
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA modified Beckmann rearrangement for the facile synthesis of amidines and imidates via imidoyl fluoride intermediates†
James A. Vogel,
Kirya F. Miller,
Eunjeong Shin,
Jenna M. Krussman and
Patrick R. Melvin *
*
Department of Chemistry, Bryn Mawr College, Bryn Mawr, Pennsylvania 19010, USA. E-mail: prmelvin@brynmawr.edu
First published on 13th October 2023
Abstract
Herein, we report a modified Beckmann rearrangement using sulfone iminium fluoride (SIF) reagents to rapidly synthesize imidoyl fluoride intermediates. Subsequently, amidine and imidate products can be formed following the introduction of amine and alcohol nucleophiles, respectively. Overall, approximately 50 amidine and imidate products have been isolated in high yields utilizing mild conditions.
Introduction
For nearly a century and a half, the Beckmann rearrangement has been an effective pathway for the formation of amides (Fig. 1A).1–3 In a traditional Beckmann rearrangement, a ketoxime is activated via strong acid which facilitates the migration of an R group from carbon to nitrogen and leads to the creation of a highly reactive nitrilium ion. Subsequent combination with water and tautomerization yields the desired amide product. The quintessential illustration of this chemistry comes from the conversion of cyclohexanone oxime to caprolactam, a precursor to Nylon 6 synthesized on a 4.5-billion-kilogram scale annually.4 | ||
| Fig. 1 (A) Standard Beckmann rearrangement; (B) previous work using sulfone iminium fluorides (SIFs); (C) application of SIFs towards a modified Beckmann rearrangement. | ||
While acidic conditions are often used to activate the ketoxime, other reagent classes are capable of facilitating this rearrangement and generating the necessary nitrilium ion.5 A recent effort has been made to utilize sulfur–fluoride reagents which can transform ketoximes to amides under mild conditions.6–8 The use of S–F reagents provides an additional transformation that activation via acid does not. With free fluoride now present in solution, combination with the nitrilium electrophile leads to imidoyl fluoride intermediates which are typically ushered on to the amide product either through water present in the reaction or during the work-up procedure.6–8
Under the right conditions, the in situ-generated imidoyl fluoride could be a highly valuable synthetic precursor for other desirable products, namely amidines and imidates. The amidine motif not only appears in various natural products, pharmaceuticals and agrochemicals,9,10 but they have also been used extensively in organic synthesis.11 Likewise, imidates play a variety of roles, including both electrophile and nucleophile,11 with more recent applications centered on their use as effective precursors in the synthesis of N-heterocycles.12 A variety of methods exist for the preparation of these important functional groups,13–17 but a protocol for the synthesis of both amidines and imidates stemming from a common intermediate would be valuable.
Recently, our group reported the design of sulfone iminium fluorides (SIFs, Fig. 1B) and their use in a series of challenging fluorination reactions.18,19 Deoxyfluorination methodologies mediated by SIF reagents are characterized by very short reaction times (60 seconds at room temperature) while still employing a practical set-up. Given the demonstrated affinity of SIFs for hydroxyl-containing functional groups, we aimed to apply these unique reagents to the formation of imidoyl fluorides through a modified Beckmann rearrangement. The rapidly generated intermediates could then be subjected to various nitrogen and oxygen nucleophiles, producing amidines and imidates, respectively (Fig. 1C). During the preparation of this work, Ohwada and co-workers demonstrated portions of this concept using a DAST–THF reagent system;8 herein, we report on the successful use of SIF reagents for the facile generation of imidoyl fluorides which has led to the significant expansion of this chemistry, both in terms of the ketoxime and nucleophile scope and the use of more favorable conditions.
