
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical reduction of CO2 into CO coupling with triethanolamine decomposition†
Zhen Li *,
Caili Yang,
Yingshi Su,
Yonghui Cheng,
Yanjia Cui,
Suyao Liu and
Yiwen Fang
*,
Caili Yang,
Yingshi Su,
Yonghui Cheng,
Yanjia Cui,
Suyao Liu and
Yiwen Fang
Department of Chemistry and Key Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province, Shantou University, Shantou 515063, China. E-mail: lizhenlicp@stu.edu.cn
First published on 30th October 2023
Abstract
In this work, the impacts of triethanolamine (TEOA) on the performance of photochemical CO2 reduction were investigated in a simple homogeneous system. We demonstrates that CO2 can be converted into CO coupling with the decomposition of triethanolamine in TEOA aqueous solution without other additives under light irradiation. About 7.5 μmol CO product is achieved within 7 h with a maximum apparent quantum yield (AQY) of 0.086% at 254 nm. The isotope labelling experiment confirms that CO product originates from the reduction of CO2 rather than the decomposition of TEOA. In addition, the photochemical system exhibits excellent stability, no obvious inactivation is observed during long-term photochemical CO2 reduction reaction. This work provides a deep understanding of the effects of TEOA on the performance of photocatalytic CO2 reduction. Upon utilizing TEOA as a sacrificial electron donor in photocatalytic system, the contribution of TEOA must be considered once evaluating the catalytic activity of catalysts.
Introduction
Artificial photochemical conversion of CO2 into fuels or value-added chemicals using solar energy is considered as a promising strategy for addressing both energy crisis and environmental issues associated with the consumption of fossil fuels.1–5 However, the CO2 reduction reaction faces significant challenges due to the thermodynamic stability and low reactivity of CO2 molecule. To overcome these challenges, researchers have been focusing on the development of photochemical systems, including homogeneous and heterogeneous systems. The homogeneous system, which involves molecular catalysts, has gained attention due to its advantages in high activity, selectivity, tunable structure, and trackable chemical reactivity.6,7Generally, a typical homogeneous photochemical system for CO2 reduction includes a photosensitizer, a sacrificial reductant, and a molecular catalyst. Designing effective molecular catalysts has been a key focus in achieving high activity and selectivity for CO2 reduction. In the past decades, various types of catalysts, such as metal pyridyl complexes,8,9 metal porphyrin catalysts,10 metal salen complexes,11,12 imidazolium salts,13 and ionic liquids,14,15 have been developed and shown excellent catalytic activity in homogeneous systems. In these photochemical systems, molecular complexes usually exhibit dual function. First, molecular catalyst can function as a photosensitizer, which converts solar light into chemical energy for CO2 reduction. While the metal center of the complex acts as an active site, transferring the photoinduced electron into the reduction reaction of CO2. In addition, it is crucial to ensure the rapid acceptance of an additional electron from sacrificial electron donors to avoid the accumulation of charge carriers, which can induce catalyst decomposition and diminish the durability of the photocatalytic system.16,17 TEOA, acting as a sacrificial reductive quencher for chromophore excited states, is commonly used as an electron donor in photochemical CO2 reduction reaction. However, it should be noted that previous reports have overlooked the potential secondary roles of TEOA in photochemical CO2 reduction performance. In other words, TEOA might directly reduce CO2 under light irradiation. Thereby, further research is needed to fully understand the potential applications and contributions of TEOA in photochemical CO2 reduction mechanisms.
Herein, the effects of TEOA on the performance of homogeneous photochemical CO2 reduction were studied in detail. The gaseous and liquid products were analyzed by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy, mass spectroscopy (MS), attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR), and gas chromatography (GC). These studies collectively provide a comprehensive understanding of the reaction mechanism during the photochemical CO2 reduction in the presence of TEOA aqueous solution without other additives.
