DOI:
10.1039/D3RA06692D
(Paper)
RSC Adv., 2023,
13, 33114-33119
Enhancing the electrocatalytic performance of SnX2 (X = S and Se) monolayers for CO2 reduction to HCOOH via transition metal atom adsorption: a theoretical investigation†
Received
2nd October 2023
, Accepted 6th November 2023
First published on 9th November 2023
Abstract
Exploring highly efficient, stable, and low-cost electrocatalysts for CO2 reduction reaction (CRR) can not only mitigate greenhouse gas emission but also store renewable energy. Herein, CO2 electroreduction to HCOOH on the surface of SnX2 (X = S and Se) monolayer-supported non-noble metal atoms (Fe, Co and Ni) was systematically investigated using first-principles calculations. Our results show that Fe, Co and Ni adsorbed on the surface of SnX2 (X = S and Se) monolayers can effectively enhance their electrocatalytic activity for CO2 reduction to HCOOH with low limiting potentials due to the decreasing energy barrier of *OOCH. Moreover, the lower free energy of the *OOCH intermediate on the surface of TM/SnX2 (X = S and Se) monolayers verifies that the electroreduction of CO2 to HCOOH prefers to proceed along the path: CO2 → *OOCH → *HCOOH → HCOOH. Interestingly, SnX2 (X = S and Se) monolayer-supported Co and Ni atoms prefer the HCOOH product with low CRR overpotentials of 0.03/0.01 V and 0.13/0.05 V, respectively, showing remarkable catalytic performance. This work reveals an efficient strategy to enhance the electrocatalytic performance of SnX2 (X = S and Se) monolayers for CO2 reduction to HCOOH, which could provide a way to design and develop new CRR catalysts experimentally in future.
1. Introduction
Nowadays, the greenhouse effect and energy shortage have become more serious with the rapid worldwide economic expansion, and threaten the survival and development of mankind.1,2 So mitigating CO2 emissions and converting CO2 into valuable products have attracted great research attention due to the potential impact on energy storage and recycling resources.3 The electrocatalytic CO2 reduction reaction (CRR) has been widely studied experimentally and theoretically,4–10 which not only reduces CO2 emissions but also simultaneously produces valuable chemicals, such as the main C1 products HCOOH, CO, CH3OH and CH4. However, it is challenging to explore efficient electrocatalysts to improve the exothermic CRR process due to the inert nature of CO2.11,12 In addition, there are some severe bottleneck issues in the process of CRR, such as various intermediates and many reduction reaction pathways, leading to poor product selectivity as well as loss of efficiency toward the competing hydrogen evolution.13–15 Therefore, it is very necessary to design and synthesize electrocatalysts with high product selectivity, low limiting potential and eminent catalytic performance for CRR.
It has been reported that Sn-based catalysts have been found with superior performance toward CRR because of their high activity and selectivity.8,9,16–25 Generally, the *OOCH intermediate is more favorable for Sn-based catalysts during CRR because CO2 prefers to form a bidentate O–Sn adsorption configuration on Sn sites, which results in the HCOOH product, corresponding to a two-electron transfer mechanism. For example, Guan et al.8,9 fabricated metal tin and tin oxides catalysts by different electrodeposition methods and revealed that these catalysts had efficient catalytic ability for CO2 electroreduction to HCOOH. Geng et al.22 demonstrated the catalytic activity of SnS2 nanosheets for CRR was obviously enhanced by surface hydrogen incorporation, which decreases the energy barrier of *OOCH intermediate formation and is in favor of HCOOH product. He et al.24 synthesized a SnSe2@CC electrocatalyst that is high selectivity and catalytic activity towards HCOOH production. All these reported works show that the catalytic performance of two-dimensional (2D) SnX2 (X = S and Se) for CRR can be modulated by surface modification.
In the other hand, single-atom catalysts (SACs) are prospective CRR electrocatalysts because SACs characterized by an isolated metal atom supported on the solid surface play important roles in activating substrate materials.26–32 For example, Xie et al.28 reported that Co atoms on the surface of atomically thin layers prefer to form *OOCH intermediate during CRR with higher activity and selectivity towards HCOOH product. Zheng and co-workers29 synthesized a low-cost carbon nanoparticle supported Ni single atoms via a simple and scalable method, which is high selective and active toward CO production. Moreover, 2D materials supported transition metal (TM) atoms have been supposed as efficient electrocatalyst for CRR due to their large surface-to-volume ratio, tunable electronic structures and strong coordinating capability.30–32 Thus, inspired by these reported works, whether TM atoms anchored on the surface of SnX2 (X = S and Se) monolayers could also improve the catalytic performance of these monolayers for CRR or not, which is necessary to gain an insight into the relationship between the TM atoms and SnX2 (X = S and Se) monolayers and screens out the optimal electrocatalysts for HCOOH production.
