
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic synthesis of renewable phenol derivatives from biobased furanic derivatives†
Adrien Ratiera,
Richail D. Moulandou-Koumba a,
Mélanie Anizana,
Sarah Behloula,
Fréderic Guegan*a,
Gilles Frappera,
Quentin Blancart Remaurya,
Karine De Oliveira Vigiera,
Jianxia Zhengb and
François Jérôme*a
a,
Mélanie Anizana,
Sarah Behloula,
Fréderic Guegan*a,
Gilles Frappera,
Quentin Blancart Remaurya,
Karine De Oliveira Vigiera,
Jianxia Zhengb and
François Jérôme*a
aInstitut de Chimie des Milieux et Matériaux de Poitiers, Université de Poitiers, CNRS, 1 rue Michel Brunet, 86073 Poitiers, France. E-mail: francois.jerome@univ-poitiers.fr; frederic.guegan@univ-poitiers.fr
bEco-Efficient Products and Process Laboratory, SOLVAY/CNRS 3966 Jin Du Rd., Xin Zhuang Industrial Zone, Shanghai 201108, China
First published on 16th October 2023
Abstract
Here, we study a sequence Diels–Alder/aromatization reaction between biobased furanic derivatives and alkynes, paving the way to renewable phenols. Guided by DFT calculations, we revealed that, in the case of dimethylfuran, the methyl group can migrate during the aromatization step, making this substrate also eligible to access renewable phenols. This reaction has been then successfully transposed to furfural and furfuryl alcohol, allowing molecular diversity and complexity to be created on phenol ring starting from two cheap biobased furanic derivatives available on large scale.
Introduction
Phenol is an important class of chemical platform from which important polymers1 such as polycarbonate, phenolic resins and a very wide range of products such as vanillin, salicylic acid, caprolactam, bisphenol A, picric acid, among many others, are produced.2,3 As a result, phenol derivatives find applications in many end-industries, including paints, adhesives, coatings, cosmetics, detergence, packaging, automotive, electronics, to name a few. Worldwide, phenol is produced at a rate of around 10 Mt per year2 from fossil feedstocks, by converting benzene mainly through the cumene process. With the awareness of our impact on the environment, the defossilization of the chemical industry has become a priority. In this context, the synthesis of phenol derivatives from renewable feedstocks has attracted much interest. Boosted by strict regulations, and also by the growing demand of consumers for biobased products, recent projections revealed that the market value for biobased phenol should increase from 12.5 to 21.3 billons USD from 2021 to 2031, respectively.3Biobased phenols are mainly obtained from lignocellulosic biomass waste, in particular by catalytic depolymerization of the lignin fraction.4 So far, different catalytic pathways have been proposed such as acid hydrolysis,5 oxidation,6 hydrogenolysis7 and pyrolysis of lignin.8 These routes generally yield a mixture of phenol derivatives such as guaiacol, syringol, vanillin (in minor proportion), and their alkylated forms. Phenol derivatives can also be produced by catalytic fast pyrolysis of vegetable oils9 or directly extracted from natural resources such as cashew nut shell10 or tannins11 for instance.
So far, most of renewable aromatics obtained from lignin are para-substituted. To complement the scope of biomass for the synthesis of renewable aromatics, the Diels–Alder reaction of biobased furanic derivatives with various dienophiles, followed by an aromatization step, have been also investigated.12 In contrast to the most common lignin-based method, this furanic-based route provides an access to ortho and meta-substituted aromatics. Among biobased furanic derivatives, furfural, and its two downstream derivatives, furfuryl alcohol and methylfuran, are cheap (1.5–2.5 € per kg) and commercially produced on large scale (300 kt year−1), making them attractive building blocks for the synthesis of renewable aromatics. We13 and others14 recently demonstrated that furfural, furfuryl alcohol and methylfuran could react with various dienophiles, paving the way to industrially relevant aromatics such as terephthalic acid or meta-xylylene diamine for instance.
With these previous results in mind, we explored here the reactivity of furfural, and its derivatives, in a sequential Diels–Alder/aromatization reaction with alkynes to get an access to renewable phenol derivatives. To date, the reaction of biobased furanic derivatives with alkynes has been sporadically explored in the literature.15 As compared to the current state of the art, we show here, through a combined theoretical and experimental study, that (1) the corresponding intermediates in the aromatization step are not Wheland intermediates per se, but epoxides, (2) in the case of biobased dimethylfuran, the methyl group can migrate during the aromatization step and (3) this reaction can be transposed to furfural which is, unlike furan or alkylated furans, usually unreactive in Diels–Alder reaction.
First, we studied methylfuran (MF) as a biobased furan to determine the optimal conditions before studying the case of furfural, which is less reactive. As a dienophile, we selected ethylpropiolate as it is one of the most abundant functionalized alkynes at an industrial scale (readily obtained from acetylene, this latter being also possibly made from biobased feedstocks).16
Preliminary tests
In a first set of experiments, MF was mixed with 5-fold-excess of ethylpropiolate (EP) and heated at 100 °C. Inspired by our previous works,13 ZnCl2 (15 mol%) has been selected as an acid catalyst. Variation of the experimental conditions is discussed later. The reaction was monitored by gas chromatography (GC) (Fig. S1–S4†) and 1H NMR. After 2 h of reaction, more than 95% of MF was consumed. By 1H NMR, typical![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH aromatic signals were detected in the 6.9–7.4 ppm window. GC analysis confirmed the nearly complete conversion of MF and the formation four main products. The coupling of GC with high resolution mass spectrometry (HRMS) revealed that these four products have the same molecular formula (C10H12O3), with a m/z = 180.0783 (±0.0004) (Fig. S5†). Purification of the crude media over silica gel, and comparison with commercial standards, confirmed that the major product was the phenol 1-o, with the methyl group in ortho position to the carbonyl group. Calibration of the GC led us to determine the yield of the phenol 1-o, i.e. 52%. The meta isomer, phenol 1-m, was also isolated, but it was formed in a much lower amount (1% yield) than the ortho, the ratio ortho/meta was 98/2 (Fig. 1).
CH aromatic signals were detected in the 6.9–7.4 ppm window. GC analysis confirmed the nearly complete conversion of MF and the formation four main products. The coupling of GC with high resolution mass spectrometry (HRMS) revealed that these four products have the same molecular formula (C10H12O3), with a m/z = 180.0783 (±0.0004) (Fig. S5†). Purification of the crude media over silica gel, and comparison with commercial standards, confirmed that the major product was the phenol 1-o, with the methyl group in ortho position to the carbonyl group. Calibration of the GC led us to determine the yield of the phenol 1-o, i.e. 52%. The meta isomer, phenol 1-m, was also isolated, but it was formed in a much lower amount (1% yield) than the ortho, the ratio ortho/meta was 98/2 (Fig. 1).
