
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and evaluation of tirbanibulin derivatives: a detailed exploration of the structure–activity relationship for anticancer activity†
Jaebeom Parkab,
Minji Kangac,
Ahyoung Lima,
Kyung-Jin Choa,
Chong Hak Chaea,
Byumseok Koh *ac and
Hongjun Jeon*ac
*ac and
Hongjun Jeon*ac
aTherapeutics & Biotechnology Division, Korea Research Institute of Chemical Technology, Daejeon 34114, Republic of Korea. E-mail: bkoh@krict.re.kr; hjeon@krict.re.kr
bCollege of Pharmacy, Chungbuk National University, Cheongju 28160, Republic of Korea
cMedicinal Chemistry & Pharmacology, University of Science & Technology, Daejeon 34113, Republic of Korea
First published on 6th December 2023
Abstract
Tirbanibulin, an FDA-approved microtubule-targeting agent (MTA) introduced in 2020, represents a pioneering treatment for precancerous actinic keratosis. Despite its failure to gain approval as an anticancer agent due to insufficient efficacy, there remains potential value in extending its application into malignancy treatment through tirbanibulin-based derivatives. Tirbanibulin possesses a distinctive dual mechanism of action involving microtubule and Src inhibition, distinguishing it from other MTAs. In spite of its unique profile, exploration of tirbanibulin's structure–activity relationship (SAR) and the development of its derivatives are significantly limited in the current literature. This study addresses this gap by synthesizing various tirbanibulin derivatives and exploring their SAR through modifications in the core amide motif and the eastern benzylamine part. Our results underscore the critical role of the pyridinyl acetamide core structure for optimal cellular potency, with favorable tolerance observed for modifications at the para position of the benzylamine moiety. Particularly noteworthy is the analogue modified with p-fluorine benzylamine, which exhibited favorable in vivo PK profiles. These findings provide crucial insights into the potential advancement of tirbanibulin-based compounds as promising anticancer agents.
Introduction
Since cell cycle dysregulation is a hallmark of cancer, and targeting mitosis remains a mainstay of anticancer therapy.1–5 The most representative drugs in this category are microtubule-targeting agents (MTAs). MTAs disrupt microtubule dynamics by stabilizing or destabilizing microtubules, leading to cell cycle arrest at the G2/M phase and, ultimately apoptosis.6–9 Paclitaxel (1, Chart 1) and ixabepilone (2) are microtubule-stabilizing agents. Vinca alkaloids, exemplified by 3 and 4, as well as eribulin (5), function as microtubule-destabilizing agents. Other mitosis-targeted drugs have been developed, such as aurora kinase,10 polo-like kinase-1,11 and motor protein inhibitors.12,13 However, owing to their limited efficacy as anticancer agents, these drugs have not yet been approved by the FDA, suggesting the effectiveness of direct targeting of microtubules. Indeed, microtubules play a critical role throughout the cell cycle, and are involved in mitosis-independent cellular functions such as intracellular trafficking, angiogenesis, and metastasis-related cell migration.5,14 Recently, MTAs have regained attention, as evidenced by the incorporation of chemically modified synthetic drugs like DM1 (6) and MMAE (7) into antibody–drug conjugates (ADCs), a promising modality for cancer treatment.15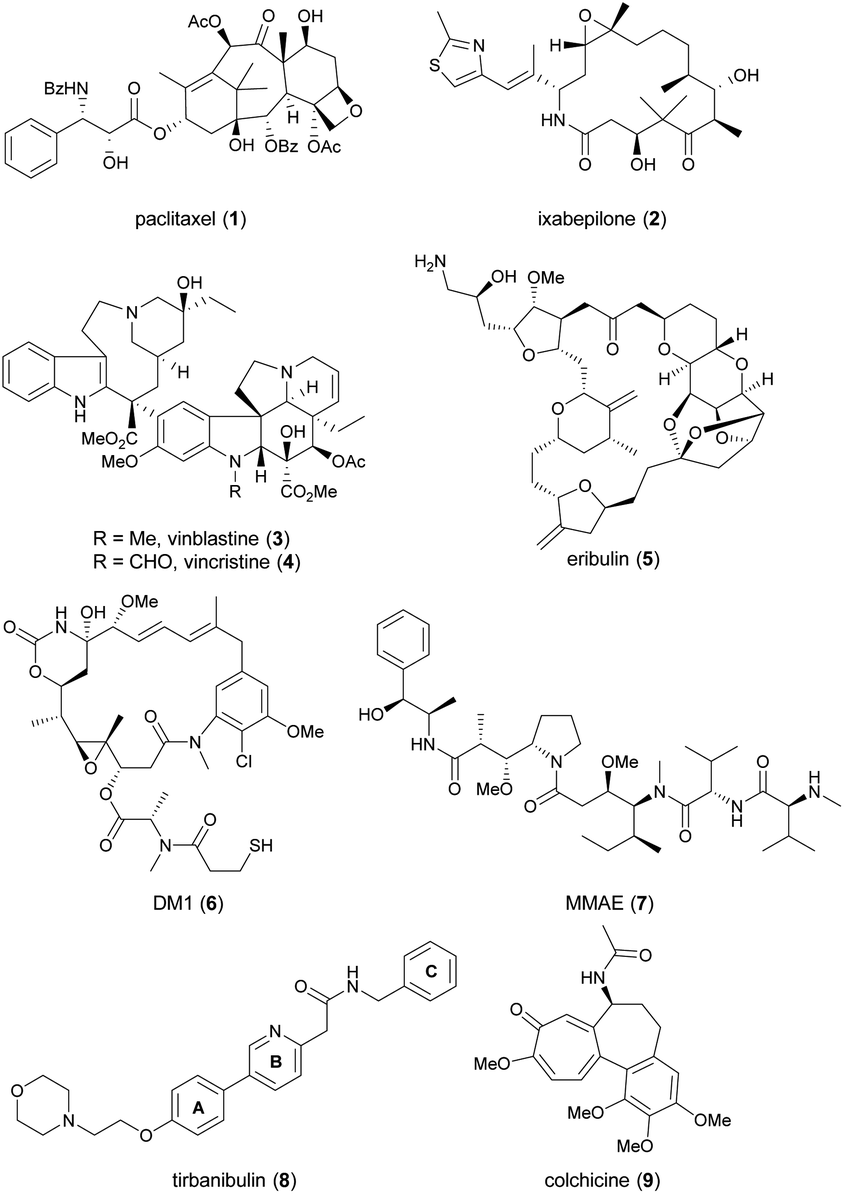 | ||
| Chart 1 The structures of microtubule-targeting agents. | ||
An emerging approach in the development of new MTAs, aiming for enhanced clinical efficacy and reduced undesired effects, is the design of dual-target inhibitors with synergistic effects.16,17 Active efforts have been made to modify MTAs to confer an additional inhibitory ability on kinases, histone deacetylases, estrogen receptor, or immune-related targets.16 The sole successful MTA in this class is tirbanibulin (8, KX-01, Chart 1), which received approval from the U.S. Food and Drug Administration in 2020 under the trade name Klisyri®.
Tirbanibulin possesses dual mechanism of action (MOA), combining Src signaling inhibition and disruption of microtubule dynamics,18–20 the latter of which was identified during a phase 2 trial.20 Initially developed as an anticancer drug for patients with bone metastatic castration-resistant prostate cancer who had not received prior chemotherapy, tirbanibulin failed to meet the efficacy endpoint in a phase 2 trial (NCT01074138).21 It was later repurposed for dermatological use, in the form of an ointment (NCT03285477 and NCT03285490), and approved by the FDA as a first-in-class medication for the treatment of pre-cancerous actinic keratosis on the face or scalp.22 Recently, indication expansion studies for tirbanibulin are under investigation for basal cell carcinoma (NCT05713760 and NCT06112522)23 and plaque-type psoriasis (NCT05522816).24
Tirbanibulin exhibits distinctive features. It shows significant in vitro antitumor activity with nanomolar IC50 values, even against tumor cell lines that have developed resistance to dasatinib, a multi-kinase inhibitor targeting Src, owing to its dual MOA.18–20 Furthermore, tirbanibulin has demonstrated low toxicity in preclinical and clinical studies, attributed to its reversible binding to the colchicine (9)-binding site of β-tubulin.25
Despite its unique antitumor MOA and low clinical toxicity, there has been limited effort towards synthesizing tirbanibulin-based derivatives or elucidating the associated structure–activity relationship (SAR). To the best of our knowledge, only two medicinal chemistry studies on tirbanibulin's structure have been reported in journals.26,27 The research reported in 2011 replaced the pyridine ring (B-ring) of tirbanibulin with thiazole, resulting in thiazolyl derivatives with only micromolar GI50 values.26 The recent study published in 2021 focused on altering the western morpholine part to enhance its antiproliferative effect.27 However, other segments of tirbanibulin, like the core amide motif and the A- or C-rings, remain unexplored. In this study, we examined structural variations in the core amide motif and the eastern benzylamine portion (C-ring) while evaluating the antitumor effects of analogs. Additionally, we performed both in vitro MOA studies and in vivo PK assessments of the selected synthetic analogs.