Results and discussion
We first optimized the performance of photochemical CO2 reduction in a 250 mL custom-built cell with different volume ratio of TEOA and H2O (Fig. S1–S3†). A 300 W Xe lamp was used as the irradiation source, and the irradiation area was 12.5 cm2. It can be observed that the generation rate of CO exhibits similar trend in three photochemical systems, which increases upon increasing irradiation time. About 1.29, 1.58 and 1.65 μmol h−1 of CO generation rates are achieved at 7 h irradiation, corresponding to 3, 5 and 10 vol% TEOA aqueous solution respectively. As a result, 5 vol% TEOA aqueous solution system was applied for the following comparison. The UV-vis absorption spectrum was performed to monitor reaction intermediate. As depicted in Fig. S4,† the original TEOA aqueous solution mainly absorb ultraviolet light below 275 nm. Upon irradiation, a new absorption band emerges, possibly attributed to the formation of carbamate species.18,19 The peak intensity gradually increases with prolonged irradiation time and reaches a maximum at 7 h irradiation. Such evolution is consistent with that of CO2 generation rate, suggesting the presence of carbamate intermediate.Fig. 1a shows the time-dependent CO generation amount during photochemical CO2 reduction reaction in 5 vol% TEOA aqueous solution under light irradiation. About 7.5 μmol CO product is achieved within the testing time frame. Note that the initial rate for CO generation is low at the first 3 h of photochemical reaction, which may be associated to an induced period.20 The quasi-linear plot of CO generation amount with irradiation time implies CO gas comes from photochemical reduction.21 Table S1† summarizes the photochemical CO2 reduction under different sets of conditions. The result suggests that the TEOA aqueous solution systems exhibit significant performance in the conversion of CO2 into CO under light irradiation. However, no CO product is detected (entry 4) after equipping with a cut-off filter (λ > 420 nm), implying that this photochemical reaction needs to be initiated by ultraviolet light, which is supported by the test of UV-vis absorption spectrum. Once CO2 is replaced with N2 (entry 5), no detectable CO is observed, suggesting that CO2 serves as the carbon source in our photochemical reaction, in other words, the CO product originates from CO2 rather than the decomposition of TEOA. In order to confirm that CO is generated exclusively from CO2, the isotope labeling experiment of 13CO2 gas was conducted as shown in Fig. S5.† The mass spectra presents one peak at m/z = 29 assigned to 13CO, verifying that the CO product comes from the reduction of CO2 molecule. The apparent quantum yield (AQY) was performed with a 300 W Xe lamp irradiation equipped with various band-pass filters (254, 275, 295, 313, 350, 365 and 380 nm) as depicted in Table S2.† The result suggests that this photochemical system exhibits low AQY value, and the maximum AQY of CO generation is calculated to be 0.086% at 254 nm. In addition, the stability of the photochemical system was performed as shown in Fig. 1b, there is no obvious inactivation during photochemical CO2 reduction process after 100 h irradiation, suggesting the robustness of this photochemical system.