Herein, the SnX2 (X = S and Se) monolayers before and after supported Fe, Co and Ni atoms for CO2 electroreduction to HCOOH were systematically investigated by first-principles calculation based on density functional theory (DFT)33,34 in the work. Considering the competition of hydrogen evolution reaction (HER) and CRR, the Gibbs free energies of *H, *COOH and *OOCH intermediates were calculated, which is plotted by ΔG*COOH/*OOCH versus ΔG*H. The results show that SnX2 (X = S and Se) monolayers supported Fe, Co and Ni atoms tend to form *OOCH intermediate, leading to the HCOOH production. Moreover, the overpotential and limiting potential calculations revealed that the electrocatalytic performance of SnX2 (X = S and Se) monolayers for CO2 reduction to HCOOH can be obviously improved by adsorption of Fe, Co and Ni atoms. Especially, Co and Ni anchored on SnX2 (X = S and Se) monolayers for HCOOH production with the lower overpotentials and limiting potentials, corresponding to the potential determining step of *+ CO2 → *CO2. Our calculated results are beneficial to synthesize more efficient CRR catalysts in future, which would provide a perspective on the phenomena observed in the experiment.
2. Computational methods
All the geometry optimization and energy calculations were performed using the Vienna ab initio simulation package (VASP)35,36 based on the DFT with the Perdew–Burke–Ernzerhof (PBE) functional for generalized gradient approximation (GGA)37–39 to characterize the electron exchange and correlation effects. The van der Waals interaction was described in Grimme's scheme (DFT-D3).40 A cutoff energy of 500 eV was used for the plane-wave basis set, and the energy and force convergence criteria were set to 10−6 eV and 0.01 eV Å−1, respectively. The vacuum space of 18 Å was chosen to avoid the interaction between the periodical images. Brillouin zone sampling is performed with Monkhorst–Pack special k-point meshes,41 and the 6 × 6 × 1 k-grid was chosen for the geometry optimization of SnX2 (X = S and Se) monolayers.
The adsorption energy (Eads) of reaction intermediate (A) on the surface of different SnX2 (X = S and Se) monolayers was defined as:
| | |
Eads = E*adsorbate − E* − Eadsorbate
| (1) |
where
E*adsorbate,
E* and
Eadsorbate are the total energy of the adsorbate on the surface of catalyst, the catalyst and the adsorbate, respectively.
Free energies of the CRR intermediates in electrochemical reaction pathways were evaluated according to the computational hydrogen electrode (CHE) model developed by Nørskov et al.42,43 using the reversible hydrogen electrode (RHE) as a reference which chemical potential of (H+ + e−) is equivalent to that of 1/2H2 at 0 V and at all pH values. In the acidic condition, the mechanism of CO2 reduction to HCOOH is generally regarded as the following steps:8,9
| | |
*+ CO2 + H+ + e− → *COOH
| (2) |
| | |
*COOH + H+ + e− → *+ HCOOH
| (3) |
| | |
*+ CO2 + H+ + e− → *OOCH
| (4) |
| | |
*OOCH + H+ + e− → *+ HCOOH
| (5) |
Thus, the Gibbs free energies of each intermediate were calculated as:42
| | |
ΔG = ΔE − ΔEZPE − TΔS + ΔGU + ΔGpH
| (6) |
where Δ
E, Δ
EZPE and
TΔ
S are the total calculated energies, zero-point energy corrections and entropy contributions at 298.15 K, respectively. Δ
GU is the free energy change of electrode potential (
U). Δ
GpH is the free energy correction relating to the solution pH (in this paper pH = 0 for acidic medium). The less negative limiting potential (
UL), the higher activity and selectivity of the catalysts, so
UL determined by the potential determining steps (PDS) is obtained from the free energy change (Δ
GMAX) by using the relation
UL = −Δ
GMAX/
ne. Moreover, the overpotential (
η) is calculated to evaluate the catalytic performance of different SnX
2 (X = S and Se) monolayers, which is the difference between the equilibrium potential (
Ue) and
UL. The vibrational frequency of intermediates for CRR by VASPKIT software that is a pre and post-processing program.