Results and discussion
Structure–activity relationship study of the core structure of tirbanibulin
According to the co-crystal structure of 8 bound to colchicine (9)-binding site at the intradimer interface of α and β tubulins (PDB code: 6KNZ), the key ligand-protein interaction involves water-mediated hydrogen bonding. Both the nitrogen and oxygen atoms in the pyridinyl acetamide core act as hydrogen bonding acceptors, forming interactions with the adjacent Glu200 through a conserved water molecule.25 Based on this and to understand the key SAR at the chemotype level, a series of tirbanibulin derivatives was initially synthesized by modification around the pyridinyl acetamide core (Scheme 1). Tirbanibulin (8) was prepared in gram scale, in two steps, from commercially available compound 10 (Scheme 1A). Amide coupling of 10 with benzylamine afforded compound 11. Subsequent use of the cross-coupling Suzuki reaction with diboron 12 led to the synthesis of tirbanibulin (8). Further α-methylation by sodium hydride successfully yielded analog 13, whereas selective N-methylation of 8 failed. Thus, N-methyl analog 14 was prepared from commercially available methyl ester 15 (Scheme 1B). The Suzuki reaction with 12 and subsequent aminolysis of the resulting compound 16, by N-methyl benzylamine, efficiently provided analog 14. The inverted amide analog 17 was prepared from amine 18 via coupling with phenylacetic acid and the Suzuki reaction of the resulting 19 with 12 (Scheme 1C). Pyrimidine analog 20 was synthesized using a similar procedure, starting from 21 and proceeding via 22 (Scheme 1D). For the synthesis of pyridone analog 23, selective N-alkylation of bromopyridone 24 was performed to yield 25, which underwent the Suzuki reaction with 12 (Scheme 1E).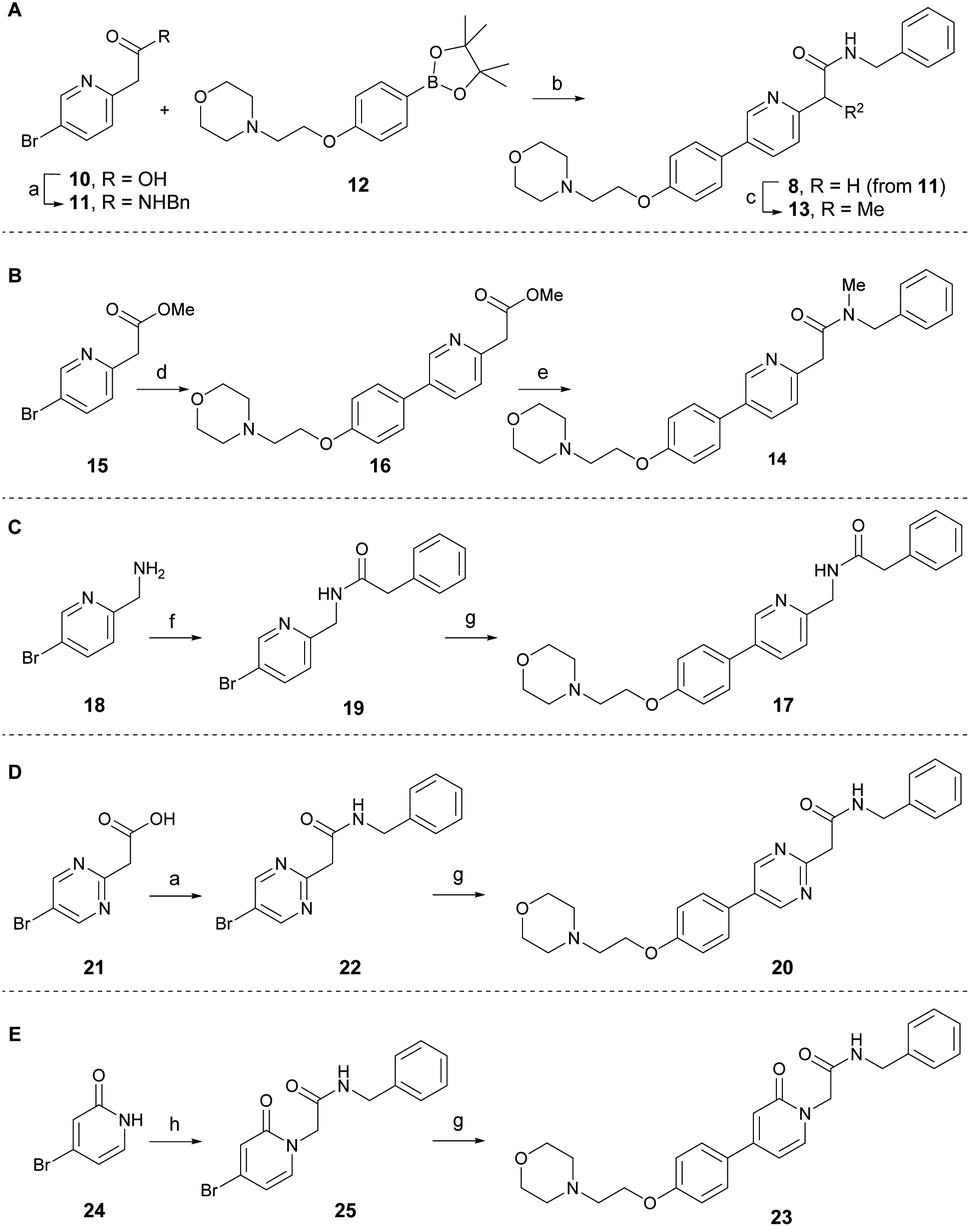 | ||
| Scheme 1 Synthesis of tirbanibulin (8) and its derivatives. (a) benzylamine, EDC·HCl, HOBt, Et3N, CH2Cl2, rt, 18 h and 77% for 11; 6 h and 51% for 22; (b) Pd(dppf)Cl2·CH2Cl2, Cs2CO3, PPh3, DMF/H2O, 100 °C, 2 h, 80%. (c) MeI, NaH, DMF, rt, 30 min, 51%. (d) 12, Pd(dppf)Cl2·CH2Cl2, KF, PPh3, DMF/H2O, 100 °C, 4 h, 99%. (e) N-methyl benzylamine, TBD, 1,4-dioxane, 60 °C, 17 h, 63%. (f) phenylacetic acid, HATU, DIPEA, DMF, rt, 13 h, 62%. (g) 12, Pd(dppf)Cl2·CH2Cl2, Cs2CO3, PPh3, DMF/H2O, 100 °C, 2 h and 82% for 17; 2 h and 36% for 20; 20 h and 47% for 23. (h) N-Benzyl-2-bromoacetamide, K2CO3, DMF, 100 °C, 3 h, 85%. EDC·HCl = N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride, HATU = 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate, TBD = 1,5,7-triazabicyclo[4.4.0]dec-5-ene, DIPEA = N,N-diisopropylethylamine. | ||
The antitumor activity of the synthesized analogs was evaluated on HeLa cells using the CCK-8 assay (Table 1). Tirbanibulin (8) exhibited potent antiproliferative activity, with an IC50 value of 44 nM. However, the introduction of an α-methyl group to analog 13 significantly reduced its activity to submicromolar levels. This result suggests that the binding pocket surrounding the amide moiety of 8 in tubulins lacks the necessary space to accommodate even a small methyl group. The N-methylated analog 14 showed very weak antiproliferative activity. The inverted amide analog 17, which possesses physicochemical properties highly similar to those of 8, was less potent than 8, indicating that a single hydrogen acceptor is probably not sufficient for optimal hydrogen bonding via water mediation. Retaining both the hydrogen acceptors, the pyridine core of 8 was replaced by alternative heteroaromatics, as exemplified by pyrimidine 20 and pyridone 23. The potencies of 20 and 23 were greater than that of a series of amide-modified analogs (13, 14, and 17). Although potency of pyridone analog 23 was lower than that of tirbanibulin (8), compound 23 possessed a lower cLogP value. These results indicate that 23 could be employed in late-stage medicinal chemistry studies to improve solubility and metabolic stability.28,29 Overall, these results demonstrate that both hydrogen acceptors in the central structure of 8 are required for optimal interaction with tubulins.
