel slope (43 mV dec−1) over the Pt-5 catalyst is close to that of Heyrovsky step, suggesting a Volmer–Heyrovsky mechanism reaction (H3O+ + e− = Hads + H2O and H+ + e− + Hads = H2) onto the surface of Pt-5 and the electrochemical desorption (Heyrovsky step) is considered as the rate-limiting step, different from a Volmer–Tafel mechanism (H3O+ + e− = Hads + H2O and Hads + Hads = H2) for commercial Pt/C (Fig. 3c).11,18,40 The exchange current density (j0, η = 0) obtained by the extrapolation of Tafel plots is used to assess the intrinsic catalytic activity.21,43 Pt-5 is found to have a larger HER j0 value of 0.98 mA cm−2, higher than 0.59 mA cm−2 of commercial Pt/C, revealing excellent catalytic activity of Pt-5. To further compare the activity of Pt-loading catalysts, the mass activity is calculated by normalizing the content of Pt. The mass activity of Pt-5 displays an approximate 2.7-fold higher than that of commercial Pt/C at the overpotential of 50 and 100 mV, respectively (Fig. 3e).
logj, where a, b, and j represent intercept, Tafel slope and current density, respectively).39–41 The Tafel slope for Pt-5 is 43 mV dec−1, which is lower than those of Pt-1 (57 mV dec−1) and MoNx (114 mV dec−1). The smaller Tafel slope demonstrates there is a fast increase of the hydrogen generation rate for Pt-5.21,41 It is accepted that HER in acidic condition goes through two processes, including electrochemical reduction step (Volmer step) with a Tafel slope of 120 mV dec−1 and the desorption of adsorbed hydrogen. The latter is either electrochemical desorption (Heyrovsky step) with a Tafel slope of 40 mV dec−1 or recombination step (Tafel step) with a Tafel slope of 30 mV dec−1.42 The Tafel slope (43 mV dec−1) over the Pt-5 catalyst is close to that of Heyrovsky step, suggesting a Volmer–Heyrovsky mechanism reaction (H3O+ + e− = Hads + H2O and H+ + e− + Hads = H2) onto the surface of Pt-5 and the electrochemical desorption (Heyrovsky step) is considered as the rate-limiting step, different from a Volmer–Tafel mechanism (H3O+ + e− = Hads + H2O and Hads + Hads = H2) for commercial Pt/C (Fig. 3c).11,18,40 The exchange current density (j0, η = 0) obtained by the extrapolation of Tafel plots is used to assess the intrinsic catalytic activity.21,43 Pt-5 is found to have a larger HER j0 value of 0.98 mA cm−2, higher than 0.59 mA cm−2 of commercial Pt/C, revealing excellent catalytic activity of Pt-5. To further compare the activity of Pt-loading catalysts, the mass activity is calculated by normalizing the content of Pt. The mass activity of Pt-5 displays an approximate 2.7-fold higher than that of commercial Pt/C at the overpotential of 50 and 100 mV, respectively (Fig. 3e).
Electrochemical impedance spectroscopy (EIS) tests were performed at the applied potential of 300 mV (Fig. 3d). The lower charge transfer resistance (Rct) of Pt-5 is lower than those of Pt-1 and MoNx, indicating a faster charge transfer and more favorable reaction kinetics for HER.26,41 Electrochemical active surface areas (ECSA) were measured using the cyclic voltammetry (CV) curves in the potential range from 0.2 to 0.4 V (versus RHE) with scan rates of 5, 10, 20, 40, and 80 mV s−1 (Fig. S8†). The double-layer capacitance (Cdl) derived from the CV curve is proportional to ECSA. Larger Cdl indicates more exposed surface active sites. As illustrated in Fig. S8d,† there is a higher Cdl for Pt-5 (12.1 mF cm−2) than those of Pt-1 (8.3 mF cms−2) and MoNx (7.2 mF cm−2).