44
3. Results and discussion
3.1 Adsorption of *H, *COOH and *OOCH
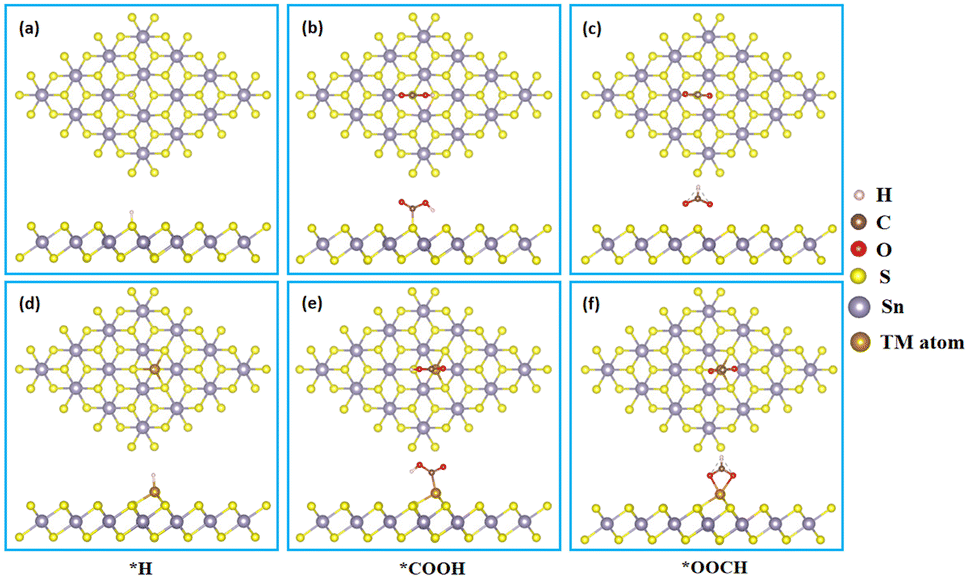
It is well known that the hydrogen evolution reaction (HER) is a competitive reaction during the process of CRR on the most of metal catalysts due to the selectivity of first protonation step.45,46 Therefore, we firstly calculated the *H, *COOH and *OOCH intermediates adsorption on the surface of SnX2 (X = S and Se) monolayers, and the corresponding optimized energy favorable structures are presented in Fig. 1 and S1.† In accordance with our previous work,47 the transition metal atoms (Fe, Co and Ni) also prefer to anchor on the six-member ring (hollow) site of SnSe2 monolayers (Fig. S1†).
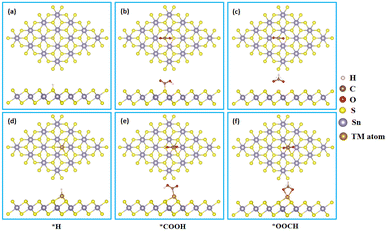 |
| | Fig. 1 Side and top views of key intermediates (*H, *COOH and *OOCH) adsorption on the surface of (a–c) pristine SnS2 and (d–f) TM/SnS2 monolayers. | |
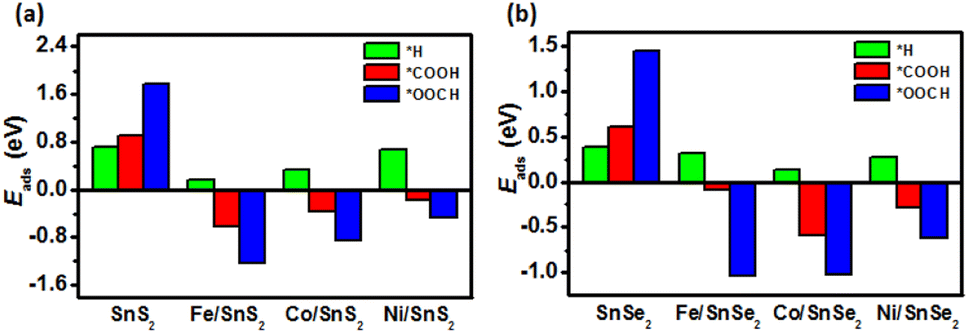
It can be noted that the Eads of *H, *COOH and *OOCH intermediates absorbed on pristine SnX2 (X = S and Se) monolayers are above zero as shown in Fig. 2, indicating unfavorable adsorption. In contrary, the adsorption of *COOH and *OOCH intermediates on the surface of TM/SnX2 (X = S and Se) monolayers are strengthened, especially the *OOCH adsorption, which reveals that the first protonation step tends to form *COOH and *OOCH intermediates for the CRR. In addition, the adsorption of CO2, H2O, CO, and HCOOH molecules on the surface of SnX2 (X = S and Se) monolayers was calculated to evaluate the interaction between these small molecules and the monolayers. As shown in Fig. S2 and Table S1,† the distinct difference in adsorption energies among CO2, H2O, CO, and HCOOH due to the presence of active site on SnX2 (X = S and Se) monolayers, indicating the selectivity of CRR.
