
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA tri(ethylene glycol)-tethered Morita–Baylis–Hillman dimer in the formation of macrocyclic crown ether-paracyclophane hybrid structures†
Mario Saletti ,
Jacopo Venditti,
Marco Paolino,
Arianna Zacchei,
Germano Giuliani,
Gianluca Giorgi,
Claudia Bonechi,
Alessandro Donati and
Andrea Cappelli*
,
Jacopo Venditti,
Marco Paolino,
Arianna Zacchei,
Germano Giuliani,
Gianluca Giorgi,
Claudia Bonechi,
Alessandro Donati and
Andrea Cappelli*
Dipartimento di Biotecnologie, Chimica e Farmacia, Università di Siena, Via A. Moro 2, 53100 Siena, Italy. E-mail: andrea.cappelli@unisi.it; Tel: +39 0577 232416
First published on 11th December 2023
Abstract
A Morita–Baylis–Hillman acetate was dimerized by a click-chemistry Copper(I)-Catalysed Azide–Alkyne Cycloaddition (CuAAC) reaction employing a tri(ethylene glycol) diazide derivative to obtain a dimeric MBHA derivative. The reaction of this dimeric MBHA derivative with n-butylamine afforded a photoisomerizable macrocyclic crown ether-paracyclophane hybrid architecture that is potentially useful in a large variety of applications as well as those already well-known for crown ethers.
Introduction
Crown ethers are macrocyclic compounds of poly(ethylene glycol) (PEG) that can exist in two forms: aliphatic ones, formed from four to ten ethylene oxide units, and the aromatic ones, in which we can find one or two benzene rings (benzocrown and dibenzocrown ethers respectively). Their name comes from the studies conducted by Charles J. Pedersen who in 1967 observed the aspect of host–guest complexes formed with metal cations. This ability is due to the conformational and chemical features of the system, which allow the creation of an electron rich cavity whose dimensions are crucial to confer a coordination selectivity of alkali, alkaline earth ions and, as recently demonstrated, non-metallic species. For these reasons, crown ethers have many applications: as phase transfer catalysts, for creating chiral stationary phases and in the construction of mechanically interlocked structures such as rotaxanes, pseudorotaxanes and catenanes. This has led to their use in the development of synthetic molecular devices activated by light, pH and redox. They also have applications in the biological field as parts of artificial intramembrane canals and to allow the translocation of ionic species.1–3The ability to control the functions of crown ethers by an on–off light switch was investigated by Shinkai and coworkers in 1987. They synthesized photoresponsive crown ethers having an intra-annular azo substituent that could interact with a metal only following photoisomerization of the intra-annular azo substituent from trans to cis.4 Also Vicens and coworkers explained an interesting system where the binding activity of the calix[4]crown ether is photosensitive: Na+ is favorably extracted by the trans-isomer of the structure, while the cis form extracts better the Rb+ and Cs+.5