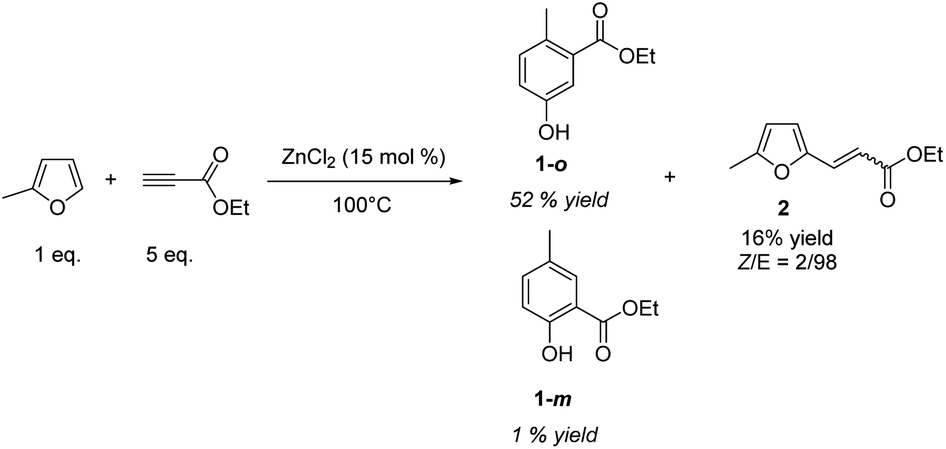 | ||
| Fig. 1 ZnCl2-catalyzed Diels–Alder/aromatization of MF with ethylpropiolate. | ||
The two other products detected by GC did not display the structural signature of a phenol derivative. To get more insights on the structures of these co-products, a sylilation of the crude medium was performed before GC analysis. While the retention times of 1-o and 1-m were logically shifted due to the sylilation of the –OH group, the retention times of the two other products did not change, indicating the absence of –OH functionality (Fig. S6†). Further purification over silica gel led us to identify by 1H and 13C NMR, including COSY, NOESY and HSQC sequences, that the product 2 resulted from the Z/E addition of the CH bond of MF on ethylpropiolate (16% yield), with a Z/E ratio of 14/86 (Fig. S7†). Besides compounds 1 and 2, other detected products by GC-HRMS resulted mainly from successive Diels–Alder reactions yielding for instance a biphenyl moiety which was detected at m/z = 244.1099 (Fig. S8†). Using equilibrium geometry at ground state in gas with density functional M062X/6-311++G(d,p) calculations, the gap between the HOMO of MF and the LUMO of the in situ formed Diels–Alder adduct (ΔE = 6.61 eV and 6.64 eV for the meta and ortho adducts, respectively) was found lower than the gap between the HOMO of MF and the LUMO of ethylpropiolate (ΔE = 7.06 eV), which explains the occurrence of successive Diels–Alder reactions.
Impact of the reaction conditions
The reaction was performed without catalyst at 100 °C (Table 1, entry 1). Over 24 h of reaction, MF was slowly consumed (60% conversion). However, no formation of phenol 1 was evidenced and MF was mainly converted through successive Diels–Alder reactions with ethylpropiolate, as corroborated by mass spectrometry analysis. This result shows that a catalyst is required not only to improve the conversion rate of MF but also to better control the selectivity to phenol 1 (i.e. promotion of the aromatization step).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Conv. (%) | Yield (%) |
| 1 | — | 24 | 60 | — |
| 2 | Amberlyst-15 | 24 | 97 | 5 |
| 3 | AcOH | 6 | 40 | < 1 |
| 4 | Isobutyric acid | 24 | 90 | < 1 |
| 5 | Triflic acid | 0.5 | 95 | <1 |
| 6 | CuCl2 | 2 | 100 | 1 |
| 7 | AlCl3 | 0.5 | 95 | 29 |
| 8 | SnCl2 | 6 | 100 | 39 |
| 9 | ZnCl2 | 2 | 100 | 54 |
| 10 | ZnI2 | 2 | 90 | 19 |

Next, the performance of ZnCl2 was compared to other catalysts, first in terms of phenol yields (Table 1). All corresponding kinetic profiles are provided in Fig. S9–S16.† Brønsted acid catalysts such as cation exchange resins (A15, purolite) or homogeneous acid catalysts (acetic, isobutyric and triflic acids) led to the formation of phenol 1-o,m\\S01vdifs\DDRIVE\DEMDATA\fr57\MYFILES\RSC\RA\d3ra06461a\CEPAGEQC\chem1hypenocommam in trace amount (<5% yield) (Table 1, entries 1–5). CuCl2 is a well-known catalyst for the activation of alkynes (e.g. Huisgen reaction) and was thus tested as a Lewis acid catalyst. Unfortunately, the yield to the desired phenol 1-o,m remained below 5% and CuCl2 mainly catalyzed the oligomerization of ethyl propiolate (Table 1, entry 6). Other metal chlorides such as AlCl3 and SnCl2 were found more efficient than CuCl2 affording the phenol 1-o,m in 29 and 39% yield, respectively (Table 1, entries 7, 8). However, the maximum yield into phenol 1-o,m was still lower than in the case of ZnCl2 (54%, Table 1, entry 9). Replacing ZnCl2 by ZnI2 led to the phenol 1-o,m in a lower yield (19% vs. 54% with ZnCl2, Table 1 entries 9,10), which can be explained by its uncomplete solubility in the reaction media. To assess if the differences of phenol 1-o,m yields were not a result of a difference of activity between all catalysts, the selectivity to phenol 1-o,m was compared for all tested catalysts at 60% conversion (Fig. 2). It definitely confirms that ZnCl2 is the most selective catalyst for the synthesis of phenol 1-o,m (56% selectivity), a result in line with our previous results on the Diels–Alder reaction of furanic derivatives with alkenes.13 Hence, in the following experiments, ZnCl2 was selected as a catalyst. Note that the ortho/meta ratio remained unchanged (98/2), whatever the catalysts tested.
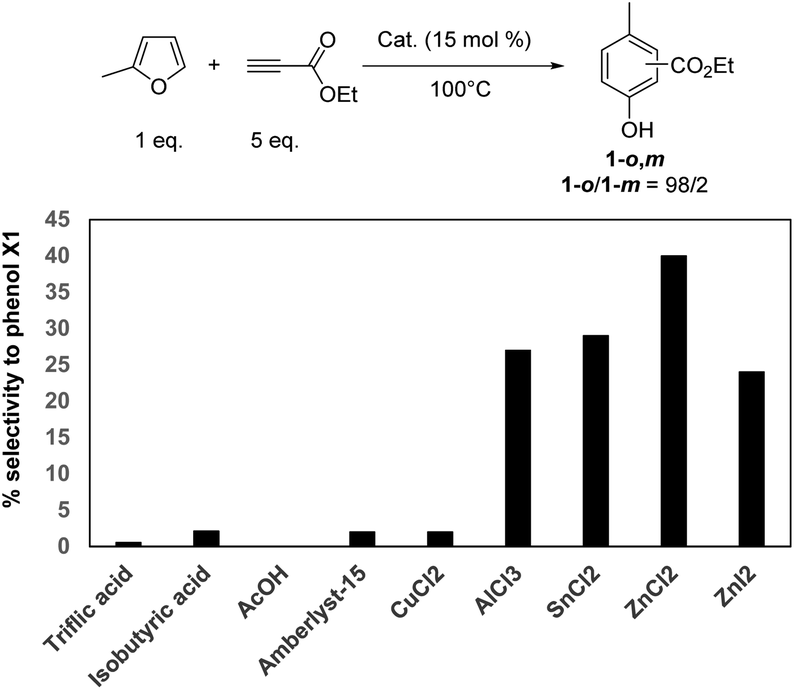 | ||
| Fig. 2 Selectivity to phenol 1-o,m at 60% conversion of MF as a function of acid catalyst (100 °C, 15 mol%). | ||
Next, the reaction was studied at different temperatures. Kinetic profiles are provided in Fig. S16–S20.† Decreasing the temperature of the reaction from 100 °C to 70 and 40 °C obviously decreased the conversion rate of MF from 12 to 2 and 0.4 mmol h−1, respectively (Fig. 3). However, whatever the conversion level, a decrease of the reaction temperature led to a decrease of the selectivity to phenol 1-o,m, for instance from 56 to 42 and 19% at 100, 70 and 40 °C, respectively, at 95% conversion of MF (Fig. 3). This result strongly suggests that the aromatization step occurred at a higher temperature than the Diels–Alder reaction. Unfortunately, an increase of the reaction temperature from 100 °C to 130 °C did not afford phenol 1-o,m with a higher selectivity (Fig. 3). Instead, we noticed that a temperature higher than 100 °C led to a degradation of phenol 1-o,m. In addition, while at temperatures below 100 °C ethyl propiolate was converted in a stoichiometric way, at temperatures higher than 100 °C, more than 1 eq. of ethyl propiolate was consumed (1.5 and 2.0 eq. at 130 and 160 °C, respectively), indicating the occurrence of side reactions involving ethylpropiolate at such temperatures (Fig. S21†). Hence, 100 °C was selected as the optimal temperature in the following experiments. Under these conditions, the excess of ethyl propiolate can be recovered by distillation at the end of the reaction and recycled.