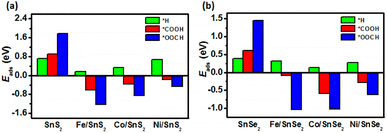 |
| | Fig. 2 The adsorption energy (Eads) of key intermediates (*H, *COOH and *OOCH) adsorption on the surface of (a) SnS2 and (b) SnSe2 monolayers. | |
3.2 Selectivity between CRR and HER
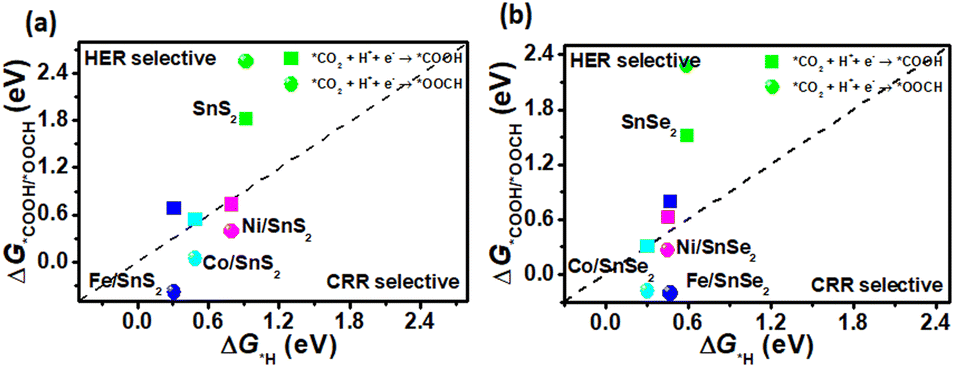
As mentioned above, there is a competition to form *COOH, *OOCH and *H intermediates in the first protonation step for CRR. In addition, it is reported that the reaction with lower free energy is assumed to be more selective with a low reaction barrier.48–51 Therefore, the Gibbs free energies of *COOH, *OOCH and *H absorbed on SnX2 (X = S and Se) monolayers were calculated and compared (shown in Fig. 3) to gain an insight into the selectivity of the catalysts for CRR and HER, respectively. As shown in Fig. 3, the Gibbs free energy change of *COOH and *OOCH (ΔG*COOH and ΔG*OOCH) versus that of *H (ΔG*H) was plotted, and usually the electrocatalysts under the dotted line are CRR selective, while above the dotted line are HER selective. For all TM/SnX2 (X = S and Se) monolayers, *OOCH is more favorable than *COOH and they are below the diagonal line, implying that TM/SnX2 (X = S and Se) monolayers prefer to form *OOCH in the first protonation step for CRR rather than *COOH and *H, and leading to the inhibition of HER. However, pristine SnX2 (X = S and Se) monolayers are above the diagonal line, meaning more selective for HER. And the free energy barriers of the formation of *COOH and *OOCH for pristine SnX2 (X = S and Se) monolayers are higher than 1 eV, revealing that the CRR is difficult. In contrary, the