
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancing nitrate reduction to ammonia: insights into mechanism, activity control, and catalyst design over Pt nanoparticle-based ZrO2†
Ayyaz Mahmood abcd,
Fouzia Perveene,
Tehmina Akramf,
Shenggui Chen*bcd,
Ahmad Irfang and
Huafu Chen*a
abcd,
Fouzia Perveene,
Tehmina Akramf,
Shenggui Chen*bcd,
Ahmad Irfang and
Huafu Chen*a
aSchool of Life Science and Technology, University of Electronic Science and Technology, Chengdu 610054, China. E-mail: chenhf@uestc.edu.cn
bSchool of Mechanical Engineering, Dongguan University of Technology, Dongguan 523808, China. E-mail: dgutchensg@163.com
cSchool of Art and Design, Guangzhou Panyu Polytechnic, Guangzhou 511483, China
dDongguan Institute of Science and Technology Innovation, Dongguan University of Technology, Dongguan 523808, China
eSchool of Interdisciplinary Engineering & Sciences (SINES), National University of Sciences and Technology (NUST), Sector H-12, Islamabad, 44000, Pakistan
fCAS Key Laboratory of Soft Matter Chemistry, Hefei National Laboratory for Physical Sciences at Microscale, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), Department of Chemistry, University of Science and Technology of China, Hefei 230026, China
gDepartment of Chemistry, College of Science, King Khalid University, P.O. Box 9004, Abha 61413, Saudi Arabia
First published on 24th November 2023
Abstract
The reduction of nitrogen oxides (NOx) to NH3, or N2 represents a crucial step in mitigating atmospheric NO3 and NO2 emissions, a significant contributor to air pollution. Among these reduction products, ammonia (NH3) holds particular significance due to its utility in nitrogen-based fertilizers and its versatile applications in various industrial processes. Platinum-based catalysts have exhibited promise in enhancing the rate and selectivity of these reduction reactions. In this study, we employ density functional theory (DFT) calculations to explore the catalytic potential of Pt nanoparticle (PtNP)-supported ZrO2 for the conversion of NO3 to NH3. The most favorable pathway for the NO3 reduction to NH3 follows a sequence, that is, NO3 → NO2 → NO → ONH → ONH2/HNOH → NH2/NH → NH2 → NH3, culminating in the production of valuable ammonia. The introduction of low-state Fe and Co dopants into the ZrO2 support reduces energy barriers for the most challenging rate-determining hydrogenation step in NOx reduction to NH3, demonstrating significant improvements in catalytic activity. The incorporation of dopants into the ZrO2 support results in a depletion of electron density within the Pt cocatalyst resulting in enhanced hydrogen transfer efficiency during the hydrogenation process. This study aims to provide insights into the catalytic activity of platinum-based ZrO2 catalysts and will help design new high-performance catalysts for the reduction of atmospheric pollutants and for energy applications.
1. Introduction
Nitrogen oxides (NOx) are significant pollutants in the atmosphere, primarily originating from the combustion of fossil fuels, including coal.1 As a major consumer of coal, China faces the challenge of substantial NOx emissions during combustion, leading to their release into the atmosphere. These emissions, combined with particulate matter, contribute to the occurrence of severe smog in northern regions of China, posing a serious threat to public health.2,3 Among the NOx gases, nitrogen dioxide (NO2) is an inorganic air pollutant that poses significant harm to human health, animals, and the environment.1,4,5 While natural events such as volcanic eruptions, lightning strikes, and forest fires also contribute to NOx emissions, the most substantial sources are human activities, including combustion processes, transportation, and industrial operations.1 When NO2 is exposed to sunlight, it undergoes photolysis, breaking down into nitrogen monoxide (NO) and atomic oxygen, O(3P). The atomic oxygen then combines with molecular oxygen to form tropospheric ozone.6 This process plays a crucial role in the formation of photochemical smog, which consists of a harmful mixture of toxic chemicals resulting from the interaction between NOx and volatile organic compounds (VOCs). Exposure to photochemical smog, including elevated levels of tropospheric ozone, can have detrimental effects on the respiratory system and overall lung health.5,7On the other hand, over the past century, the widespread use of artificial nitrogen fertilizers has significantly increased food production, but it has also led to environmental challenges on a global scale. Eutrophication and acidification of water resources are among the issues caused by the excessive presence of high-valence-state nitrate, which is highly toxic to both humans and the environment.8,9 To mitigate these problems, the World Health Organization (WHO) has set strict guidelines recommending a maximum nitrate concentration of 50 mg dm−3 in drinking water.10 Unfortunately, the contamination of underground water sources due to the excessive emission of nitrate ions has become a widespread issue caused by factors such as fertilizer runoff, stock farming, and industrial wastewater.11,12 To combat this problem, there is a growing need to develop efficient methods for converting toxic nitrate into valuable ammonia. Ammonia, with its low-valence-state, is not only an essential component for nitrogen-based fertilizers but also serves as a key chemical in the production of artificial fibers.13–15 Currently, the synthesis of ammonia involves the energy-intensive Haber–Bosch process, which requires high pressures and temperatures.16 In contrast, the conversion of toxic nitrate into ammonia in a catalytic reduction process holds significant scientific and practical importance. By developing a catalytic system for the reduction of nitrate, it becomes possible to convert harmful nitrate compounds into valuable ammonia. This process offers several benefits, including its potential contribution to the ecosystem, improvement of human health, and facilitation of sustainable social development.
Photocatalysis offers a promising and sustainable approach to facilitate the process.17,18 Various photocatalysts, including TiO2, SrTiO3, and BiOCl, have been investigated for their ability to catalyze the reduction of NO3− into NH3.19–21 Furthermore, the deposition of metal particles, such as Cu, Au, Ag, Pt, and Pd, has been extensively employed to enhance both the catalytic performance and visible light absorption characteristics of Catalysts/TiO2.22–25 This strategy effectively suppresses electron–hole recombination and promotes photocatalytic activity. For example, Yamauchi et al. successfully achieved high NH3 selectivity of approximately 78% in the photocatalytic reduction of NO3− by depositing alloyed CuPd nanoparticles on TiO2.25 However, the electron-rich nature of TiO2 tends to promote the undesired oxidation of NO2 to NO3− species rather than the desired reduction to nitrogen-containing compounds. This is due to the ability of TiO2 to stabilize intermediate NO2 adsorbates and facilitate their oxidation through a bifunctional charge exchange mechanism. The stabilization of surface-NO3− and NO3− ion pair configurations on TiO2 further supports the notion that TiO2 may be more favorable for NO2 oxidation reactions rather than its reduction to nitrogen-containing compounds.
Multicomponent catalyst systems, which comprise two or more interdependent components, have gained recognition as promising materials for achieving higher catalytic activities. One approach involves the utilization of oxide-supported metals, which operate through the interfacial effect at the interfaces between the oxide and metal components.26–29 Significant efforts have been devoted to the exploration of active metals for these multicomponent catalyst systems, ranging from precious platinum-group metals to nonprecious transition metals. The objective is to identify suitable catalysts that can effectively promote the desired chemical reactions. Meanwhile, there is a wide range of oxide supports available for catalyst systems, including ZnO, Al2O3, ZrO2, SiO2, TiO2, CeO2, Ga2O3, MgO, Cr2O3, and Nb2O5. In these systems, metals are commonly recognized as the active sites. The properties of metals, such as particle size, dispersion, specific surface area, and alloy components, along with their interactions with the supports, have been extensively discussed in various reviews and perspective articles. These studies aim to establish correlations between these metal and support properties and the catalytic performance of the systems.30–37
Zirconium dioxide (ZrO2) is highly valued as both a catalyst and support material for various reactions due to its exceptional thermal stability, excellent redox properties, and the presence of acid–base sites on its surface. It has been successfully utilized in reactions such as CO2 methanation,38,39 water–gas shift,40,41 and NO2 selective catalytic reduction.42,43 Particularly noteworthy is the powerful catalytic activity exhibited by transition metal oxides dispersed on the surface of ZrO2 in NO reduction reactions. Studies have shown that Cu/ZrO2 catalysts demonstrate high NO conversion to N2 at low temperatures, utilizing a nitrous oxide (N2O) intermediate in the NO–CO reaction.44 ZrO2, as a support material, outperforms CeO2 in reducing the energy barrier for NO dissociation.45 Additionally, ZrO2 offers ample sites for NOx adsorption, which is beneficial for NO reduction.46,47 Notably, a study by Koga et al. discovered that a c-ZrO2 (110) ultrathin film covering a Cu surface exhibited remarkable activity in NOx reduction.48
Among metal supports, Pt is widely recognized as a versatile catalyst and finds extensive use in both experimental studies and industrial applications. It serves as a cocatalyst in various photocatalytic reduction reactions, including water splitting for hydrogen production,49–52 CO2 reduction,53–55 and NO3− reduction processes.56–58 Additionally, Pt is extensively employed in electrocatalytic NO3− reduction processes aimed at producing NH3. Previous studies by Dima and Figueiredo, as well as their colleagues, demonstrated the catalytic activity of pure Pt electrodes in the reduction of NO3− in acidic solutions, proposing adsorbed NO as the reactive intermediate in NH3 formation.57,58 Moreover, Cerron-Calle et al. achieved a remarkably high NH3 selectivity of 94% by utilizing alloyed CuPt catalysts for electrocatalytic NO3− reduction.56 Considering Pt's excellent ability to provide surface-active H* in photocatalysis and catalyze NO3− reduction in electrocatalysis, it is reasonable to speculate that Pt/ZrO2 would serve as an excellent model system for investigating the photocatalytic reduction of NO3− to NH3.