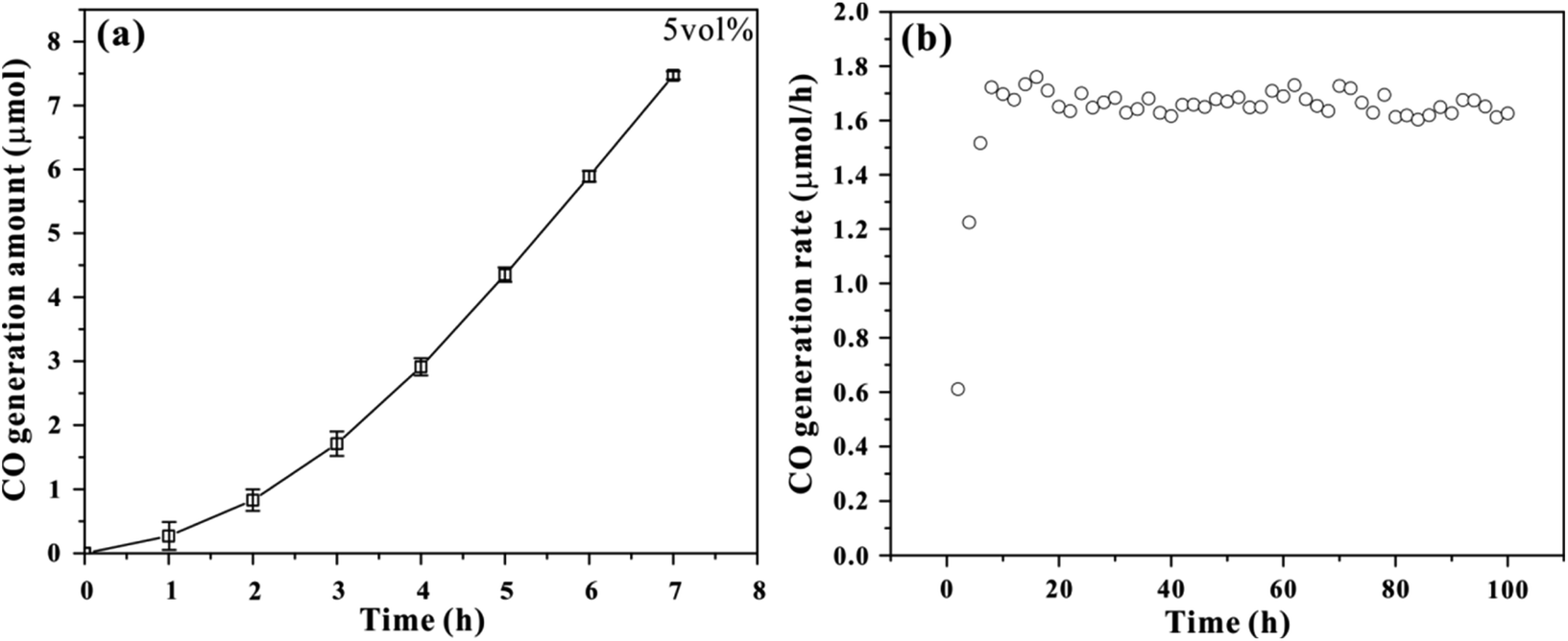 | ||
| Fig. 1 (a) Time-dependent performance of photochemical CO2 reduction in 5 vol% TEOA aqueous solution under light irradiation. The error bars represent the standard deviation of three separate measurements. (b) The stability investigation during photochemical CO2 reduction in 5 vol% TEOA aqueous solution under light irradiation. | ||
Note that the sources of protons and electrons are required during CO2 reduction reaction because they play a vital role in enabling the deoxygenation process of CO2 molecule. The primary source of the protons can be attributed to H2O or TEOA in our photochemical system. However, the electron can only come from TEOA molecule rather than H2O since no oxygen has been detected during CO generation. In particular, TEOA can donate two protons and two electrons during its decomposition (see Scheme 1).22–25 Therefore, we speculate that the electrons from TEOA decomposition participate in CO2 reduction reaction.
 | ||
| Scheme 1 The decomposition pathway of TEOA upon monoelectronic oxidation. | ||
In order to confirm this point, 1H and 13C NMR spectra of pure TEOA, TEOA after 12 h irradiation and TEOA after 100 h irradiation were recorded on a Bruker 600 MHz spectrometer as shown in Fig. 2. Prior to test, the solution after irradiation was freeze-dried for 48 h to remove residual water. In Fig. 2a, three 1H NMR signals are observed for pure TEOA, the peaks centred at 4.73, 3.55 and 2.60 ppm are attributed to OH, O–CH2 and N–CH2 groups according to previous reports.26–29 After 12 h irradiation under CO2 bubbling condition, two new signals are monitored, one peak at 3.58 ppm is assigned to O–CH2 group, while the other at 2.68 ppm is assigned to N–CH2 groups in diethanolamine (DEOA) molecules. However, the OH signal of DEOA disappears due to its overlaps with TEOA, as supported by the area integration analysis and the inserted figure in Fig. 2a. After 100 h irradiation under CO2 bubbling condition, the peak intensity of DEOA is significantly enhanced. The results indicate the presence of DEOA in the solution after irradiation. Furthermore, the content of DEOA increases once prolonging the irradiation time. This observation suggests that the photochemical CO2 reduction reaction leads to the decomposition of TEOA. It is also confirmed by 13C NMR spectra shown in Fig. 2b, two 13C NMR signals are detected for pure TEOA, the peaks centred at 58.82 and 55.62 ppm are attributed to N–CH2 and O–CH2 groups of TEOA molecules, respectively.30–32 After 12 h irradiation under CO2 bubbling condition, two new signals are observed, one peak at 59.49 ppm is assigned to O–CH2 group, while the other at 49.62 ppm is assigned to N–CH2 groups in DEOA molecules. Similarly, the peak intensity of DEOA is significantly enhanced after 100 h irradiation under CO2 bubbling condition.