free energy barriers of *OOCH and *COOH are obvious decreased for TM/SnX2 (X = S and Se) monolayers, especially the free energy of *OOCH is 0.04 eV, which exhibits that TM atoms (Fe, Co and Ni) anchored on the surface of SnX2 (X = S and Se) monolayers is an effective strategy to improve the catalytic activity for CRR.
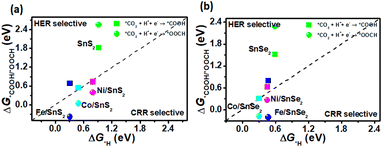 |
| | Fig. 3 The free energy change of the first protonation step in the CRR and HER on the surface of (a) SnS2 and (b) SnSe2 monolayers. | |
3.3 Electrocatalytic activity
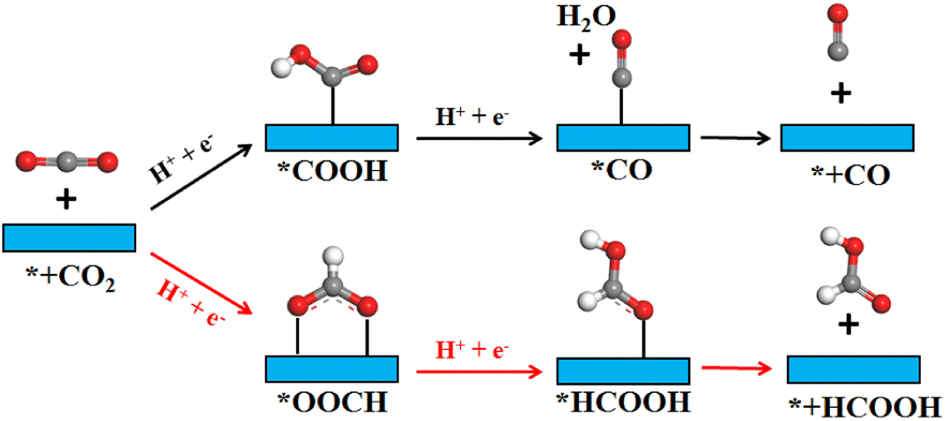
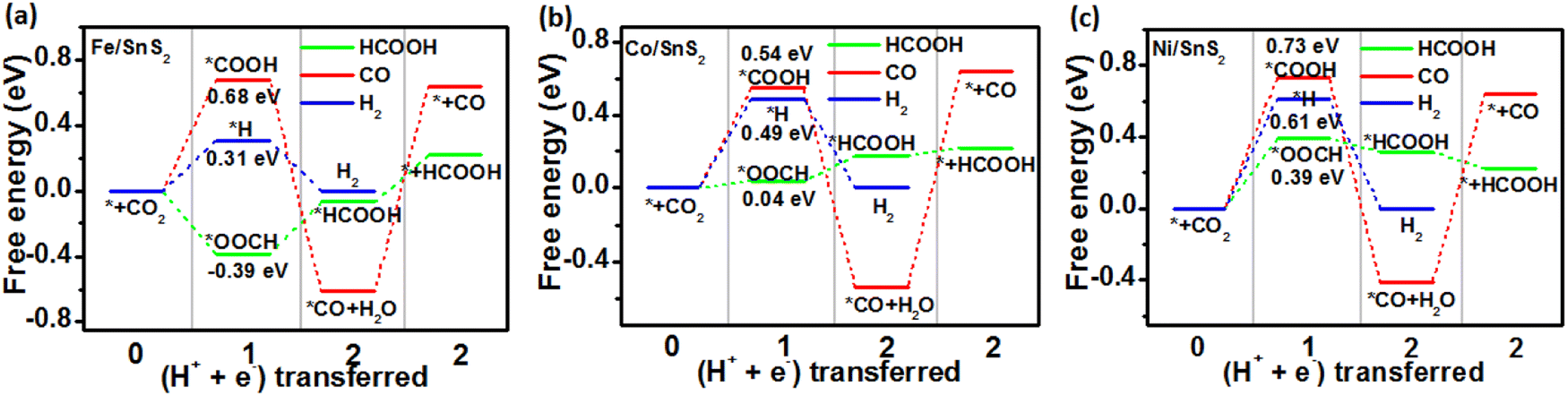
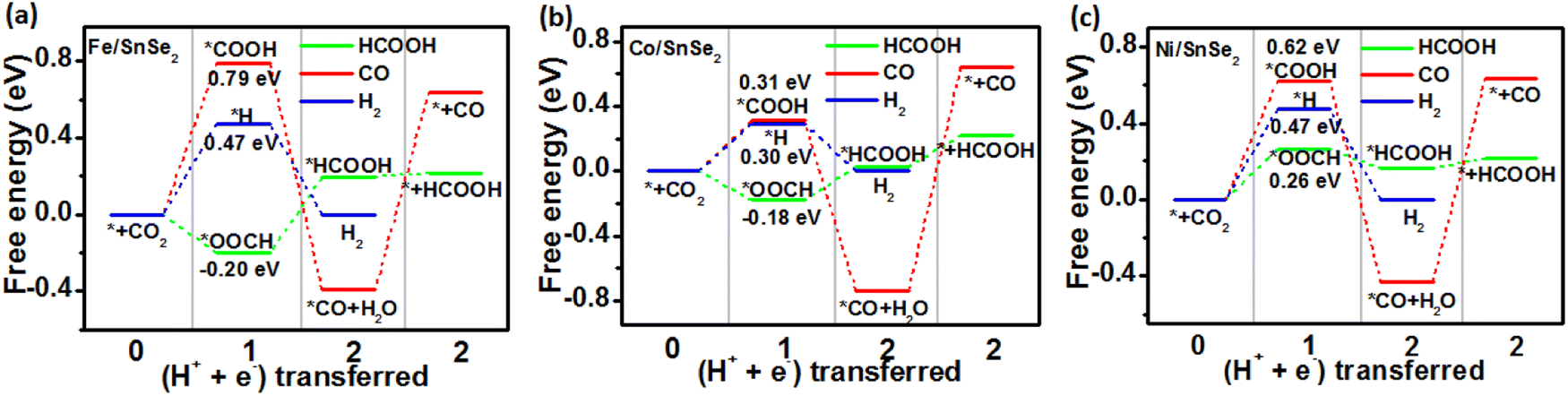
In order to gain a clearer understanding of the electrocatalytic performance and mechanism of TM/SnX2 (X = S and Se) monolayers for CO2 reduction to HCOOH, the Gibbs free energies of all possible intermediates for CRR were systematically calculated and the corresponding configurations presented in Fig. S3 and S4.† The possible reaction pathways of CRR with two electrons transfers are illustrated in Fig. 4, which have been widely accepted for the tin-based electrocatalysts.8,9,21–25 And the lower free energy of *OOCH intermediate on the surface of TM/SnX2 (X = S and Se) monolayers also verifies the electroreduction of CO2 to HCOOH prefers to proceed along CO2 → *OOCH → *HCOOH → HCOOH (Fig. 5 and 6). However, there are high free energy barrier of *COOH for CO2 reduction to HCOOH and CO over the pristine SnX2 (X = S and Se) monolayers (as shown in Fig. S5†), indicating the unfavorable electrocatalytic performance for CRR.