| Compound | HeLa IC50a,b (μM) | cLogPc |
|---|---|---|
| a Data represent the mean of three independent determinations from triplicate samples.b IC50 values were determined using the CCK-8 assay in HeLa cells.c cLogP values were calculated using online ALOGPS 2.1 program. | ||
| 8 | 0.044 ± 0.011 | 3.3 |
| 13 | 0.95 ± 0.35 | 3.7 |
| 14 | >1.0 | 3.2 |
| 17 | 0.97 ± 0.23 | 3.3 |
| 20 | 0.37 ± 0.10 | 2.8 |
| 23 | 0.24 ± 0.06 | 2.6 |
Synthesis and evaluation of amide-modified analogs
To develop new and potent microtubule inhibitors based on tirbanibulin (8), the bioactive conformation of 8 was considered. In the co-crystal structure (PDB code: 6KNZ),25 8 exists in a cis-amide conformation, which is energetically unfavorable. We hypothesized that the introduction of chemical modifications to mimic the cis-amide conformation would reduce the energy required for optimal ligand-protein binding, consequently enhancing the affinity and potency of the compound as a microtubule-targeting agent.To incorporate the cis-amide bioisosteres in 8 while retaining the two hydrogen bonding acceptors, several analogs were designed and synthesized as shown in Scheme 2.30 The representative bioisosteres for the cis-amide are 1,5-tetrazole or 1,5-triazole, both of which have been incorporated into various approved drugs.30 The 1,5-tetrazole analog 26 was directly synthesized by the treatment of DPPA with inverted amide 17 (Scheme 2A).31 The 1,5-triazole analog 27 in Scheme 2B was designed to have a hydroxy group at the benzylic position to induce additional interactions with adjacent Val238. An attempt to achieve the regioselective formation of 1,5-triazole from azide 28 (ref. 32) and α-ethynylbenzyl alcohol using ruthenium-catalyzed cycloaddition conditions yielded, only unidentified side products and no detectable desired product.33 As an alternative approach, thermal azide–alkyne cycloaddition reactions were conducted in the absence of catalyst, leading to a separable mixture of regioisomers (29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 = 1:1) with a combined yield of 68%. The desired intermediate 30 was subsequently transformed into triazole analog 27 via the Suzuki reaction. The structure of 30 was indirectly confirmed by exclusively obtaining the 1,4-regioisomer 29 through a regiospecific Cu(I)-catalyzed 1,3-dipolar cycloaddition of 28 with α-ethynylbenzyl alcohol.34 To enhance the molecular conformational rigidity, a sulfonyl urea analog 31 was designed to possess intramolecular hydrogen bonding, which aimed to mimic the cis-amide conformation; with lower N–H proton acidity for improved hydrogen bonding interactions (Scheme 2C). Aminopyridine 32 reacted with benzenesulfonyl isocyanate to produce 33, which upon Suzuki reaction was converted to analog 31. Urea analog 34 was synthesized from 32 through the intermediate 35. The N-hydroxy amide 36 was also designed to possess intramolecular hydrogen bonding. It was prepared from acid 10 through several synthetic steps: amide coupling with hydroxylamine, a Suzuki reaction with the resulting amide 37, and a subsequent debenzylation of 38 (Scheme 2D). The cis-amide bioisostere analogs, 1,5-tetrazole 26 and 1,5-triazole 27, exhibited no antitumor activity, even at the maximum concentration of 4.0 μM (Table 2). Sulfonylurea 31, designed to induce rigidity through intramolecular hydrogen bonding, was ineffective against cancer cells, partly owing to the planarity of the urea moiety, as is evident from the reduced potency of the urea analog 34. N-hydroxy amide analog 36 displayed only modest antiproliferative activity at a micromolar level. Consequently, amide modification using other isosteres did not offer sufficient potency to justify further investigation.
:30 = 1:1) with a combined yield of 68%. The desired intermediate 30 was subsequently transformed into triazole analog 27 via the Suzuki reaction. The structure of 30 was indirectly confirmed by exclusively obtaining the 1,4-regioisomer 29 through a regiospecific Cu(I)-catalyzed 1,3-dipolar cycloaddition of 28 with α-ethynylbenzyl alcohol.34 To enhance the molecular conformational rigidity, a sulfonyl urea analog 31 was designed to possess intramolecular hydrogen bonding, which aimed to mimic the cis-amide conformation; with lower N–H proton acidity for improved hydrogen bonding interactions (Scheme 2C). Aminopyridine 32 reacted with benzenesulfonyl isocyanate to produce 33, which upon Suzuki reaction was converted to analog 31. Urea analog 34 was synthesized from 32 through the intermediate 35. The N-hydroxy amide 36 was also designed to possess intramolecular hydrogen bonding. It was prepared from acid 10 through several synthetic steps: amide coupling with hydroxylamine, a Suzuki reaction with the resulting amide 37, and a subsequent debenzylation of 38 (Scheme 2D). The cis-amide bioisostere analogs, 1,5-tetrazole 26 and 1,5-triazole 27, exhibited no antitumor activity, even at the maximum concentration of 4.0 μM (Table 2). Sulfonylurea 31, designed to induce rigidity through intramolecular hydrogen bonding, was ineffective against cancer cells, partly owing to the planarity of the urea moiety, as is evident from the reduced potency of the urea analog 34. N-hydroxy amide analog 36 displayed only modest antiproliferative activity at a micromolar level. Consequently, amide modification using other isosteres did not offer sufficient potency to justify further investigation.