In the field of the synthesis and study of mechanically interlocked molecules (MIMs), the concept of “click” chemistry is very relevant. In 2001 Sharpless, Finn, and Kolb, proposed that chemists could generate substances by linking small modular units in reactions with high yield, high chemoselectivity and stereospecificity, high thermodynamic driving force that could lead to the formation of irreversible bonds and ultimately under mild reaction conditions with few or no by-products. For these reasons, the best-known click reaction, the Copper(I)-Catalised Azide–Alkyne Cycloaddition (CuAAC) was quickly embraced by the MIM community as soon as it appeared. Many syntheses of MIMs are based on click-type reactions, such as the formation of amide, ureidic, carbamate, or SN2-type bonds and cycloaddition reactions to stop the interlocked structure.
The mild and chemoselective nature of the CuAAC reaction was the first quality to be highlighted in practice: Stoddart described the synthesis of rotaxanes and catenanes using the CuAAC reaction together with their iconic “blue box” viologen macrocycle.6
The Morita–Baylis–Hillman (MBH) reaction is very important in the field of organic chemistry because it afford to create new C–C bonds. In particular, MBH Adducts (MBHA) arise from a reaction between a carbon electrophile (for example an aldehyde) and an activated alkene, catalysed by a tertiary amine or phosphine, to provide an allylic alcohol.7 This can be processed into a great leaving group by acetylation. In case an activated imine reacts instead of the aldehyde, it is called aza-Morita–Baylis–Hillman reaction. These reactions allow to obtain versatile and chiral products using chiral substrates or organic chiral catalysts; this makes them an excellent example of organocatalysis for green chemistry.8
Some years ago, we have developed MBHA derivatives capable of reacting with imidazole to afford cinnamic derivatives showing fluorogenic properties.9 Moreover, the structures of these compounds were subsequently elaborated to obtain a number of MBHA derivatives potentially useful in a wide range of different applications in protein functionalization (Fig. 1).10–14
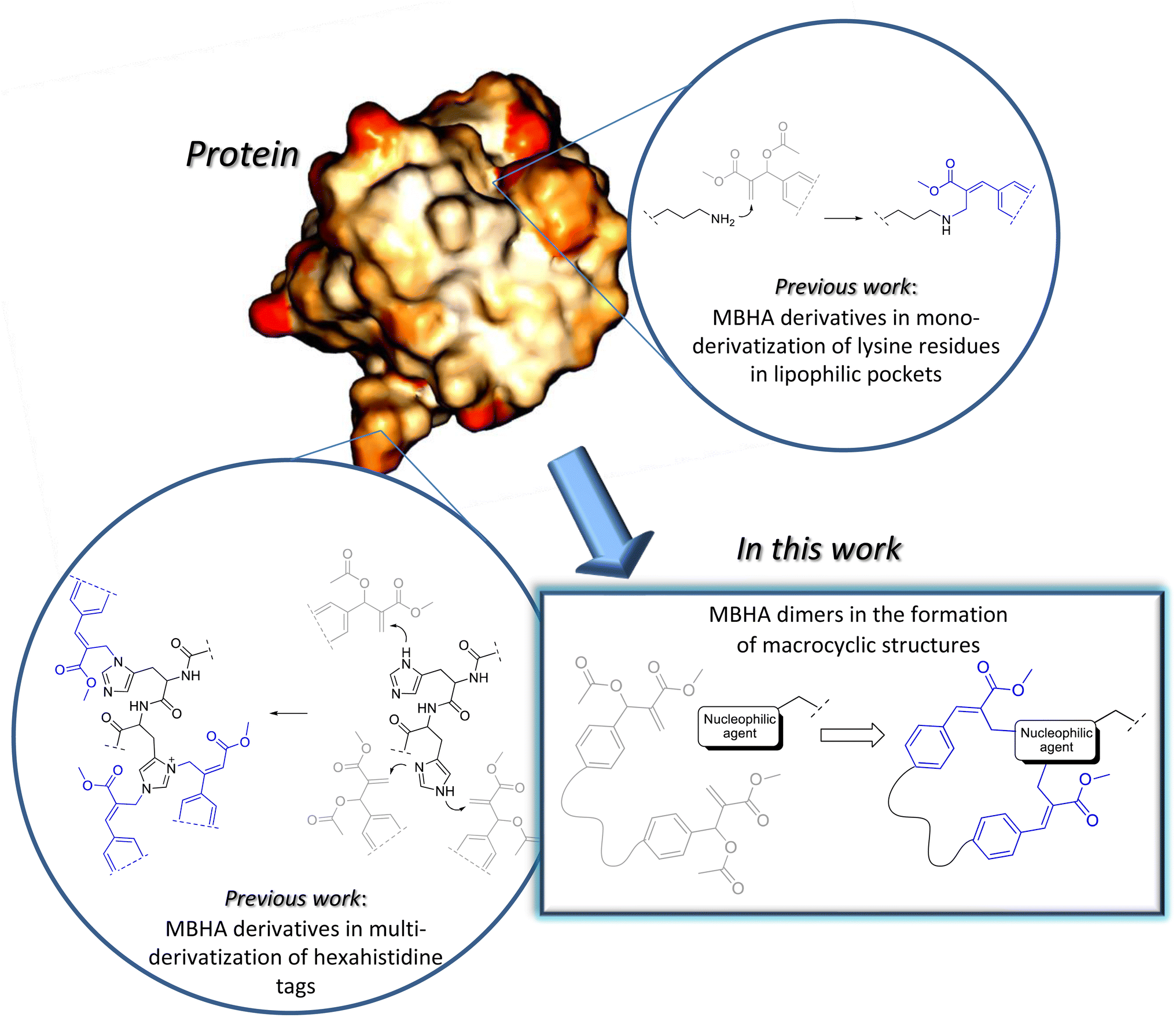 | ||
| Fig. 1 Applications of MBHA derivatives to protein functionalization. | ||