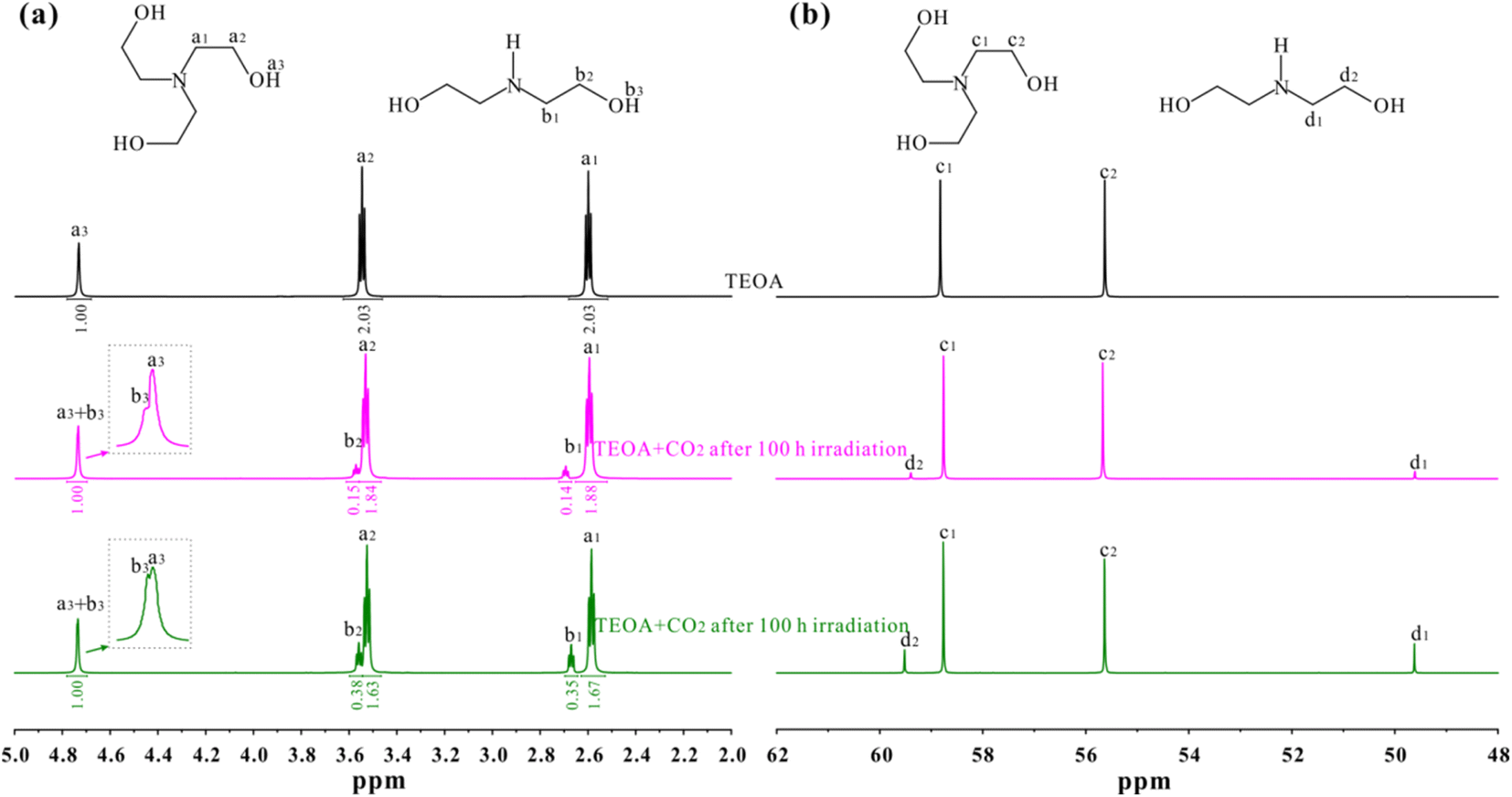 | ||
| Fig. 2 (a) 1H and (b) 13C NMR spectra of pure TEOA, TEOA after 12 or 100 h irradiation under CO2 bubbling condition. The solution consists of 100 μL TEOA and 500 μL D2O. The TEOA after photochemical reaction in 5 vol% TEOA aqueous solution is achieved by freeze-drying for 48 h to remove residual water. | ||
The ATR-FTIR spectra were also performed to study the decomposition of TEOA as shown in Fig. S6.† For pure TEOA, it shows the typical vibration modes of TEOA molecules, consistent with previous reports.33 After 12 h irradiation under CO2 bubbling condition, two new peaks centred at 1647 and 1544 cm−1 are observed, which derives from the vibration modes of N–H deformation in DEOA molecules and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching in NCOO− group34–36 that generated from the adsorption of CO2 by amine species.29 While their intensity increases as irradiation proceeds, proving that the formation process of DEOA is caused by photochemical reaction. In addition, the ESI-MS spectra were applied to further confirm the formation of DEOA from the decomposition of TEOA as shown in Fig. S7–S9.† For pure TEOA, the signals at m/z = 150 and 172 correspond to TEOA, while the new signal at m/z = 106 was attributed to DEOA after light irradiation.
O stretching in NCOO− group34–36 that generated from the adsorption of CO2 by amine species.29 While their intensity increases as irradiation proceeds, proving that the formation process of DEOA is caused by photochemical reaction. In addition, the ESI-MS spectra were applied to further confirm the formation of DEOA from the decomposition of TEOA as shown in Fig. S7–S9.† For pure TEOA, the signals at m/z = 150 and 172 correspond to TEOA, while the new signal at m/z = 106 was attributed to DEOA after light irradiation.
