DOI:
10.1039/D3RA06387A
(Paper)
RSC Adv., 2023,
13, 34239-34248
Design, synthesis and in silico evaluation of newer 1,4-dihydropyridine based amlodipine bio-isosteres as promising antihypertensive agents†
Received
19th September 2023
, Accepted 13th November 2023
First published on 22nd November 2023
Abstract
Hypertension remains a major global health concern, prompting ongoing research into innovative therapeutic approaches. This research encompasses the strategic design, synthesis, and computational assessment of a novel series of 1,4-dihydropyridine based scaffolds with the objective of developing promising antihypertensive agents as viable alternatives to the well-established dihydropyridine based drugs such as amlodipine, felodipine, nicardipine, etc. The crystal structure of the lead compound determined using X-ray crystallography offers crucial insights into its 3D-conformation and intermolecular interactions. In silico molecular docking experiments conducted against the calcium channel responsible for blood pressure regulation revealed superior docking scores for all the bioisosteres P1–P14 than the standard amlodipine, indicating their potential for enhanced therapeutic efficacy. Extensive ADMET profiling and structure–activity relationship (SAR) elucidated favourable pharmacokinetic properties and essential structural modifications influencing antihypertensive effectiveness. Specifically, P6–P10, P12 and P14 hybrids were found in accordance with Lipinski rules and exhibited druglikeliness attributes, involving high GI absorption and no BBB permeance. In particular, P7 was found to be crystalline in nature having the highest binding affinity with the concerned calcium channels with excellent ADMET profile. The findings highlight the significance of the presence of triazole tethered aryl/heteroaryl ring in the synthesized hybrids, providing a foundation for further preclinical and clinical translation as antihypertensive medications.
1. Introduction
Hypertension, characterized by elevated blood pressure levels, is an enduring and pervasive global health concern, impacting a substantial proportion of the world's population.1 It represents a key risk factor for cardiovascular diseases, strokes, and renal complications, contributing significantly to global morbidity and mortality rates.2,3 Consequently, the management of hypertension constitutes a paramount concern in contemporary medical treatment, necessitating the ongoing quest for innovative and effective antihypertensive drugs.
Among the arsenal of medications employed to manage hypertension, calcium channel blockers (CCBs) have proven to be effective and are widely prescribed by healthcare professionals.4 Amlodipine, a 1,4-dihydropyridine based calcium channel blocker, has established itself as a frontline antihypertensive agent by efficiently lowering the blood pressure levels by exerting vasodilatory effects on smooth muscle cells.5,6 Nevertheless, even though amlodipine demonstrates impressive antihypertensive capabilities, it is not devoid of certain limitations, including side effects and varied therapeutic responses among patients.7 Therefore, there is an unmet demand for innovative novel analogs of amlodipine that can surmount these challenges enhancing its overall pharmacological characteristics.
In the contemporary era, the concept of bioisosterism has gained prominence in the field of pharmaceutical industry as a strategic approach to drug design. Bioisosteres are the structurally different compounds that mimic the essential pharmacophoric characteristics of a reference compound to display similar biological actions.8 This concept has been extremely valuable in facilitating the development of safer and more effective pharmaceutical agents.9 Exploration of bioisosteres in the context of well-established antihypertensive drug like amlodipine offers an intriguing opportunity to fine-tune its structural and pharmacological characteristics.
1,4-Dihydropyridine based drugs such as amlodipine are the Ca2+ antagonists10 that are widely used in the treatment of cardiovascular disorders. These drugs allosterically interact with the receptor sites on CaV1.2 channels, the most pre-dominant voltage gated Ca2+ (CaV) channel type in cardiac and vascular smooth muscle myocytes. Specifically, 1,4-dihydropyridines are primarily used to treat hypertension and are considered to act as allosteric modulators that influence L-type voltage-dependent Ca2+ channels activation.11 Binding of 1,4-dihydropyridines to receptors in L-type voltage dependent Ca2+ channel inhibits the entry of Ca2+ ions through voltage-gated Ca2+ channels into both the cardiac and vascular smooth muscles.12 The target proteins CavAb complex and calmodulin were procured from the X-ray crystallographic data as reported by Tang et al.13 and Johnson C. N. et al.,14 respectively to successfully establish the in silico efficiency of our candidates as promising anti-hypertensive agents.