In the present work, the structure of our MBHA derivatives was manipulated in the aim of obtaining a tri(ethylene glycol)-tethered MBHA dimer 1 (Fig. 2) potentially useful in the functionalization of materials containing reactive basic (i.e. amino or imidazole) groups with the formation of amphiphilic loops potentially provided with a wide range of intriguing properties such as those shown by both crown ethers and paracyclophane derivatives. We found that the tri(ethylene glycol)-tethered MBHA dimer 1 was capable of reacting with primary amines (i.e. n-butylamine) leading to the formation of macrocyclic crown ether-paracyclophane hybrid structures 2 that could be modulated by light.
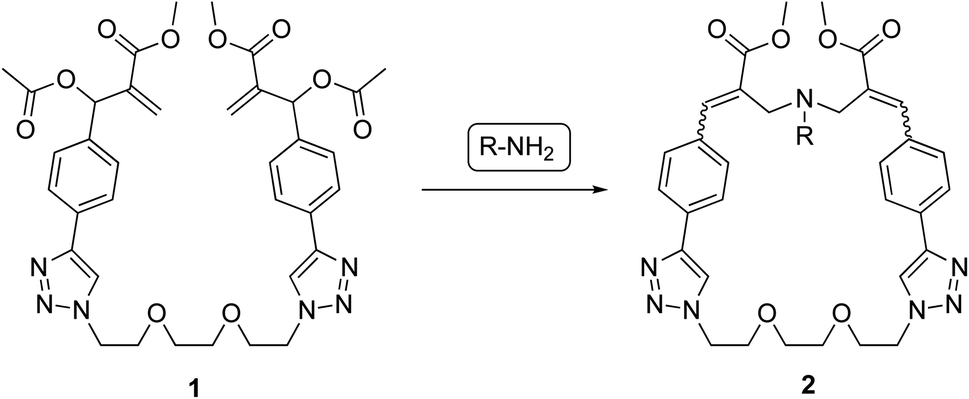 | ||
| Fig. 2 Reaction of MBHA dimer 1 with primary amines leading to macrocyclic crown ether-paracyclophane hybrid structures 2. | ||
Results and discussion
Synthesis and structural characterization
The synthesis of dimeric MBHA derivative 1 was carried out by the convergent procedure shown in Scheme 1.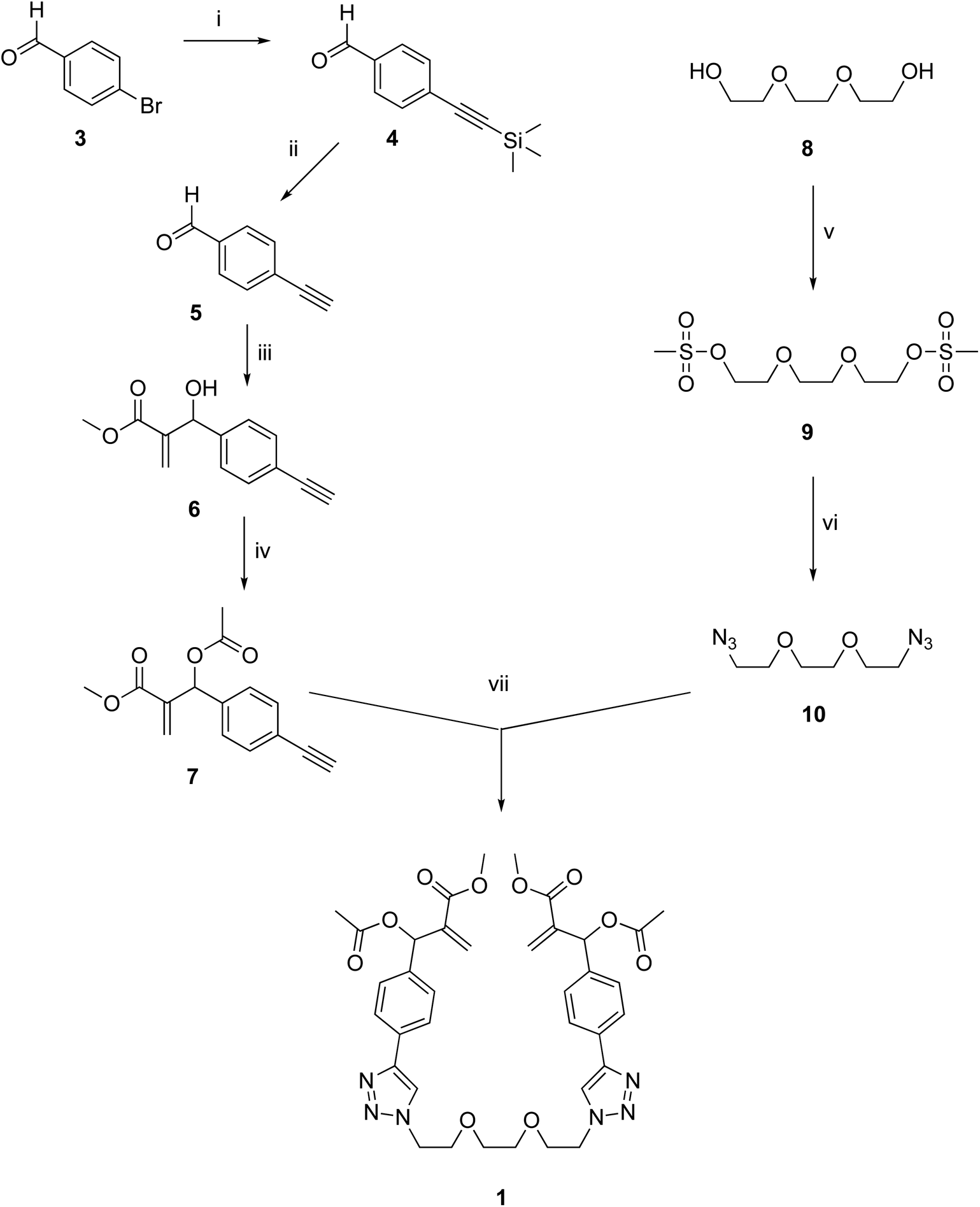 | ||
| Scheme 1 Convergent procedure for the preparation of dimeric MBHA derivative 1. Reagents: (i) trimethylsilylacetylene, Pd(PPh3)2Cl2, CuI, TEA, THF; (ii) K2CO3, MeOH; (iii) DABCO, methyl acrylate, MeOH; (iv) CH3COCl, TEA, CH2Cl2; (v) CH3SO2Cl, TEA, CH2Cl2; (vi) NaN3, DMF, CH3CN; (vii) CuBr(I), DIPEA, CH3CN. | ||
The MBHA component 7 of the convergent synthesis was prepared from 4-bromobenzaldehyde 3, which was used in a Sonogashira reaction with trimethylsilylacetylene in the presence of Pd(PPh3)2Cl2, CuI, and TEA in THF as the solvent to obtain trimethylsilyl derivative 4,15 which was promptly desilylated with potassium carbonate in methanol. The resulting aldehyde 5 (ref. 16) was used in a Morita–Baylis–Hillman reaction with methyl acrylate in the presence of DABCO and methanol to afford MBHA derivative 6, which was promptly acetylated with acetyl chloride in the presence of triethylamine as the base in dry DCM, with a satisfactory yield of 7 (i.e. 82%). The diazide component 10 of the convergent synthesis was prepared starting from the commercially available tri(ethylene glycol) 8, which was activated by reaction with mesyl chloride to the corresponding mesylate 9 and then made to react with sodium azide in DMF-acetonitrile. The final coupling step was performed in the conditions of a click chemistry Copper(I)-Catalised Azide–Alkyne Cycloaddition (CuAAC) reaction in acetonitrile in the presence of CuBr(I) as the catalyst and DIPEA as the base to obtain the tri(ethylene glycol)-tethered MBHA dimer 1.
Dimeric MBHA derivative 1 was found to react with n-butylamine in refluxing chloroform to afford the mixture of macrocyclic diastereomers 2a,b in good yield (Scheme 2).