To identify the photochemical process is stoichiometric in TEOA aqueous solution, the amount of TEOA was monitored during photochemical CO2 reduction by 1H NMR technique. As a result, the standard curves for different amount of TEOA was quantified by the peak area as shown in Fig. S10.† Fig. 3 presented the decomposition amount of TEOA, which is determined by subtracting the remaining TEOA quantity from the initial total TEOA content within the system. As we can see, about 63.7 μmol TEOA is consumed within the testing time frame. This value is much higher than that of CO generation amount as shown in Fig. 1, suggesting the photochemical CO2 reduction reaction is nonstoichiometric in TEOA aqueous solution. This is possibly caused by the self-decomposition of excited TEOA molecules.37–39 Nonetheless, it exhibited similar trend with the CO generation amount, implying the photochemical reduction of CO2 into CO coupled with TEOA decomposition.
 | ||
| Fig. 3 The time-depended consumption during photochemical CO2 reduction in 5 vol% TEOA aqueous solution under light irradiation. | ||
According to the previous reports40–43 and our experimental results, a possible mechanism for photochemical CO2 reduction in our system is proposed in Scheme 2. TEOA firstly adsorbs CO2 molecules to form carbamate, which subsequently converts into a positively charged aminyl radical (TEOA˙+) and carbon dioxide radical anions (CO2˙−) by a monoelectronic transfer under UV light irradiation condition. Then the deprotonation of the TEOA˙+ transfers into a carbon centered radical via rearrangement,44 which will contribute to the deoxygenation of CO2˙− to generate CO product. Subsequently, the iminium species further degrades into glycolaldehyde and DEOA in the present of H2O.