 | ||
| Fig. 3 Impact of the reaction temperature on the selectivity (at 95% conv. of MF) to phenol 1 and duration of the reaction (MF (1 eq.), ethylpropiolate (5 eq.), 100 °C, 15 mol% of ZnCl2). | ||
To support the promotion effect of ZnCl2 on the reaction rate and phenol 1-o,m selectivity, the amount of ZnCl2 was varied. Without ZnCl2, MF was converted at a rate of 0.6 mmol h−1 (Fig. S22†). An incremental addition of ZnCl2 into the reaction media led to a linear increase of the conversion rate of MF (Fig. S23–S26†). For instance, using 5, 10, 15, 25 and 50 mol% of ZnCl2, the conversion rate of MF linearly increased from 1.5 to 48 mmol h−1. Similarly, an incremental addition of ZnCl2 linearly increased the formation rate of phenol 1-o,m from 2.5 to 30 mmol h−1 at 5 and 50 mol% of ZnCl2, respectively.
However, the selectivity to phenol 1-o,m significantly varied as a function of the amount of ZnCl2. Fig. 4 shows the selectivity to phenol 1-o,m, determined at 95% conversion of MF, as a function of the ZnCl2 amount. Interestingly, the selectivity to phenol 1-o,m increased from 23 to 56% when the ZnCl2 was increased from 5 to 15 mol%, while the ortho/meta ratio remained unchanged (98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). This result strongly supports that ZnCl2 speeds up the aromatization rate, to the detriment of unwanted successive Diels–Alder reactions with ethylpropiolate as typically observed under catalyst-free conditions. A similar conclusion can be drawn when data were compared at 60% conversion of MF (Fig. S27†). Note that a further increase of the ZnCl2 amount from 15 to 50 mol% did not result in a further increase of the selectivity to phenol 1-o,m. Instead, it led to an overconsumption of ethylpropiolate (e.g. 2 eq. at 50 mol% of ZnCl2). Hence, the optimal amount of ZnCl2 was kept at 15 mol%, which is a good compromise to reach the highest selectivity to phenol 1-o,m. Under these conditions, phenol 1-o,m was obtained with 54% yield after 1 h of reaction, which corresponds to a space time yield of 165 kg m−3 h−1, a result in line with the industrial expectation of the field.
:2). This result strongly supports that ZnCl2 speeds up the aromatization rate, to the detriment of unwanted successive Diels–Alder reactions with ethylpropiolate as typically observed under catalyst-free conditions. A similar conclusion can be drawn when data were compared at 60% conversion of MF (Fig. S27†). Note that a further increase of the ZnCl2 amount from 15 to 50 mol% did not result in a further increase of the selectivity to phenol 1-o,m. Instead, it led to an overconsumption of ethylpropiolate (e.g. 2 eq. at 50 mol% of ZnCl2). Hence, the optimal amount of ZnCl2 was kept at 15 mol%, which is a good compromise to reach the highest selectivity to phenol 1-o,m. Under these conditions, phenol 1-o,m was obtained with 54% yield after 1 h of reaction, which corresponds to a space time yield of 165 kg m−3 h−1, a result in line with the industrial expectation of the field.
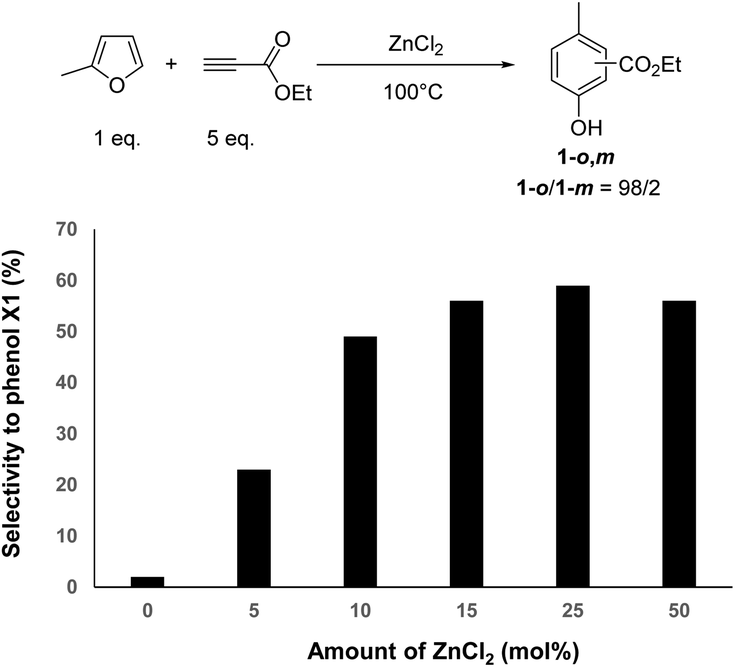 | ||
| Fig. 4 Impact of the amount of ZnCl2 on the selectivity to phenol 1 (at 95% conv. of MF) (MF (1 eq.), ethylpropiolate (5 eq.), 100 °C). | ||
Note that the variation of the ethyl propiolate/MF molar ratio from 5 to 20 did not impact significantly the selectivity to phenol 1-o,m, but it led to an overconsumption of ethyl propiolate from 1 eq. at a 1:1 ratio to 5 eq. at a 20:1 ratio (Fig. S28†).
Impact of substituents
The selectivity to phenol could be significantly improved using a disubstituted alkyne such as diethyl acetylene dicarboxylate (DEAD) which is also readily obtained from acetylene. Indeed, in this case, the side Z/E addition of theCH bond of MF on the triple bond is much less favourable. In addition, in contrast to what was observed with EP, the gap between the HOMO of MF and the LUMO of DEAD (ΔE = 6.81 eV) was lower than the gap between the HOMO of the in situ produced Diels Alder adduct and the LUMO of DEAD (ΔE = 7.65 eV), which should prevent successive Diels–Alder reactions. In this context, MF was reacted with 5-fold excess of DEAD and heated at 100 °C in the presence of ZnCl2 (15 mol%). In perfect agreement, after 2 h of reaction, the corresponding phenol 3 was obtained with 92% yield (Fig. 5).