Results and discussion
Initial investigations centered on determining the efficacy of the SIF reagents for transforming ketoximes to imidoyl fluorides. Using acetophenone oxime as the model substrate, we first utilized conditions that had been most effective for the deoxyfluorination of alcohols and carboxylic acids: 2 equivalents of SIF along with 2 equivalents of DBU as the base in dichloromethane. Following 60 seconds of reaction time at room temperature, we were delighted to observe the formation of imidoyl fluoride in a 92% yield by 19F NMR spectroscopy. As with previous SIF optimization endeavors, we also found that several other nitrogen bases were effective, with triethylamine providing quantitative conversion (Scheme 1). Likewise, various solvents provided access to imidoyl fluoride, albeit none as successfully as DCM or acetonitrile (see ESI† for full optimization details). Using these optimized conditions, various ketoximes could be converted to the corresponding imidoyl fluoride, with the vast majority providing quantitative yields by 19F NMR (see ESI†). Unfortunately, attempts at isolating imidoyl fluorides with aliphatic R groups proved challenging due to their propensity to slowly decompose to the corresponding amide product. However, those imidoyl fluorides bearing aryl substituents for both R and R′ were stable enough to be isolated; in particular, benzophenone oxime was quantitatively converted to the imidoyl fluoride by 19F NMR and isolated in a 95% yield following chromatography (Scheme 1). | ||
| Scheme 1 Initial conversions of ketoximes to imidoyl fluorides. | ||
With an efficient pathway to imidoyl fluorides, we next turned to utilizing these reactive intermediates for the formation of amidines and imidates via substitution reactions with amines and alcohols, respectively. Morpholine proved to be an excellent nitrogen nucleophile for this process; the addition of two equivalents of the secondary amine to the in situ-generated imidoyl fluoride led to a 92% isolated yield of the amidine product after just 10 minutes at room temperature (Fig. 2, 2a-a). Importantly, no acetanilide was observed in the crude reaction mixture, signifying that the amidine products are significantly more likely to form compared to the amide. These conditions also represent a substantial upgrade on previously published iterations of this chemistry,8 which used a large excess (>20 equivalents) of amine in addition to longer reaction times (>2 hours). When switching the additional nucleophile to 4-methoxyphenol, similar yields of imidate (90%, 2a-i) could be achieved, albeit with slightly modified conditions. An additional two equivalents of NEt3 were included with the phenol and reaction times were extended to 24 hours to allow for optimal conversion.