Although significant experimental progress has been made in the field of nitrate reduction to ammonia, there are still gaps in our understanding of the underlying mechanistic details, rate-determining steps, and catalyst regulation strategies. One particular area of debate revolves around the conversion of nitric oxide (NO) into ammonia (NH3), which is considered a crucial intermediate in the process. Clayborne et al. proposed a pathway where NH3 is formed through the intermediate NOH*. According to their suggested mechanism, the favorable sequence involves the conversion of NO to NOH, followed by N, NH, NH2, and finally NH3.59 In contrast, Koper et al. presented an alternative perspective, suggesting that ammonia is produced through the intermediates HNO* and H2NO*. Their proposed pathway starts with the conversion of NO to HNO, then H2NO, and finally NH3.60 Additionally, Cuesta et al. put forward the idea that the intermediate HNOH* plays a pivotal role in NO reduction, suggesting a pathway of NO to NOH (HNO), then HNOH, and ultimately NH3.61
Clearly, there is ongoing debate and multiple proposed pathways for the conversion of NO to NH3 in nitrate reduction. To improve the efficiency of this process, it is imperative to dedicate further efforts towards understanding the complete reaction mechanism, including the energetics and intermediates involved. Advancements in this area will contribute to refining the catalyst design and optimization strategies, ultimately leading to enhanced nitrate reduction efficiency and the production of valuable ammonia.
Thus, in this context, we employed density functional theory (DFT) calculations to systematically investigate the process of NO3− reduction to NH3 on a Pt nanoparticle (PtNP)/ZrO2 composite model. Our previous research (under review) involved extensive ab initio molecular dynamics (AIMD) simulations, which allowed us to determine the PtNP/ZrO2(111) composite structures (Fig. S2†).62 Based on the stability, hydrogen evolution reaction (HER) and water-splitting reaction (WSR) activities, Pt11/ZrO2 was chosen for current study. First, we examined the reaction energetics of various elementary steps within different potential pathways of NO3− reduction. Through this analysis, we identified the most favorable pathway and determined the rate-determining steps. Next, we explored the effect of charge density modulation on the NO3− reduction activity by manipulating the PtNP's charge density on the ZrO2 support. By understanding the trend in activity variation, we proposed a strategy to enhance the catalytic performance of PtNP/ZrO2 by doping ZrO2 with a suitable element, such as Fe and Co. This proposed modification was supported by our calculated results, which indicated a significant reduction in reaction barriers and an increase in energy gains. Overall, our study provides valuable insights into the NO3− reduction mechanism on PtNP/ZrO2 catalysts, offering a potential avenue for improving catalytic performance through targeted modifications. The findings from this study will contribute to the understanding and development of advanced catalyst materials for efficient NO3 reduction, with potential applications in automotive emission control systems and industrial processes. Ultimately, the utilization of Pt nanoparticle-based ZrO2 catalysts holds promise for mitigating the adverse effects of NO3/NO2 pollution and promoting cleaner and healthier environments.
2. Methods
In this study, computational simulations were performed using the BAND module of the Amsterdam Modeling Suite (AMS) developed by Software for Chemistry & Materials BV (SCM).63,64 The simulations employed the linear combination of atomic orbitals (LCAO) approach65 and the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional within the framework of density functional theory (DFT).66,67 To describe the atomic structures, a combination of valence triple-zeta polarized (TZP) Slater-type orbitals and Herman–Skillman numerical atomic orbitals (NAOs) with a small frozen core was utilized. The van der Waals (vdW) interactions were accounted by using Grimme's third-generation dispersion correction together with Becke–Johnson damping function (D3-BJ).68 The Pt11/ZrO2 structure was chosen for this investigation based on the results presented in our previous study which explored the effect of size of nanoparticle size on the catalytic activity determining Pt11 to be the optimal nanoparticle size.62 The size of nanoparticles can significantly influence catalyst activity. Smaller nanoparticles may have fewer active sites, potentially limiting their catalytic efficiency, while larger nanoparticles might suffer from decreased charge transfer efficiency, potentially hindering their performance due to increased surface recombination. Our findings suggest that Pt11/ZrO2 strikes a balance between these factors. It provides an adequate surface area with numerous active sites, facilitates efficient charge transfer, and minimizes surface recombination, yielding the best catalyst activity.For geometry optimizations, a maximum gradient threshold of 0.03 eV Å−1 was employed. The convergence criteria for the electronic self-consistent loop were 10−6 eV which is benchmark in many studies. Relativistic effects, including spin–orbit coupling, were considered using the zero-order regular approximation (ZORA) method. Periodic boundary conditions were applied to represent the repeating unit cell structure, with a (111) surface model constructed using a 3 × 3 supercell containing two ZrO2 trilayers (O–Zr–O layer). A vacuum spacing of 15 Å was set between the slabs, and a Monkhorst–Pack scheme with a 6 × 6 × 1 k-point sampling was used to sample the Brillouin zone.69 Dipole correction was applied in all calculations. The bottom ZrO2 trilayer was held fixed at its bulk positions, while the top ZrO2 trilayer and the adsorbents were allowed to fully relax to reach their equilibrium structures. To determine the stable representative configuration of Pt11 cluster on ZrO2, we employed AIMD simulations. The simulations utilized Nosé–Hoover thermostats in the canonical (NVT) ensemble for over 20 ps, achieving equilibrium around 10 ps. Structural configurations were selected at 1 ps intervals from the equilibrated trajectory and fully optimized to minimize all forces. Out of 10 potential structural candidates, we determined optimal Pt11/ZrO2 structures based on their lowest energy.
Transition states associated with the water-splitting reactions were determined by systematically exploring the potential energy surface (PES) along the relevant reaction coordinates. The PES was scanned to identify the highest energy point, which was then optimized to obtain the transition state structure.70 The forces threshold for the transition state search was set to 0.05 eV Å−1. To validate the transition states, it was ensured that they exhibited only one imaginary frequency along the reaction coordinate.71,72 The vibrational modes were calculated using similar methods as for optimizations. They were specifically computed for the cluster and reacting species on the cluster, excluding the ZrO2 substrate for computational efficiency. The finite differences method was employed to calculate the vibrational frequencies, utilizing a step length of 0.005 Å. The Gibbs free energies of reaction (ΔGr) and Gibbs activation energy barriers (Δ‡G) for the water-splitting reaction were calculated as the differences in free energy between the involved species, as described by eqn (1) and (2), respectively.
| ΔGr = ΔGProducts − ΔGreactants | (1) |
| Δ‡G = ΔGTS − ΔGreactants | (2) |
3. Results and discussion
We chose Pt11/ZrO2 composite as the substrate to represent the Pt nanoparticle-based ZrO2 catalyst which was obtained from ab initio molecular dynamics (AIMD) simulations (Fig. S2†) presented in our previous study which investigates the size effect of Pt nanoparticles on the catalytic activity.62 To maintain a neutral charge in our simulation, we introduced a NaNO3 molecule into the system to represent the NO3− ion. We positioned the Na+ cation of the NaNO3 molecule uniformly on the opposite (bottom) layer of the ZrO2 slab and allowed it to relax (Fig. 1 and S1†). Bader charge calculations (Table S1†) reveal that the Pt11 cluster possesses an overall positive charge of +0.72 |e|, with an average charge of +0.067 |e| on each Pt atom within the cluster. When negatively charged NO3− species are introduced, the total charge decreases by 0.48 |e|, leading to a net charge of −0.22 |e| on the cluster. This charge is subsequently neutralized by the presence of Na+ ions, restoring the total charge to +0.72 |e|. In essence, the cluster's charge is initially reduced by the introduction of NO3−, but it returns to its original positive state due to the compensating effect of Na+ ions. Based on the adsorption energies and stability of various adsorption configurations of NO3− on different adsorption sites, we identified the most stable adsorption structure for NO3− (Eads (NO3−) = −1.52 eV) relative to a desorbed NO3− in the gas phase, with Na+ positioned on the opposite layer which is presented in Fig. 1.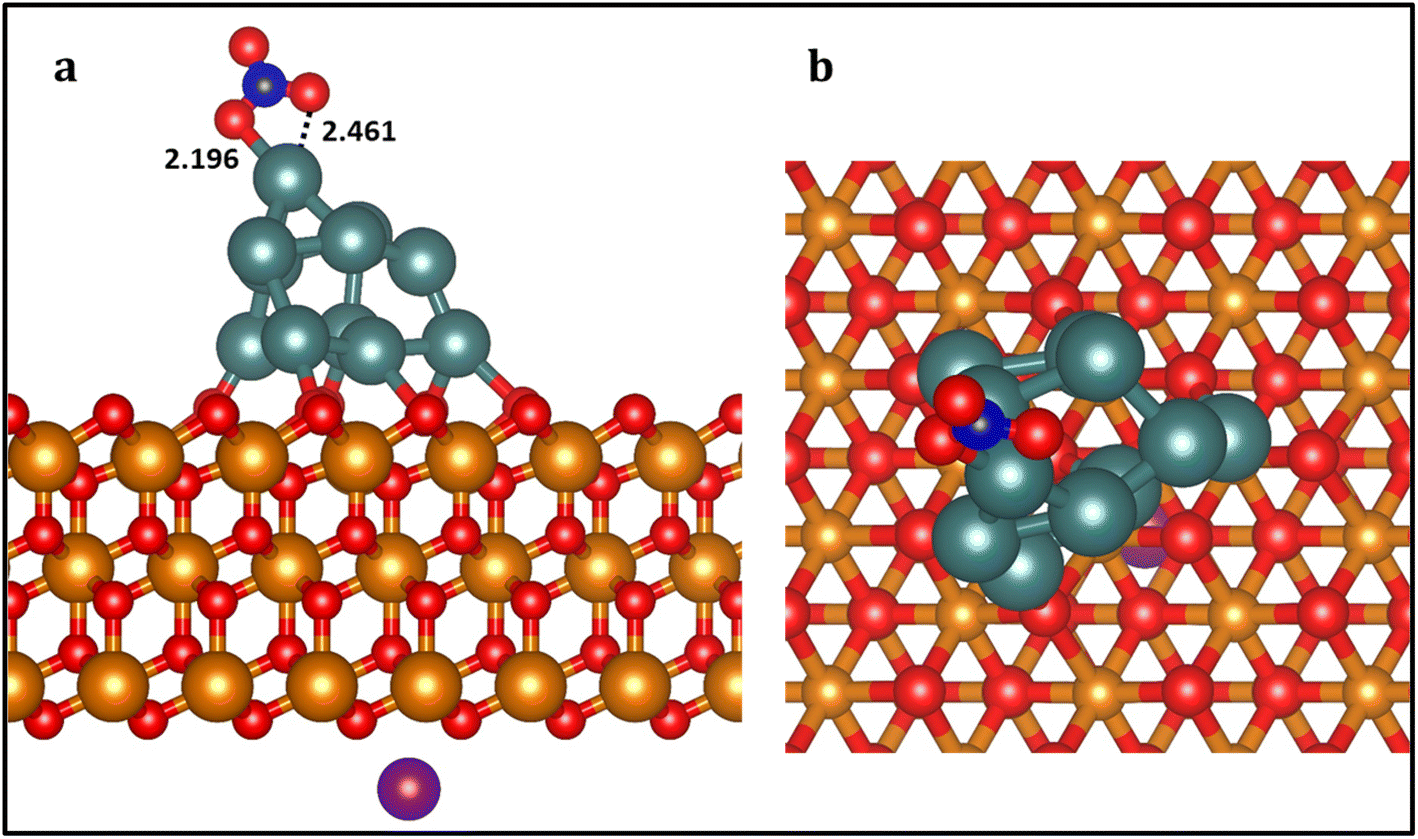 | ||
| Fig. 1 Side (a) and top (b) view of the calculated most stable structure of NO3 on Pt11/ZrO2 composite. Bond distances values in Angstrom (Å). Zr: orange; O: red; Pt: dark cyan; N: dark blue; Na+: purple. This color scheme is used throughout the paper. The radius of Zr atoms in the top view is reduced for clarity. | ||
In this adsorption configuration, NO3− forms specific bonds with Zr atoms on the ZrO2 surface. Two O–Pt (O1–Pt1 and O2–Pt1) bonds were formed with the same Pt atom, with bond lengths of 2.196 Å and 2.461 Å, respectively. Furthermore, considering the unique properties of the ZrO2 surface, such as its high thermal stability and acid–base characteristics, we simulated the subsequent reduction of NO3− under conditions where reactive hydrogen species (H*) are adequately available on the ZrO2 surface in the form of water molecules which release protons.