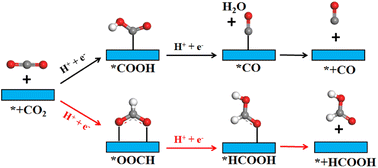 |
| | Fig. 4 Schematic illustration of CO2 reduction to HCOOH and CO products on the surface of SnX2 (X = S and Se) monolayers. The gray, red and white atoms represent C, O and H, respectively. | |
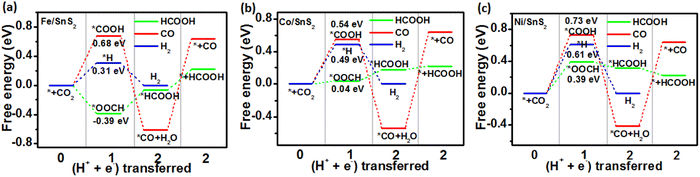 |
| | Fig. 5 Free energy diagram for electroreduction of CO2 to HCOOH (green), CO (red) and H2 (blue) on the surface of (a) Fe/SnS2, (b) Co/SnS2 and (c) Ni/SnS2 monolayers. | |
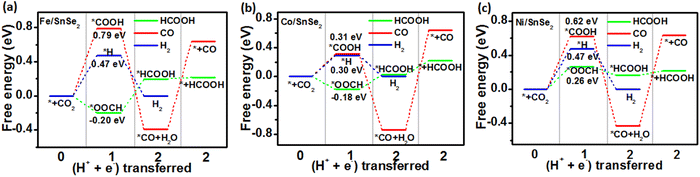 |
| | Fig. 6 Free energy diagram for electroreduction of CO2 to HCOOH (green), CO (red) and H2 (blue) on the surface of (a) Fe/SnSe2, (b) Co/SnSe2 and (c) Ni/SnSe2 monolayers. | |
As shown in Fig. 5 and 6, the calculated energy barrier of *OOCH is lower than those of *H and *COOH, revealing the selectivity of TM/SnX2 (X = S and Se) monolayers for CO2 electroreduction to HCOOH. It can be noted that Fe, Co and Ni atoms anchored on the surface of SnX2 (X = S and Se) monolayers for CRR favor to form *OOCH intermediate with a much smaller free energies compared to form *COOH and *H intermediates in the first protonation step. For the second protonation step, the reaction proceeds from *OOCH to *HCOOH for Ni/SnX2 (X = S and Se) monolayers is exothermic, indicating a spontaneous process. In contrast, this protonation process for other monolayers is endothermic, which reveal that this step cannot spontaneously occur. Interestingly, the activation energy was found to be 0.04 eV for CO2 → *OOCH on Co/SnS2, and the subsequent step of *OOCH → *HCOOH and *HCOOH → HCOOH also owns small barrier of about 0.21 and 0.02 eV, respectively. However, the energy barrier of CO2 to *OOCH and *COOH on the pristine SnX2 (X = S and Se) monolayers is above 1.5 eV (Fig. S5†), which is not beneficial for the formation of HCOOH. Moreover, the free energy of *H for HER on TM/SnX2 (X = S and Se) is much higher than that of *OOCH for CRR, confirms the selectivity of CRR over HER. The Fig. S6 and S7† also exhibit that Fe, Co and Ni atoms anchored on the surface of SnX2 (X = S and Se) monolayers can obviously narrow the free energy barrier of the first protonation step (* + CO2 + H+ + e− → *OOCH) in CRR, which is beneficial to improve the electrocatalytic activity for CO2 reduction to HCOOH. Meanwhile, Our calculated results also show that the free energy barrier (from *CO + H+ + e− to *CHO) is high in this protonation step during CRR for TM/SnX2 (X = S and Se) monolayers (shown in Fig. S8†), indicating unfavorable formation of formaldehyde and methanol product.
3.4 Limiting potential and product selectivity
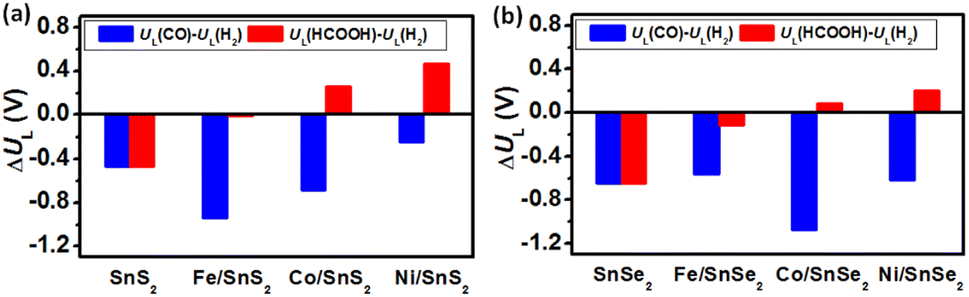
It has been reported that the limiting potential difference (i.e., UL(CO2) − UL(H2), where UL = −ΔGmax/e) can be used as a reasonable selectivity descriptor for CRR and HER.52–54 Usually, the more positive UL(CO2) − UL(H2) value corresponds to the more favorable CRR selectivity. Therefore, to further evaluate the CRR and HER selectivity on different SnX2 (X = S and Se) monolayers, the limiting potential difference (ΔUL) at zero applied potential and acidic condition was calculated and compared. As clearly seen in Fig. 7, the ΔUL for CO2 to HCOOH is positive on the surface of SnX2 (X = S and Se) monolayers supported Co and Ni atoms, signifying a higher HCOOH selectivity for CRR. In contrast, the pristine SnX2 (X = S and Se) monolayers exhibit worse CRR selectivity without applied potential (U = 0.0 V versus RHE). As the Fe atom anchored on SnX2 (X = S and Se), the ΔUL for HCOOH route becomes nearly zero, suggesting the obvious effect of suppressing HER. In addition, the calculated limiting potential difference of UL(CO)−UL(H2) is negative for all SnX2 (X = S and Se) monolayers, which indicates worse CRR selectivity for CO production.