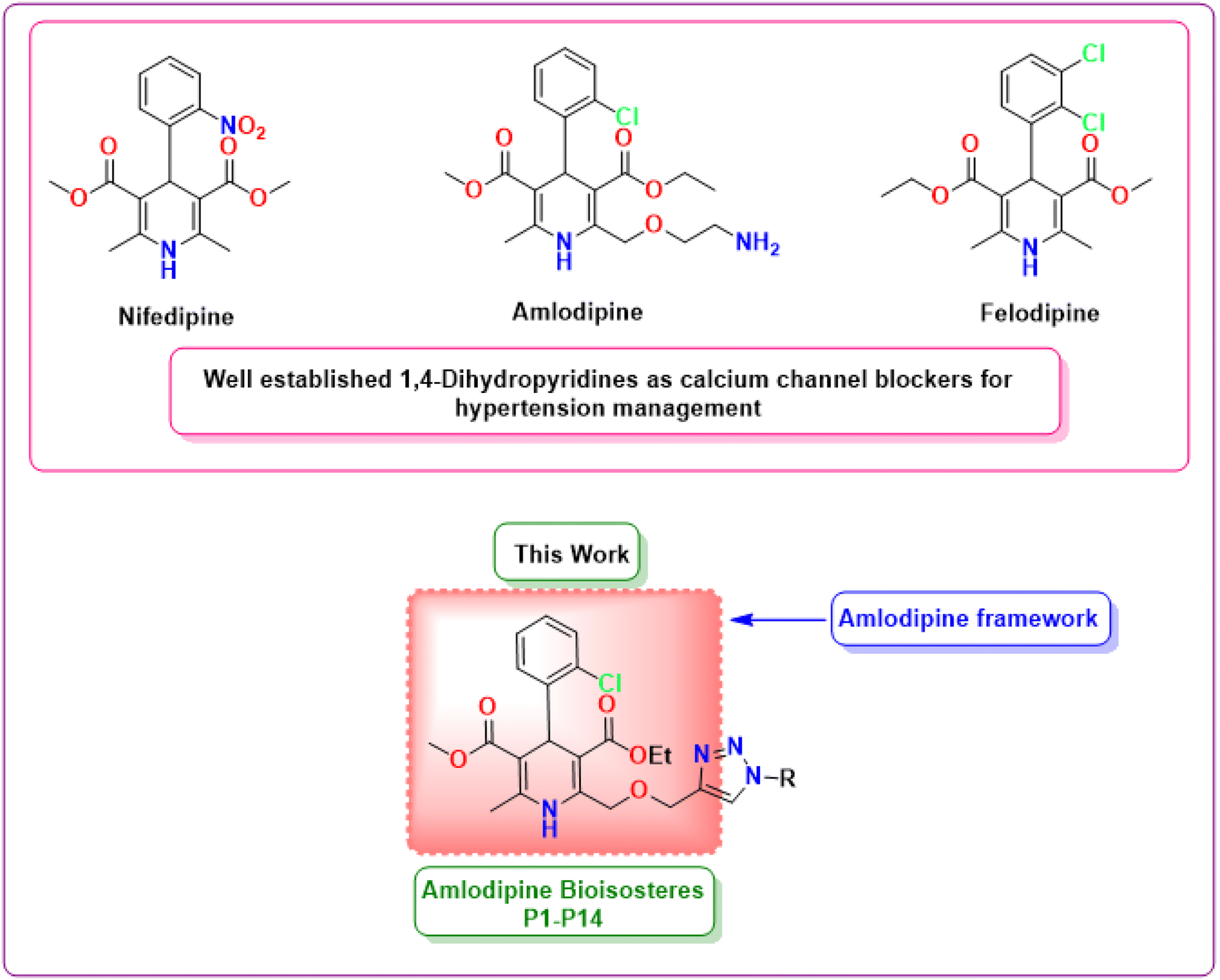
The present study undertakes a comprehensive exploration of newer 1,4-dihydropyridine based amlodipine bioisosteres incorporating molecular design, synthetic methodologies, and advanced computational analysis (Fig. 1). In particular, the in silico evaluation, the fundamental aspect of this study plays a pivotal role in analyzing the critical features of the synthesized hybrids, including their binding interactions with target proteins, pharmacokinetic characteristics and potential therapeutic efficacy.15 This approach not only accelerates the drug discovery process but also reduces the risks and resource expenditures involved with traditional trial-and-error methods. This study holds the potential to significantly advance the field of cardiovascular medicine by introducing a newer category of antihypertensive drugs that could redefine the standard therapy for hypertension, thereby advancing the frontiers of medicinal chemistry.
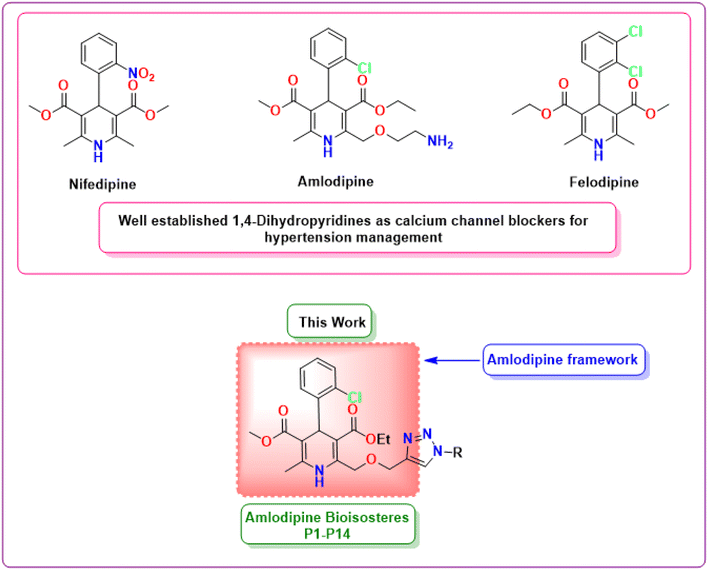 |
| | Fig. 1 Chemical structures of well-established 1,4-dihydropyridines as antihypertensive drugs and the framework of amlodipine bioisosteres synthesized in this work. | |
2. Methodology
2.1 Materials and methods
All the reagents and solvents (analytical grade) were procured from the commercial providers and employed directly for the synthesis without any further purifications. Thin layer chromatographic (TLC) plates precoated with silica gel were used to monitor the reaction optimizations. Melting points were determined using an uncorrected Buchi instrument (M-560). KBr disks were utilized to record infrared spectra on a SHIMADZU IR Affinity 1S spectrophotometer. The JEOL, ECX-400P Spectrometer USA was used to record 1H-NMR and 13C NMR spectra in deuterated DMSO-d6 at 400 MHz and 100 MHz, respectively with TMS (tetramethylsilane) as internal reference standard. The mass spectrometry measurements of all the compounds were acquired using a 6530 Accurate-Mass Q-TOF LC/MS spectrometer.