3.1. *NO3− reduction to *NO2
We investigated the process of *NO3− reduction to *NO2 by analyzing three possible pathways for breaking the first N–O bond in NO3−* (as shown in Fig. 2). The first pathway is direct dissociation (referred to as direct NO3− dis., represented by the red curve). In this pathway, *NO3− dissociates directly into *NO2 and *O by overcoming an energy barrier of 0.76 eV, with a significant exothermic reaction energy of −0.67 eV. The second pathway, denoted as O2N–OH* dissociation (HO–NO2 dis., represented by the blue curve), involves the capture of a hydrogen specie (H*) by *NO3 to form *O2N–OH on the surface of the catalyst. Importantly, the first hydrogenation steps are mostly characterized by an extremely low energy requirement, rendering it spontaneous. Subsequently, the formed O2N–OH* species dissociates into NO2* and OH* with a barrier of 0.62 eV, accompanied by a substantial energy release of −0.94 eV.
dissociation (referred to as direct NO3− dis., represented by the red curve). In this pathway, *NO3− dissociates directly into *NO2 and *O by overcoming an energy barrier of 0.76 eV, with a significant exothermic reaction energy of −0.67 eV. The second pathway, denoted as O2N–OH* dissociation (HO–NO2 dis., represented by the blue curve), involves the capture of a hydrogen specie (H*) by *NO3 to form *O2N–OH on the surface of the catalyst. Importantly, the first hydrogenation steps are mostly characterized by an extremely low energy requirement, rendering it spontaneous. Subsequently, the formed O2N–OH* species dissociates into NO2* and OH* with a barrier of 0.62 eV, accompanied by a substantial energy release of −0.94 eV.
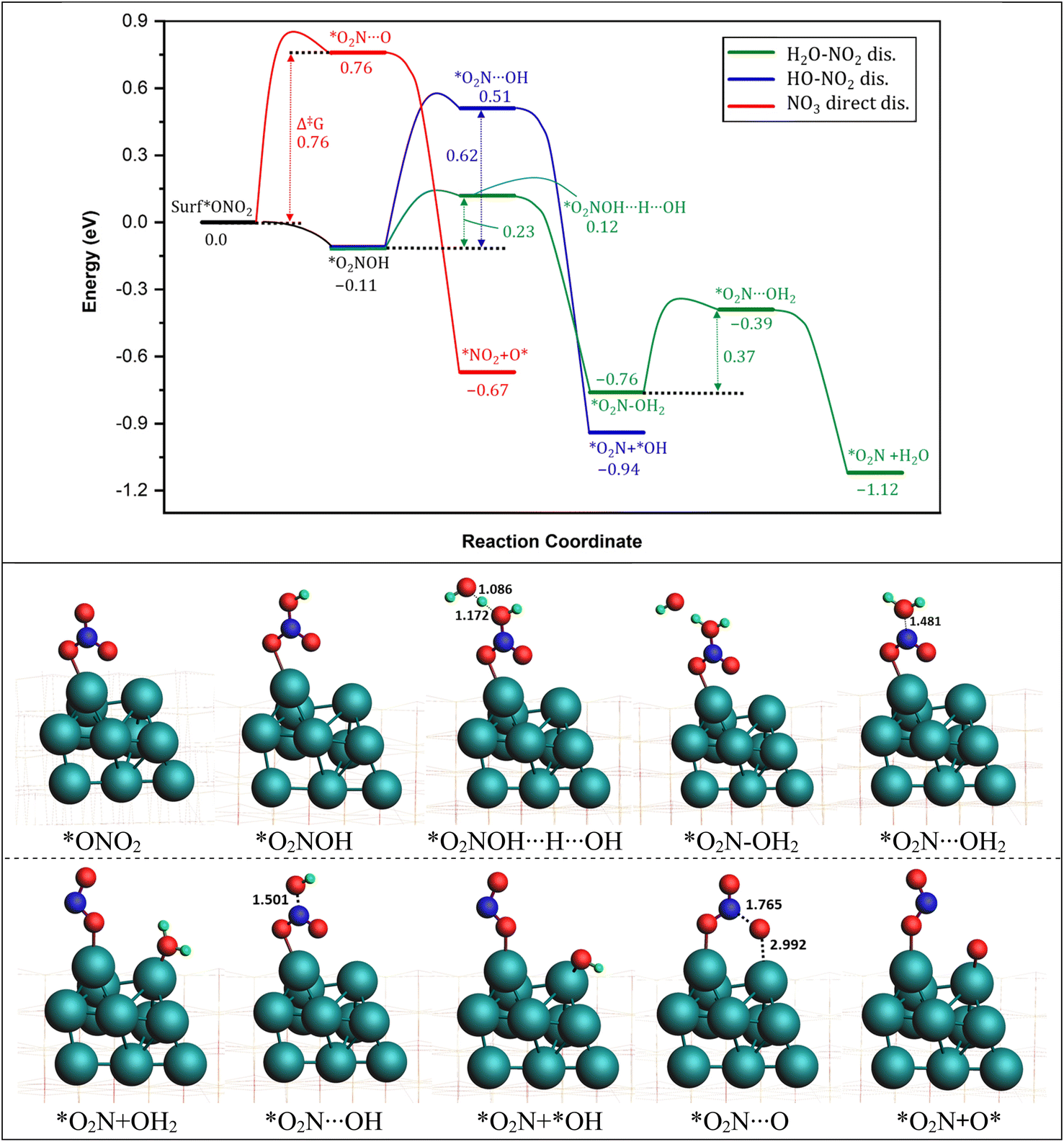 | ||
| Fig. 2 (Top) reaction energy profile for the conversion pathway of NO3− into NO2. Green: H2O–NO2 dissociation, blue: HO–NO2 dissociation, red: NO3− direct dissociation. The energy of the entering or leaving species was discounted from the total energy of the system to calculate the relative energy. The energy of the H was calculated as 1/2 of the energy of H2. (Bottom) optimized structures of various intermediates and transition states involved in the reaction pathways. ZrO2 layers are represented by wireframes for clarity. Energy values in eV. Bond distances in Å. Similar colors were used for intermediates names. | ||
A third pathway involves the capture of a 2nd proton by the O2N–OH (Δ‡G 0.23) formed in the 2nd pathway to form the water molecule *O2N–OH2 specie (H2O–NO2 dis., represented by the green curve). In the next step, water molecule (H2O) detaches with a reaction energy barrier of 0.37 eV passing through the transition state *O2N⋯H2O. This step is calculated to be exothermic with an energy release of 0.36 eV with an overall energy release in this pathway to be −1.12 eV, indicating the most exothermic pathway for the conversion of *NO3 into *NO2.