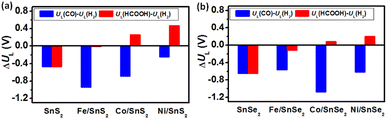 |
| | Fig. 7 Limiting potential differences (UL(CO) − UL(H2) and UL(HCOOH) − UL(H2)) between CRR and HER on the surface of different (a) SnS2 and (b) SnSe2 monolayers at 0 V vs. RHE. | |
The potential determining steps (PDS), limiting potentials (UL) and overpotentials (η) for CO2 reduction to HCOOH and CO were summarized in Tables 1 and S2,† respectively. It can be noted that the free energy changes of PDS for pristine SnS2 and SnSe2 are obvious higher than those of TM/SnX2 (X = S and Se), and the high overpotentials for CO2 reduction to HCOOH and CO reveal poor CRR selectivity. In contrast, the electrocatalytic activity of SnX2 (X = S and Se) monolayers supported Fe, Co and Ni atoms for CO2 to HCOOH production is distinctly enhanced with lower overpotentials, but for CO formation displaying inferior catalytic activity. Especially, the Ni/SnX2(X = S and Se) monolayers are supposed to be superior in catalyzing CO2 to HCOOH based on UL and η calculated from free energy changes of PDS, where *+ CO2 → *CO2 is predicted to be the PDS. All these results illustrate that the adsorption of Fe, Co and Ni atoms on the surface of SnX2 (X = S and Se) monolayers can effectively tune the catalytic activity for CRR, which would provide an insight to the selective electroreduction of CO2 based on the materials in the future experiment.
Table 1 The potential determining steps (PDS), limiting potentials (UL = −ΔGmax/e) and overpotentials (η = Uequ − UL, Uequ is the equilibrium potential) for CO2 reduction to HCOOH on the different surface of SnX2 (X = S and Se) monolayers
| Surface |
PDS |
UL/V |
η/V |
| Pristine SnS2 |
*CO2 → *COOH |
–1.39 |
1.19 |
| Fe/SnS2 |
*COOH → *HCOOH |
–0.32 |
0.12 |
| Co/SnS2 |
*+ CO2 → *CO2 |
–0.23 |
0.03 |
| Ni/SnS2 |
*+ CO2 → *CO2 |
–0.33 |
0.13 |
| Pristine SnSe2 |
*CO2 → *COOH |
–1.24 |
1.04 |
| Fe/SnSe2 |
*+ CO2 → *CO2 |
–0.59 |
0.39 |
| Co/SnSe2 |
*OOCH → *HCOOH |
–0.21 |
0.01 |
| Ni/SnSe2 |
*+ CO2 → *CO2 |
–0.25 |
0.05 |
4. Conclusions
In summary, the electrocatalytic activity of pristine and TM atoms (Fe, Co and Ni) anchored on SnX2 (X = S and Se) monolayers for CO2 reduction to HCOOH were systematically investigated by first-principles calculation method. The calculated free energies of *COOH, *OOCH and *H reveal that it is easier to form *OOCH intermediate in the first protonation step of CRR on the SnX2 (X = S and Se) monolayers supported Fe, Co and Ni atoms. Meanwhile, the electrocatalytic activity of SnX2 (X = S and Se) monolayers for CO2 reduction to HCOOH can be obviously promoted by TM atoms (Fe, Co and Ni) adsorption, which confirms the selectivity of CRR over HER. Moreover, the calculated limiting potential differences (ΔUL) display that the adsorption of Co and Ni on SnX2 (X = S and Se) monolayers prefers a higher HCOOH selectivity for CRR with lower overpotentials, while pristine SnX2 (X = S and Se) monolayers exhibit worse CRR selectivity with higher overpotentials both for HCOOH and CO production. All these results demonstrate that TM atoms (Fe, Co and Ni) own great potential of enhancing the electrocatalytic performance of SnX2 (X = S and Se) monolayers for CO2 reduction to HCOOH, which can provide a theoretical reference for designing high-efficiency SnX2 (X = S and Se)-based single atom catalysts experimentally in future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21703087).
Notes and references
- D. R. Yang, B. Ni and X. Wang, Adv. Energy Mater., 2020, 10, 2001142 CrossRef CAS.
- R. Ahmed, G. J. Liu, B. Yousaf, Q. Abbas, H. Ullah and M. U. Ali, J. Cleaner Prod., 2020, 242, 118409 CrossRef CAS.
- A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700759 CrossRef.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- Z. Y. Sun, T. Ma, H. C. Tao, Q. Fan and B. X. Han, Chem, 2017, 3, 560–587 CAS.
- X. W. An, S. S. Li, A. Yoshida, T. Yu, Z. D. Wang, X. G. Hao, A. Abudula and G. Q. Guan, ACS Appl. Mater. Interfaces, 2019, 11, 42114–42122 CrossRef CAS PubMed.
- X. W. An, S. S. Li, A. Yoshida, Z. D. Wang, X. G. Hao, A. Abudula and G. Q. Guan, ACS Sustainable Chem. Eng., 2019, 7, 9360–9368 CrossRef CAS.