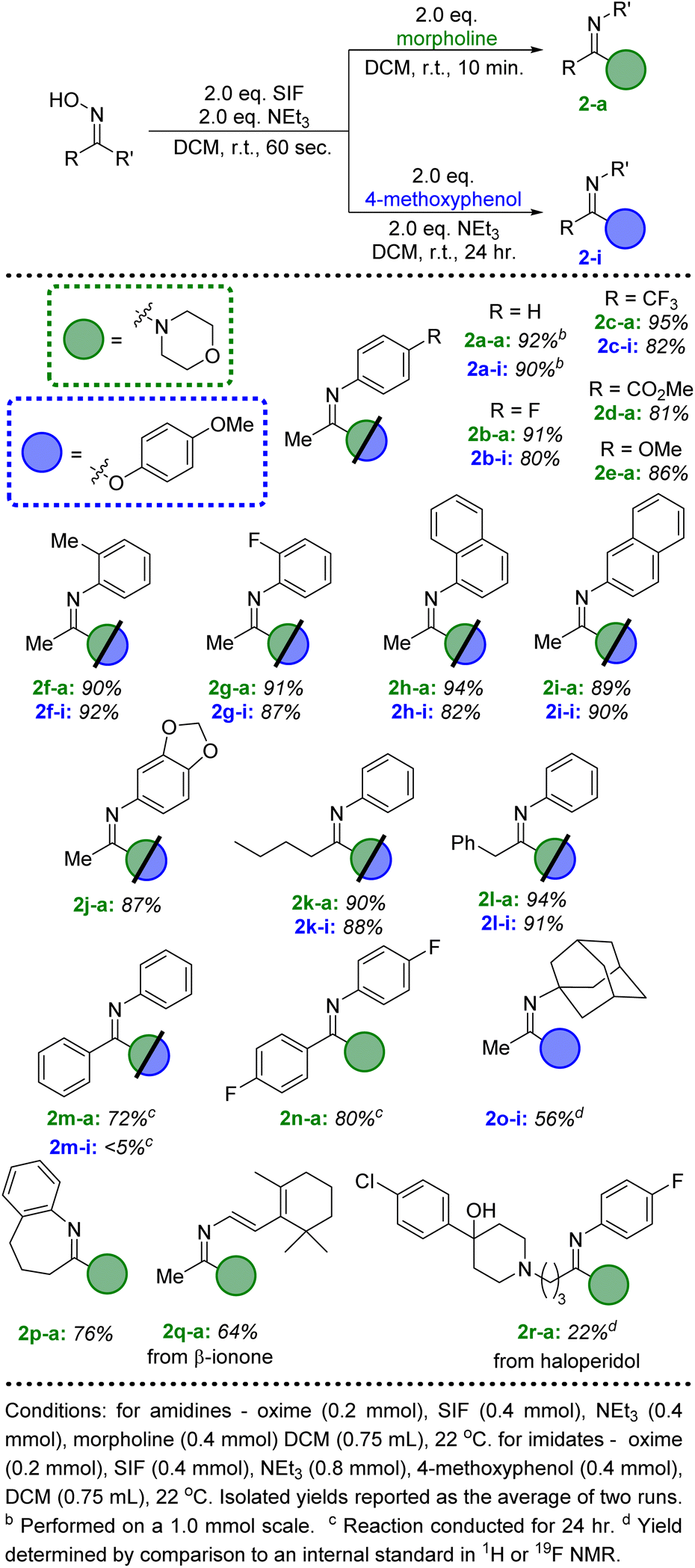 | ||
| Fig. 2 Substrate scope for the conversion of ketoximes to amidine and imidate products via SIF reagents. | ||
We then looked to expand beyond acetophenone oxime and explore the scope of the ketoxime component of this reaction (Fig. 2). Unsurprisingly, several substituted acetophenone oxime derivatives were highly successful at generating the amidine and imidate products in excellent yields using the standard conditions described above. Both electron withdrawing and donating groups at the para position of the phenyl ring were well tolerated (2b–2e); more importantly, ortho substitution did not lead to any significant decrease in the isolated yields (2f–2h). The methyl substituent of the ketoxime could also be altered to include more sterically hindered groups, such as butyl (2k) and benzyl (2l). Unfortunately, the use of a larger substituent, such as cyclohexyl or tert-butyl, did not lead to any significant conversion to the amidine or imidate products, despite the formation of the corresponding imidoyl fluorides after SIF addition. Next, we investigated benzophenone oxime derivatives; while longer reaction times were needed, good yields of the amidine products were achieved after 24 hours at room temperature (2m-a, 2n-a). The same results were achieved from either the in situ-generated imidoyl fluoride or from the isolated compound. The combination of the less reactive benzophenone derived imidoyl fluoride with 4-methoxyphenol did not lead to significant conversion to the imidate product (2n-i), even when using elevated temperatures with acetonitrile as the solvent. Ring expansion could also be achieved when using tetralone oxime, successfully generating a 7-membered ring imidoyl fluoride which efficiently formed the corresponding amidine (2p-a).
Moving beyond morpholine, we next investigated other secondary and primary amines for their efficacy in forming amidine products generated from acetophenone oxime (Fig. 3). Several cyclic, secondary amines were ideal candidates for this transformation, providing amidines 3a–3h in good to excellent isolated yields. These reactions were once again conducted with only 2 equivalents of the required amine and reached full conversion in just 10 minutes at room temperature. In particular, imidazole, 2-methylimidazole and benzimidazole (3f–3h) proved to be excellent nucleophiles to pair with the imidoyl fluoride intermediate. An acyclic, secondary amine (3i) could also be employed without any significant decrease in the yield. Due to the lessened nucleophilic character, primary anilines produced diminished yields (3j, 3k) of amidines under optimal conditions. A chiral primary amine with steric hindrance proximal to the nitrogen delivered the corresponding amidine with an isolated yield of 78% (3m). Overall, this methodology demonstrates that a variety of amidines can be quickly and efficiently synthesized when using SIF reagents to generate the necessary imidoyl fluoride intermediate.