These results indicate that all three pathways are feasible for the reduction of *NO3 to *NO2. The NO3 direct dissociation pathway involves a higher energy barrier (0.76 eV) but offers a larger energy release upon completion. In contrast, the HO–O2N dissociation pathway involves a lower energy barrier (0.37 eV) but a slightly smaller energy release. The bond lengths associated with the breaking N–O bond in the NO3 direct dissociation pathway and the HO–O2N dissociation pathway were analyzed (as shown in Fig. 1). It was observed that the distance of the breaking N–O bond in the direct NO3 direct dissociation pathway (*O2N⋯O: 1.765 Å) is significantly longer compared to that in the HO–NO2 dissociation pathway (*O2N⋯OH2: 1.481 Å). Based on both the reaction energetics and the transition-state structures, it can be concluded that the H*-assisted HO–O2N dissociation pathway is more favorable (0.37 vs. 0.76 eV) than the direct NO3 dissociation pathway on the ZrO2 surface. The calculated difference in the activation energies between both pathways is probably due to the fact that the transition states (*O2N⋯OH2: 1.481 Å) found in O2N–OH dissociation pathway are early transition states because they resemble more the reactants and according to the Hammond postulate the early transition states are generally characteristic of fast exothermic reactions.73 On the other hand, the transition states (*O2N⋯O: 1.765 Å) found in the NO3 direct dissociation pathway are late transition states as they resemble more the products, and thus are characteristic of slow endothermic reactions. This finding is consistent with previous reports indicating the preference for the H*-assisted pathway in the reduction of *NO3.74,75
3.2. *NO2 reduction to *NO
The pathways for the dissociation of the second N–O bond in NO2 were investigated next. Three possible routes were considered: the NO2 direct dissociation pathway, and two H*-assisted dissociation pathways, as depicted in Fig. 3. In the NO2 direct dissociation pathway (red curve), breaking the N–O bond requires overcoming a substantial energy barrier of 1.17 eV. Although the formation of *NO and O* releases energy (−0.28 eV), the initial bond breaking step is energetically demanding. In the *H-assisted dissociation pathways, we explored the involvement of H* in facilitating the dissociation process. It was observed that the O-end of NO2* readily captures H* and forms *NO–OH on the Pt surface with a small energy change of 0.09 eV. Subsequently, the dissociation of *NO–OH into *NO and *OH occurs with a large barrier of 0.68 eV and a significant energy release of −0.56 eV. The overall pathway is calculated to be exothermic with a large free energy of the reaction of −0.65 eV.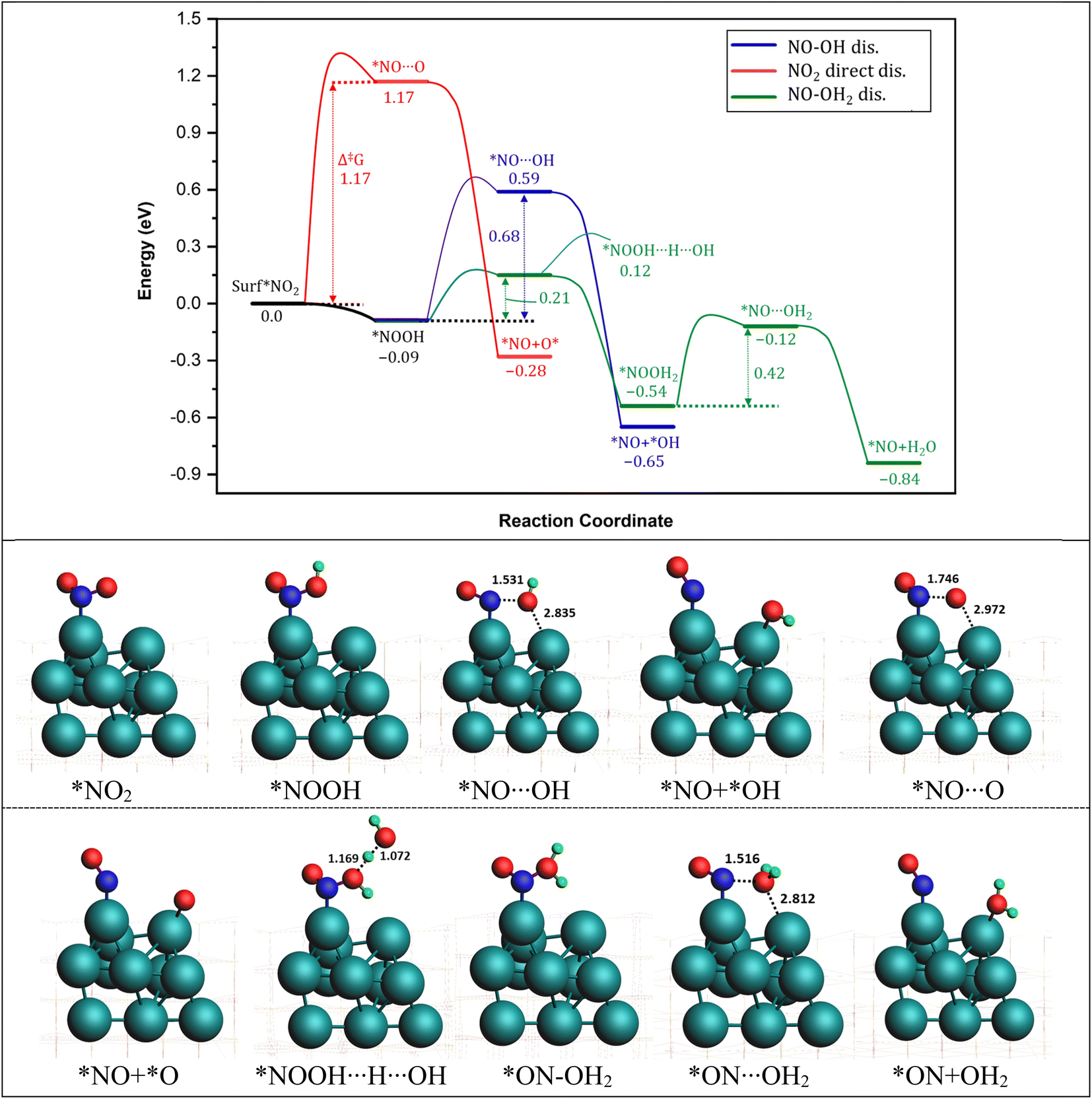 | ||
| Fig. 3 (Top) reaction energy profile for the conversion pathway of NO2 into NO. Green: NO–OH2 dissociation, blue: NO–OH dissociation, red: NO2 direct dissociation. The energy of the entering or leaving species was discounted from the total energy of the system to calculate the relative energy. The energy of the H was calculated as 1/2 of the energy of H2. (Bottom) optimized structures of various intermediates and transition states involved in the reaction pathways. ZrO2 layers are represented by wireframes for clarity. Energy values in eV. Bond distances in Å. | ||
A third possible pathway involves the capture of a second proton by the *NO–OH species that is formed in the second pathway, leading to the formation of a water molecule called *NO–OH2. In the subsequent step, the water molecule (H2O) undergoes detachment, with a reaction energy barrier of 0.42 eV, as it passes through the transition state *NO⋯H2O. This step is calculated to be exothermic, releasing an energy of 0.30 eV. Overall, this pathway exhibits a significant energy release of −0.84 eV, indicating that it is the most energetically favorable pathway for the conversion of *NO2 into *NO. Based on the energy profiles in Fig. 3, it is evident that the most favorable pathway for the reduction of *NO2 involves the sequential steps of *NO2 + *H2 → *ON–OH2 + * → *NO + *OH2. This pathway, which generates NO* as a key intermediate, is consistent with experimental findings reported by Chen et al.76