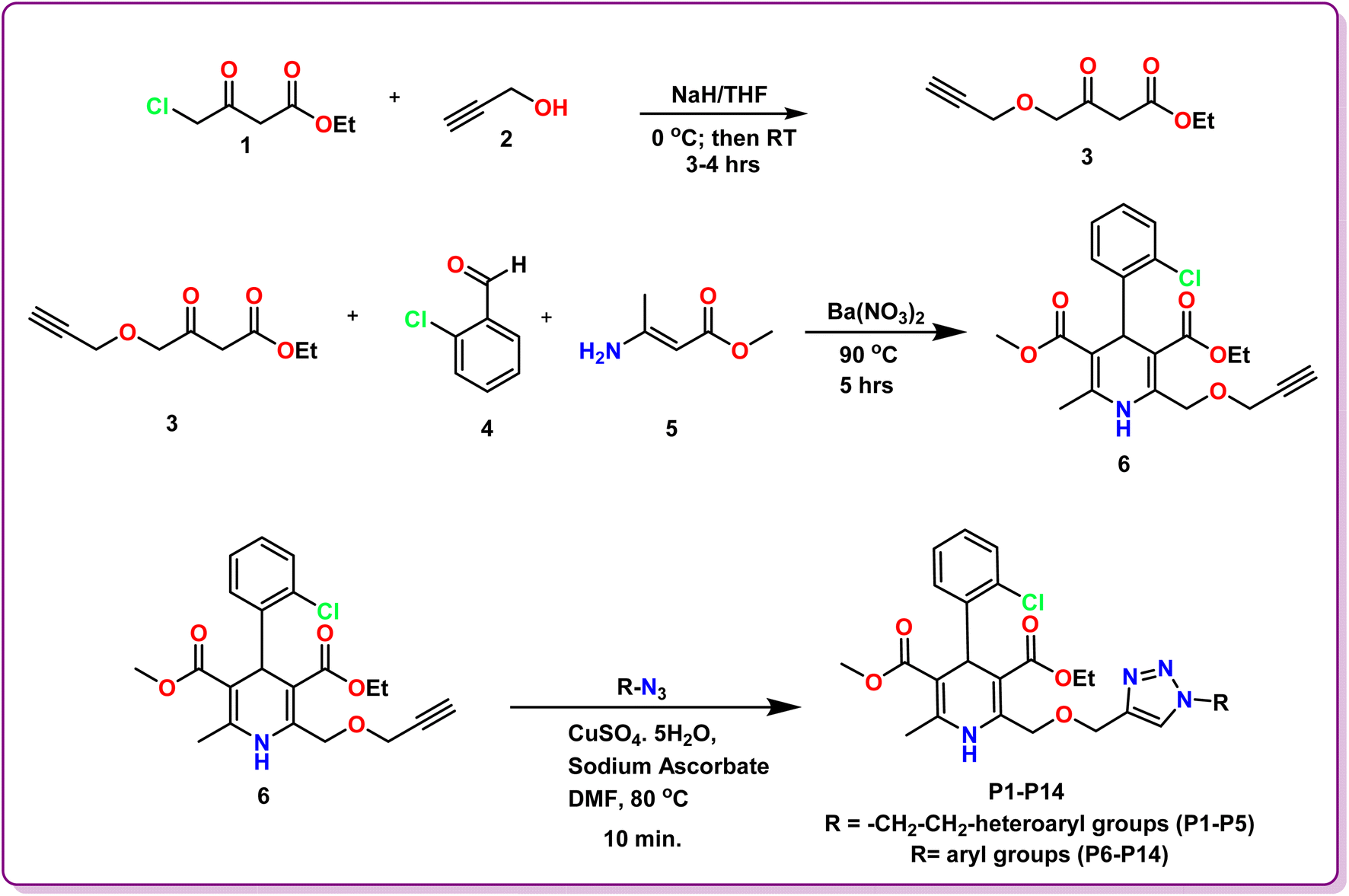
2.1.1 General procedure for the synthesis of ethyl 3-oxo-4-(prop-2-yn-1-yloxy)butanoate (3). Sodium hydride (2 mmol) was firstly added to THF at ice cold condition and was stirred vigorously for 15 min. After this, ethyl 4-chloroacetoacetate (1; 1 mmol) and propargyl alcohol (2; 1 mmol) were added to it dropwise. The reaction mixture was then stirred for around 3–4 h at room temperature and the completion of reaction was monitored via thin layer chromatography. After the completion of reaction, distilled water was added to the reaction mixture and was extracted using ethyl acetate. Anhydrous Na2SO4 was further added to remove any water content and the separated ethyl acetate layer was then evaporated under reduced pressure. The obtained product was purified via column chromatography to obtain pure dark yellow colored liquid 3 in 72% yield.
2.1.2 General procedure for the synthesis of 3-ethyl 5-methyl 4-(2-chlorophenyl)-6-methyl-2-((prop-2-yn-1-yloxy)methyl)-1,4-dihydropyridine-3,5-dicarboxylate (6). In a round bottom flask, a mixture of ethyl 3-oxo-4-(prop-2-yn-1-yloxy)butanoate (3; 1 mmol), 2-chlorobenzaldehyde (4; 1 mmol), methyl 3-aminocrotonate (5; 1 mmol), and barium nitrate (0.1 mmol) was heated at 90 °C for around 5 hours (Scheme 1). The reaction's progress was monitored by TLC at various time intervals. After the reaction was completed, the mixture was allowed to settle at room temperature before being extracted with ethyl acetate and any water content was removed using anhydrous Na2SO4. The solution was evaporated at low pressure, and the crude product was purified using column chromatography to procure 6 as a yellow solid in 65% yield.