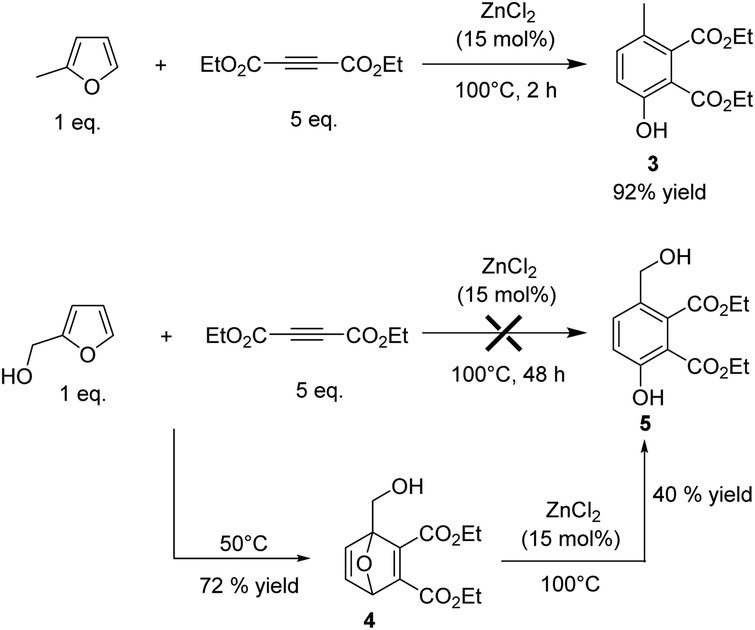 | ||
| Fig. 5 Synthesis of phenol derivatives from MF and furfurylalcohol. | ||
Next, the reactivity of furfuryl alcohol and furfural was explored with the aim of creating molecular complexity and diversity. Furfuryl alcohol is known to be highly reactive and usually quickly polymerizes under acid conditions. As expected, when furfuryl alcohol was mixed either with ethylpropiolate or DEAD in the presence of ZnCl2 (15 mol%), only degradation products (black tar materials) were formed. To circumvent this problem, the reaction was investigated at 50 °C and in two steps. First, the reaction was conducted without ZnCl2. Under catalyst free conditions, the Diels–Alder reaction was not selective using ethylpropiolate as a dienophile, leading to a messy mixture of unidentified chemicals, presumably due to successive Diels–Alder reactions as mentioned above. As expected on the simple consideration of HOMO–LUMO gap, successive Diels–Alder reaction is less likely to occur with DEAD. As anticipated, using DEAD as a dienophile, the Diels–Alder adduct 4 was successfully formed with 72% yield after 48 h of reaction (Fig. 5). NMR characterizations of the Diels–Alder adduct 4 are provided in the ESI.† Then, 15 mol% of ZnCl2 were added into the solution and the resulting mixture was heated at 100 °C. After 24 h of reaction, the corresponding phenol 5 was formed with 40% yield (overall yield Diels Alder + aromatisation = 29% yield, determined by 1H NMR) (Fig. 5).
The direct Diels Alder of furfural remains a challenging task due to the presence of the electron withdrawing –CHO group, which inhibits the reactivity of the furanic ring. We recently demonstrated that the reversible protection of furfural with ethylene glycol (i.e. ketalization of furfural) restored the reactivity of the furanic ring, thus increasing its reactivity in Diels–Alder reaction.13 It should be noted that furfural can be also activated by reversible derivatization of the –CHO group with hydrazine.14 As expected, no reaction was observed from furfural. Hence, furfural was derivatized either with ethylene glycol or 1,2-ethanethiol (ESI†). As for the case of furfuryl alcohol, the reaction was explored in two steps (Diels–Alder and then aromatization) using DEAD as a dienophile. While unsatisfactory results were obtained with the ketal of furfural due to its partial in situ deprotection (trace of water was present), the furfural dithiane 6 (more stable) led to the formation of the Diels Alder adduct 7 with 52% yield after 48 h of reaction (Fig. 6). Then, 15 mol% of ZnCl2 were added and the mixture was heated at 100 °C. Unfortunately, no aromatization reaction was observed. We strongly suspect that ZnCl2 promoted the in situ deprotection of the dithio group (strong affinity of Zn for sulfur),17 leading to degradation reactions, as corroborated by the rapid formation of a sticky gel-type material at the end of the reaction. Cyclic dithiane derivatives are much more robust than cyclic ketal and cannot be simply deprotected in the presence of a Brønsted acid.18 In this context, ZnCl2 was replaced by Amberlyst-15 (A15, 15 mol% of H+), a cation exchange resin bearing –SO3H. To our delight, after 3 h of reaction at 100 °C, the Diels–Alder adduct 7 was aromatized into the phenol 8 which was formed in 82% yield (Fig. 6).
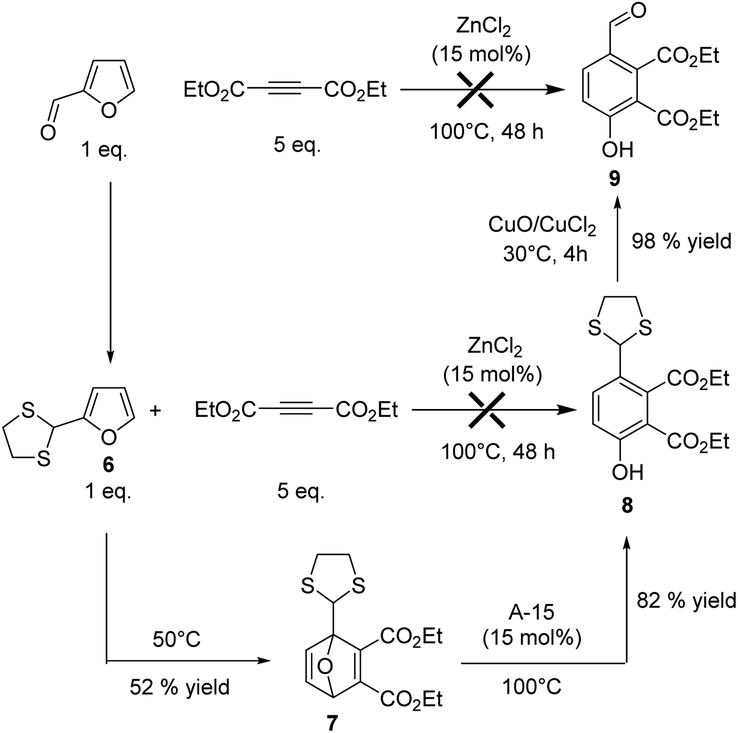 | ||
| Fig. 6 Synthesis of a phenol derivatives from furfural. | ||
Altogether, these results demonstrate that functionalized and industrially relevant renewable phenol derivatives could be obtained through this route. One should note that, in the case of furfural, the –CHO group of 8 can be conveniently recovered using the deprotection procedure described by Narasaka,19 leading to the phenol 9 in 98% yield (Fig. 6)
In the course of our study, we firstly hypothesized that methyl migrations could occur during the aromatization step (1,2-methyl shifts on the associated Wheland intermediates).20 By means of DFT calculations, we found that the energy barrier for such methyl shifts can indeed be quite low (less than 10 kcal mol−1 in the case of dimethylfuran, see below). Although this migration was not experimentally observed starting from MF, it prompted us to explore the reactivity of dimethylfuran (DMF), a biobased furanic derivative obtained by hydrogenolysis of 5-hydroxymethylfurfural, the “sleeping giant” of the sugar chemistry. DMF is normally not eligible for the synthesis of aromatics as the second –CH3 group prevents the aromatization step. To our delight, when DMF (1 eq.) was mixed with ethylpropiolate (5 eq.) and heated at 100 °C in the presence of 15 mol% of ZnCl2, a mixture of three phenol derivatives were obtained in 20, 16 and 8% yield (i.e. 44% overall yield), showing that the methyl group has migrated during the aromatization steps (Fig. 7). Formation of these three phenol derivatives was first confirmed by a GC-HRMS analysis which revealed a same exact mass at m/z = 194.0928 ± 0.0004 for the three detected products (Fig. S29†). In addition, the retention times on GC were in a similar region than phenols obtained from methyl furan (15–18 min) while, as discussed above, a silylation of the crude reaction with trimethylchlorosilane, before GC analysis, shifted the retention times of these three products to 18-19 min, an observation consistent with the presence of an –OH group (Fig. S30†).