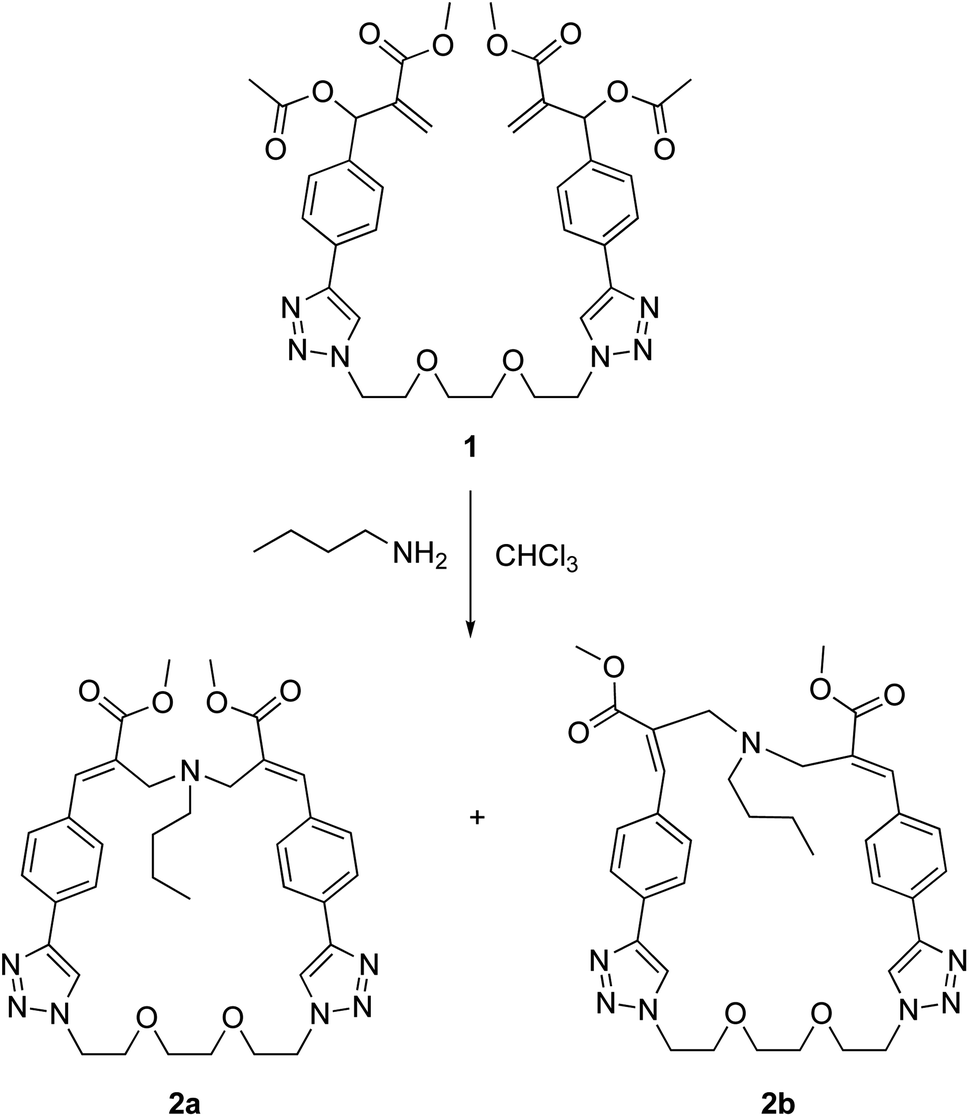 | ||
| Scheme 2 Reaction of MBHA dimer 1 with n-butylamine leading to macrocyclic crown ether-paracyclophane hybrid structures 2a,b. | ||
Fortunately, these isomers showed suitable physicochemical features to be easily separated by flash chromatography with ethyl acetate–methanol (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) as the eluent. In particular, compound 2b was obtained as a white crystalline solid (yield 17%) as the least polar fraction of the chromatographic purification, whereas compound 2a was obtained as a yellowish-white crystalline solid (yield 35%) as the most polar fraction of the chromatographic purification. Moreover, after purification by flash chromatography, compound 2a was recrystallized from ethyl acetate by slow evaporation to obtain single crystals suitable for X-ray diffraction studies, which allowed the structure to be confirmed by crystallographic studies (Fig. 3).
:5) as the eluent. In particular, compound 2b was obtained as a white crystalline solid (yield 17%) as the least polar fraction of the chromatographic purification, whereas compound 2a was obtained as a yellowish-white crystalline solid (yield 35%) as the most polar fraction of the chromatographic purification. Moreover, after purification by flash chromatography, compound 2a was recrystallized from ethyl acetate by slow evaporation to obtain single crystals suitable for X-ray diffraction studies, which allowed the structure to be confirmed by crystallographic studies (Fig. 3).
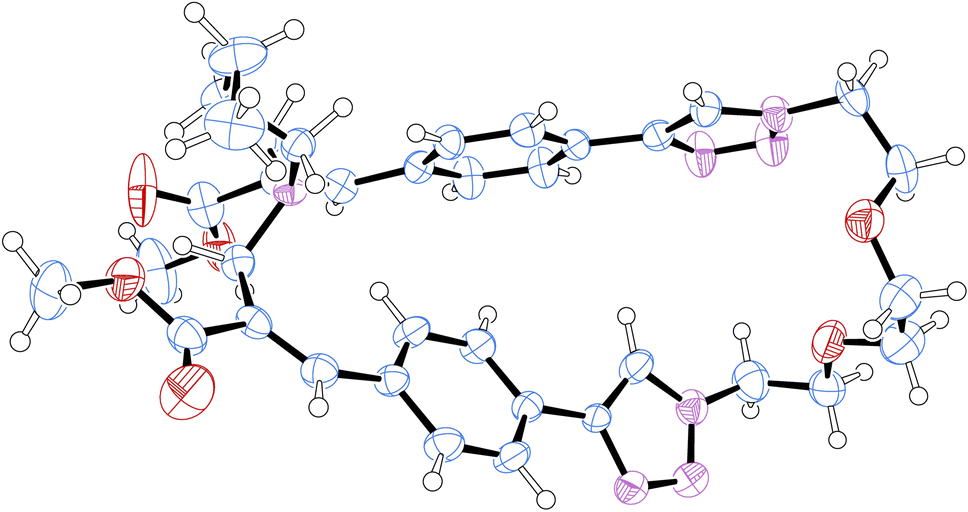 | ||
| Fig. 3 Structure of compound 2a obtained by crystallographic studies. Ellipsoids enclose 50% probability. | ||
The crystallographic structure of (E,E) diastereomer 2a was considered to constitute a good starting point for the structural characterization of 2b. Thus, the 1H and 13C NMR spectra of 2a were assigned and compared with those of the corresponding (E,Z) diastereomer 2b (Fig. 4 and 5).