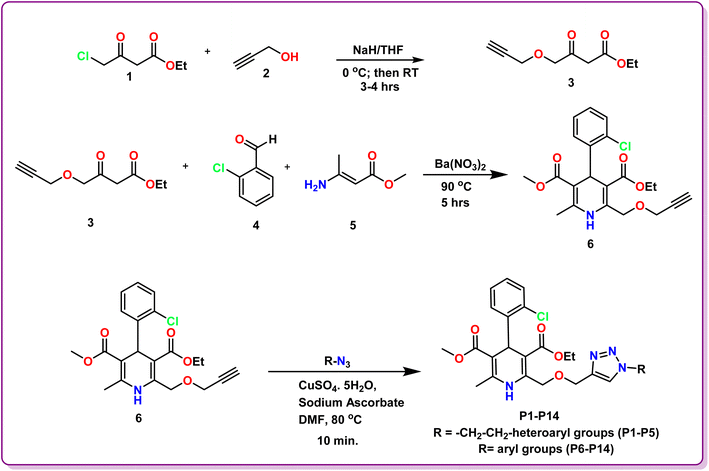 |
| | Scheme 1 Schematic route for the synthesis of 1,4-dihydropyridine based amlodipine bioisosteres P1–P14. | |
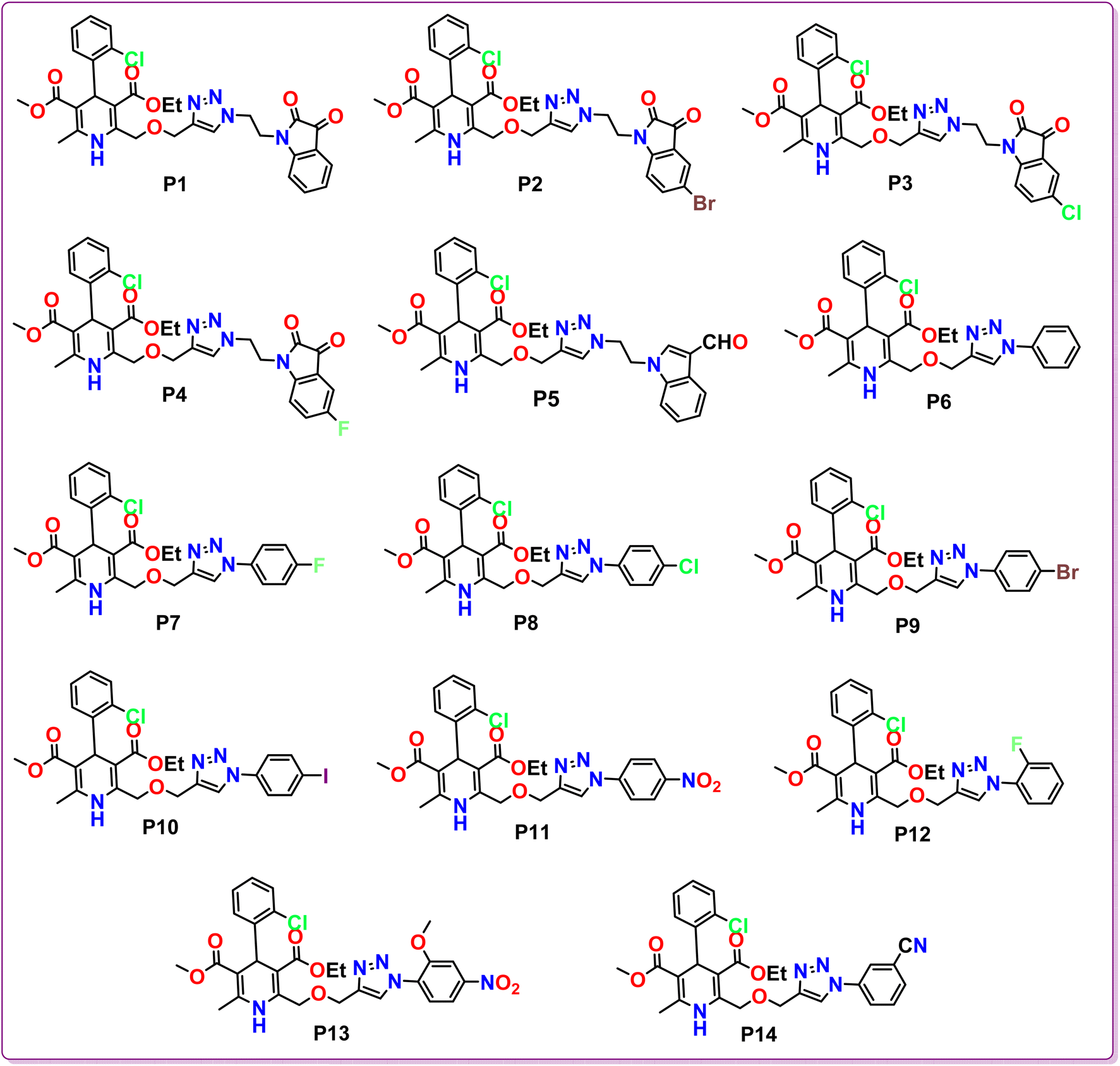
2.1.3 General procedure for the synthesis of 1,4-dihydropyridine based amlodipine bioisosteres (P1–P14). 3-Ethyl 5-methyl 4-(2-chlorophenyl)-6-methyl-2-((prop-2-yn-1-yloxy)methyl)-1,4-dihydropyridine-3,5-dicarboxylate (6; 1 mmol) and different aryl and heteroaryl azides (1 mmol) were dissolved in DMF (10 ml) in a round bottom flask. Following this, a solution of sodium ascorbate (0.4 mmol) and copper sulphate (0.2 mmol) in H2O was introduced to the reaction mixture and was refluxed for 10 min (Scheme 1). TLC was employed to track the progress of the reaction. Following the completion of the reaction, water was added to the reaction mixture and the product was extracted using ethyl acetate. Subsequently, anhydrous Na2SO4 was used to eliminate any water content in the organic layer and the solvent was evaporated under reduced pressure. Crude products obtained were then purified using column chromatography to obtain pure hybrids P1–P14 in good yields (Fig. 2).