To explore the origin of the higher HER activity of Pt-5, the surface elements and valence state were investigated by X-ray photoelectron spectroscopy (XPS). The survey spectrum verified the existence of N, Mo, and Pt (Fig. S7a†). The Mo 3d of high-resolution XPS spectrum could be deconvoluted into three doublet peaks with separated spin–orbit components (Δ ≈ 3.15 eV) for three states of Moσ+, Mo4+, and Mo6+ on the Mo surface (Fig. 4a). For bare MoNx, the peaks at the binding energy (BE) of 229.04 and 232.19 eV for Mo 3d5/2 and 3d3/2, respectively, are originated from Moσ+ (σ = 2 or 3) of Mo–N in MoNx.44–46 The peaks located at 230.08 and 233.23 eV are attributed to Mo 3d5/2 and Mo 3d3/2 of Mo4+ for MoO2.47 And the doublet peaks of 232.82 and 235.97 eV are ascribed to Mo 3d5/2 and Mo 3d3/2 of Mo6+ for MoO3. The inactive MoO2 and MoO3 are derived from surface oxidation of MoNx, which is commonly inevitable upon air exposure.41,48 Compared to the bare MoNx, the Mo 3d peaks of Pt-5 and Pt-1 exhibit obvious red-shifts (Fig. S7b and Table S2†). The positions of Moσ+ peaks shift to lower binding energy implying that there are more electrons surrounding the Mo atoms.26,36 The enriched electrons around Mo can improve HER activity by decreasing the density of empty d-orbitals and optimizing Mo–H binding energy.46 As confirmed by the results, Pt doping could modulate the electronic feature of Mo. This is coincident with the literature reports, the electron structure of Mo could be tuned via doping transition metals.17,26 Considering the partial overlap of N 1s and Mo 3p,46 the N 1s spectrum can be fitted into three peaks (Fig. 4c). The peaks located at 394.85 and 397.72 eV correspond to N 1s and Mo 3p of Mo–N, respectively. The remaining peak at 399.97 eV is attributed to Mo–O of MoOx for Mo 3p.32,49 The XPS results of N 1s, Mo 3d, and Mo 3p verify the formation of the MoNx. The XPS spectrum of Pt 4f can be deconvoluted into two doublet peaks (Fig. 4b). The peaks at 71.55 and 74.85 eV are attributed to metallic Pt, and the peaks at 72.52 and 76.86 eV are assigned to Pt2+.42
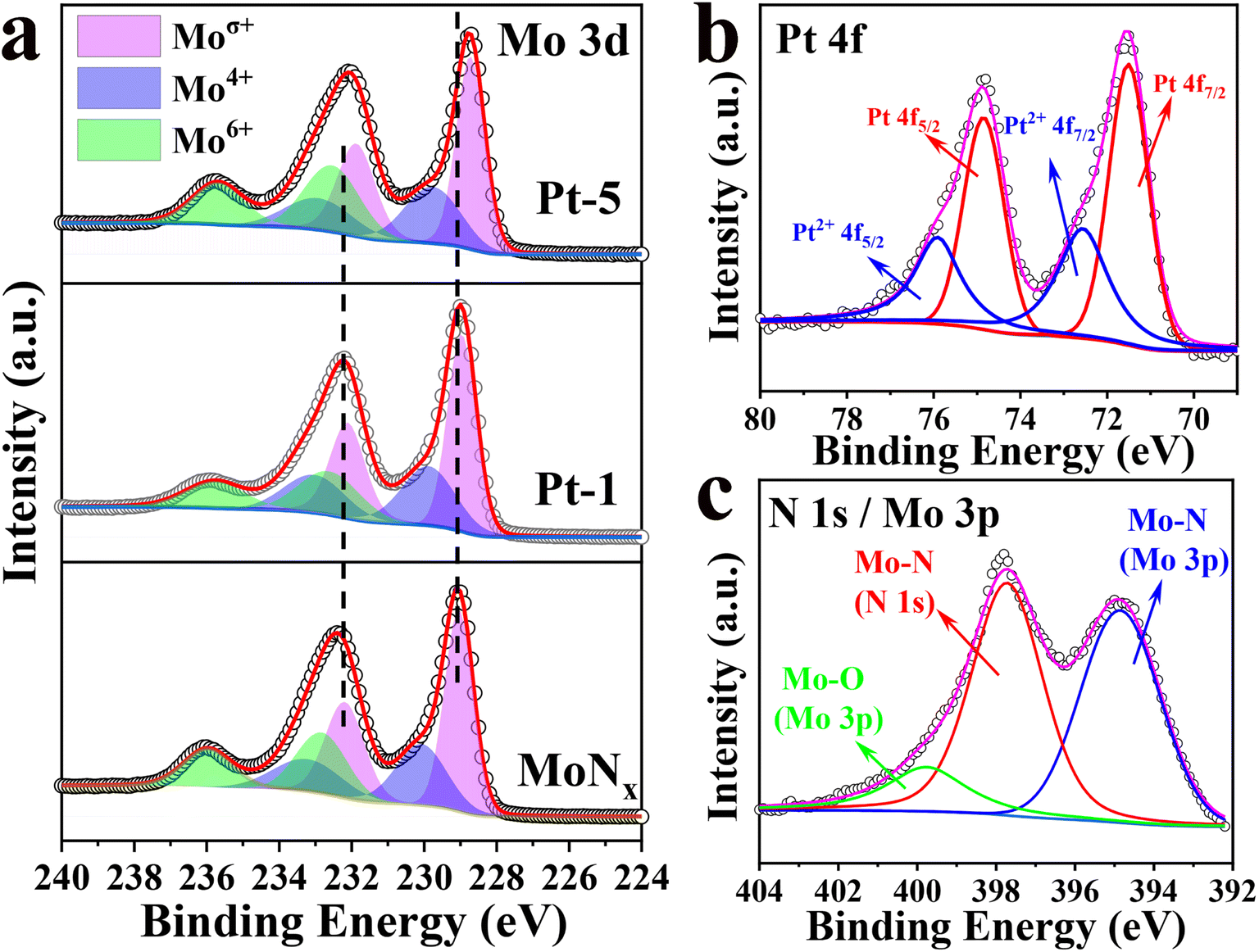 | ||
| Fig. 4 High-resolution XPS spectra of Mo 3d (a), Pt 4f (b) and N 1s/Mo 3p (c). | ||