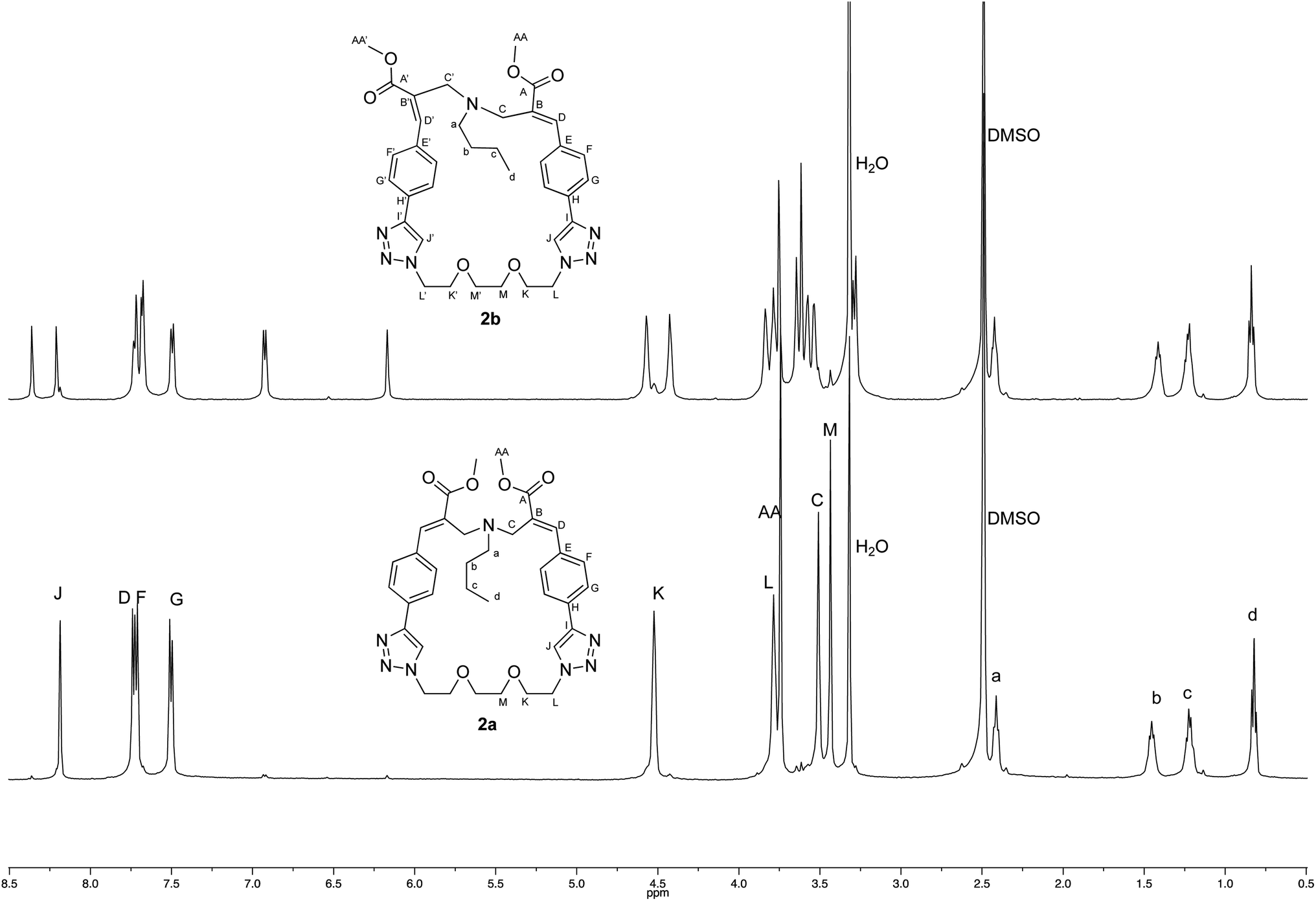 | ||
| Fig. 4 Comparison of the 1H NMR spectra recorded (500 MHz, DMSO-d6) with crystalline samples of 2a and 2b. | ||
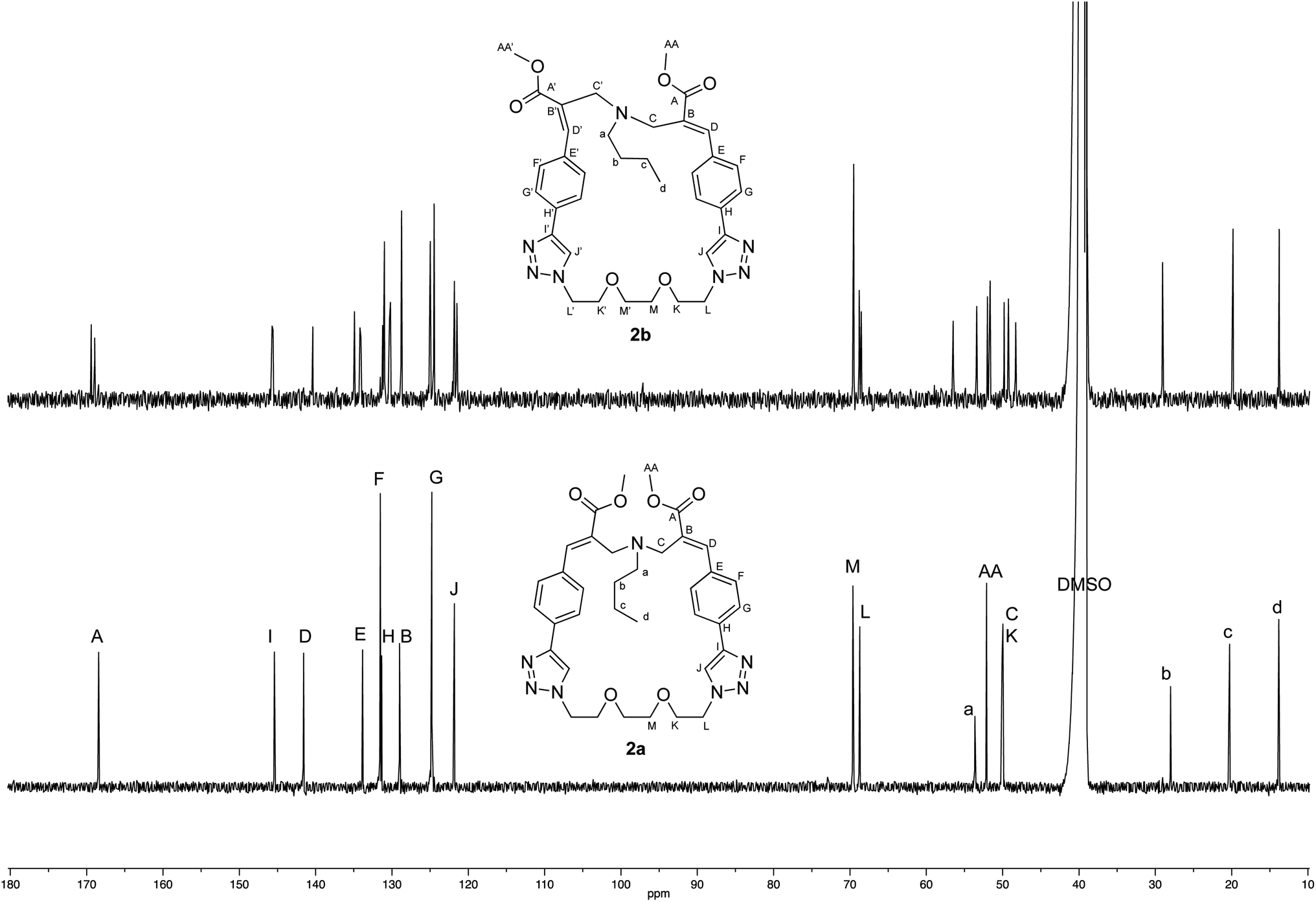 | ||
| Fig. 5 Comparison of the 13C NMR spectra recorded (125 MHz, DMSO-d6) with crystalline samples of 2a and 2b. | ||
The comparison stressed the symmetric structure of 2a (as supported by the crystallographic studies) with respect to the relative lack of symmetry of (E,Z) diastereomer 2b, which showed two distinct sets of signals in almost all the regions of its NMR spectra with the exception if the signals of the butylamine moiety. Thus, these results supported the structure of diastereomer 2b.