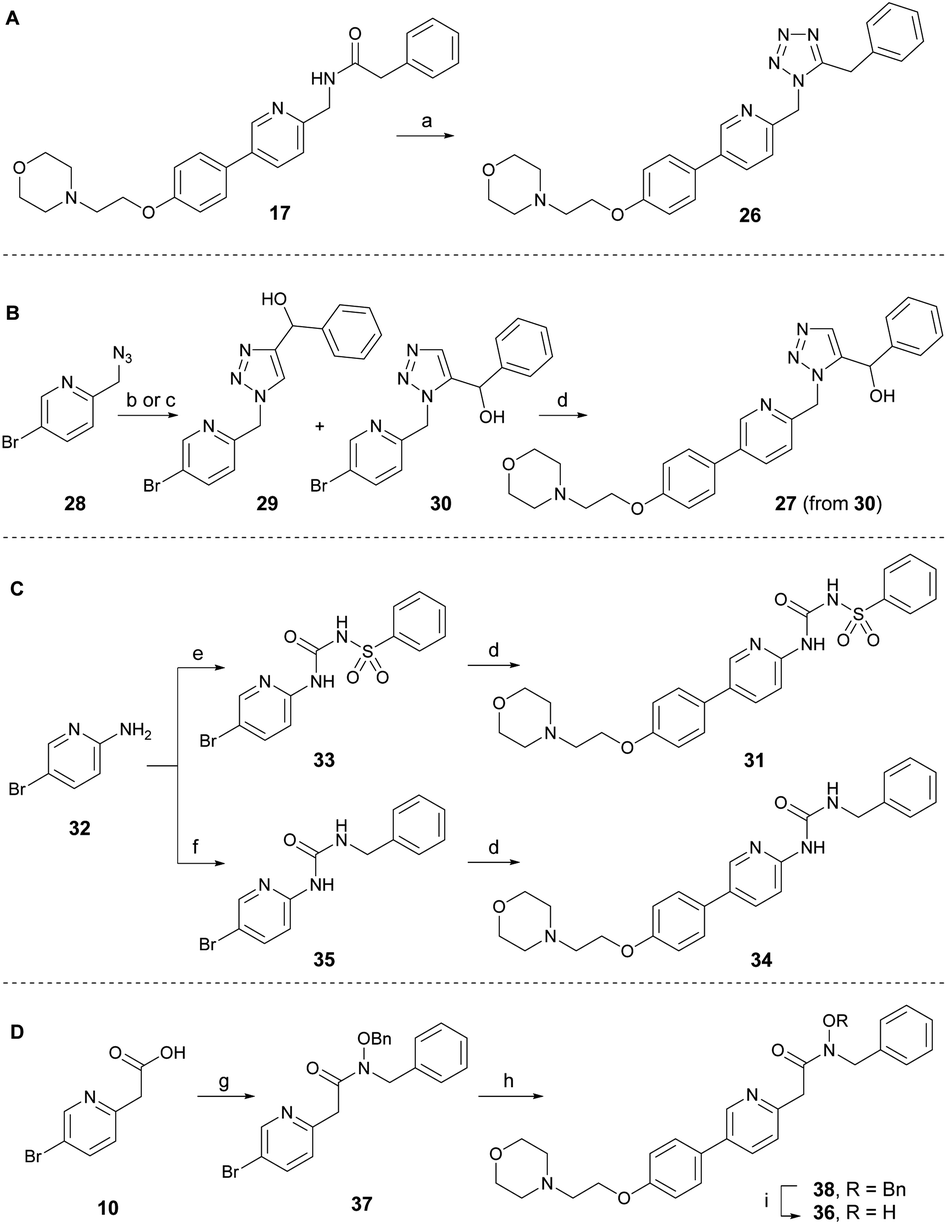 | ||
| Scheme 2 Synthesis of amide-modified analogs. (a) DPPA, 4-picoline, 100 °C, 25 h, 68%. (b) phenylprop-2-yn-1-ol, toluene, 18 h, 68% (29 and 30). (c) CuSO4, ascorbic acid, CH3Cl/H2O (3:1), rt, 14 h, 94% for 29. (d) 12, Pd(dppf)Cl2·CH2Cl2, Cs2CO3, PPh3, DMF/H2O, 100 °C, 1 h and 79% for 27; 40 min and 57% for 31; 2 h and 37% for 34. (e) benzenesulfonyl isocyanate, CH2Cl2, rt, 10 min, 76%; (f) benzyl isocyanate, THF, reflux, 6 h, 83%. (g) N,O-dibenzylhydroxylamine, EDC·HCl, HOBt, Et3N, CH2Cl2, rt, 6 h, 78%; (h) 12, Pd(dppf)Cl2·CH2Cl2, KF, PPh3, DMF/H2O, 100 °C, 16 h, 22%. (i) Pd/C, H2(g), MeOH, rt, 6 h, 84%. DPPA = diphenyl phosphoryl azide, EDC·HCl = N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride. | ||
| Compound | HeLa IC50a,b (μM) | cLogPc |
|---|---|---|
| a Data represent the mean of three independent determinations from triplicate samples.b IC50 values were determined using the CCK-8 assay in HeLa cells.c cLogP values were calculated using online ALOGPS 2.1 program. | ||
| 8 | 0.044 ± 0.011 | 3.3 |
| 26 | >4.0 | 2.9 |
| 27 | >4.0 | 2.9 |
| 31 | >4.0 | 2.9 |
| 34 | 0.78 ± 0.20 | 3.5 |
| 36 | 1.1 ± 0.4 | 3.4 |
Synthesis and evaluation of benzylamine-modified analogs
Based on the SAR study, our efforts shifted towards modifying the benzylamine moiety while preserving the core amide structure, connected with either pyridine or pyridone. Co-crystal structural analysis revealed a limited binding site around the benzylamine moiety of 8, making it unsuitable for large substituents. The binding site consisted of several polar amino acid residues (Tyr52, Gln136, and Asn167). Therefore, the design of analogs ruled out the use of large substituents and prioritized small or polar substituents as well as heteroaromatics. As shown in Scheme 3, the aminolysis of 16 with diverse amines afforded the designed benzylamine-modified analogs.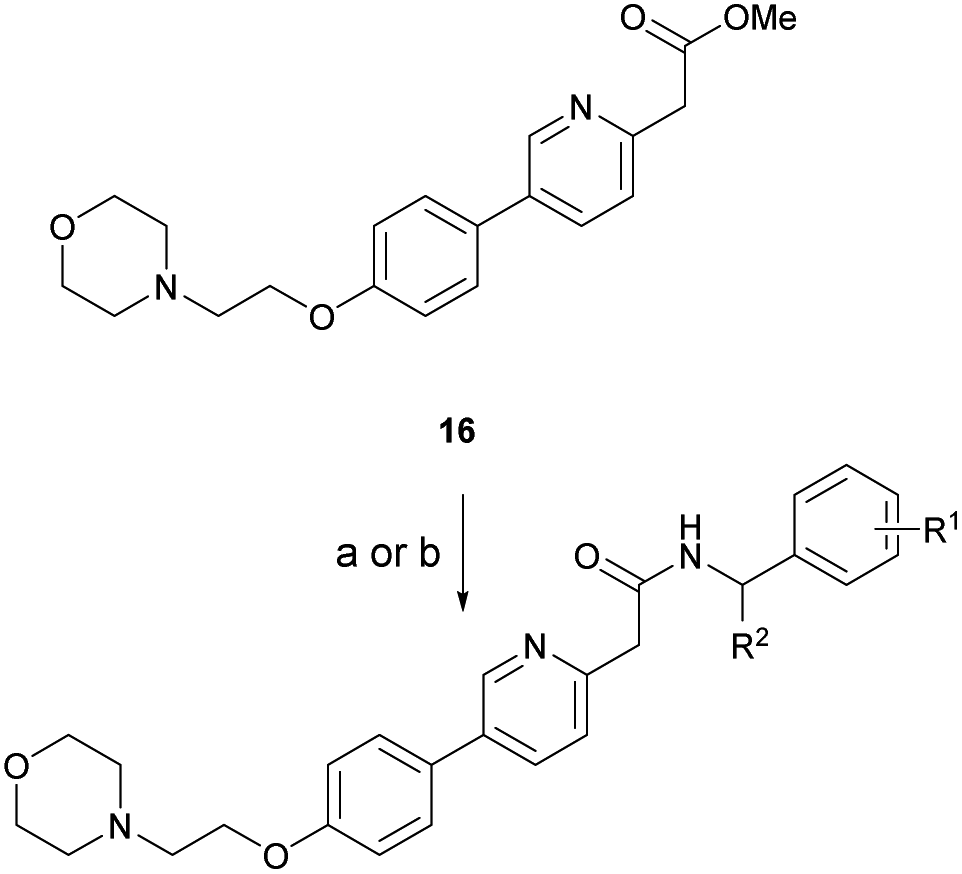 | ||
| Scheme 3 Synthesis of benzylamine-modified analogs via ester aminolysis. (a) amines, DBU, 1,4-dioxane, microwave, 180–200 °C, 2 h. (b) amines, TBD, 1,4-dioxane, 60 °C, 15 h–4 d. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, TBD = 1,5,7-triazabicyclo[4.4.0]dec-5-ene. | ||
Heteroaromatic analogs 39 and 40, including pyridine and pyrimidine, respectively, exhibited weak cellular activity only at a micromolar level (Table 3). Phenol 41 was designed for the additional hydrogen bonding interaction between the hydroxy group of 41 and Glu200 in the binding pocket. However, a micromolar IC50 value was observed. The ortho- and meta-methylated 42 and 43 were ineffective. However, para-methylated 44 was equipotent to tirbanibulin (8) (44: IC50 = 27 nM vs. 8: IC50 = 44 nM), indicating that the para-substituent was tolerable. A similar trend was observed in a series of fluorinated analogs (45–47), where para-fluorinated 47 showed a potency comparable to 8 with an IC50 value of 40 nM. Analog 48 with a para-CF3 group, with a stronger electron-withdrawing effect than fluoride, was 7-fold less potent than compound 47, despite having a higher cLogP value which potentially indicates high cell permeability. The potency of para-methoxy analog 49 was reduced to micromolar levels, whereas methylamino analog 50 exhibited significant activity with an IC50 value of 0.15 μM. Compound 50 was the most potent in a series of related analogs such as aniline 51 and N,N-dimethyl analog 52. Saturation of the benzyl moiety (analog 53) had a negative impact on potency, indicating the importance of the π-interaction of the benzene ring with surrounding amino acid residues. Furthermore, the addition of a small methyl group at the amino-benzylic position of 8, which would be beneficial for hydrophobic interactions with the surrounding residues, was evaluated. We presumed that the addition of (S)-methyl group would be more efficient than (R)-methyl group since the pro-(R) hydrogen already interacts with the backbone carbonyl oxygen of Val238 in the co-crystal structure. As expected, (S)–Me compound 54 was more potent than the (R)–Me compound 55 (54: IC50 = 0.45 μM vs. 55: IC50 > 4.0 μM), although the growth inhibition activity of 54 was still insufficient. In addition, para-methyl and fluorine substituents that elicited potency equal to that of tirbanibulin were incorporated into pyridone analogs 56 and 57. The methylated analog 56 showed the potency improvement compared to the pyridone analog 23 (56: IC50 = 0.25 μM vs. 23: IC50 = 0.24 μM), while the cellular potency was similar for the fluorinated compound 57.