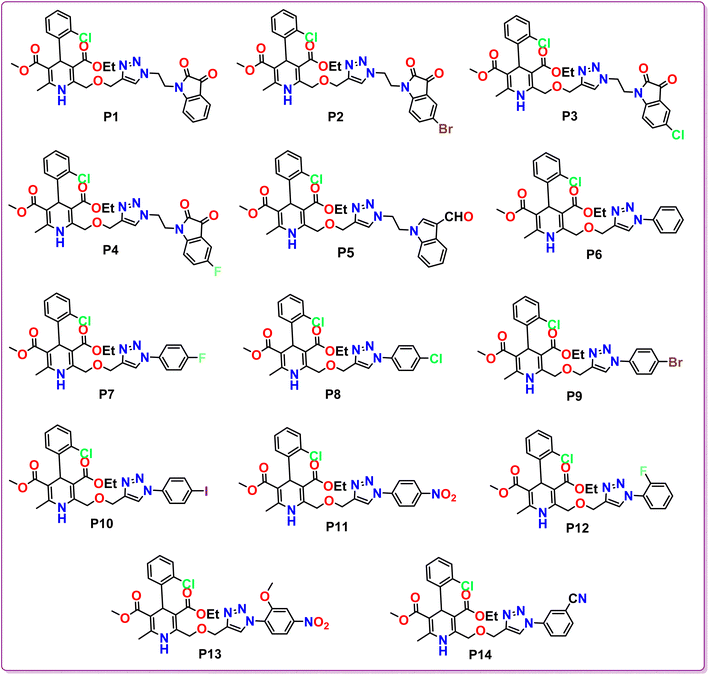 |
| | Fig. 2 Chemical structures of the synthesized 1,4-dihydropyridine based amlodipine bioisosteres P1–P14. | |
2.2 Single crystal X-ray structure
Fine crystals of ligand P7 were grown by slow evaporation of saturated mother solvent methanol & chloroform which was layered by hexane and diffused by ether at room temperature for SC-XRD studies. Good quality rectangular shaped yellowish crystals were taken and exposed to X-rays on a Bruker diffractometer employing a graphite monochromatized Mo/Kα radiation (λ = 0.71073 Å) at temperature 107 K. The crystal data was reduced using CrysAlis pro software available with the diffractometer. The structure was solved by direct methods using SHELXL-2016/4 and refined by the full-matrix least-squares method on Olex2.refine 1.5.16 All calculations were carried out using the OLEX2 package of the crystallographic programs.17 For the molecular graphics, the program Mercury (2022.3.0) was used.18
2.3 Molecular docking studies
Molecular docking examinations were executed using AutoDock Vina to evaluate the binding potential and molecular interactions of the synthesized hybrids P1–P14 with the proteins (PDB ID: 5KMD and 6M7H).19 The 3-D structures of 5KMD and 6M7H were procured from the protein data bank. The protein preparation process comprised of multiple steps, including the removal of co-factors, water molecules, and co-crystallized ligands from the protein structure. Subsequently, polar hydrogen atoms and kollmann charges were introduced to the protein prepared using AutoDock Tools 1.5.6. Following this, docking was executed separately for each ligand with different proteins by setting the grid size according to the active binding sites of the receptors. Best docked pose with lowest binding affinity value was recorded for each compound. The protein–ligand interactions were further visualized using BIOVIA Discovery Studio 2021.
2.4 In silico ADMET analysis
Effective drug discovery requires superior pharmacokinetics and minimal toxicity, making them more drug-like. In silico techniques for screening potential candidates for Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) play a crucial role in mitigating the risk of late-stage mortality and refining the selection of most promising candidates. Consequently, in this study, all the synthesized hybrids P1–P14 underwent screening for their druglikeliness, oral bioavailability and pharmacokinetic characteristics using the SwissADME20,21 and preADMET tools.
3. Result and discussion
3.1 Chemistry
The methodology for the development of novel 1,4-dihydropyridine based amlodipine bioisosteres P1–P14 was established in three steps. The precursor ethyl 3-oxo-4-(prop-2-yn-1-yloxy)butanoate 3 was prepared according to the reported method in literature using ethyl 4-chloroacetoacetate and propargyl bromide in the presence of NaH in dry THF solvent. Further, ethyl 3-oxo-4-(prop-2-yn-1-yloxy)butanoate 3, 2-chlorobenzaldehyde 4 and methyl 3-aminocrotonate 5 were employed to obtain novel 3-ethyl 5-methyl 4-(2-chlorophenyl)-6-methyl-2-((prop-2-yn-1-yloxy)methyl)-1,4-dihydropyridine-3,5-dicarboxylate 6 by modified Hantzsch synthesis as reported procedure in literature. These synthesized 1,4-dihydropyridine scaffolds were then tethered with various aryl and heteroaryl azides via Cu-catalyzed 1,3-dipolar cycloaddition click reaction under different reaction conditions to procure the desired amlodipine bioisosteres P1–P14 in excellent yields. The structures of the synthesized scaffolds were substantiated by various spectroscopic techniques, including Nuclear Magnetic Resonance (1H NMR and 13C NMR), Fourier-transform infrared (FT-IR), High Resolution Mass Spectrometry (HRMS), and Single Crystal XRD.