The structures of macrocyclic compounds 2a,b were investigated also by several different mass spectrometry techniques such as trapped ion mobility mass spectrometry (TIMS), tandem mass spectrometry (MSn), and energy resolved tandem mass spectrometry. The results of TIMS characterization suggested the presence of two different ion populations due to different protonation sites in the amine and triazole nitrogen atoms or to two different conformations. However, the MS/MS spectra of the two ion populations were superimposable, thus supporting the latter hypothesis rather than the former one. For both compounds fragmentation pathways show the loss of 28 u, due to N2 from the triazole moiety, and of C4H9N from the butylamine moiety. Moreover, energy resolved tandem mass spectrometry showed that diastereomer (E,E) 2a appeared to be more stable than the corresponding (E,Z) diastereomer 2b in agreement with the most abundant formation in the reaction of 1 with n-butylamine.
Photophysical and photochemical characterizations
The photophysical features of compounds 2a,b were investigated in terms of absorption spectra in methanol (see ESI†). The absorption spectra of these two cinnamic derivatives showed very similar low energy peaks in the UV-B range, with absorption maximum at ca. 300 nm for the (E,E) diastereomer 2a and a slight blue shifted peak at ca. 290 nm for the (E,Z) diastereomer 2b. The photochemical features of the macrocyclic compounds were investigated by 1H NMR spectroscopy studies starting from (E,E) diastereomer 2a owing to its well-established (i.e. by crystallography) symmetric structure, the simplicity of its 1H NMR spectrum, and its solubility in deuterated solvents such as methanol and DMSO. Thus, compound 2a was dissolved in deuterated methanol into a 5 mm NMR tube and the resulting solutions were exposed to UV-B light into an appropriate photoreactor (Multirays, Helios Quartz). 1H NMR spectra were registered at regular time intervals, elaborated, and compared (see ESI†). The comparison of the 1H NMR spectra recorded with the solution of 2a in deuterated methanol exposed to UV-B irradiation for increasing times (i.e. from 5 to 30 min) supported the liability of this (E,E) diastereomer to undergo photoisomerization with the formation of an almost insoluble compound, which precipitated from the reaction mixture. In fact, after 15 min exposition to UV-B irradiation, the formation of a white precipitate was observed, and the amount of this material appeared to increase in the time with the UV-B exposition. After storing the NMR tube containing the mixture at room temperature for three weeks, the spectrum suggested that negligible amounts of materials remained in the solution. Thus, a spectrum was recorded after dissolving the white precipitate with deuterated DMSO, and this spectrum supported the formation of (E,Z) diastereomer 2b by photoisomerization with UV-B light of (E,E) diastereomer 2a in methanol. Apparently, the low solubility of the light-induced (E,Z) diastereomer 2b in methanol could affect this photoisomerization reaction, but further experiments in different solvents are granted to explore the potential existence of different reaction pathways.Computational studies
To further support the experimental data obtained, we calculated the relative energies of the two isomers using density functional theory (DFT) methodology (Fig. 6). Calculations were performed using B3LYP as functional and 6-31G* as basis set of the GAUSSIAN package (version 16).17 The Polarized Continuum Model (PCM) simulating the solvent environment and Grimme dispersion D3 correction for long range (van der Waals) interactions were used. The two diastereomers were fully optimized in DMSO as implicit solvent (ε = 46.826), calculating the energy gap (at 298 K) between the conformations. The results confirm the higher stability of the (E,E) diastereomer 2a compared to the (E,Z) one 2b, with an energy gap of 6.2 kcal mol−1. Further computational studies are in progress in order to rationalize the photoisomerization features of these interesting macrocyclic derivatives.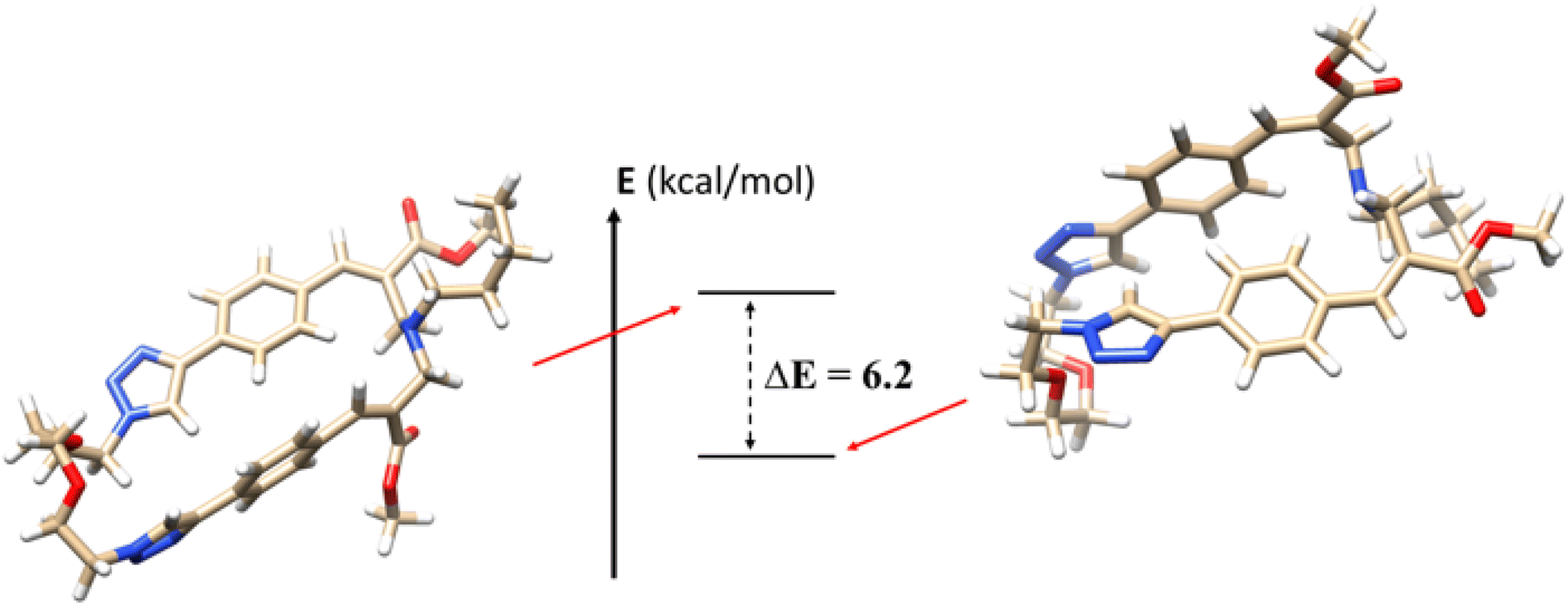 | ||
| Fig. 6 Comparison of the 3D structures of 2a (right) and 2b (left). | ||
Conclusions