The stability of the catalyst plays an important role in the practical electrocatalysis process. The stability of as-synthesized catalyst was evaluated by continuous CV cycling tests and the chronopotentiometric curve. As shown in Fig. 3f, there is a slight decay with an overpotential of 5 mV for Pt-5 after 2000 CV cycles between −0.5 and 0.2 V (versus RHE). Moreover, multi-current steps chronopotentiometry curve of Pt-5 was recorded with current densities of 10, 50, and 100 mA cm−2. During each constant current period, the potential change is negligible. All these results demonstrate Pt-5 has a higher durability for HER in acidic electrolyte. Furthermore, the structural stability of Pt-5 was further confirmed by XRD and TEM after long-term operation (Fig. S9†). According to the results of XRD patterns and TEM images, the crystalline structure and morphology of Pt-5 are well preserved after continuous CV cyclings and chronopotentiometric tests.
Conclusion
In summary, Pt-doping molybdenum nitrides electrocatalysts were fabricated by hydrothermal-ammoniation, followed with wetness impregnation-reduction process for efficient hydrogen evolution reaction. It is found that introducing an appropriate amount of Pt can enrich the electron around Mo, which leads to weakening of Mo–H bonding energy, thereby enhancing HER activity. As a result, Pt (5 wt%)–MoNx exhibited excellent performance with a lower overpotential (η10 = 47 mV) and Tafel slope (43 mV dec−1), comparable to most of the reported Mo-based catalysts. This work may provide a strategy to develop transition metal nitrides with modifiable electronic structure for electrocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Research Funds of Shanxi Institute of Science and Technology (2023011) and Science and Technology Innovation Program for Higher Education Institutions of Shanxi Province (2023L443).References
- W. Yuan, T. Jiang, X. Fang, Y. Fan, S. Qian, Y. Gao, N. Cheng, H. Xue and J. Tian, Chem. Eng. J., 2022, 439, 135743 CrossRef CAS.
- J. Xu, C. Zhang, H. Liu, J. Sun, R. Xie, Y. Qiu, F. Lü, Y. Liu, L. Zhuo, X. Liu and J. Luo, Nano Energy, 2020, 70, 104529 CrossRef CAS.
- H. Wei, Q. Xi, X. Chen, D. Guo, F. Ding, Z. Yang, S. Wang, J. Li and S. Huang, Adv. Sci., 2018, 5, 1700733 CrossRef.
- Y. Li, X. Wei, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 19550–19571 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. Hou, Y. Wu, B. Zhang, S. Cao, Z. Li and L. Sun, Adv. Funct. Mater., 2019, 29, 1808367 CrossRef.
- J. He, F. Liu, Y. Chen, X. Liu, X. Zhang, L. Zhao, B. Chang, J. Wang, H. Liu and W. Zhou, Chem. Eng. J., 2022, 432, 134331 CrossRef CAS.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- J. Liu, C. Tang, Z. Ke, R. Chen, H. Wang, W. Li, C. Jiang, D. He, G. Wang and X. Xiao, Adv. Energy Mater., 2022, 12, 2103301 CrossRef CAS.