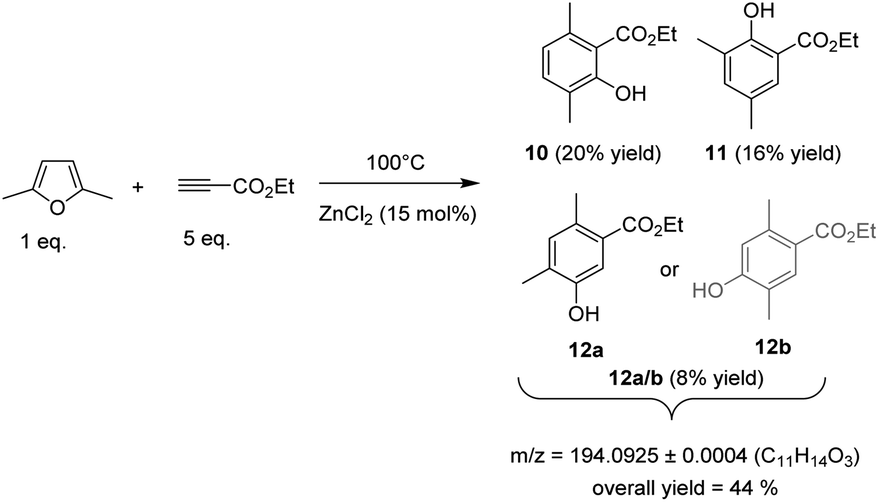 | ||
| Fig. 7 Mixture of phenol derivatives formed from dimethyl furan and ethylpropiolate. | ||
To get more information on the chemical structures of these three phenols derivatives, further inspections by 1H NMR were performed. To this end, the phenol fraction was first purified over silica gel using a 95/5 cyclohexane/ethylacetate mixture as an eluent. Two phenol derivatives, with a retention time corresponding to the two major peaks observed by GC, were isolated and analysed by 1H NMR. Analysis of the third phenol (minor one) is discussed later.
The first phenol derivative, obtained with 20% yield, was characterized by the presence of two aromatic CH groups at 6.79 and 7.00 ppm, respectively. The –OH group was observed at 7.58 ppm. Two methyl substituents groups were visible at 1.76 and 1.85 ppm. A COSY sequence proved that the two –CH3 groups are in para position each other (Fig. S31†). Furthermore, a NOESY sequence did not reveal any spatial coupling between the –CH3 groups, confirming that they are not in an adjacent position on the phenyl ring. This claim was further supported by an HMBC sequence (Fig. S32†) and also by a coupling constant between the two CH protons of 8.1 Hz, a value consistent with a 3JH–H coupling (i.e. CH groups are in ortho position each other). Hence, we assume that the major phenol formed is ethyl 2-hydroxy-3,6-dimethylbenzoate (10).
The second purified phenol derivative, and obtained with 16% yield, was also characterized by two aromatic CH groups at 7.14 and 7.49 ppm, and a phenol –OH group at 10.91 ppm. The –OH group was also supported by FT-IR analysis which revealed a broad band at 3100-3350 cm−1. The two methyl substituents groups appeared at 2.23 and 2.25 ppm. As observed above with phenol 10, no spatial coupling was observed between the two –CH3 groups, ruling out a substitution of the phenyl ring by two adjacent –CH3 groups. In addition, the two CH protons coupled each other with a coupling constant of 3.1 Hz, a value consistent with a 4JH–H coupling (i.e. CH groups are in meta position each other).21 Finally, an HMBC sequence confirmed that the –CH3 group are in meta position each other (Fig. S33†). Hence, we suggest that the second most abundant phenol derivative is the ethyl 2-hydroxy-3,5-dimethylbenzoate (11).
Unfortunately, we failed in isolating a fraction rich in the third (minor) phenol derivative (12a/b). Hence, its structure was tentatively deduced from 1H NMR by eliminating the peaks corresponding to phenols 10 and 11. This phenol was characterized by two CH groups at 6.88 and 7.30 ppm. Two methyl groups were also distinguished around 2.10–2.20 ppm. Again, no spatial coupling between the two –CH3 groups was observed by a NOESY sequence, indicating that the phenyl ring was not substituted by two adjacent –CH3 groups. In contrast to phenols 10 and 11, no coupling constant was observed between the two CH groups (only two singlets were observed) suggesting that these two CH groups are in para position each other. Two possible structures could fit with these characterizations: (1) ethyl 5-hydroxy-2,4-dimethylbenzoate (12a) and (2) the ethyl 4-hydroxy-2,5-dimethylbenzoate (12b). It is however noteworthy that a 3JHC coupling between both –CH2-H and the aromatic CH group seems existing based on a HMBC sequence, suggesting that the two –CH3 are in meta position each other (Fig. S35†). In addition, the HMBC sequence also seems supporting a coupling between one –CH2–H and the aromatic C bearing the –CO2Et group and the second –CH2–H with the aromatic C bearing the –OH group (Fig. S34†). Altogether, the HMBC sequence strongly suggests that 12a is more likely to be the correct phenolic compounds. However, at this stage of our investigation, we prefer keeping safe in the discrimination between 12a and 12b because we failed in isolating this phenol in its pure form. The preferential formation of 12a has been finally supported below using DFT calculations.
By re-analysing the crude reaction media by 1H NMR, a similar 10/11/12 distribution that the one claimed by GC was observed. However, as discussed above for MF, analysis of the crude reaction also revealed that the side Z/E addition of dimethyl furan on ethylpropiolate still occurred, leading to the alkylated furanic derivative in 8% yield. On the basis of the 3JHH coupling constant determined by 1H NMR (12.5 Hz), the trans isomer is again more likely to be the major one. More information is provided in Fig. S35.†
Mechanism investigations
From this NMR analysis it appears that three major phenol derivatives can be synthesized following our experimental conditions, although it is worth noticing that traces of other aromatic compounds can be proposed on the basis of very small peaks in theCH aromatic region of 1H NMR spectra. At this stage, the cause of this selectivity remains unclear. DFT calculations helped understanding this selectivity and supported the structural assignation of products 10/11/12 (Scheme 1).