 | ||
| Scheme 2 Proposed mechanism for CO2 reduction coupling with TEOA decomposition. | ||
Conclusion
In summary, our work demonstrates that CO2 can be converted into CO in a TEOA aqueous solution under light irradiation without other additives. The isotope labelling experiment confirms the CO product originates from CO2 rather than the decomposition of TEOA. This photochemical system exhibits excellent stability during long-term photochemical CO2 reduction reaction. In addition, the decomposition of TEOA was also analyzed by 1H and 13C NMR, MS and ATR-FTIR. At last, a possible mechanism is proposed for CO2 reduction coupling with TEOA decomposition. We believe that this work will provide a deep understanding of the effects of TEOA on the performance of photocatalytic CO2 reduction. When TEOA is served as a sacrificial electron donor in photocatalytic system, the investigation of intrinsic activity for catalyst needs to take TEOA contributions into consideration.Author contributions
C. L. Yang: investigation, validation, data analysis, writing. Y. S. Su, Y. H. Cheng, Y. J. Cui: investigation. S. Y. Liu, Y. W. Fang: supervision. Z. Li: review, editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of Guangdong Province (2022A1515012359), National Natural Science Foundation of China (21902121), STU Scientific Research Foundation for Talents (NTF21020), the 2020 Li Ka Shing Foundation Cross-Disciplinary Research Grant (2020LKSFG09A) and the Department of Education of Guangdong Province (2021KCXTD032).References
- T. Wang, L. Zhang, J. Liu, X. X. Li, L. Yuan, S. L. Li and Y. Q. Lan, Chem. Commun., 2022, 58, 7507–7510 RSC
.
- C. C. Lin, T. R. Liu, S. R. Lin, K. M. Boopathi, C. H. Chiang, W. Y. Tzeng, W. H. C. Chien, H. S. Hsu, C. W. Luo, H. Y. Tsai, H. A. Chen, P. C. Kuo, J. Shiue, J. W. Chiou, W. F. Pong, C. C. Chen and C. W. Chen, J. Am. Chem. Soc., 2022, 144, 15718–15726 CrossRef CAS PubMed
- Q. Y. Ge, Y. Y. Liu, K. J. Li, L. F. Xie, X. J. Ruan, W. Wang, L. Q. Wang, T. Wang, W. B. You and L. W. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304189 CrossRef CAS PubMed
- A. Sole-Daura, Y. Benseghir, M. H. Ha-Thi, M. Fontecave, P. Mialane, A. Dolbecq and C. Mellot-Draznieks, ACS Catal., 2022, 12, 9244–9255 CrossRef CAS
- L. K. Putri, B. J. Ng, W. J. Ong, S. P. Chai and A. R. Mohamed, Adv. Energy Mater., 2022, 12, 2201093 CrossRef CAS
- B. Ma, M. Blanco, L. Calvillo, L. J. Chen, G. Chen, T. C. Lau, G. Drazic, J. Bonin, M. Robert and G. Granozzi, J. Am. Chem. Soc., 2021, 143, 8414–8425 CrossRef CAS PubMed
- D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed
- A. C. Tsipis and A. A. Sarantou, Dalton Trans., 2021, 50, 14797–14809 RSC
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC
- C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134–138 CrossRef CAS
- M. North, S. C. Z. Quek, N. E. Pridmore, A. C. Whitwood and X. Wu, ACS Catal., 2015, 5, 3398–3402 CrossRef CAS
- C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed
- F. D. Bobbink, D. Vasilyev, M. Hulla, S. Chamam, F. Menoud, G. Laurenczy, S. Katsyuba and P. J. Dyson, ACS Catal., 2018, 8, 2589–2594 CrossRef CAS
- M. H. Anthofer, M. E. Wilhelm, M. Cokoja, M. Drees, W. A. Herrmann and F. E. Kühn, ChemCatChem, 2015, 7, 94–98 CrossRef CAS
- Z. Z. Yang, Y. F. Zhao, G. P. Ji, H. Y. Zhang, B. Yu, X. Gao and Z. M. Liu, Green Chem., 2014, 16, 3724–3728 RSC