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R | HeLa IC50a,b (μM) | cLogPc | Compound | R | HeLa IC50a,b (μM) | cLogPc |
| a Data represent the mean of three independent determinations from triplicate samples.b IC50 values were determined using the CCK-8 assay in HeLa cells.c cLogP values were calculated using online ALOGPS 2.1 program. | |||||||
| 8 | 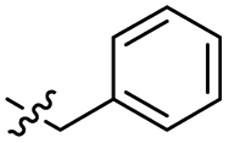 |
0.044 ± 0.011 | 3.3 | 48 | 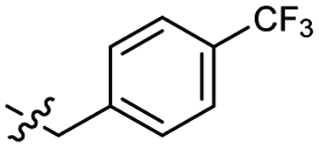 |
0.27 ± 0.07 | 4.0 |
| 39 | 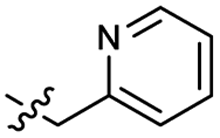 |
>4.0 | 2.8 | 49 | 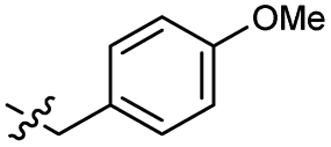 |
2.0 ± 0.5 | 3.2 |
| 40 | 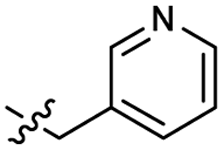 |
>4.0 | 2.8 | 50 | 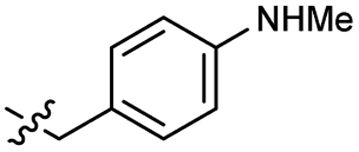 |
0.15 ± 0.04 | 2.7 |
| 41 | 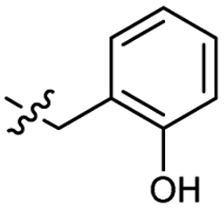 |
2.1 ± 0.6 | 3.0 | 51 | 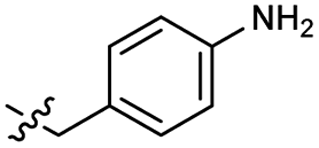 |
0.63 ± 0.07 | 2.0 |
| 42 | 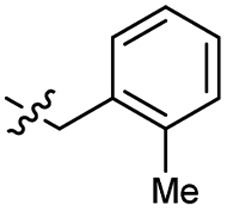 |
2.2 ± 0.5 | 3.5 | 52 | 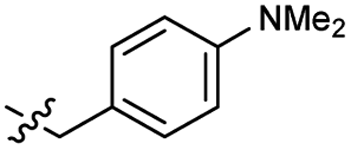 |
0.48 ± 0.06 | 3.4 |
| 43 | 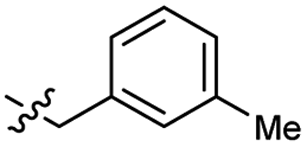 |
0.97 ± 0.22 | 3.5 | 53 | 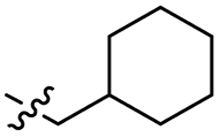 |
>4.0 | 4.2 |
| 44 | 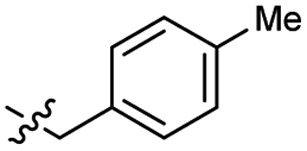 |
0.027 ± 0.004 | 3.5 | 54 |  |
0.45 ± 0.10 | 3.6 |
| 45 | 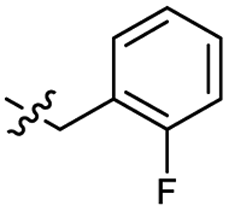 |
0.26 ± 0.06 | 3.4 | 55 | 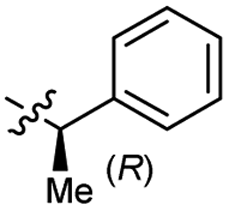 |
>4.0 | 3.6 |
| 46 | 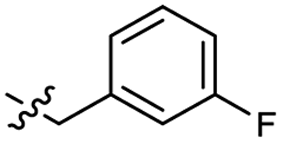 |
0.53 ± 0.15 | 3.4 | 56 | 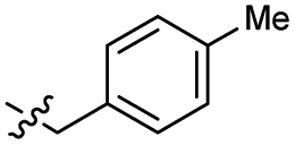 |
0.25 ± 0.06 | 3.6 |
| 47 | 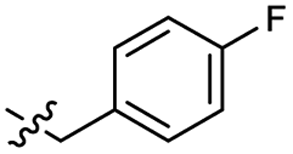 |
0.040 ± 0.009 | 3.4 | 57 | 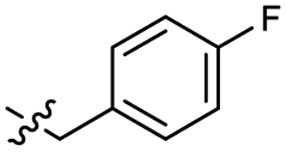 |
0.34 ± 0.11 | 3.3 |
Molecular docking simulation
To further investigate the binding modes of the selected compounds, a molecular modeling study was performed. The ligand binding modes of compounds 44 and 47 were almost identical to those of tirbanibulin (Fig. 1). The main binding affinities were water-mediated hydrogen bonds and direct interactions with the B-chain of tubulin. Specifically, the amide carbonyl group and nitrogen atom in pyridine, both of which reside in compounds 44 and 47, participate in water-mediated hydrogen bonding with Glu200, whereas the amide carbonyl group forms an additional direct hydrogen bond with Glu200. Notably, the addition of para-substituents to the benzene moiety did not change the binding mode. Instead, methyl and fluorine groups were positioned in the hydrophobic pocket formed by Ile4, Gln136, Phe169, and Tyr202, implying that these substituents could be further utilized to design a molecule with improved affinity with tubulins. Additionally, the replacement of pyridine in 8 with pyridone in 23 showed an almost identical binding mode, according to this docking model.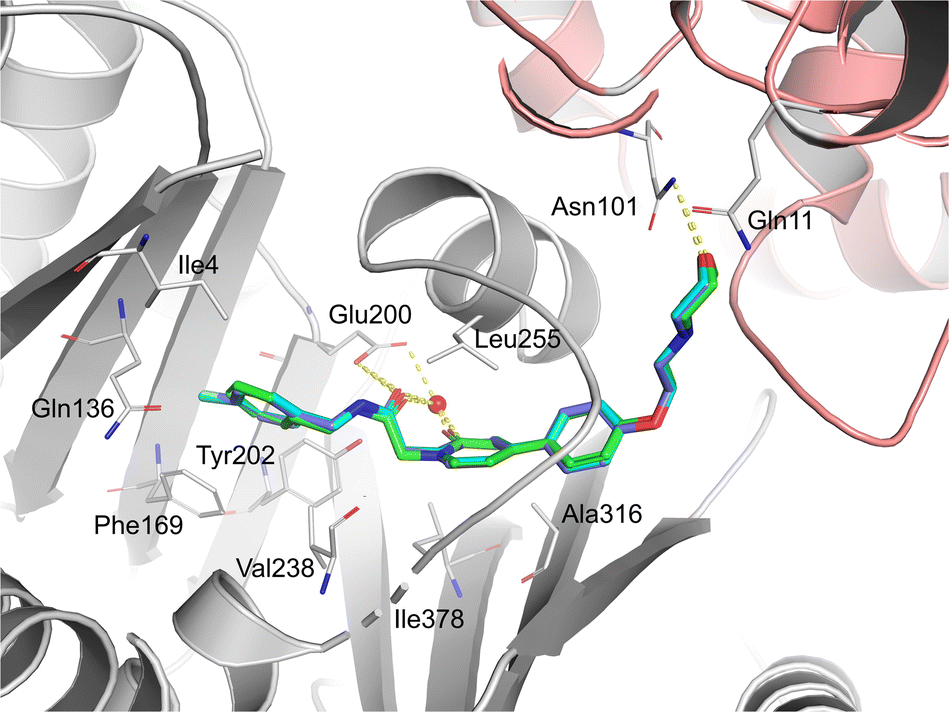 | ||
| Fig. 1 Predicted and superimposed binding poses of pyridone analog 23 (green), p-Me analog 44 (cyan) and p-F analog 47 (blue) in the colchicine-binding site of tubulin (A-chain: salmon, B-chain: gray). | ||
In vitro ADME and in vivo PK
As equipotent molecules to 8, compounds 44, 47, and 56 were further evaluated by in vitro ADME, including microsomal stability and plasma protein binding. Tirbanibulin (8) showed good microsomal stability in both mice and humans, with over 50% remaining at 30 min. The para-methylated analog 44 was found to be very unstable in liver microsomes, likely because of oxidative liability at the benzylic position of the toluene moiety. Exchange of Me with F, as shown in compound 47, resulted in improved microsomal stability. Furthermore, the metabolic stability of the para-methylbenzene moiety in 44 was improved by changing the pyridine core to pyridone in 56, likely due to a decrease in lipophilicity. The percentage of plasma protein binding of all compounds was within acceptable ranges.Further evaluation of the para-substituted compounds 44 and 47 was conducted using animal PK studies. Both compounds exhibited moderate clearance (Table 5). Compound 44, however, showed an oral bioavailability of less than 25%, resulting in a low oral AUC and Cmax. This was likely due to the first-pass effect, which is supported by the microsomal instability of compound 44 (Table 4). In contrast, a high oral AUC value of the fluorinated 47 was observed at 4.0 μg h mL−1; 4-fold higher than that of compound 44, despite a shorter half-life (Table 5). In addition, compound 47 showed an excellent oral bioavailability (F = 116%). These findings suggest that fluorine can be effectively utilized for further chemical modification of the benzylamine moiety of 8, considering its effects on potency and PK profiles.