During the NO3− reduction process, the hydrogenation of *NO2 plays a crucial role in the overall reaction. This hydrogenation occurs through an interesting mechanism known as the hydrogen shuttling model (HSM), wherein hydrogen is transferred via a nearby water molecule, acting as a bridge between *NO2 and surface H*. This unique mechanism, allows for efficient hydrogenation reactions, however, in our case, the H is adsorbed on the surface quite strongly and hence can't be abstracted easily by the water molecule. Thus, the transition states could not be located for HSM mechanism. Therefore, we adopted the direct hydrogenation mechanism as the preferred approach for calculating the reaction energetics of hydrogenation steps in our work. This choice allows for a more accurate understanding of the hydrogenation reactions involved in the NO3− reduction on the ZrO2 surface, enhancing our insights into the overall reaction mechanism and aiding in the development of efficient catalysts for nitrate reduction.
3.3. NO* reduction to *NH3
In order to elucidate the mechanism and address the conflicting views regarding the conversion of NO* to NH3 in experiments, we conducted comprehensive density functional theory (DFT) calculations to explore all possible pathways of NO* reduction to NH3 with an aim to gain insights into the intricate pathways involved in this reduction process and determine the most favorable sequence of reactions. The reduction of NO* to NH3 primarily entails two fundamental types of reactions: deoxygenation, involving the removal of oxygen, and nitrogen hydrogenation. The key challenge lies in determining the most favorable sequence of deoxygenation and hydrogenation steps. In Fig. 4, we present a summarized diagram illustrating the calculated energetics of the potential elementary steps, and the corresponding structures are presented in Fig. 5. Each of the four possible pathways is discussed below.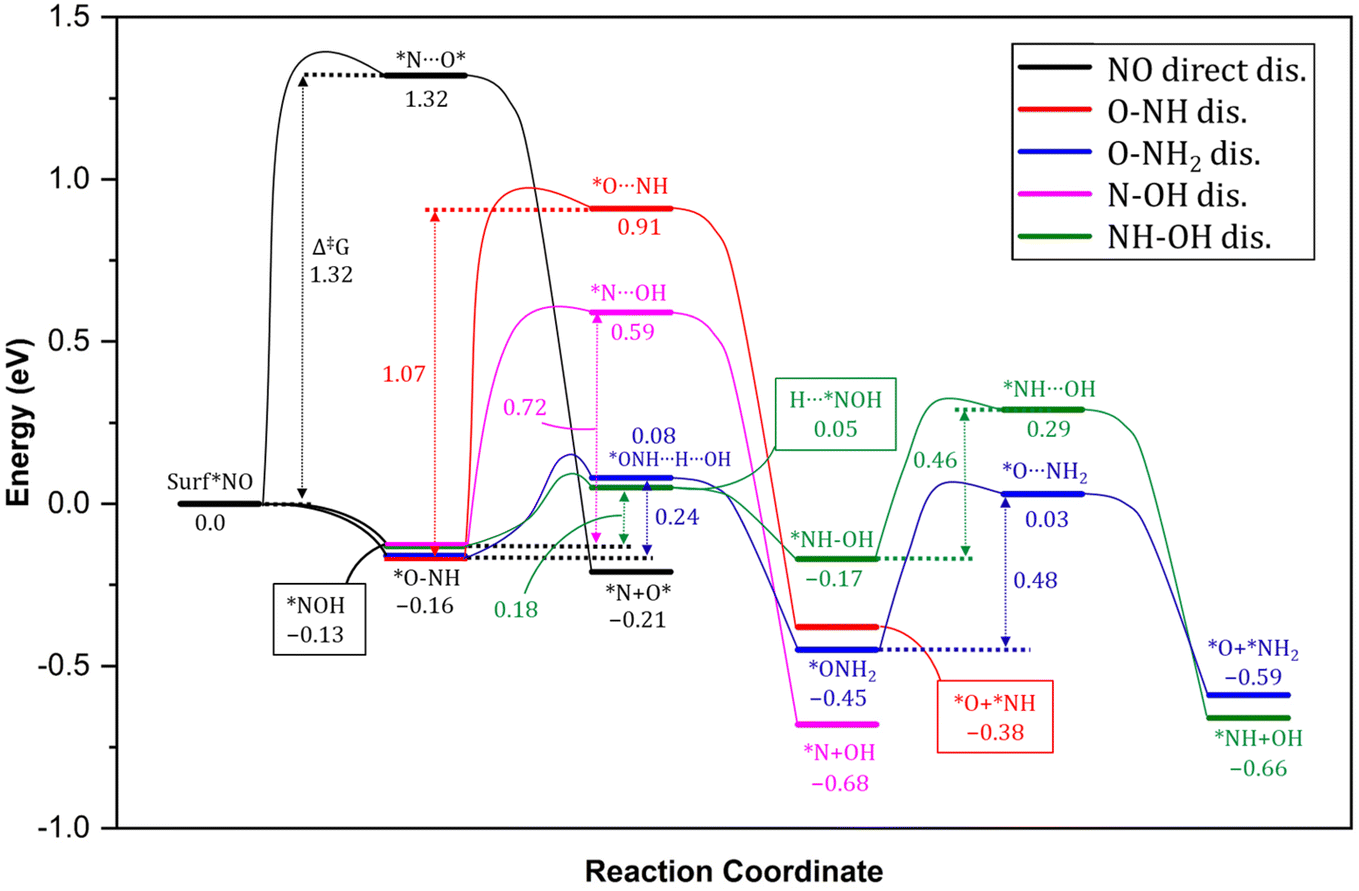 | ||
| Fig. 4 Reaction energy profile for the conversion pathway of *NO into *N + O*. Green: NO–OH2 dissociation, blue: NO–OH dissociation, red: NO2 direct dissociation. The energy of the entering or leaving species were discounted from the total energy of the system to calculate the relative energy. The energy of the H was calculated as 1/2 of the energy of H2. ZrO2 layers are represented by wireframes for clarity. Energy values in eV. Bond distances in Å. | ||
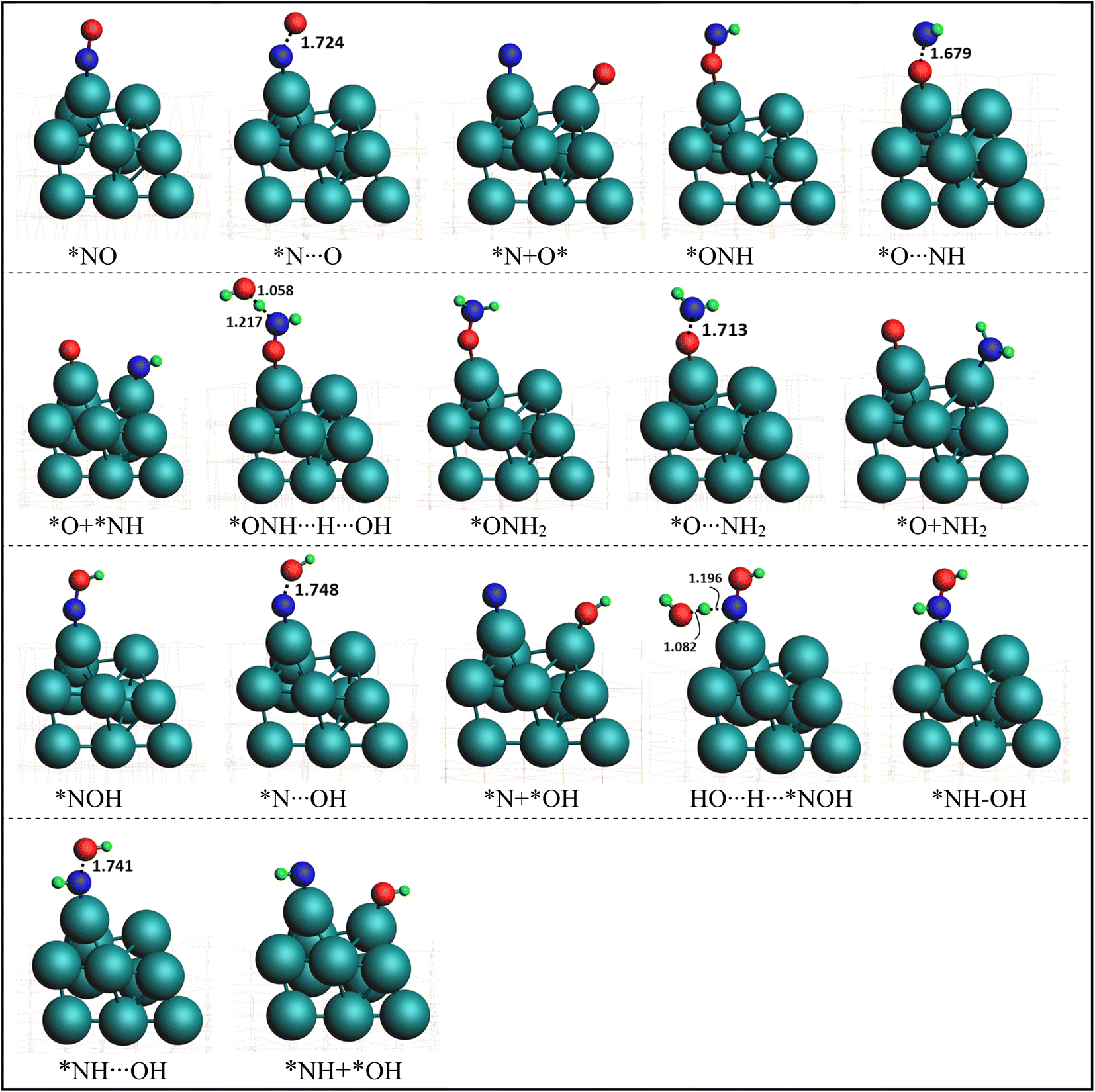 | ||
| Fig. 5 Optimized geometries of various intermediate, transition states, and products involved in NO splitting into N and O. Geometries presented in the same order (with matching color names) as of reactions presentation in Fig. 4. | ||
Thus, based on the above discussion and comparing the relative stability of intermediates, and products and the reaction energy barriers, the most favorable pathway for the cleavage of N–O bond and conversion on PtNP/ZrO2 is either *NO → *NOH → HNOH* → HN + OH* or NO* → *ONH → H2NO* → H2N* + O*. The two calculated pathways compete with each requiring activation energy of 0.46 and 0.48 eV, respectively with comparable reaction exothermicity (0.66 vs. 0.59 eV). The calculated pathways are consistent with the nitrate reduction mechanism reported on the Pd surface by Shin et al.77
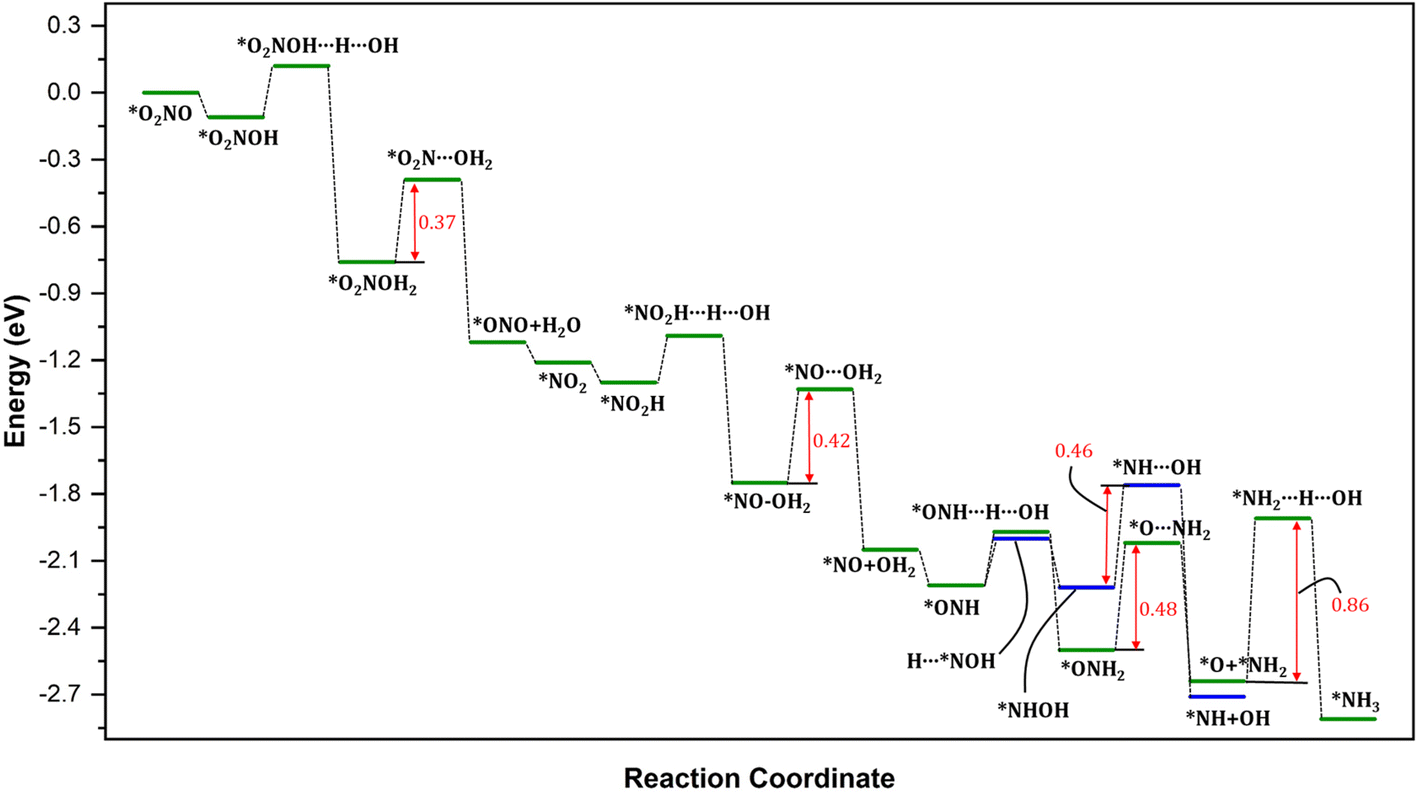 | ||
| Fig. 6 Reaction energy profile for the overall pathway for the conversion of NO3− into NH3 on the catalyst surface. Energy values in eV. The red vertical arrows represent the activation energy barriers for the rate-determining steps involved in the process. The energy of the entering or leaving species was discounted from the total energy of the system to calculate the relative energy. The energy of the H was calculated as 1/2 of the energy of H2. | ||
To provide a comprehensive understanding of the NO3− reduction mechanism to NH3 on the catalyst surface, we present the complete pathway in Fig. 6. It is evident that the overall process is highly exothermic, indicating that the reaction releases a significant amount of energy. Throughout the mechanism, we observe four notable energy barriers, as indicated by the red arrows.