- M. Takahashi, T. Asatani, T. Morimoto, Y. Kamakura, K. Fujii, M. Yashima, N. Hosokawa, Y. Tamaki and O. Ishitani, Chem. Sci., 2023, 14, 691–704 RSC
- M. Pschenitza, S. Meister and B. Rieger, Chem. Commun., 2018, 54, 3323–3326 RSC
- H. J. Kwon, J. R. Cha and M. S. Gong, Polymer, 2017, 113, 4313–4322 Search PubMed
- K. H. Chae and Y. H. Kim, Adv. Funct. Mater., 2007, 17, 3470–3476 CrossRef CAS
- S. X. Min and G. X. Lu, J. Phys. Chem. C, 2011, 115, 13938–13945 CrossRef CAS
- Z. G. Liu, J. Y. Li, Z. Y. Chen, M. Y. Li, L. Z. Wang, S. Q. Wu and J. L. Zhang, Appl. Catal., B, 2023, 326, 122338 CrossRef CAS
- P. W. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726–7727 CrossRef CAS PubMed
- K. Kalyanasundaram, J. Kiwi and M. Grätzel, Helv. Chim. Acta, 1978, 61, 2727–2730 CrossRef
- E. D. Cline, S. E. Adamson and S. Bernhard, Inorg. Chem., 2008, 47, 10378–10388 CrossRef CAS
- D. G. Whitten, Acc. Chem. Res., 1980, 13, 83–90 CrossRef CAS
- Y. S. Ge, G. Cheng and H. Z. Ke, J. CO2 Util., 2022, 57, 101873 CrossRef CAS
- A. García-Abuín, D. Gomez-Díaz, A. B. Lopez, J. M. Navaza and A. Rumbo, Ind. Eng. Chem. Res., 2013, 52, 13432–13438 CrossRef
- C. Chatterjee and A. Sen, J. Mater. Chem. A, 2015, 3, 5642–5647 RSC
- J. H. Choi, S. G. Oh, Y. E. Kim, Y. I. Yoon and S. C. Nam, Environ. Eng. Sci., 2012, 29, 328–334 CrossRef CAS
- A. U. Maheswari and K. Palanivelu, J. CO2 Util., 2014, 6, 45–52 CrossRef
- G. Szalontai, G. Kiss and L. Bartha, Spectrochim. Acta, Part A, 2003, 59, 1995–2007 CrossRef PubMed
- P. V. Kortunov, L. S. Baugh, M. Siskin and D. C. Calabro, Energy Fuels, 2015, 29, 5967–5989 CrossRef CAS
- S. Ahmed, A. Ramli and S. Yusup, Int. J. Greenhouse Gas Control, 2016, 51, 230–238 CrossRef CAS
- G. G. Qi, Y. B. Wang, L. Estevez, X. N. Duan, N. Anako, A. H. A. Park, W. Li, C. W. Jones and E. P. Giannelis, Energy Environ. Sci., 2011, 4, 444–452 RSC
- A. Zhao, A. Samanta, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2013, 52, 6480–6491 CrossRef CAS
- H. He, C. Liu, M. E. Louis and G. H. Li, J. Mol. Catal. A: Chem., 2014, 395, 145–150 CrossRef CAS
- Q. Y. Li, Z. L. Jin, Z. G. Peng, Y. X. Li, S. B. Li and G. X. Lu, J. Phys. Chem. C, 2007, 111, 8237–8241 CrossRef CAS
- X. J. Zhang, Z. L. Jin, Y. X. Li, S. B. Li and G. X. Lu, J. Phys. Chem. C, 2009, 113, 2630–2635 CrossRef CAS
- Q. Y. Li, L. Chen and G. X. Lu, J. Phys. Chem. C, 2007, 111, 11494–11499 CrossRef CAS
- A. Miyaji and Y. Amao, New J. Chem., 2021, 45, 5780–5790 RSC
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS
- B. H. Zhao, Y. Huang, D. L. Liu, Y. F. Yu and B. Zhang, Sci. China: Chem., 2020, 63, 28–34 CrossRef CAS
- Y. X. Pan, Y. You, S. Xin, Y. T. Li, G. T. Fu, Z. M. Cui, Y. L. Men, F. F. Cao, S. H. Yu and J. B. Goodenough, J. Am. Chem. Soc., 2017, 139, 4123–4129 CrossRef CAS PubMed
- P. J. Delaive, T. K. Foreman, C. Giannotti and D. G. Whitten, J. Am. Chem. Soc., 1980, 102, 5627–5631 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Photochemical CO2 reduction performance, UV-vis absorption spectra, photochemical reaction under different sets of conditions, mass spectra, AQY measurements, ATR-FTIR observation, standard curve of TEOA for 1H NMR analysis. See DOI: https://doi.org/10.1039/d3ra06585e |
| This journal is © The Royal Society of Chemistry 2023 |