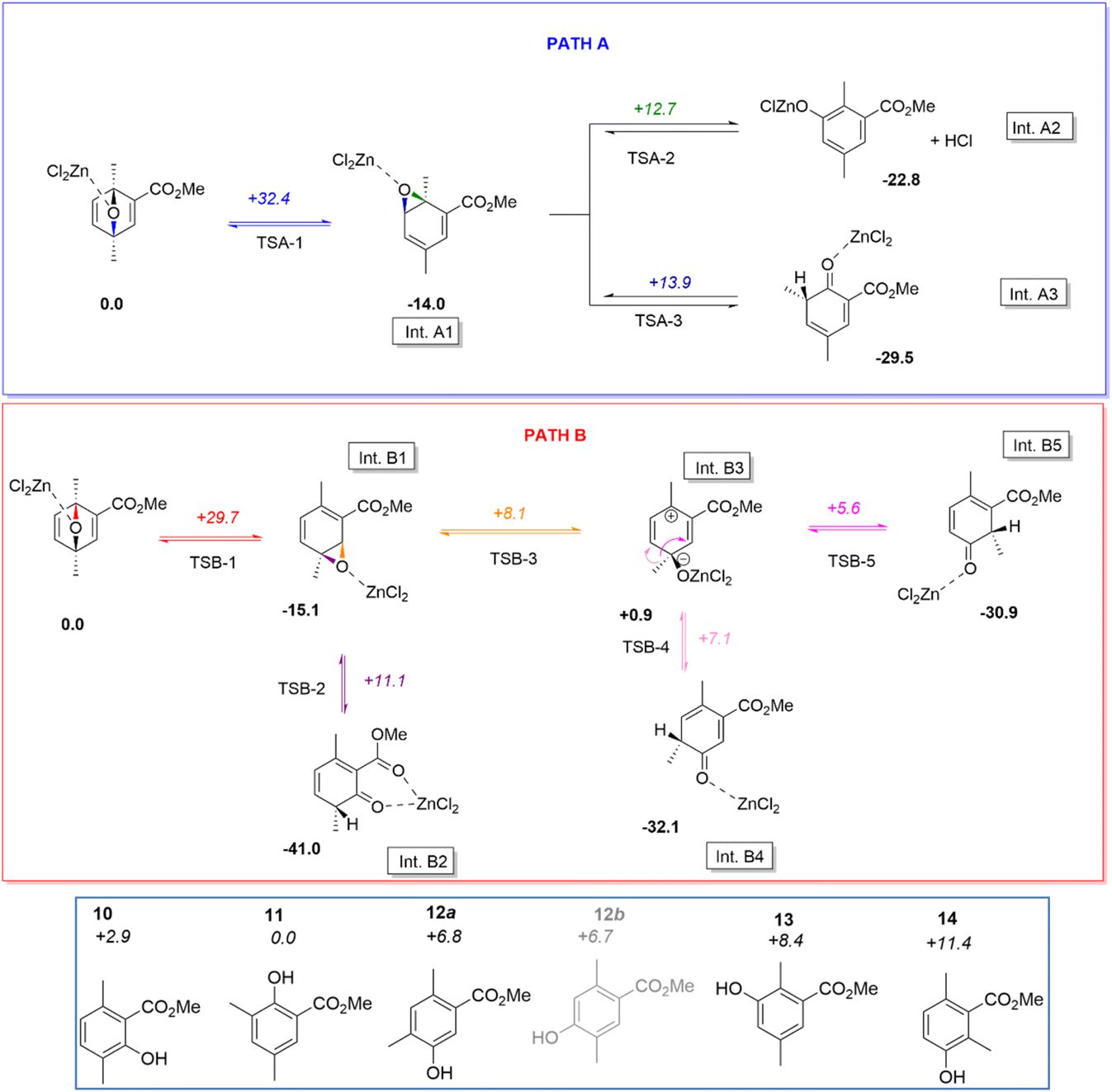 | ||
| Scheme 1 Top: DFT proposed reaction paths for the aromatization of the Diels–Alder adduct formed by the reaction of dimethylfuran on methylpropiolate in presence of ZnCl2. The structures of the reagent, intermediates and products are displayed. Bottom: Structure and relative energies (E+ZPE) of all considered phenolic products. Relative energies (E+ZPE) are given in kcal mol−1 (as computed at the M062X/6-311++G(d,p) level of theory). Phenol 12b is coloured in gray to remind that no reaction route was found to lead to this product. | ||
Starting from a simplified model (ethyl group being replaced by a methyl, and the catalyst being modelled by a single ZnCl2 moiety), it can be shown that the ring opening of the cycloadduct can indeed proceed in two different ways, associated to the cleavage of the C–O bond involving either the carbon atom in ortho or meta position with respect to the carboxyl function (TSA-1 and TSA-2). The associated activation barriers for these two steps are quite high (ca. 32.4 and 29.7 kcal mol−1, respectively, for transition states TSA-1 and TSB-1), in line with the need to heat to observe the aromatization reaction. It may be noted that the corresponding intermediates Int.A1 and Int.B1 produced by this first reaction step are not Wheland intermediates per se, but epoxides. This can be explained from resonance Lewis structures: the expected Wheland intermediates would indeed be zwitterionic, bearing an endocyclic positive and alkoxide negative charges, and thus be quite destabilised compared to an epoxide.
From intermediate Int.A1, two epoxide ring opening can then be proposed, corresponding to the cleavage of the C–O in ortho or meta position with respect to the carboxylate. As can be seen from Scheme 1, the relative transition state (TS) energies are quite comparable +12.7 and +13.9 kcal mol−1 with respect to the starting reagent, respectively, for TSA-2 and TSA-3, and significantly lower than the first transition state. Hence, these steps are not expected to be kinetically limiting. It may additionally be noted that this reaction step is more complex than a mere C–O bond breaking in both cases. Indeed, in the case of TSA-2 one observes a subsequent deprotonation of the carbon in meta position with respect to the carboxylate by one chloride ligand, leading to the release of one HCl molecule (and the formation of a chlorophenolate zinc salt). In the case of TSA-3, the ring opening triggers a 1,2-methyl shift from the ortho to meta position with respect to the carboxylate. It may be noted that the resulting intermediate Int.A3 could then yield phenol 11 upon subsequent prototropy and ZnCl2 decoordination. Conversely, Int.A2 would yield phenol 13 (Scheme 1, bottom), which was not experimentally proposed on the basis of NMR analysis.
Focusing now on intermediate Int.B1, two epoxide ring opening reactions can once again be proposed, corresponding to the cleavage of the C–O bond involving the carbon atom in meta (TSB-2) or ortho (TSB-3) position with respect to the carboxyl. Both transition states are here also found at comparable energies (+11.1 and +8.1 kcal mol−1 with respect to the starting reagent, respectively, for TSB-2 and TSB-3), and at a lower energy than TSB-1. Hence, here again, this second reaction step is not kinetically limiting. Transition state TSB-2 directly affords intermediate Int.B2, which is stabilised by intramolecular coordination (chelate effect) and which could yield phenol 10 by a mere prototropy and ZnCl2 decoordination. On the other hand, TSB-3 produces a genuine Wheland intermediate (Int.B3). This intermediate is quite stable as it does not localise the electron vacancy in ipso position to the carboxylate, but it is plain to see it cannot directly yield a phenol by a similar process as the one proposed for Int.B2.
However, calculations reveal that two further transition states can be located for this intermediate, corresponding to 1,2 methyl shift reactions from Int.B3. TSB-4 is associated to the transfer of the methyl group to the carbon atom in para position with respect to the carboxyl, while TSB-5 is associated to the transfer in ortho position. Both are found at low energy (+7.1 and +5.6 kcal mol−1, respectively, with respect to the starting reagent), and the produced intermediates are significantly stabilised (relative energies of −32.1 and −30.9 kcal mol−1, respectively, for Int.B4 and Int.B5). These two intermediates could then yield phenol 12a (one of the two proposed structures in Fig. 6), and phenol 14 (not observed experimentally).