| Compound | i.v.a (2 mg kg−1) | p.o.b (10 mg kg−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| AUClast (μg h mL−1) | t1/2 (h) | CL (L h−1 kg−1) | Vss (L kg−1) | AUClast (μg h mL−1) | Cmax (μg mL−1) | t1/2 (h) | F % | |
| a Dosed at 2 mg kg−1 for i. v. administration of ICR mouse (n = 3, mean ± SD) in 5% DMSO/40% PEG400/55% DW (5 mL kg−1).b Dosed at 10 mg kg−1 for p. o. administration of ICR mouse (n = 3, mean ± SD) in 5% DMSO/40% PEG400/55% DW (5 mL kg−1). | ||||||||
| 44 | 0.77 ± 0.04 | 7.05 ± 1.92 | 2.58 ± 0.15 | 1.89 ± 0.31 | 0.94 ± 0.16 | 1.54 ± 0.33 | 5.45 ± 1.48 | 24 |
| 47 | 0.84 ± 0.11 | 1.69 ± 1.09 | 2.39 ± 0.32 | 3.23 ± 0.81 | 4.88 ± 2.07 | 2.88 ± 1.22 | 2.30 ± 0.23 | 116 |
| Compound | Microsomal stability (%)a | Plasma protein binding rate (%) | ||
|---|---|---|---|---|
| Mouse | Human | Mouse | Human | |
| a Percentage of test compounds remaining after 30 min in mouse and human liver microsomes in the presence of NADPH (phase I stability). | ||||
| 8 | 67.7 ± 5.6 | 50.7 ± 3.2 | 84.8 ± 3.4 | 81.3 ± 2.0 |
| 44 | 15.7 ± 0.4 | 1.4 ± 0.1 | 94.3 ± 0.6 | 93.5 ± 0.6 |
| 47 | 38.6 ± 0.6 | 21.8 ± 1.4 | 89.3 ± 1.0 | 87.4 ± 2.4 |
| 56 | 50.6 ± 1.2 | 20.2 ± 2.3 | 96.0 ± 0.2 | 82.9 ± 0.5 |
Confirmation of mechanism of action and biocompatibility
Next, we aimed to confirm the MOA of the representative analog 47 using several in vitro assays. We performed a cell cycle assay using HeLa cells to analyze the proportion of the cell population in each phase. Similar to tirbanibulin (8), analog 47 induced G2/M phase arrest in a concentration-dependent manner, which is a key MTA characteristic (Fig. 2A). The results of the tubulin polymerization assay indicated that, like tirbanibulin (8), analog 47 is microtubule destabilizers; with greater depolymerization observed with increasing chemical concentration from 0 to 8 μM (Fig. 2B). These findings suggest that para-fluorinated 47 targets microtubules and causes an antiproliferative effect by disrupting tubulin polymerization.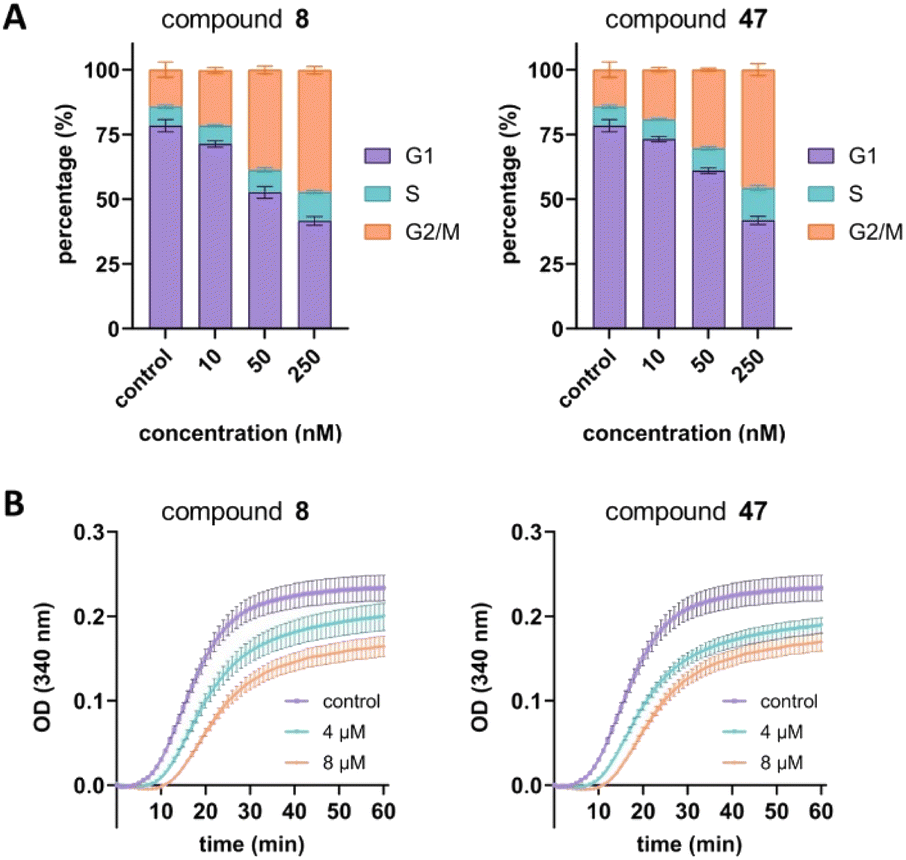 | ||
| Fig. 2 Confirmation of mechanism of action. (A) Cell cycle assay conducted in HeLa cells for compound 8 and 47. The p values of G2/M percentages for both compounds 8 and 47 are <0.0001 (one-way ANOVA). (B) Tubulin polymerization assay of compound 8 and 47. The p values for both compounds 8 and 47 are <0.0001 (two-way ANOVA). | ||
Src inhibition is an additional MOA of tirbanibulin.18–20 Tirbanibulin disrupts the Src signaling pathway by binding to the peptide substrate-binding site, which is distinct from conventional ATP-competitive kinase inhibitors. Considering that Src is strongly associated with various malignancies,35,36 it would be beneficial if the designed analog could retain its Src inhibitory capability. We investigated the inhibition of target protein phosphorylation in HeLa cells exposed to compound 47. When compound 47 was treated to the HeLa cells, a reduction in p-Src and p-FAK levels was observed even at a low concentration (50 nM), while total Src and FAK levels remained unchanged (Fig. 3A). Notably, compound 47 demonstrated an enhanced inhibitory ability for the phosphorylation of Src and FAK compared to compound 8 (Fig. 3B). This suggests that para fluorination on the benzylamine moiety could improve the inhibitory effect on Src by preserving the molecule's binding to the peptide substrate site of Src.