In conclusion, a tri(ethylene glycol)-tethered MBHA dimer (1) was synthesized and found to react with n-butylamine leading to the formation of macrocyclic crown ether-paracyclophane hybrid structures 2 that could be modulated by light. Thus, reactive MBHA dimer 1 could find applications in the functionalization of materials containing reactive basic groups with the formation of amphiphilic loops provided with a wide range of intriguing properties such as those shown by both crown ethers and paracyclophane derivatives. Deeper investigations are in progress to characterize the complex photoisomerization features of this interesting new family of macrocyclic derivatives, also in light of the little information on similar structures reported in the literature.18Experimental section
Chemistry
All chemicals used were of reagent grade. Yields refer to purified products and are not optimized. Melting points were determined in open capillaries on a Gallenkamp apparatus and are uncorrected. Merck silica gel 60 (230–400 mesh) was used for flash chromatography purifications. Merck TLC plates, silica gel 60 F254 were used for TLC. NMR spectra were recorded with a Bruker DRX-400 AVANCE or a Bruker DRX-500 AVANCE spectrometer in the indicated solvents (TMS as internal standard): the values of the chemical shifts are expressed in ppm and the coupling constants (J) in Hz. Mass spectra were recorded on an Agilent 1100 LC/MSD operating with an electrospray source.X-ray crystallography
A single crystal of 2a was submitted to X-ray data collection on an Oxford-Diffraction Xcalibur Sapphire 3 diffractometer with a graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at 293 K. The structure was solved by direct methods implemented in SHELXS-97 program.19 The refinement was carried out by full-matrix anisotropic least-squares on F2 for all reflections for non-H atoms by means of the SHELXL-97 program.20 The structure crystallizes in the monoclinic crystal system and space group P21/a with cell parameters: a = 10.9638(9) b = 22.3683(18) c = 14.3096(13)Å, β = 102.531(8)°. Crystallographic data for this structure have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 2286050. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; (fax: + 44 (0) 1223 336 033; or e-mail: E-mail: deposit@ccdc.cam.ac.uk).Mass spectrometry measurements
Each compound was dissolved in methanol (Sigma, HPLC Grade) to obtain a final concentration of about 1 × 10−5 M and injected in the electrospray source via flow injection at a flow rate of 5 μL min−1.Two different mass spectrometers have been used: an LCQ-Deca ion trap (IT, Thermo Finnigan, Bremen, D) and a timsTOF trapped ion mobility QTOF (Bruker, Bremen, D), both of them operating with an electrospray ionization source (ESI) in positive ion mode.
Operating conditions for the ESI sources were as follows: IT: spray voltage 4.5 kV; capillary temperature 200 °C; sheath gas (nitrogen) flow rate ca. 0.75 L min−1; tims TOF: end plate offset 500 V; spray voltage 4.5 kV; capillary temperature 180 °C; sheath gas (nitrogen) flow pressure 5.8 psi; dry gas flow rate 4.0 L min−1.
The calibration of the timsTOF mass spectrometer was carried out using Na Formate: 10 mM NaOH in a solution of isopropanol/H2O (50/50) with 0.2% of formic acid.
MSn product ion experiments carried out inside the ion trap were done by isolating the precursor ion and then by applying a supplementary potential for collision induced dissociations; collision gas: He; collision energy: 20–40% arbitrary units.
MS2 product ion experiments carried out inside the timsTOF were done by isolating the precursor ion, by the quadrupole analyzer, and submitting it to collision induced dissociation reactions inside the collision cell with nitrogen and making production ion analysis in high resolution mode inside the time of flight analyzer.
In all MSn experiments the isolation window of the precursor ion was 1 or 2 u.
For ion mobility MS experiments, the timsTOF instrument has been used: gas inside the TIMS cell was N2, and the temperature of the cell was 305 K. The TIMS mode was set on “custom” with 1/K0 starting on 1.23 V‧s cm−2 and 1/K0 ending on 1.27 V‧s cm2; ramp time was set on 600.0 ms with lock duty cycle at 100%. The calibration was carried out manually for each calibrant, using ESI-L Low Concentration Tuning Mix (Agilent Technologies). Before the calibration, the calibrant ion at m/z 622 was set on 132.0 ± 1.0 V through the setting of the gas flow on the instrument.