Evaluation of 1H NMR of P1 revealed the characteristic peaks at δ 8.57 and δ 8.23 as singlets for one proton each signifying the existence of –NH of 1,4-dihydropyrindine ring and –CH of the triazole ring, respectively. A prominent singlet peak of 1 proton at δ 5.29 ppm corresponds to the –CH of the chiral centre of 1,4-dihydropyridine at 4-position. The 13C NMR spectrum of this compound depicted the presence of characteristic peak for both the carbonyl groups of the isatin ring at δ 183.37 ppm and δ 158.59 ppm. Further, the presence of two ester groups at 1,4-dihydropyridine ring is confirmed by the prominent characteristic peaks at δ 167.60 and δ 166.72 ppm. Additionally, the HRMS data of P1 demonstrated the calculated mass ion (M + H)+ as 620.1834, satisfying the observed mass of 620.1922.
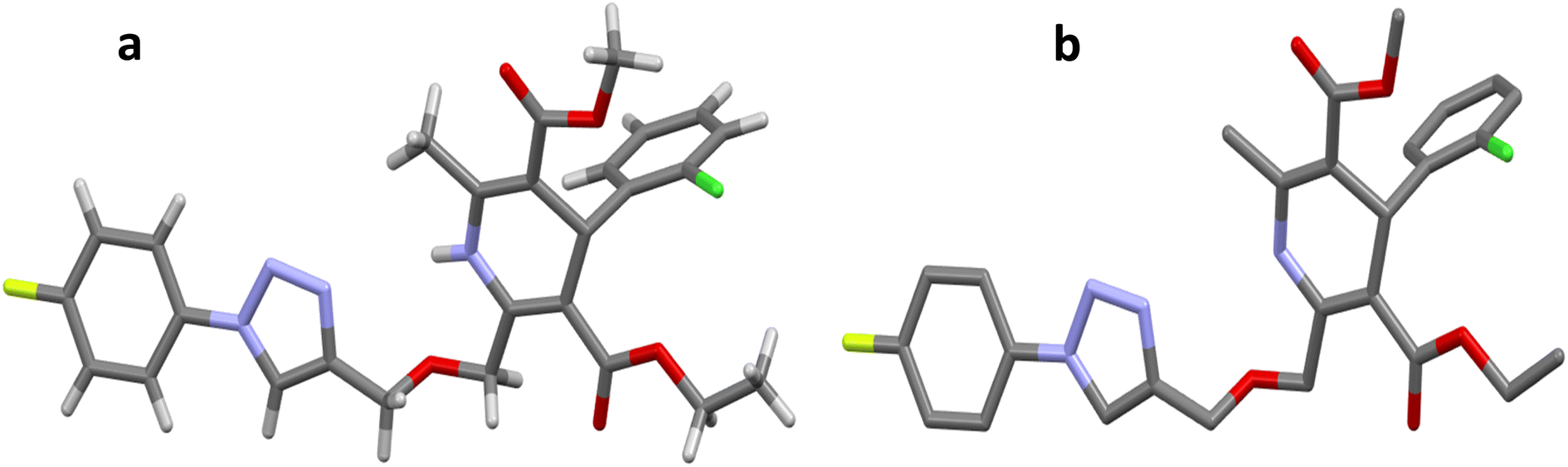
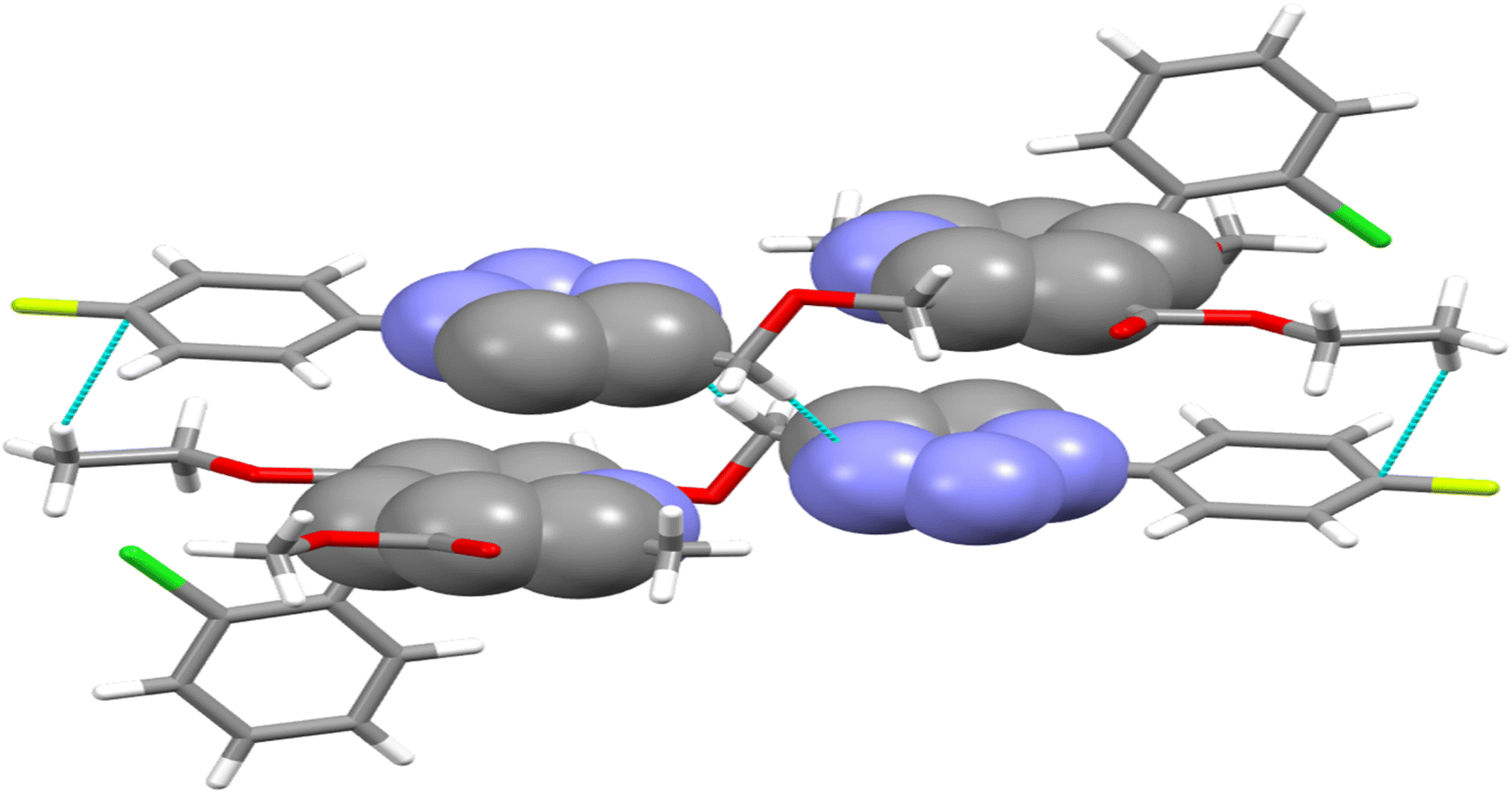
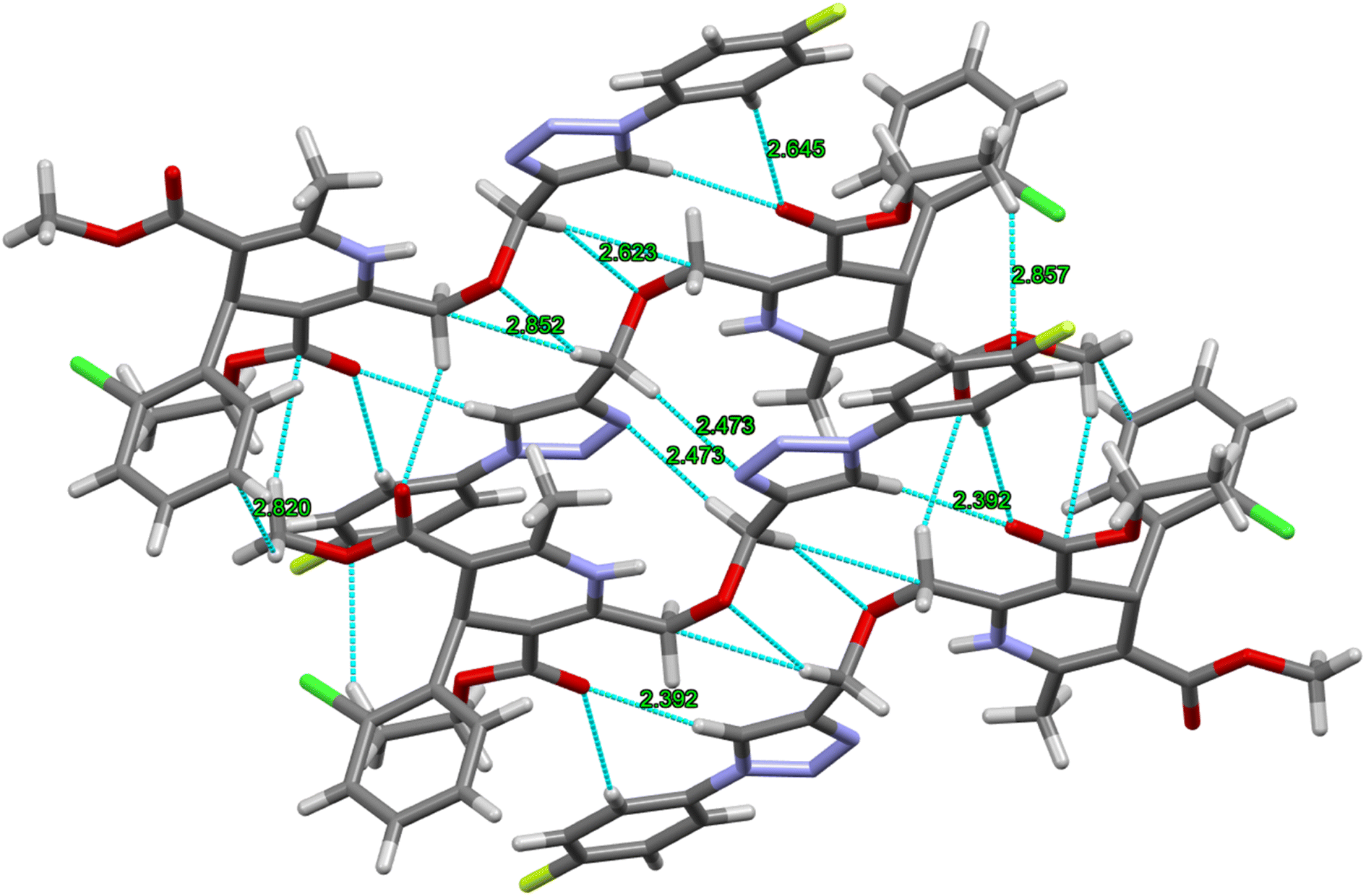
The crystal structure of ligand P7 was determined by Single Crystal X-ray diffraction method which revealed that the molecular structure of compound consists of a fluorophenyl substituted triazole ring. It further revealed the presence of vertical appended chlorobenzene on the top of the dihydropyridine