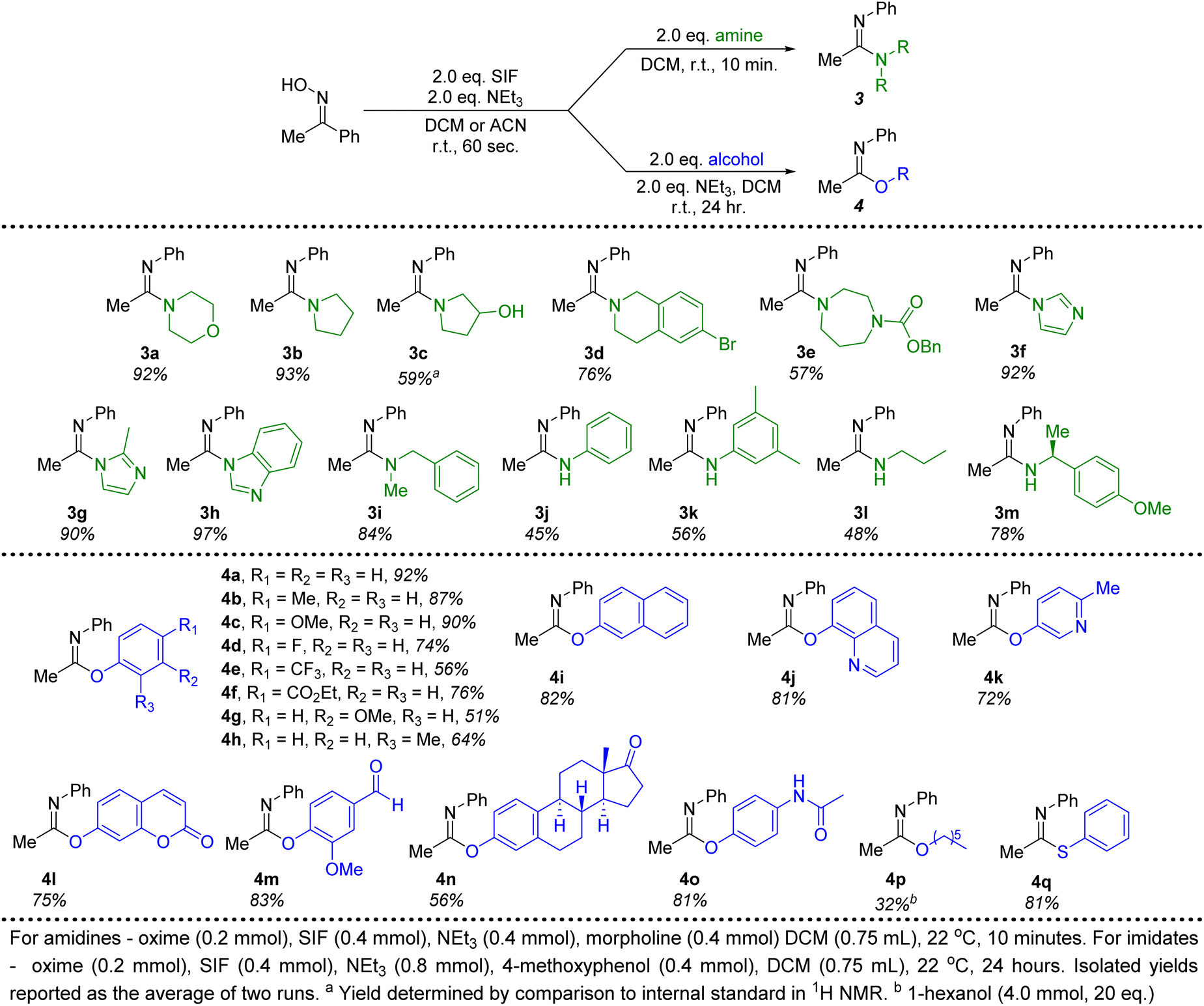 | ||
| Fig. 3 Amine and alcohol substrate scope for the conversion of acetophenone oxime to amidine and imidate products. | ||
Switching to alcohol nucleophiles, a wide selection of imidate products stemming from phenols were possible. A variety of electronically-diverse, para-substituted phenols were viable candidates for this methodology, including several electron-withdrawing moieties (4d–4f). Likewise, meta- (4g, 4i, 4l, 4n) and ortho-substituents (4h, 4j, 4m) were well tolerated. Heterocyclic phenols could also be included, with 8-hydroxyquinoline (4j, 81%), 3-hydroxy-6-methylpyridine (4k, 72%) and umbelliferone (4l, 75%) all providing efficient access to imidate products when combined with the SIF-generated imidoyl fluoride. Furthermore, the use of vanillin as the phenol (4m) demonstrates that aldehydes are unaffected in this chemistry. Attempts to translate this methodology to aliphatic alcohols were not as successful. Even the use of increased amounts (20 equivalents) of 1-hexanol led to poor conversions to the corresponding imidate product (4p, 32%). However, when switching to the sulfur derivative, thiophenol, an excellent isolated yield of the thioimidate was obtained (4q). All told, close to 30 different amidine and imidate products could be isolated in good to excellent yields, all stemming from the rapid generation of the imidoyl fluoride of acetophenone oxime.
Finally, ketones are common moieties in many natural products and pharmaceuticals. Therefore, we desired to show how the chemistry described herein could be executed using several of these relevant molecules. To this end, the ketoxime derivatives of β-ionone and haloperidol were synthesized and proved to be excellent candidates for this transformation. Under the ideal conditions described above, morpholine was used in conjunction with both ketoximes (2q and 2r), demonstrating that this methodology could be an effective way to diversify common drug motifs. Likewise, phenols are prevalent functional groups in a variety of important molecules.20 We selected estrone and acetaminophen as two promising candidates to act as the hydroxyl-containing species alongside acetophenone oxime (Fig. 3, 4n and 4o). Both pharmaceutically relevant phenols were effective partners in this chemistry, providing access to novel imidate products in 56% and 81% yield, respectively.