- J. Zeng, L. Zhang, Q. Zhou, L. Liao, Y. Qi, H. Zhou, D. Li, F. Cai, H. Wang, D. Tang and F. Yu, Small, 2022, 18, 2104624 CrossRef CAS.
- C. Wu, D. Liu, H. Li and J. Li, Small, 2018, 14, 1704227 CrossRef.
- J. Wang, Y. Sun, Y. Qi and C. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 57392–57402 CrossRef CAS.
- K. Wang, C. Liang, S. Li, J. Li, Z. Yi, F. Xu, Y. Wang, L. Lei, M. Zhu, S. Li, L. Zhuang and Z. Xu, Sci. China Mater., 2023, 66, 2662–2671 CrossRef CAS.
- M. Kuang, J. Zhang, D. Liu, H. Tan, K. N. Dinh, L. Yang, H. Ren, W. Huang, W. Fang, J. Yao, X. Hao, J. Xu, C. Liu, L. Song, B. Liu and Q. Yan, Adv. Energy Mater., 2020, 10, 2002215 CrossRef CAS.
- Z. Yin, X. Liu, S. Chen, H. Xie, L. Gao, A. Liu, T. Ma and Y. Li, Mater. Today Nano, 2022, 17, 100156 CrossRef CAS.
- H. Tabassum, R. Zou, A. Mahmood, Z. Liang and S. Guo, J. Mater. Chem. A, 2016, 4, 16469–16475 RSC.
- X. Lang, M. A. Qadeer, G. Shen, R. Zhang, S. Yang, J. An, L. Pan and J.-J. Zou, J. Mater. Chem. A, 2019, 7, 20579–20583 RSC.
- H. J. Song, M. C. Sung, H. Yoon, B. Ju and D. W. Kim, Adv. Sci., 2019, 6, 1802135 CrossRef.
- J. Miao, Z. Lang, X. Zhang, W. Kong, O. Peng, Y. Yang, S. Wang, J. Cheng, T. He, A. Amini, Q. Wu, Z. Zheng, Z. Tang and C. Cheng, Adv. Funct. Mater., 2019, 29, 1805893 CrossRef.
- P. Wang, J. Qi, C. Li, X. Chen, T. Wang and C. Liang, ChemElectroChem, 2020, 7, 745–752 CrossRef CAS.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, 1802880 CrossRef.
- Y. Xu, T. Chen, T. Wang, J. Yang, Y. Zhu and W. Ding, Nanoscale, 2018, 10, 21944–21950 RSC.
- F. G. Baddour, E. J. Roberts, A. T. To, L. Wang, S. E. Habas, D. A. Ruddy, N. M. Bedford, J. Wright, C. P. Nash, J. A. Schaidle, R. L. Brutchey and N. Malmstadt, J. Am. Chem. Soc., 2020, 142, 1010–1019 CrossRef CAS.
- H. Jiang and J. Caro, Chem, 2017, 3, 209–210 CAS.
- Y. Ma, G. Guan, C. Shi, A. Zhu, X. Hao, Z. Wang, K. Kusakabe and A. Abudula, Int. J. Hydrogen Energy, 2014, 39, 258–266 CrossRef CAS.
- Y. Ma, M. Chen, H. Geng, H. Dong, P. Wu, X. Li, G. Guan and T. Wang, Adv. Funct. Mater., 2020, 30, 2000561 CrossRef CAS.
- S. Kim, C. Choi, J. Hwang, J. Park, J. Jeong, H. Jun, S. Lee, S.-K. Kim, J. H. Jang, Y. Jung and J. Lee, ACS Nano, 2020, 14, 4988–4999 CrossRef CAS.
- S. Yao, L. Lin, W. Liao, N. Rui, N. Li, Z. Liu, J. Cen, F. Zhang, X. Li, L. Song, L. Betancourt De Leon, D. Su, S. D. Senanayake, P. Liu, D. Ma, J. G. Chen and J. A. Rodriguez, ACS Catal., 2019, 9, 9087–9097 CrossRef CAS.