ring at 124.9° angle. Ethyl ester and methyl ester groups are attached at 3rd and 5th position of 1,4-dihydropyridine ring at angles 117.8° and 118.91°, respectively, confirming the streamlined shape of the compound which is clearly shown in Fig. 3. The chair form geometry when the two adjacent molecules encapsulate the hexane solvent via –CH–Pi interactions in the compound from chlorobenzene to the fluorobenzene ring via triazole ring is confirmed by the bond angles analysis as depicted in Fig. 4. The bond distances of adjacent atoms from chlorine to fluorine lie in 1.355(2)–1.749(2) Å whereas inter and intra molecular hydrogen bond lengths lie close to (2.471–2.853 Å). These weaker coordinating interactions form the core of crystal engineering and are therefore essential. Interestingly, crystal structure of P7 shown in the present study display a variety of non-covalent interactions such as a network of inter-molecular H-bonding and π–π stacking arrangements in the crystal structures (Fig. 5).
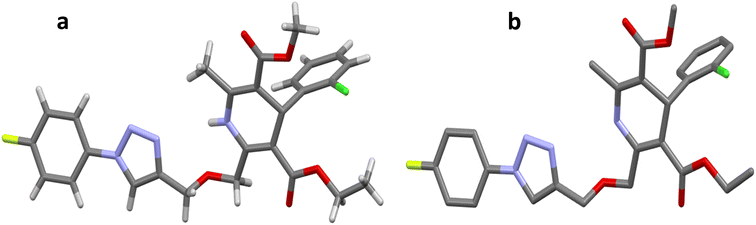 |
| | Fig. 3 Molecular structures (a and b) of compound P7. H atoms are omitted in (b) for clarity. C = gray, N = blue, F = yellow, Cl = green, O = red, H = gray. | |