Overall, we found reaction paths affording phenols 10, 11, and 12a. Interestingly, no route could be delineated for the other phenolic candidate 12b proposed in Fig. 6, thus suggesting 12 is indeed methyl-5-hydroxy-2,4dimethylbenzoate. It may however be noted that two additional phenols (13 and 14) were found by the DFT calculations (Scheme 1), which were not reported experimentally. Their absence in experiments cannot be explained by kinetic selectivity; for instance, the reaction route to phenol 14 is lower in energy than that of 11, and only the latter is observed. If we now evaluate the thermodynamic selectivity, by calculating the relative energies of all phenols (Scheme 1, bottom), we notice the perspective is quite different. Indeed, phenols should then order, from lowest to largest yield, according to 14 < 13 < 12a ≈ 12b < 10 < 11. It may be noted that the thermodynamic selectivity seems to comply more with the experimental observations. Because no route to phenol 12b could be found, we may indeed withdraw this compound from our ordering, resulting in phenols 10 to 12a being expected as major products, as observed experimentally. The fact that phenol 11 is not associated with the highest yield can further be explained: among all phenols, it is associated with the largest activation barriers. Hence some extent of kinetic blockage could be expected in that case, accounting for the deviation from ideal thermodynamic selectivity.
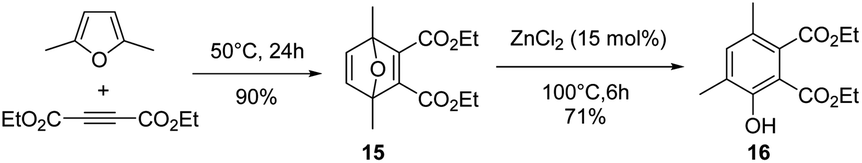
Inspired by these results, as migration of the methyl group during the aromatization step is possible, the reaction of DMF with 5 eq. of DEAD was finally explored (15 mol% ZnCl2, 100 °C). After 6 h of reaction, the in situ formed Diels–Alder adduct 15 was not completely aromatized to the phenol 16 (Fig. 7). Phenol 16 is characterized by two methyl group in meta position each other, as suggested by an HMBC NMR sequence (Fig. S36†). Unfortunately, extending the time of the reaction up to 48 h mainly led to a decomposition of the phenol 16, indicating that, at high conversion, the degradation rate of phenol 16 became the dominant reaction (Fig. 8). Hence, as discussed above, the reaction was also performed in two steps to optimize the phenol 16 selectivity. DMF was first reacted with 5 eq. of DEAD at 50 °C without any catalyst for 24 h, affording the Diels–Alder adduct 15 in 90% yield. Then, ZnCl2 (15 mol%) was added into the solution. Within 2 h, 75% of the Diels–Alder adduct 15 was converted and the phenol 16 was formed in 65% yield, i.e. 90% selectivity. Extending the time of the reaction to 6 h improved the yield in phenol 16 to 71% and the conversion of the Diels–Alder adduct 15 to 82%, i.e. 86% selectivity (Fig. 9).
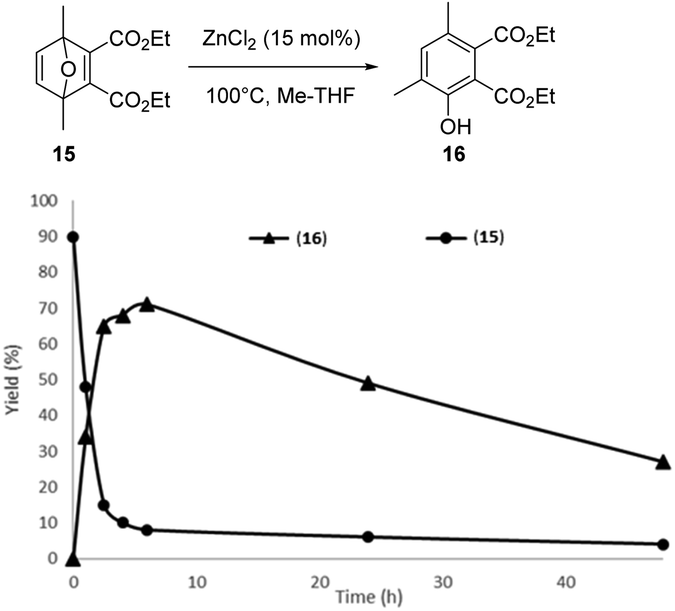 | ||
| Fig. 8 Conversion of the Diels–Alder adduct 15 obtained from DMF and DEAD and yield in phenol 16 as a function of the reaction time (15 mol% ZnCl2, 100 °C). | ||
 | ||
| Fig. 9 Reaction of MF with 5 eq. DEAD in a two-step process. | ||
However, gradually extending the time of the reaction from 6 to 48 h resulted in a gradual drop of the phenol 16 selectivity probably due to its degradation. It is noteworthy that, here again, only the phenol derivative with the methyl group in ortho position to the –OH was observed, reinforcing the hypothesis that, once the first migration of the methyl group has occurred, the aromatization reaction is a very fast process.
Conclusions
Here we propose a bio-based route to functionalized phenol derivatives from furfural and its derivatives. The strategy is based on a sequential Diels–Alder reaction of biobased furans with alkynes, followed by an aromatization reaction. Advantageously, in contrast to the classical Diels–Alder/aromatization of biobased furans with alkenes, no water is released during the aromatization step. In addition, this reaction involves cheap and industrially available biobased furans as organic building blocks. Through combined experimental and theoretical investigations, we revealed that methyl-functionalized furanic derivatives can also take part in this reaction, undergoing 1,2 methyl shift during the aromatization reaction, thus opening the possibility of using disubstituted furanic derivatives, a mean to further improve the chemical diversity and complexity which can be created through this route.In contrast to biobased lignin-derived phenol derivatives which are often para-substituted, this route yields phenol substituted in ortho or meta position (relative to the –OH), thus complementing the scope of biomass for producing phenolic compounds.
One should note that the fact that alkynes are nowadays 100% petrobased is not an issue. The debate is not so much whether this route opens the way to 100% biobased phenols or not, but rather whether it is more sustainable or not than current routes. For this reason, a complete life cycle assessment will be the topic of future investigations to carefully check the sustainability of this biobased route, as compared to the fossil ones.
Author contributions
Adrien Ratier, Mélanie Anizan and Sarah Behloul did the experiments while François Jérôme and Karine De Oliveira Vigier supervised the work at the University of Poitiers. Frederic Guégan, Gilles Frapper and Richail D. Moulandou-Koumba performed DFT calculations. Quentin Blancart Remaury was in charge of the phenol characterizations and Jianxia Zheng supervised the work for SOLVAY.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors are grateful to the CNRS and the University of Poitiers for the funding of these researches. Authors also strongly acknowledge the Région Nouvelle-Aquitaine and SOLVAY for the funding of the postdoctoral grant of Adrien Ratier and the ANR CATALFUR (ANR-21-CE43-0005) for the PhD grant of Sarah Behloul.Notes and references
- M. Decostanzi, R. Auvergne, B. Boutevina and S. Caillol, Green Chem., 2019, 21, 724–747 RSC.
- https://www.chemanalyst.com/industry-report/phenol-market-184, Consulted on June 21st, 2023.
- https://www.transparencymarketresearch.com/bio-based-phenol-market.html, Consulted on June 21st, 2023.
- (a) C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115(21), 11559–11624 CrossRef CAS PubMed; (b) A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyma, Science, 2014, 344(6185), 1246843 CrossRef PubMed; (c) J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110(6), 3552–3599 CrossRef CAS PubMed; (d) S. Van den Bosch, S.-F. Koelewijn, T. Renders, G. Van den Bossche, T. Vangeel, W. Schutyser and B. F. Sels, Top. Curr. Chem., 2018, 376, 36 CrossRef CAS PubMed; (e) M. Al-Naji, F. Brandi, M. Drieß and F. Rosowski, Chem. Ing. Tech., 2022, 94(11), 1611–1627 CrossRef CAS; (f) R. E. Key and J. J. Bozell, ACS Sust. Chem. Eng., 2016, 4(10), 5123–5135 CrossRef CAS; (g) J. S. Mahajan, R. M. O'Dea, J. B. Norris, L. T. J. Korley and T. H. Epps III, ACS Sust. Chem. Eng., 2020, 8(40), 15072–15096 CrossRef CAS; (h) R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55(29), 8164–8215 CrossRef CAS PubMed; (i) X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, ChemSusChem, 2020, 13(17), 4296–4317 CrossRef CAS PubMed.