The initial hydrogenation of N is energetically favorable without any barriers. The process of breaking the N–O in *Nx–OHx (x = 0, 1, 2) intermediates leading to the formation of both *NHx and *OHx (x = 0, 1, 2) species requires significantly lower barriers. The resulting NHx (x = 0, 1, 2) intermediate then undergoes hydrogenation to ultimately yield ammonia (*NH3). Thus, considering the small reaction barriers and the large thermodynamic driving force, the production of *NH is expected to be a facile process. However, the subsequent hydrogenation step, *NH2 + H → *NH3, however, requires energy barriers of 0.86 eV. The large energy barrier for the hydrogenation leading to the formation of *NH3 corresponds to the most energetically demanding steps in the entire mechanism and is, therefore, considered the rate-determining step (Fig. 6) in the formation of NH3 after the cleavage of N–O bond. These findings suggest that the hydrogenation of NH2* on the Pt surface is challenging, consistent with previous reports in the literature.78,79 Breaking the N–O bond in NO on metal surfaces is known to have a notably high activation barrier, around 1.7 eV.80–82 This means that under typical electro- and photochemical conditions, it is practically impossible to initiate this dissociation process. However, when NO is partially hydrogenated to form NOH*, the activation barrier to break the N–O bond is significantly lower, at approximately 0.9 eV. As further hydrogenation occurs, the activation barrier for dissociating HNOH* is even more reduced, reaching about 0.7 eV.59,80–83 In simpler terms, the process becomes easier as you add hydrogen atoms. Notably, our calculated results demonstrate significantly lower activation energy barriers compared to those reported in the literature. Precisely, our calculations indicate that the direct N–O dissociation requires approximately 0.76–1.32 eV, and as the degree of hydrogenation increases, the activation energy barriers decrease further to approximately 0.62–0.98 eV for NH–OH intermediates and 0.37–0.42 eV for NO–OH2 intermediates. These results are in line with those reported in literature, including the initial hydrogenation steps requiring very little energy.59,80–84
3.4. The rational design of the PtNP/ZrO2 catalyst with metal doping
The design of highly efficient PtNP/ZrO2 catalysts involves careful consideration of the metal-support interaction between the deposited Pt metal and the ZrO2 support. This interaction plays a critical role in determining the catalytic properties of the catalyst.85–87 Metal/ZrO2 systems are known for their strong metal-support interaction, resulting in charge redistribution at the metal-support interface. This charge redistribution significantly impacts the catalytic performance of the deposited Pt metal.88,89 To modulate this metal-support interaction and alter the charge density of the Pt particles, we propose the promising approach of doping the ZrO2 support with low-valence-state metals, that is, iron (Fe) and cobalt (Co).By substituting Zr4+ cations with Fe2+ and Co2+ ions in the ZrO2 lattice, we can effectively modify the charge distribution in the PtNP/ZrO2 system. This strategic modification aims to induce electron density depletion in both the ZrO2 support and the Pt cocatalyst, ultimately enhancing charge transfer and separation processes, which are crucial for improved catalytic activity. Experimental studies have demonstrated the feasibility and effectiveness of this doping approach in enhancing the catalytic performance of ZrO2-based catalysts.90–94 Introducing Fe or Co into the ZrO2 lattice promotes electron density depletion in the Pt cocatalyst, leading to more efficient charge transfer and enhanced catalytic activity. Bader charge analyses indicate that the introduction of Fe2+ and Co2+ dopant ions cause the Pt11 cluster to gain a charge of +0.54 |e| and +0.46 |e|, respectively, compared to the undoped Pt11/ZrO2 (See Table 1 in SI).
| Major reactions | Pt11 neutral | Fe#1–Pt/ZrO2 | Co#1–Pt/ZrO2 | |||
|---|---|---|---|---|---|---|
| Δ‡G | ΔGr | Δ‡G | ΔGr | Δ‡G | ΔGr | |
| O2N–O diss. | 0.37 | −0.36 | 0.39 | −0.37 | 0.36 | −0.38 |
| ON–O diss. | 0.42 | −0.30 | 0.41 | −0.33 | 0.44 | −0.31 |
| N–O diss. | 0.46 | −0.14 | 0.45 | −0.17 | 0.45 | −0.15 |
| HN–OH diss. | 0.48 | −0.49 | 0.49 | −0.47 | 0.47 | −0.51 |
| NH2 → NH3 | 0.87 | −0.17 | 0.69 | −0.24 | 0.71 | −0.21 |
To investigate the impact of Fe and Co doping on the PtNP/ZrO2 catalyst, we constructed model catalysts, denoted as Fe#1–Pt/ZrO2 and Co#1–Pt/ZrO2, where Fe or Co was introduced as dopants, presented in Fig. 7. The dopants were placed away from the Pt cluster to focus solely on charge density regulation without introducing other effects. The dopants were placed at two other positions as well denoted as Fe/Co#2–Pt/ZrO2 and Fe/Co#3–Pt/ZrO2 and so on, as presented in Fig. S3,† however, this change of doping position has a negligible effect on the energetics.
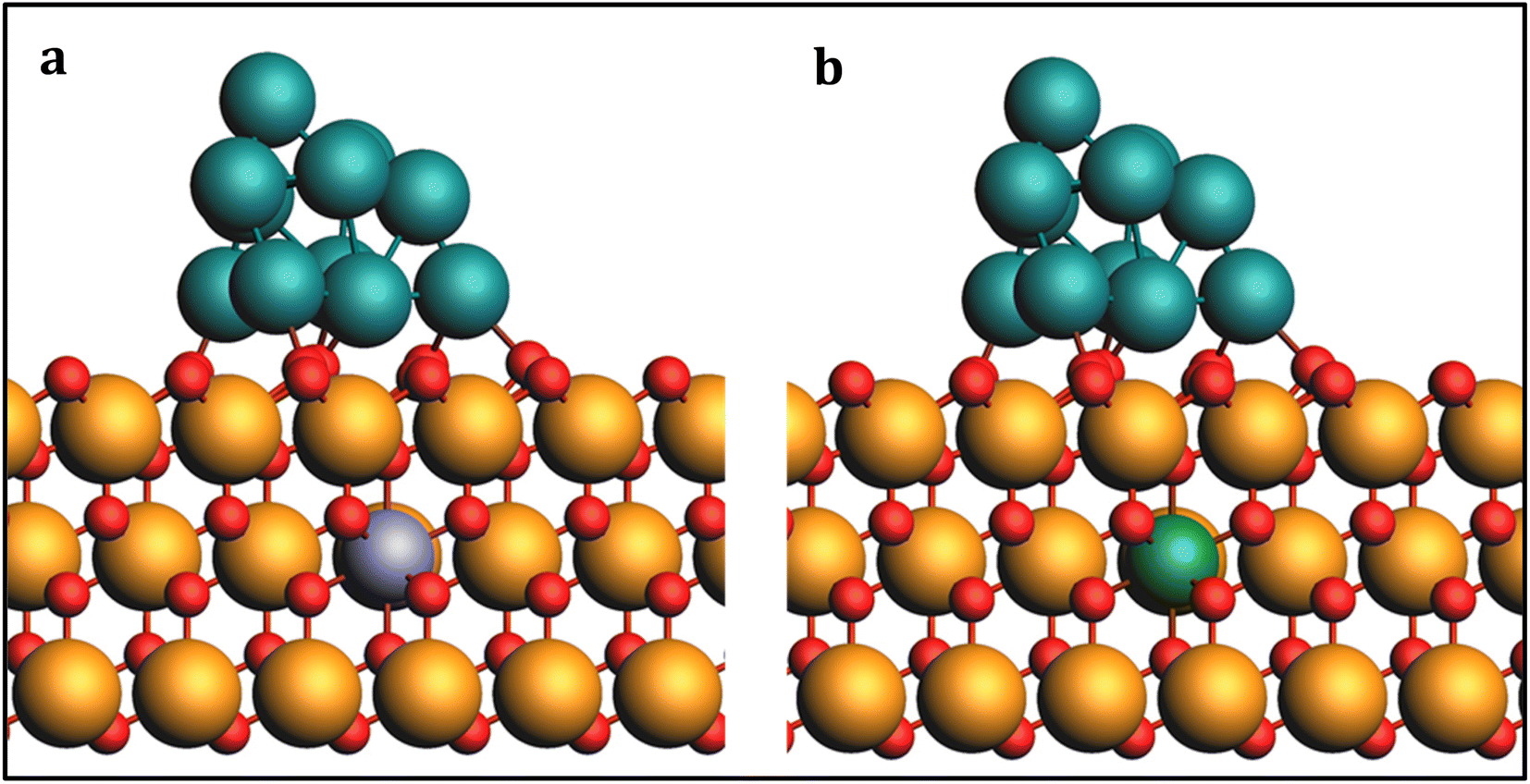 | ||
| Fig. 7 Optimized geometries of (a) Fe and (b) Co-doped Pt11/ZrO2 structures. Light blue: Fe; green: Co. | ||
To validate the feasibility of our approach, we conducted a thorough investigation of the energetics associated with key steps in the catalytic cycle, specifically focusing on the *NO3 dissociation, and *NH2 hydrogenation processes. We compared the results for the undoped Pt11 catalyst with those of the doped catalysts, Fe#1–Pt/ZrO2 and Co#1–Pt/ZrO2. Our analysis revealed that the *NO3 dissociation over Fe#1–Pt/ZrO2 and Co#1–Pt/ZrO2 catalysts exhibited similar energetics to the undoped Pt11 catalyst. This implies that the introduction of Fe or Co dopants had minimal impact on the *NO3 dissociation step. However, a significant improvement in the reaction barriers and energies was observed for the *NH2 hydrogenation steps, which are the rate-determining steps in the catalytic cycle, upon Fe and Co doping. The incorporation of Fe or Co dopants led to a promotion of approximately 0.20 eV in the energetics of the *NH2 hydrogenation steps, resulting in a more favorable and efficient catalytic process.