Optical properties
UV-vis absorption spectra are obtained with a Specord 210 Analytik Jena spectrometer.Author contributions
The manuscript was written through the contributions of all authors. In particular: Mario Saletti, Jacopo Venditti, Marco Paolino, Arianna Zacchei, and Germano Giuliani were involved in the synthesis and the preliminary characterization; Claudia Bonechi and Alessandro Donati performed the NMR studies; Gianluca Giorgi performed the crystallographic studies and the mass spectrometry characterization; Andrea Cappelli designed the compounds and organized the research activities. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
M. P. and A. C. acknowledge the MUR for the financial support under the project CN00000041 – “National Center for Gene Therapy and Drugsbased on RNA Technology” – CUP B63C2200061 0006 Mission 4 Component 2 (M4C2) – investment 1.4 [CN3] of the National Recovery and Resilience Plan (PNRR) funded by the European Union “Next Generation EU”. M. P. acknowledge the University of Siena for the financial support of the project Chromo-GEnUP through the F-CUR2022 funding line (2265-2022-PM-CONRICMIUR_PC-FCUR2022_003). Thanks are due also to Prof. Lluis Blancafort for providing access to computational resources.References
- F. Nicoli, M. Baroncini, S. Silvi, J. Groppi and A. Credi, Direct synthetic routes to functionalised crown ethers, Org. Chem. Front., 2021, 8, 5531–5549 RSC.
- D. B. Amabilino and J. Fraser Stoddart, Interlocked and intertwined structures and superstructures, Chem. Rev., 1995, 95, 2725–2828 CrossRef CAS.
- J. Y. Sung, S.-M. Jin, S. Lee, S.-Y. An and J. S. Jin, Unusual enantiomeric separation due to residual amines in chiral crown ether stationary phase linked by long alkyl chain, Talanta, 2021, 235, 122739 CrossRef CAS PubMed.
- S. Shinkai, K. Miyazaki and O. Manabe, Photoresponsive crown ethers. Part 18. Photochemically ‘switched-on’ crown ethers containing an intra-annular azo substituent and their application to membrane transport, J. Chem. Soc., Perkin Trans. 1 (1972–1999), 1987, 449–456 RSC.
- R. H. El Halabieh, O. Mermut and C. J. Barrett, Using light to control physical properties of polymers and surfaces with azobenzene chromophores, Pure Appl. Chem., 2004, 76(7–8), 1445–1465 CrossRef CAS.
- A. Saady and S. M. Goldup, Triazole formation and the click concept in the synthesis of interlocked molecules, Chem, 2023, 9, 2110–2127 CAS.
- W. P. Juma, D. Nyoni, D. Brady and M. L. Bode, The Application of biocatalysis in the preparation and resolution of Morita-Baylis-Hillman Adducts and their derivatives, ChemBioChem, 2022, 23 Search PubMed.
- H. Pellissier, Recent developments in the asymmetric organocatalytic Morita−Baylis−Hillman reaction, Tetrahedron, 2017, 73, 2831–2861 CrossRef CAS.
- V. Razzano, M. Paolino, A. Reale, G. Giuliani, R. Artusi, G. Caselli, M. Visintin, F. Makovec, A. Donati, F. Villafiorita-Monteleone, C. Botta and A. Cappelli, Development of imidazole-reactive molecules leading to a new aggregation-induced emission fluorophore based on the cinnamic scaffold, ACS Omega, 2017, 2, 5453–5459 CrossRef CAS PubMed.
- V. Razzano, M. Paolino, A. Reale, G. Giuliani, A. Donati, G. Giorgi, R. Artusi, G. Caselli, M. Visintin, F. Makovec, S. Battiato, F. Samperi, F. Villafiorita-Monteleone, C. Botta and A. Cappelli, Poly-histidine grafting leading to fishbone-like architectures, RSC Adv., 2018, 8, 8638–8656 RSC.
- M. Paolino, A. Reale, V. Razzano, G. Giuliani, A. Donati, C. Bonechi, G. Caselli, M. Visintin, F. Makovec, C. Scialabba, M. Licciardi, E. Paccagnini, M. Gentile, L. Salvini, F. Tavanti, M. C. Menziani and A. Cappelli, Nanoreactors for the multi-functionalization of poly-histidine fragments, New J. Chem., 2019, 43, 6834–6837 RSC.
- M. Paolino, M. Visintin, E. Margotti, M. Visentini, L. Salvini, A. Reale, V. Razzano, G. Giuliani, G. Caselli, F. Tavanti, M. C. Menziani and A. Cappelli, Functionalization of protein hexahistidine tags by functional nanoreactors, New J. Chem., 2019, 43, 17946 RSC.
- G. Tassone, M. Paolino, C. Pozzi, A. Reale, L. Salvini, G. Giorgi, M. Orlandini, F. Galvagni, S. Mangani, X. Yang, B. Carlotti, F. Ortica, L. Latterini, M. Olivucci and A. Cappelli, Xanthopsin-like systems via site-specific click-functionalization of a retinoic acid binding protein, ChemBioChem, 2022, 23, e202100449 CrossRef CAS.
- M. Saletti, M. Paolino, J. Venditti, C. Bonechi, G. Giuliani, A. Boccia, C. Botta and A. Cappelli, Synthesis, Photophysical and Photochemical features of a Morita-Baylis-Hillman Adduct Derivative Bearing a Triphenylamine Moiety, Dyes Pigm., 2023, 219, 111571 CrossRef CAS.
- K. Worm-Leonhard and M. Meldal, Green Catalysts: Solid-phase peptide carbene ligands in aqueous transition-metal catalysis, Eur. J. Org Chem., 2008, 31, 5244–5253 CrossRef.
- G. Pelletier, W. S. Bechara and A. B. Charette, Controlled and chemoselective reduction of secondary amides, J. Am. Chem. Soc., 2010, 132, 12817–12819 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Akabori and S. Tsuchiya, The Preparation of crown ethers containing dicinnamoyl groups and their complexing abilities, Bull. Chem. Soc. Japan, 1990, 63, 1623–1628 CrossRef CAS.
- G. M. A. Sheldrick, Short History of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis and the characterization of compounds 1, 2a,b and their intermediates. CCDC 2286050. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra06792k |
| This journal is © The Royal Society of Chemistry 2023 |