- C. W. Lahive, P. J. Deuss, C. S. Lancefield, Z. Sun, D. B. Cordes, C. M. Young, F. Tran, A. M. Z. Slawin, J. G. de Vries, P. C. J. Kamer, N. J. Westwood and K. Barta, J. Am. Chem. Soc., 2016, 138(28), 8900–8911 CrossRef CAS PubMed.
- (a) G. F. De Gregorio, R. Prado, C. Vriamont, X. Erdocia, J. Labidi, J. P. Hallett and T. Welton, ACS Sust. Chem. Eng., 2016, 4(11), 6031–6036 CrossRef; (b) C. Cabral Almada, A. Kazachenko, P. Fongarland, D. Da Silva Perez, B. N. Kuznetsov and L. Djakovitch, Biomass Convers. Biorefin., 2022, 12, 3795–3808 CrossRef CAS; (c) H. Lange, S. Decina and C. Crestini, Eur. Polym. J., 2013, 49(6), 1151–1173 CrossRef CAS; (d) C. Liu, S. Wu, H. Zhang and R. Xiao, Fuel Process. Technol., 2019, 191, 181–201 CrossRef CAS.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- (a) C. Liu, J. Hu, H. Zhang and R. Xiao, Fuel, 2016, 182, 864–870 CrossRef CAS; (b) C. Peng, G. Zhang, J. Yue and G. Xu, Fuel Process. Technol., 2014, 24, 212–221 CrossRef; (c) Z. Dong, H. Yang, P. Chen, Z. Liu, Y. Chen, L. Wang, X. Wang and H. Chen, Energy Fuels, 2019, 33(10), 9934–9941 CrossRef CAS.
- (a) S. Liu, Y.-Q. Guo, P.-Y. Sun, S.-H. Zhang and Z. Long Yao, J. Fuel Chem. Technol., 2021, 49(12), 1911–1920 CrossRef CAS; (b) L. Melia Terry, C. Li, J. J. Chew, A. Aqsha, B. S. How, A. C. M. Loy, B. L. F. Chin, D. S. Khaerudini, N. Hameed, G. Guan and J. Sunarso, Carbon Res. Conv., 2021, 4, 239–250 Search PubMed; (c) F. Abnisa, W. M. A. Wan Daud and J. N. Sahu, Environ. Prog. Sustain., 2014, 33(3), 1026–1033 CrossRef CAS; (d) M. Bertero, et al., Pyrolysis Products from Residues of Palm Oil Industry, in Sustainable Technologies for the Management of Agricultural Wastes. Applied Environmental Science and Engineering for a Sustainable Future, ed. Z. Zakaria, Springer, Singapore, 2018 Search PubMed.
- (a) M. Yuliana, B. T. Nguyen-Thi, S. Faika, L. Huong Huynh, F. E. Soetaredjo and Y.-H. Ju, J. Taiwan Inst. Chem. Eng., 2014, 45(5), 2187–2193 CrossRef CAS; (b) P. Phani Kumar, R. Paramashivappa, P. J. Vithayathil, P. V. Subba Rao and A. Srinivasa Rao, J. Agric. Food Chem., 2002, 50(16), 4705–4708 CrossRef CAS PubMed; (c) M. M. Ranarijaona, N. A. H. R. Rakotomena, M. T. Andrianjafy, F. D. Ramiharimanana, L. C. Herinirina, N. H. Ramarosandratana, B. Briou, P. Fajardie, P. Mavingui, E. Métay, V. V. Ramanandraibe and M. Lemaire, Molecules, 2021, 26(24), 7625 CrossRef CAS PubMed.
- (a) A. K. Das, M. N. Islam, M. O. Faruk, M. Ashaduzzaman and R. Dungani, S. Afr. J. Bot., 2020, 135, 58–70 CrossRef CAS; (b) Y. Shirmohammadli, D. Efhamisisi and A. Pizzi, Ind. Crops Prod., 2018, 126, 316–332 CrossRef CAS; (c) P. V. Dhawale, S. K. Vineeth, R. V. Gadhave, J. Fatima, M. V. Supekar, V. K. Thakur and P. Raghavan, Mater. Adv., 2022, 3, 3365–3388 RSC; (d) A. Arbenza and L. Avérous, Green Chem., 2015, 17, 2626–2646 RSC.
- (a) K. I. Galkin and V. P. Ananikov, Int. J. Mol. Sci., 2021, 22(21), 11856 CrossRef CAS PubMed; (b) R. C. Cioc, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, Angew. Chem., Int. Ed., 2022, 61(17), e202114720 CrossRef CAS PubMed; (c) R. C. Cioc, M. Lutz, E. A. Pidko, M. Crockatt, J. C. van der Waald and P. C. A. Bruijnincx, Green Chem., 2021, 23, 367–373 RSC; (d) Z. Li, Y. Jiang, Y. Li, H. Zhang, H. Lia and S. Yang, Catal. Sci. Technol., 2022, 12, 1902–1921 RSC.
- (a) I. Scodeller, S. Mansouri, D. Morvan, E. Muller, K. de Oliveira Vigier, R. Wischert and F. Jérôme, Angew. Chem., Int. Ed., 2018, 57(33), 10510–10514 CrossRef CAS PubMed; (b) I. Scodeller, K. De Oliveira Vigier, E. Muller, C. Ma, F. Guégan, R. Wischert and F. Jérôme, ChemSusChem, 2021, 14(1), 313–323 CrossRef PubMed.
- (a) R. C. Cioc, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, ChemSusChem, 2022, 15(18), e202201139 CrossRef CAS PubMed; (b) S. Higson, F. Subrizi, T. D. Sheppard and H. C. Hailes, Green Chem., 2016, 18, 1855–1858 RSC.
- (a) A. W. Mcculloch and A. G. McInnes, Can. J. Chem., 1971, 49(19), 3152–3157 CrossRef CAS; (b) A. F. Oleinik, E. V. Adamskaya, K. Y. Novitskii, N. P. Soloveva and E. M. Peresleni, Chem. Heterocycl. Compd., 1979, 15, 13–16 CrossRef.
- P. Jiang, G. Zhao, H. Zhang, T. Ji, L. Mu, X. Lu and J. Zhu, Green Energy Environ., 2022 DOI:10.1016/j.gee.2022.12.004.
- A. Vakalopoulos and H. M. R. Hoffmann, Org. Lett., 2001, 3(14), 2185–2188 CrossRef CAS PubMed.
- T. E. Burghardt, J. Sulfur Chem., 2005, 26(4–5), 411–427 CrossRef CAS.
- N. Koichi, S. Takeshi and M. Teruaki, Bull. Chem. Soc. Jpn., 1972, 45(12), 3724 CrossRef.
- D. A. McCaulay and A. P. Lien, J. Am. Chem. Soc., 1952, 74(24), 6246–6250 CrossRef CAS.
- M. Zanger, Org. Magn. Reson., 1972, 4(1), 1–25 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06461a |
| This journal is © The Royal Society of Chemistry 2023 |