- S. Li, J. Liu, Z. Yin, P. Ren, L. Lin, Y. Gong, C. Yang, X. Zheng, R. Cao, S. Yao, Y. Deng, X. Liu, L. Gu, W. Zhou, J. Zhu, X. Wen, B. Xu and D. Ma, ACS Catal., 2019, 10, 907–913 CrossRef.
- J. Xie, S. Li, X. Zhang, J. Zhang, R. Wang, H. Zhang, B. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 RSC.
- L. Ma, L. R. L. Ting, V. Molinari, C. Giordano and B. S. Yeo, J. Mater. Chem. A, 2015, 3, 8361–8368 RSC.
- X. Xiao, H. Yu, H. Jin, M. Wu, Y. Fang, J. Sun, Z. Hu, T. Li, J. Wu, L. Huang, Y. Gogotsi and J. Zhou, ACS Nano, 2017, 11, 2180–2186 CrossRef CAS.
- J. Xiong, W. Cai, W. Shi, X. Zhang, J. Li, Z. Yang, L. Feng and H. Cheng, J. Mater. Chem. A, 2017, 5, 24193–24198 RSC.
- G. Liu, H. Bai, Y. Ji, L. Wang, Y. Wen, H. Lin, L. Zheng, Y. Li, B. Zhang and H. Peng, J. Mater. Chem. A, 2019, 7, 12434–12439 RSC.
- F. Li, X. Zhao, J. Mahmood, M. S. Okyay, S.-M. Jung, I. Ahmad, S.-J. Kim, G.-F. Han, N. Park and J.-B. Baek, ACS Nano, 2017, 11, 7527–7533 CrossRef CAS.
- H. Lin, N. Liu, Z. Shi, Y. Guo, Y. Tang and Q. Gao, Adv. Funct. Mater., 2016, 26, 5590–5598 CrossRef CAS.
- J. Wang, X. Yan, Z. Zhang, H. Ying, R. Guo, W. Yang and W. Q. Han, Adv. Funct. Mater., 2019, 29, 1904819 CrossRef.
- T. Wang, Z. Kou, S. Mu, J. Liu, D. He, I. S. Amiinu, W. Meng, K. Zhou, Z. Luo, S. Chaemchuen and F. Verpoort, Adv. Funct. Mater., 2018, 28, 1705048 CrossRef.
- H. C. Yang, C. H. Wang, F. Hu, Y. J. Zhang, H. Lu and Q. B. Wang, Sci. China Mater., 2017, 60, 1121–1128 CrossRef CAS.
- X. Yan, H. Li, J. Sun, P. Liu, H. Zhang, B. Xu and J. Guo, Carbon, 2018, 137, 405–410 CrossRef CAS.
- X. Zhang, J. Wang, T. Guo, T. Liu, Z. Wu, L. Cavallo, Z. Cao and D. Wang, Appl. Catal. B, 2019, 247, 78–85 CrossRef CAS.
- H. Yuan, L. Zhao, B. Chang, Y. Chen, T. Dong, J. He, D. Jiang, W. Yu, H. Liu and W. Zhou, Appl. Catal. B, 2022, 314, 121455 CrossRef CAS.
- J. Ji, Y. Zhang, L. Tang, C. Liu, X. Gao, M. Sun, J. Zheng, M. Ling, C. Liang and Z. Lin, Nano Energy, 2019, 63, 103849 CrossRef CAS.
- H. Yan, Y. Xie, Y. Jiao, A. Wu, C. Tian, X. Zhang, L. Wang and H. Fu, Adv. Mater., 2018, 30, 1704156 CrossRef.
- L. Zhu, L. Sun, H. Zhang, D. F. Yu, H. Aslan, J. G. Zhao, Z. L. Li, M. Yu, F. Besenbacher and Y. Sun, Nano Energy, 2019, 57, 842–850 CrossRef CAS.
- X. Shi, A. Wu, H. Yan, L. Zhang, C. Tian, L. Wang and H. Fu, J. Mater. Chem. A, 2018, 6, 20100–20109 RSC.
- C. Bin, H. Yuan, L. Li, J. Yu, X. Liu, W. Yu, B. Wang, L. Zhao, X. Liu, S. Sun, H. Liu and W. Zhou, Appl. Catal. B, 2023, 320, 121777 CrossRef.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef.
- Y. Zhu, G. Chen, X. Xu, G. Yang, M. Liu and Z. Shao, ACS Catal., 2017, 7, 3540–3547 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06954k |
| This journal is © The Royal Society of Chemistry 2023 |