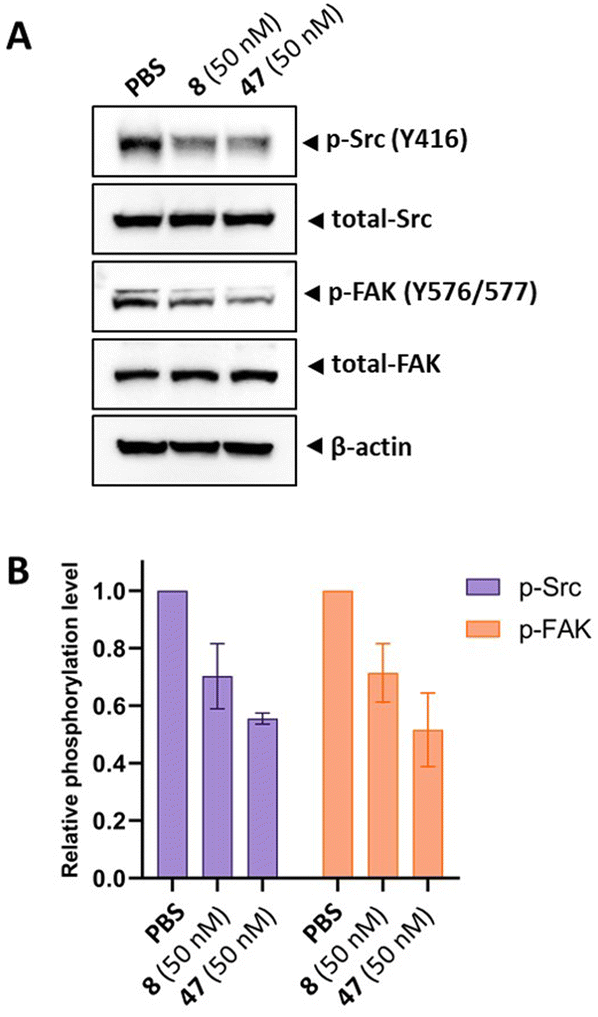 | ||
| Fig. 3 Western blot analysis of p-Src and p-FAK in the protein samples obtained from HeLa cells treated with PBS, 8 (50 nM), and 47 (50 nM) for 6 h. (A) Western blot image. (B) Relative phosphorylation level of Src and FAK. | ||
Additionally, compound 47's biocompatibility was evaluated through in vitro cytotoxicity assays using three normal cell lines (MCF-10A, CCD-18Co, and BJ). Treatment with concentrations below 4 μM for compound 47 demonstrated no significant cytotoxic effects on these non-tumorigenic cells, and their cell morphologies remained mostly unaltered (Fig. S5 in the ESI†).37,38 This outcome suggests the safety of compound 47, considering its double-digit nanomolar IC50 value (47's IC50 = 40 nM) against HeLa cancer cells.
Conclusions
In summary, we designed and synthesized a series of tirbanibulin-based analogs, including the amide- and benzylamine-modified versions. Initial SAR study, conducted for the efficient design of derivatives, highlighted the importance of the core pyridinyl acetamide structure with two hydrogen bonding acceptors. Amide-modified analogs designed to mimic the bioactive cis-amide conformation were ineffective in terms of cytotoxicity. In contrast, para substitution of the benzylamine moiety provided several equipotent derivatives, represented by para-fluorinated 47. This analog also displayed favorable in vivo PK properties, with an excellent oral bioavailability. The mechanism of action was identified to involve both the microtubule-destabilizing effect and Src inhibition. Notably, the Src inhibitory effect of compound 47 was observed to be improved compared to that of tirbanibulin, showing another advantage of para fluorination of the benzylamine part of tirbanibulin. Overall, these results provide guidance for future optimization and development of new tirbanibulin-based molecules as anticancer agents. While this study focused on the structural variation of tirbanibulin in the amide motif and C-ring, the results from our medicinal chemistry program, which encompasses the systemic variation of tirbanibulin, including derivatization of A/B-ring, will be reported in due course.Experimental
General information
The most reagents used in this study were purchased from Sigma-Aldrich (St. Louis, MO, USA), TCI (Tokyo, Japan), Alfa Aesar (Auburn Hills, MI, USA), BLDpharm (Telangana, India), and so forth, and used without further purification. The progress of the reaction was confirmed by thin layer chromatography (TLC) using TLC silica gel 60 F254, 0.25 mm (Merck, St, Louis, MO, USA). Separation processes were carried out using either medium-pressure liquid chromatography (CombiFlash NEXTGEN 300+ [Teledyne ISCO, Lincoln, NE, USA]) using a column cartridge (4–120 g, RediSepⓇRf) or high-performance liquid chromatography (HPLC) (YL9100 Semi-prep HPLC system with YL9101S Vacuum Degasser, YL9111S Binary Pump, and YL9120S UV/Vis Detector) and Agilent 5 Prep-C18 × 21.2 mm column cartridge. Mass spectroscopy of the products was identified by Agilent 6130 Quadrupole liquid chromatography-mass spectrometer (LC/MS). High-resolution mass spectrometry of the compounds was obtained using either the FAB or EI mode, with a resolution of 10000, employing the JEOL JMS-700 High-Resolution Mass Spectrometer (HRMS). Nuclear magnetic resonance (NMR) spectra for the structural analysis of the products were measured by Bruker UltraShield 300 MHz, 400 MHz, and 500 MHz NMR spectrometers. Most of NMR solvents were purchased from Cambridge Isotope Laboratories such as chloroform-d, methanol-d4, and dimethylsulfoxide-d6.
Cell viability assay
Cells were purchased from ATCC (Manassas, VA). Cells were cultured in RPMI-1640 medium (Gibco, Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Gibco) at 37 °C and 5% CO2. One day prior to drug treatment, 10000 cells of each cell line were seeded into 96-well plates in 100 μL of RPMI-1640 medium. On the following day, the cells were treated with test compounds solubilized in dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO). After 48 h of drug treatment, 10 μL of CCK-8 reagent was added to each well, and the absorbance at 450 nm was measured using a SpectraMax iD5 spectrophotometer (Molecular Devices, San Jose, CA). The absorbance signal at 450 nm is proportional to the number of viable cells.
Cell cycle assay
To study the effects of test compounds on the cell cycle, a Muse cell cycle kit (Luminex, Austin, TX) was used. The experimental protocol followed the manufacturer's instructions. First, 1 × 106 cells were grown in 6-well cell culture plates (Corning, Corning, NY). The cells were then treated with compounds for 48 hours. After treatment, the cells were collected and fixed with 70% pure ethanol. The fixed cells were then frozen at −20 °C for 12 h and stained with a cell cycle reagent for 30 min at room temperature in the dark. Finally, the fixed and stained cells were analyzed with a Muse cell analyzer (Luminex).Tubulin polymerization assay
A tubulin polymerization assay kit (Cytoskeleton Inc., Denver, CO) was used to evaluate the activity of test compounds. The assay protocol strictly followed the manufacturer's instructions. In brief, test compounds were mixed with tubulin in the general tubulin buffer provided by the manufacturer. Tubulin polymerization reactions were then monitored for 1 h at 37 °C in a SpectraMax iD5 spectrophotometer (Molecular Devices, San Jose, CA) with an absorbance wavelength of 340 nanometers.Western blot assay
To investigate the effect of the compound on the target signaling pathway, western blot assay was conducted. HeLa cells were treated either with compounds (50 nM) or PBS (control group) for 6 hours and then, proteins were extracted. Equal amounts of proteins were resolved SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking with buffer, the blot was incubated with primary antibodies, and then incubated with the appropriate secondary antibodies.Molecular modelling
The Maestro software in the Schrodinger Suite was utilized in the molecular modelling studies.39 The crystal structure of tubulins was obtained from the Protein Data Bank (PDB code: 6KNZ), and any structural defects were rectified using the Protein Preparation Workflow module. The structures of selected compounds were docked against the colchicine binding site using the Glide docking module, following standard protocols. The reproduction of crystal ligand poses with a good root mean square displacement of 0.3 Å demonstrated the robustness of our docking protocol. The best-docked pose, characterized by the lowest Glide score, was selected for the subsequent binding pose analysis.Statistical analysis
Statistical analysis was performed using GraphPad Prism 10 software (GraphPad Software, San Diego, CA, USA) and Origin 8.5 (OriginLab Corporation, Northampton, MA, USA). The values are expressed as the mean ± standard error of means. The significance between means was tested using one-way or two-way ANOVA. The Dunnett test was conducted to compare every mean to a control mean. A value of P < 0.05 was considered significant.Author contributions
HJ conceived the study, wrote the manuscript, and obtained the necessary funding. JP, MK, and AL were responsible for compound synthesis, purification, and characterization. BK conducted the in vitro assays, including the cell viability assay, cell cycle assay, and tubulin polymerization assay. KC performed the western blotting assay. CC was responsible for computational modeling and docking studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2022R1C1C1004145). It also received support from Korea Research Institute of Chemical Technology (KK2331-20).Notes and references
- A. Serrano-del Valle, C. Reina-Ortiz, A. Benedi, A. Anel, J. Naval and I. Marzo, Biochem. Pharmacol., 2021, 190, 114655 CrossRef CAS PubMed.