The observed improvements in the *NH2 hydrogenation step upon Fe and Co doping provide strong evidence for the effectiveness of our rational design. The introduction of Fe or Co dopants into the ZrO2 support induces electron density depletion in the Pt cocatalyst, facilitating the delivery of hydrogen to H-acceptor intermediates during the hydrogenation process, thereby promoting the overall catalytic activity. Importantly, similar reductions in hydrogen adsorption energy have been observed over other positively charged metals, such as Co, Ni, Cu, Pd, and Rh, corroborating the general principle for enhancing hydrogenation activity through charge density modulation.95 It is crucial to note that due to the promotional effects of Fe and Co doping, the rate-determining *NH2 hydrogenation step exhibits a relatively reduced energy barrier of 0.69 eV. This reduction underscores the effectiveness of doping in facilitating efficient hydrogenation during the nitrate-to-ammonia conversion over Pt-based catalysts. These findings emphasize the potential for enhanced catalytic performance and more sustainable ammonia production through the strategic use of doping techniques.
In the realm of catalysis, it is established that the coverage effect holds the potential to modify the bonding characteristics through interactions between adsorbed molecules. This phenomenon can notably influence the overall catalytic activity, particularly when dealing with materials that possess a substantial coverage of intermediate species.96 In our study, we have also investigated the potential impact of the coverage effect on nitrate reduction, particularly focusing on the key rate-determining intermediate *NH2. To assess the influence of adjacent *NH2 on the surface, we calculated the reaction energetics of crucial processes involving dissociation and hydrogenation. By comparing the energetics obtained in the absence and presence of coadsorbed *NH2, to explore the possible consequences of the coverage effect. The results, as shown in Table S2,† revealed several important findings. Firstly, for both *NO3 and *NO2 dissociation, the optimal pathway remains unchanged, occurring via the intermediates *NO2–H2O and *NO–OH rather than through direct dissociation. Secondly, although *NH2 hydrogenation is mildly inhibited by the presence of neighboring *NH2, the critical rate-determining *NH2 hydrogenation step exhibits a notable effect, with a reduced barrier height of 0.11 eV and an increase in reaction energy by −0.13 eV (Table S2†). These findings suggest that the reaction mechanism may not be significantly altered when considering the coverage effect, and moreover, improved activity can be anticipated due to the enhanced energetics of the rate-determining step at high coverage. Regarding the formation of byproducts, our results indicate that the key intermediates *NO2 and *NO are unlikely to be released due to their strong adsorption energies of −1.21 and −2.05 eV (Fig. 6), respectively, which is consistent with experimental observations.19
In summary, our study employed systematic density functional theory calculations to investigate the mechanism of nitrate reduction to ammonia over a Pt11/ZrO2(111) catalyst. The following major conclusions were drawn:
(I) We identified the most favorable pathway for H*-assisted NO3 reduction on PtNP/ZrO2 as *NO3 → *NO2 → *NO → *ONH → *ONH2/HNOH → *NH2/NH → *NH2 → *NH3. Among the various reaction dissociation pathways, the cleavage of N–O bond and *NH2 hydrogenation were found to have the highest barriers of 0.45 and 0.69 eV upon metal doping, respectively, indicating that N–O cleavage and *NH2 hydrogenation are the rate-determining steps of the overall pathway.
(II) We observed distinct trends in the reaction energetics of dissociation and hydrogenation steps significantly influenced by metal-support interaction, driving charge redistribution at the interface. In light of this, we proposed a novel approach to modulate this metal-support interaction and to deplete the electron density of Pt by introducing low-valence-state metals, namely iron (Fe) and cobalt (Co), into the ZrO2 support.
(III) We validated the effectiveness of the design strategy by demonstrating a significant promotion of approximately 0.20 eV in both the reaction barriers and energies of the rate-determining *NH2 hydrogenation steps for Fe–Pt/ZrO2 or Co–Pt/ZrO2 compared to undoped PtNP/ZrO2. This was attributed to the depletion of electron density within the Pt cocatalyst resulting in enhanced delivery of hydrogen to H-acceptor intermediates during the hydrogenation process, contributing to improved catalytic activity.
This study provides valuable insights into the catalytic activity of platinum-based ZrO2 catalysts and their potential for reducing atmospheric pollutants, particularly NO3, to valuable ammonia. The proposed doping approach holds the potential for the rational design of high-performance Pt-based catalysts for the reduction reactions on ZrO2 support. While our study provides valuable insights into the catalytic mechanism of nitrate reduction over Pt nanoparticle-based ZrO2, we acknowledge that the intricacies of charge density transfer to/from Na+ ions and its nuanced effects on the interaction between Pt clusters and reaction intermediates represent aspects that may benefit from a more comprehensive investigation in future studies. These considerations are pivotal for refining our understanding of the complex interplay between catalyst composition, charge density variations, and catalytic activity. Nonetheless, our study not only contributes to the understanding of catalytic activity of platinum-based ZrO2 catalysts but also offers a novel strategy for catalyst design to address atmospheric pollutant reduction. The presented results, insights, and proposed doping approach hold implications for advancing sustainable environmental protection and fostering cleaner and healthier environments.
Author contributions
Ayyaz Mahmood: conceptualization, writing – original draft, simulation, data collection, compilation, and discussion of results. Fouzia Perveen: manuscript revisions, funding acquisition. Tehmina Akram: writing and revising the manuscript. Shenggui Chen: formal analysis, project administration, funding acquisition. Ahmad Irfan: manuscript revisions, funding acquisition. Huafu Chen: formal analysis, project administration, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research is financially supported by the Key scientific research platforms and projects of Guangdong universities (2022ZDZX3064), the research project of Guangzhou Panyu Polytechnic (2023KJ04), the research platform of Guangzhou Panyu Polytechnic (2022KYPT03), the Guangzhou higher education teaching quality and teaching reform project (2022JSJXCXTD005 and 2022CXCYJH002). A. Irfan extends his appreciation to the Deanship of Scientific Research at King Khalid University for funding through large research groups program under grant number R.G.P.2/267/44.References
- J. Ângelo, L. Andrade, L. M. Madeira and A. Mendes, J. Environ. Manage., 2013, 129, 522–539 CrossRef PubMed.
- R. Li, X. Mei, L. Chen, L. Wang, Z. Wang and Y. Jing, Remote Sens., 2020, 12, 1041 CrossRef.
- S. Zhao, B. Hu, W. Gao, L. Li, W. Huang, L. Wang, Y. Yang, J. Liu, J. Li, D. Ji, R. Zhang, Y. Zhang and Y. Wang, Atmos. Res., 2020, 241, 104977 CrossRef CAS.
- N. Serpone, Catalysts, 2018, 8, 553 CrossRef.
- J. Lasek, Y.-H. Yu and J. C. S. Wu, J. Photochem. Photobiol., C, 2013, 14, 29–52 CrossRef CAS.
- P. Warneck, Chemistry of the Natural Atmosphere, Academic Press, 1999, 2nd edn, vol. p. 71 Search PubMed.
- J. A. Last, W. M. Sun and H. Witschi, Environ. Health Perspect., 1994, 102, 179–184 CAS.
- N. Gruber and J. N. Galloway, Nature, 2008, 451, 293–296 CrossRef CAS PubMed.
- K. P. Cantor, Cancer, Causes Control, 1997, 8, 292–308 CrossRef CAS PubMed.
- Guidelines for Drinking-Water Quality, Third Edition, incorporating first and second addenda - World | ReliefWeb, https://reliefweb.int/report/world/guidelines-drinking-water-quality-third-edition-incorporating-first-and-second-addenda, accessed July 8, 2023 Search PubMed.
- A. Menció, J. Mas-Pla, N. Otero, O. Regàs, M. Boy-Roura, R. Puig, J. Bach, C. Domènech, M. Zamorano, D. Brusi and A. Folch, Sci. Total Environ., 2016, 539, 241–251 CrossRef PubMed.
- Y.-Y. Yang and G. S. Toor, Water Res., 2017, 112, 176–184 CrossRef CAS PubMed.
- R. Raja, G. Sankar and J. M. Thomas, J. Am. Chem. Soc., 2001, 123, 8153–8154 CrossRef CAS PubMed.
- H. Liu, X. Cao, L.-X. Ding and H. Wang, Adv. Funct. Mater., 2022, 32, 2111161 CrossRef CAS.
- V. Smil, Nature, 1999, 400, 415 CrossRef CAS.
- J. M. McEnaney, A. R. Singh, J. A. Schwalbe, J. Kibsgaard, J. C. Lin, M. Cargnello, T. F. Jaramillo and J. K. Nørskov, Energy Environ. Sci., 2017, 10, 1621–1630 RSC.
- G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425–3437 RSC.
- Y. Shiraishi and T. Hirai, J. Photochem. Photobiol., C, 2008, 9, 157–170 CrossRef CAS.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, ACS Catal., 2017, 7, 3713–3720 CrossRef CAS.
- H. Li, J. Shang, J. Shi, K. Zhao and L. Zhang, Nanoscale, 2016, 8, 1986–1993 RSC.
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., Int. Ed., 2014, 53, 9802–9805 CrossRef CAS PubMed.
- A. Takai and P. V. Kamat, ACS Nano, 2011, 5, 7369–7376 CrossRef CAS PubMed.
- N. Chandrasekharan and P. V. Kamat, J. Phys. Chem. B, 2000, 104, 10851–10857 CrossRef CAS.
- R. Kato, M. Furukawa, I. Tateishi, H. Katsumata and S. Kaneco, ChemEngineering, 2019, 3, 49 CrossRef CAS.
- M. Yamauchi, R. Abe, T. Tsukuda, K. Kato and M. Takata, J. Am. Chem. Soc., 2011, 133, 1150–1152 CrossRef CAS PubMed.
- G. Prieto, J. Zečević, H. Friedrich, K. P. de Jong and P. E. de Jongh, Nat. Mater., 2013, 12, 34–39 CrossRef CAS.
- S. Kattel, W. Yu, X. Yang, B. Yan, Y. Huang, W. Wan, P. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2016, 55, 7968–7973 CrossRef CAS PubMed.
- K. Larmier, W.-C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Copéret, Angew. Chem., Int. Ed., 2017, 56, 2318–2323 CrossRef CAS PubMed.
- K. W. Ting, T. Toyao, S. M. A. H. Siddiki and K. Shimizu, ACS Catal., 2019, 9, 3685–3693 CrossRef CAS.
- B. C. Sempuga and Y. Yao, J. CO2 Util., 2017, 20, 34–42 CrossRef CAS.