Conclusions
Overall, we have demonstrated that sulfone iminium fluorides are a powerful reagent class for the rapid conversion of ketoximes to imidoyl fluorides. A wide range of ketoximes can be utilized in this methodology, including several with pharmaceutical relevance. Following the 60 seconds required to form imidoyl fluoride intermediates in situ, the introduction of amines and phenols leads to excellent yields of amidines and imidates, respectively. Using practical, simple conditions, we have represented over 50 different examples of this methodology, demonstrating the diverse power that this transformation holds in the synthesis of amidines and imidates. Current work is aimed at expanding this chemistry beyond ketoxime substrates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. R. M. thanks the National Science Foundation (Grant CHE-2247109) and the Charles E. Kaufman Foundation (KA2021-121931) for their funding support. The authors acknowledge the National Science Foundation for a Major Research Instrumentation Award (CHE-0958996), which funded the acquisition of the NMR spectrometer used in this work. P. R. M. gratefully acknowledges Prof. Glenn Sammis of the University of British Columbia for valuable discussions. We would also like to acknowledge Lucy Miller for her early contributions to this work.Notes and references
- E. Beckmann, Ber. Dtsch. Chem. Ges., 1886, 19, 988–993 CrossRef.
- A. H. Blatt, Chem. Rev., 1933, 12, 215–260 CrossRef CAS.
- R. E. Gawley, in Organic Reactions, 2004, pp. 1–420, DOI:10.1002/0471264180.or035.01.
- J. Tinge, M. Groothaert, H. op het Veld, J. Ritz, H. Fuchs, H. Kieczka and W. C. Moran, in Ullmann's Encyclopedia of Industrial Chemistry, 2018, pp. 1–31, DOI:10.1002/14356007.a05_031.pub3.
- K. Kaur and S. Srivastava, New J. Chem., 2020, 44, 18530–18572 RSC.
- J. Gurjar and V. V. Fokin, Chem.–Eur. J., 2020, 26, 10402–10405 CrossRef CAS PubMed.
- Y. Cui, Y. Zhao, J. Shen, G. Zhang and C. Ding, RSC Adv., 2022, 12, 33064–33068 RSC.
- Y. Lu, A. Kasahara, T. Hyodo, K. Ohara, K. Yamaguchi, Y. Otani and T. Ohwada, Org. Lett., 2023, 25, 3482–3486 CrossRef CAS PubMed.
- J. V. Greenhill and P. Lue, Prog. Med. Chem., 1993, 30, 203–326 CrossRef CAS PubMed.
- T. D. Suja, K. V. L. Divya, L. V. Naik, A. Ravi Kumar and A. Kamal, Bioorg. Med. Chem. Lett., 2016, 26, 2072–2076 CrossRef CAS PubMed.
- S. E. Patai and Z. Rappoport, The Chemistry of Amidines and Imidates, Wiley, New York, 1991, vol. 2 Search PubMed.
- R. Thakur, Y. Jaiswal and A. Kumar, Org. Biomol. Chem., 2019, 17, 9829–9843 RSC.
- Z. Alassad, A. AboRaed, M. S. Mizrachi, M. H. Pérez-Temprano and A. Milo, J. Am. Chem. Soc., 2022, 144, 20672–20679 CrossRef CAS PubMed.
- C. G. Saluste, R. J. Whitby and M. Furber, Angew. Chem., Int. Ed., 2000, 39, 4156–4158 CrossRef CAS PubMed.
- X. Liu, H. Yue, J. Jia, L. Guo and M. Rueping, Chem.–Eur. J., 2017, 23, 11771–11775 CrossRef CAS PubMed.
- S. Caron, L. Wei, J. Douville and A. Ghosh, J. Org. Chem., 2010, 75, 945–947 CrossRef CAS PubMed.
- K. Popov and P. Somfai, J. Org. Chem., 2016, 81, 3470–3472 CrossRef CAS PubMed.
- J. A. Vogel, R. Hammami, A. Ko, H. Datta, Y. N. Eiben, K. J. Labenne, E. C. McCarver, E. Z. Yilmaz and P. R. Melvin, Org. Lett., 2022, 24, 5962–5966 CrossRef CAS PubMed.
- L. P. Miller, J. A. Vogel, S. Harel, J. M. Krussman and P. R. Melvin, Org. Lett., 2023, 25, 1834–1838 CrossRef CAS PubMed.
- D. Lin, M. Xiao, J. Zhao, Z. Li, B. Xing, X. Li, M. Kong, L. Li, Q. Zhang, Y. Liu, H. Chen, W. Qin, H. Wu and S. Chen, Molecules, 2016, 21, 1374 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06561h |
| This journal is © The Royal Society of Chemistry 2023 |