- J. Tischer and F. Gergely, J. Cell Biol., 2018, 218, 10–11 CrossRef PubMed.
- M. Haschka, G. Karbon, L. L. Fava and A. Villunger, EMBO Rep., 2018, 19, e45440 CrossRef PubMed.
- C. Dominguez-Brauer, K. L. Thu, J. M. Mason, H. Blaser, M. R. Bray and T. W. Mak, Mol. Cell, 2015, 60, 524–536 CrossRef CAS PubMed.
- K. S. Chan, C. G. Koh and H. Y. Li, Cell Death Dis., 2012, 3, e411 CrossRef PubMed.
- L. Wordeman and J. J. Vicente, Cancers, 2021, 13, 5650 CrossRef CAS PubMed.
- V. Čermák, V. Dostál, M. Jelínek, L. Libusová, J. Kovář, D. Rösel and J. Brábek, Eur. J. Cell Biol., 2020, 99, 151075 CrossRef PubMed.
- M. O. Steinmetz and A. E. Prota, Trends Cell Biol., 2018, 28, 776–792 CrossRef CAS PubMed.
- E. Mukhtar, V. M. Adhami and H. Mukhtar, Mol. Cancer Ther., 2014, 13, 275–284 CrossRef CAS PubMed.
- A. C. Borisa and H. G. Bhatt, Eur. J. Med. Chem., 2017, 140, 1–19 CrossRef CAS PubMed.
- X. Liu, Transl. Oncol., 2015, 8, 185–195 CrossRef PubMed.
- I. Garcia-Saez and D. A. Skoufias, Biochem. Pharmacol., 2021, 184, 114364 CrossRef CAS PubMed.
- A. A. El-Arabey, S. A. Salama and A. R. Abd-Allah, Life Sci., 2018, 208, 192–200 CrossRef CAS PubMed.
- A. L. Parker, M. Kavallaris and J. A. McCarroll, Front. Oncol., 2014, 4, 153–172 Search PubMed.
- C. Dumontet, J. M. Reichert, P. D. Senter, J. M. Lambert and A. Beck, Nat. Rev. Drug Discovery, 2023, 22, 641–661 CrossRef CAS PubMed.
- W. Shuai, G. Wang, Y. Zhang, F. Bu, S. Zhang, D. D. Miller, W. Li, L. Ouyang and Y. Wang, J. Med. Chem., 2021, 64, 7963–7990 CrossRef CAS PubMed.
- K. E. Arnst, S. Banerjee, H. Chen, S. Deng, D.-J. Hwang, W. Li and D. D. Miller, Med. Res. Rev., 2019, 39, 1398–1426 CrossRef PubMed.
- Y. Gilaberte and M. T. Fernández-Figueras, Actas Dermo-Sifiliogr., 2022, 113, T58–T66 CrossRef.
- S. Kim, A. Min, K.-H. Lee, Y. Yang, T.-Y. Kim, J. M. Lim, S. J. Park, H.-J. Nam, J. E. Kim, S.-H. Song, S.-W. Han, D.-Y. Oh, J. H. Kim, T.-Y. Kim, D. Hangauer, J. Y.-N. Lau, K. Im, D. S. Lee, Y.-J. Bang and S.-A. Im, Cancer Res. Treat., 2017, 49, 643–655 CrossRef CAS PubMed.
- M. P. Smolinski, Y. Bu, J. Clements, I. H. Gelman, T. Hegab, D. L. Cutler, J. W. S. Fang, G. Fetterly, R. Kwan, A. Barnett, J. Y. N. Lau and D. G. Hangauer, J. Med. Chem., 2018, 61, 4704–4719 CrossRef CAS PubMed.
- E. S. Antonarakis, E. I. Heath, E. M. Posadas, E. Y. Yu, M. R. Harrison, J. Y. Bruce, S. Y. Cho, G. E. Wilding, G. J. Fetterly, D. G. Hangauer, M.-F. R. Kwan, L. M. Dyster and M. A. Carducci, Cancer Chemother. Pharmacol., 2013, 71, 883–892 CrossRef CAS PubMed.
- A. Blauvelt, S. Kempers, E. Lain, T. Schlesinger, S. Tyring, S. Forman, G. Ablon, G. Martin, H. Wang, D. L. Cutler, J. Fang and M.-F. R. Kwan, N. Engl. J. Med., 2021, 384, 512–520 CrossRef CAS PubMed.
- A. Moore, K. Hurley and S. A. Moore, JAAD Case Rep, 2022, 28, 11–13 CrossRef PubMed.
- J.-B. Hong, P.-Y. Wu, A. Qin, Y.-W. Huang, K.-C. Tseng, C.-Y. Lai, W.-K. Chan, J. Fang, D. L. Cutler and T.-F. Tsai, Pharmaceutics, 2022, 14, 2159 CrossRef CAS PubMed.
- L. Niu, J. Yang, W. Yan, Y. Yu, Y. Zheng, H. Ye, Q. Chen and L. Chen, J. Biol. Chem., 2019, 294, 18099–18108 CrossRef CAS PubMed.
- A. Fallah-Tafti, A. Foroumadi, R. Tiwari, A. N. Shirazi, D. G. Hangauer, Y. Bu, T. Akbarzadeh, K. Parang and A. Shafiee, Eur. J. Med. Chem., 2011, 46, 4853–4858 CrossRef CAS PubMed.
- L. Wang, Y. Zheng, D. Li, J. Yang, L. Lei, W. Yan, W. Zheng, M. Tang, M. Shi, R. Zhang, X. Cai, H. Ni, X. Ma, N. Li, F. Hong, H. Ye and L. Chen, J. Med. Chem., 2021, 64, 8127–8141 CrossRef CAS PubMed.
- Y. Zhang and A. Pike, Bioorg. Med. Chem. Lett., 2021, 38, 127849 CrossRef CAS PubMed.
- D. F. V. Lewis and M. Dickins, Drug Metab. Rev., 2003, 35, 1–18 CrossRef CAS PubMed.
- S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CrossRef CAS PubMed.
- K. Ishihara, T. Shioiri and M. Matsugi, Org. Lett., 2020, 22, 6244–6247 CrossRef CAS PubMed.
- B. Guo, H. Fan, Q. Xin, W. Chu, H. Wang, Y. Huang, X. Chen and Y. Yang, J. Med. Chem., 2013, 56, 2642–2650 CrossRef CAS PubMed.
- J. R. Johansson, T. Beke-Somfai, A. Said Stålsmeden and N. Kann, Chem. Rev., 2016, 116, 14726–14768 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- S. Martellucci, L. Clementi, S. Sabetta, V. Mattei, L. Botta and A. Angelucci, Cancers, 2020, 12, 1448 CrossRef CAS PubMed.
- S. G. Pelaz and A. Tabernero, Oncogene, 2022, 41, 4917–4928 CrossRef CAS PubMed.
- M. Anbalagan, A. Ali, R. K. Jones, C. G. Marsden, M. Sheng, L. Carrier, Y. Bu, D. Hangauer and B. G. Rowan, Mol. Cancer Ther., 2012, 11, 1936–1947 CrossRef CAS PubMed.
- M. Anbalagan, L. Carrier, S. Glodowski, D. Hangauer, B. Shan and B. G. Rowan, Breast Cancer Res. Treat., 2012, 132, 391–409 CrossRef CAS PubMed.
- Schrödinger Release 2023-3: Maestro, Schrödinger, LLC, New York, NY, 2023 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06790d |
| This journal is © The Royal Society of Chemistry 2023 |