- J. Wambach, A. Baiker and A. Wokaun, Phys. Chem. Chem. Phys., 1999, 1, 5071–5080 RSC.
- M. M.-J. Li and S. C. E. Tsang, Catal. Sci. Technol., 2018, 8, 3450–3464 RSC.
- K. Jalama, Catal. Rev., 2017, 59, 95–164 CrossRef CAS.
- M. D'Arino, F. Pinna and G. Strukul, Appl. Catal., B, 2004, 53, 161–168 CrossRef.
- Y. Zhang, J. Fei, Y. Yu and X. Zheng, Energy Convers. Manage., 2006, 47, 3360–3367 CrossRef CAS.
- V. I. Bogdan, A. E. Koklin, S. A. Nikolaev and L. M. Kustov, Top. Catal., 2016, 59, 1104–1109 CrossRef CAS.
- T. Phongamwong, U. Chantaprasertporn, T. Witoon, T. Numpilai, Y. Poo-arporn, W. Limphirat, W. Donphai, P. Dittanet, M. Chareonpanich and J. Limtrakul, Chem. Eng. J., 2017, 316, 692–703 CrossRef CAS.
- J. Lin, C. Ma, Q. Wang, Y. Xu, G. Ma, J. Wang, H. Wang, C. Dong, C. Zhang and M. Ding, Appl. Catal., B, 2019, 243, 262–272 CrossRef CAS.
- M. Romero-Sáez, A. B. Dongil, N. Benito, R. Espinoza-González, N. Escalona and F. Gracia, Appl. Catal., B, 2018, 237, 817–825 CrossRef.
- C. Chen, C. Ruan, Y. Zhan, X. Lin, Q. Zheng and K. Wei, Int. J. Hydrogen Energy, 2014, 39, 317–324 CrossRef CAS.
- M. M. Kauppinen, M. M. Melander, A. S. Bazhenov and K. Honkala, ACS Catal., 2018, 8, 11633–11647 CrossRef CAS.
- X. Yao, L. Chen, J. Cao, Y. Chen, M. Tian, F. Yang, J. Sun, C. Tang and L. Dong, Chem. Eng. J., 2019, 369, 46–56 CrossRef CAS.
- S. Liu, H. Wang, Y. Wei, R. Zhang and S. Royer, ACS Appl. Mater. Interfaces, 2019, 11, 22240–22254 CrossRef CAS.
- Y. Okamoto and H. Gotoh, Catal. Today, 1997, 36, 71–79 CrossRef CAS.
- H. Koga, A. Hayashi, Y. Ato, K. Tada, S. Hosokawa, T. Tanaka and M. Okumura, Catal. Today, 2019, 332, 236–244 CrossRef CAS.
- M.-fei Luo, Yi-jun Zhong, Bo Zhu, X.-xin Yuan and X.-ming Zheng, Appl. Surf. Sci., 1997, 115, 185–189 CrossRef CAS.
- C. Sun, J. Zhu, Y. Lv, L. Qi, B. Liu, F. Gao, K. Sun, L. Dong and Y. Chen, Appl. Catal., B, 2011, 103, 206–220 CrossRef CAS.
- H. Koga, K. Tada, A. Hayashi, Y. Ato and M. Okumura, Catal. Lett., 2017, 147, 1827–1833 CrossRef CAS.
- J. Xing, Y. H. Li, H. B. Jiang, Y. Wang and H. G. Yang, Int. J. Hydrogen Energy, 2014, 39, 1237–1242 CrossRef CAS.
- D. Wang, Z.-P. Liu and W.-M. Yang, ACS Catal., 2018, 8, 7270–7278 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- Q.-H. Zhang, W.-D. Han, Y.-J. Hong and J.-G. Yu, Catal. Today, 2009, 148, 335–340 CrossRef CAS.
- C. Dong, C. Lian, S. Hu, Z. Deng, J. Gong, M. Li, H. Liu, M. Xing and J. Zhang, Nat. Commun., 2018, 9, 1252 CrossRef PubMed.
- Y. Ji and Y. Luo, ACS Catal., 2016, 6, 2018–2025 CrossRef CAS.
- G. A. Cerrón-Calle, A. S. Fajardo, C. M. Sánchez-Sánchez and S. Garcia-Segura, Appl. Catal., B, 2022, 302, 120844 CrossRef.
- G. E. Dima, A. C. A. de Vooys and M. T. M. Koper, J. Electroanal. Chem., 2003, 554–555, 15–23 CrossRef CAS.
- M. C. Figueiredo, J. Souza-Garcia, V. Climent and J. M. Feliu, Electrochem. Commun., 2009, 11, 1760–1763 CrossRef CAS.
- A. Clayborne, H.-J. Chun, R. B. Rankin and J. Greeley, Angew. Chem., 2015, 127, 8373–8376 CrossRef.
- V. Rosca, G. L. Beltramo and M. T. M. Koper, Langmuir, 2005, 21, 1448–1456 CrossRef CAS PubMed.
- A. Cuesta and M. Escudero, Phys. Chem. Chem. Phys., 2008, 10, 3628–3634 RSC.
- A. Mahmood, T. Akram, C. Shenggui and C. Huafu, Int. J. Hydrogen Energy, 2023 Search PubMed , under review.
- M. Franchini, P. H. T. Philipsen, E. van Lenthe and L. Visscher, J. Chem. Theory Comput., 2014, 10, 1994–2004 CrossRef CAS PubMed.
- P. H. T. Philipsen, G. te Velde, E. J. Baerends, J. A. Berger, P. L. de Boeij, M. Franchini, J. A. Groeneveld, E. S. Kadantsev, R. Klooster, F. Kootstra, P. Romaniello, M. Raupach, D. G. Skachkov, J. G. Snijders, C. J. O. Verzijl, J. A. Celis Gil, J. M. Thijssen, G. Wiesenekker and T. Ziegler, BAND202 SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2023 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- A. Mahmood, T. Akram, M. Kiani, X. Tian and Y. Sun, New J. Chem., 2023, 46, 16593–16602 RSC.
- A Mahmood, E. S. Teixeira and R. L. Longo, J. Org. Chem., 2015, 80(16), 8198–8205 CrossRef CAS PubMed.
- A. Mahmood and R. L. Longo, Phys Chem Chem Phys, 2016, 18(25), 17062–17070 RSC.
- S. Hammond, J. Am. Chem. Soc., 1955, 77, 334 CrossRef.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F.-Y. Chen, J. Y. (Timothy) Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- H. Shin, S. Jung, S. Bae, W. Lee and H. Kim, Environ. Sci. Technol., 2014, 48, 12768–12774 CrossRef CAS PubMed.
- Y. Wang, H. Li, W. Zhou, X. Zhang, B. Zhang and Y. Yu, Angew. Chem., Int. Ed., 2022, 61, e202202604 CrossRef CAS PubMed.
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D.-H. Nam, C.-S. Tan, Y. Ding, J. Wu, Y. Lum, C.-T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS PubMed.
- Z.-H. Zeng, J. L. F. Da Silva, H.-Q. Deng and W.-X. Li, Phys. Rev. B, 2009, 79, 205413 CrossRef.
- M. Gajdoš, J. Hafner and A. Eichler, J. Phys.: Condens. Matter, 2005, 18, 41 CrossRef.
- C. A. Farberow, J. A. Dumesic and M. Mavrikakis, ACS Catal., 2014, 4, 3307–3319 CrossRef CAS.
- C. A. Casey-Stevens, H. Ásmundsson, E. Skúlason and A. L. Garden, Appl. Surf. Sci., 2021, 552, 149063 CrossRef CAS.
- T. Hu, C. Wang, M. Wang, C. M. Li and C. Guo, ACS Catal., 2021, 11, 14417–14427 CrossRef CAS.
- M. Ali, E. Pervaiz, U. Sikandar and Y. Khan, Int. J. Hydrogen Energy, 2021, 46, 18257–18283 CrossRef CAS.
- Z. Li, L. Zhai, T. Ma, J. Zhang and Z. Wang, Catalysts, 2022, 12, 1385 CrossRef.
- A. M. Ruppert, M. Niewiadomski, J. Grams and W. Kwapiński, Appl. Catal., B, 2014, 145, 85–90 CrossRef CAS.
- R. Wang, K. Wang, H. Wang, Q. Wang, J. Key, V. Linkov and S. Ji, Int. J. Hydrogen Energy, 2013, 38, 5783–5788 CrossRef CAS.
- A. Perechodjuk, Y. Zhang, V. A. Kondratenko, U. Rodemerck, D. Linke, S. Bartling, C. R. Kreyenschulte, G. Jiang and E. V. Kondratenko, Appl. Catal., A, 2020, 602, 117731 CrossRef CAS.
- Q. Cheng, W. Yang, Q. Chen, J. Zhu, D. Li, L. Fu and L. Zhou, Appl. Surf. Sci., 2020, 530, 147291 CrossRef CAS.
- P. Ghigna, G. Spinolo, U. Anselmi-Tamburini, F. Maglia, M. Dapiaggi, G. Spina and L. Cianchi, J. Am. Chem. Soc., 1999, 121, 301–307 CrossRef CAS.
- H. Song, J. A. Hong, H. Lee and K. Lim, Sci. Rep., 2019, 9, 10965 CrossRef PubMed.
- W. Ahmed and J. Iqbal, Ceram. Int., 2020, 46, 25833–25844 CrossRef CAS.
- F. G. Pinto, M. A. Paulin, A. G. Leyva and J. Sacanell, Mater. Res. Express, 2018, 5, 066103 CrossRef.
- D. Wang, Z.-P. Liu and W.-M. Yang, ACS Catal., 2017, 7, 2744–2752 CrossRef CAS.
- A. C. Lausche, A. J. Medford, T. S. Khan, Y. Xu, T. Bligaard, F. Abild-Pedersen, J. K. Nørskov and F. Studt, J. Catal., 2013, 307, 275–282 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures of optimized geometries of various intermediates and transition states involved in reactions on Pt11/ZrO2 catalyst, AIMD trajectory and Tables for energy barrier with coverage effect and Bader charge analyses. The supplementary material is available free of charge on the journal website as pdf file. See DOI: https://doi.org/10.1039/d3ra06449b |
| This journal is © The Royal Society of Chemistry 2023 |