- L. L. Gong, D. T. Zhang, C. Y. Lin, Y. H. Zhu, Y. Shen, J. Zhang, X. Han, L. P. Zhang and Z. H. Xia, Adv. Energy Mater., 2019, 44, 1902625 CrossRef.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- O. Melchaeva, P. Voyame, V. C. Bassetto, M. Prokein, M. Renner, E. Weidner, M. Petermann and A. Battistel, ChemSusChem, 2017, 10, 3660–3670 CrossRef CAS PubMed.
- R. J. Lim, M. S. Xie, M. A. Sk, J.-M. Lee, A. Fisher, X. Wang and K. H. Lim, Catal. Today, 2014, 233, 169–180 CrossRef CAS.
- C. Zhao and J. Wang, Chem. Eng. J., 2016, 293, 161–170 CrossRef CAS.
- L. Fan, Z. Xia, M. J. Xu, Y. Y. Lu and Z. J. Li, Adv. Funct. Mater., 2018, 28, 1706289 CrossRef.
- J. Gu, F. Héroguel, J. Luterbacher and X. Hu, Angew. Chem., 2018, 130, 2993–2997 CrossRef.
- F. Li, L. Chen, M. Xue, T. Williams, Y. Zhang, D. R. MacFarlane and J. Zhang, Nano Energy, 2017, 31, 270–277 CrossRef CAS.
- Y. Luo, Y. Cui, M. Y. Li, X. L. Zhang, Y. X. Dai, C. Y. Ling and Y. C. Huang, ACS Appl. Nano Mater., 2021, 4, 2760–2767 CrossRef CAS.
- J. Xu, S. H. Lai, M. Hu, S. M. Ge, R. C. Xie, F. Li, D. D. Hua, H. Xu, H. Zhou, R. Wu, J. T. Fu, Y. Qiu, J. He, C. Li, H. X. Liu, Y. F. Liu, J. Q. Sun, X. J. Liu and J. Luo, Small Methods, 2020, 4, 2000567 CrossRef CAS.
- A. Zhang, Y. X. Liang, H. P. Li, S. L. Wang, Q. X. Chang, K. Y. Peng, Z. G. Geng and J. Zeng, Nano Lett., 2021, 21, 7789–7795 CrossRef CAS PubMed.
- Y. S. Yao, W. J. Yin, Z. K. Tang, Y. M. Xu, J. Y. Guo, J. X. Cao, H. Q. Wang and X. L. Wei, J. Phys. Chem. C, 2022, 126, 1271–1280 CrossRef CAS.
- B. He, L. Jia, Y. Cui, W. Zhou, J. Sun, J. Xu, Q. Wang and L. Zhao, ACS Appl. Energy Mater., 2019, 2, 7655–7662 CrossRef CAS.
- H. Yang, H. Liu, X. Liu, Z. Zhao and J. Luo, Catal. Sci. Technol., 2018, 8, 5428–5433 RSC.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- C. Gao, S. M. Chen, Y. Wang, J. W. Wang, X. S. Zheng, J. F. Zhu, L. Song, W. K. Zhang and Y. J. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed.
- S. Gao, Y. Lin, X. C. Jiao, Y. F. Sun, Q. Q. Luo, W. H. Zhang, D. Q. Li, J. L. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed.
- T. T. Zheng, K. Jiang, N. Ta, Y. F. Hu, J. Zeng, J. Y. Liu and H. T. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- C. Guo, T. Zhang, X. Deng, X. Liang, W. Guo, X. Lu and C. M. L. Wu, ChemSusChem, 2019, 12, 5126–5132 CrossRef CAS PubMed.
- S. Back, J. Lim, N.-Y. Kim, Y.-H. Kimb and Y. Jung, Chem. Sci., 2017, 8, 1090–1096 RSC.
- Z. X. Wang, J. X. Zhao and Q. H. Cai, Phys. Chem. Chem. Phys., 2017, 19, 23113–23121 RSC.
- P. Hohenberg and W. Kohn, Phys. Rev. B: Condens. Matter Mater. Phys., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- B. Hammer, K. W. Jacobsen and J. K. Nørskov, Phys. Rev. Lett., 1993, 70, 3971–3974 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- V. Wang, N. Xu, J. C. Liu, G. Tang and W. T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- S. Popovic, M. Smiljanic, P. Jovanovic, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef CAS PubMed.
- F. F. Xia and F. L. Yang, Energy Fuels, 2022, 36, 4992–4998 CrossRef CAS.
- Z. Z. Chen, X. Zhang and G. Lu, Chem. Sci., 2015, 6, 6829–6835 RSC.
- G. Luo, Y. Jing and Y. F. Li, J. Mater. Chem. A, 2020, 8, 15809–15815 RSC.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1938, 34, 11–24 RSC.
- J. N. Bronsted, Chem. Rev., 1928, 5, 231–338 CrossRef CAS.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu and C. Wu, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- W. Bi, X. Li, R. You, M. Chen, R. Yuan, W. Huang, X. Wu, W. Chu, C. Wu and Y. Xie, Adv. Mater., 2018, 30, 1706617 CrossRef PubMed.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Nørskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Qing Xu,
Fengli Yang,
Li Shu and
Yingpin Wen
,
Qing Xu,
Fengli Yang,
Li Shu and
Yingpin Wen