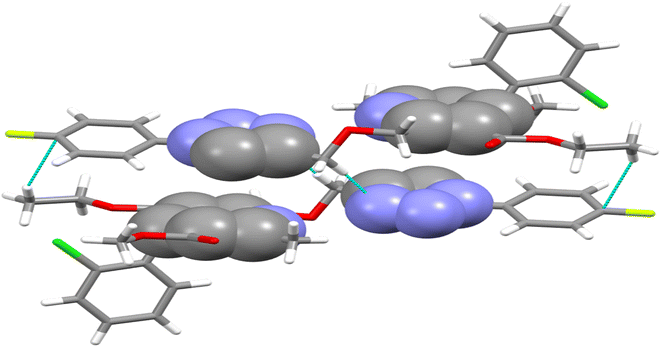 |
| | Fig. 4 Double decker π-stacked arrangement of triazole and dihydropyridine ring and hydrogen bonding between the adjacent P7 molecules. | |
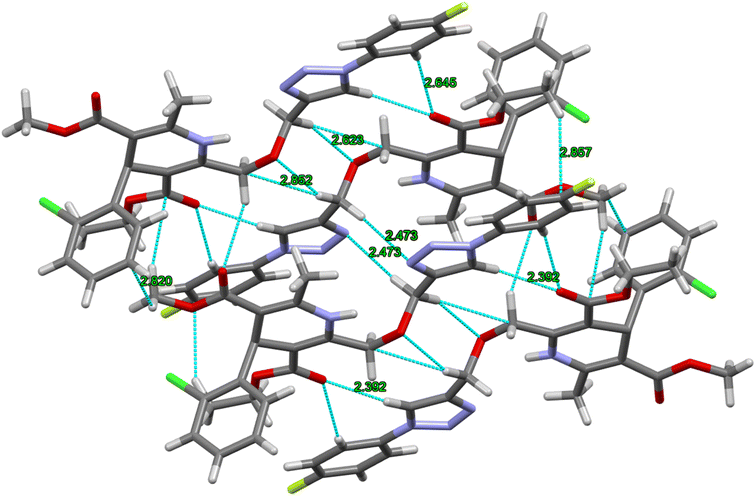 |
| | Fig. 5 A network of P7 via intra- and intermolecular H-bonding between the adjacent molecules. | |
3.2 Molecular docking evaluation
Exploration of interactions between proteins and ligands is critical in the process of structure-based drug design. Molecular docking studies were conducted to elucidate the key pharmacophores responsible for calcium channel blocker activity or anti-hypertensive activity. This investigation examined the interactions between a set of molecule ligands (amlodipine bioisosteres) and their respective protein targets 5KMD and 6M7H. The molecular docking analysis of all the synthesized hybrids P1–P14 revealed excellent docking scores and remarkable interactions with the essential amino acid residues located within the receptor's binding pocket of 5KMD and 6M7H, as compared to the standard amlodipine (Tables S3 and S4 in ESI† file). A stronger binding affinity with the receptor is indicated by the more negative binding affinity value in the docking results.
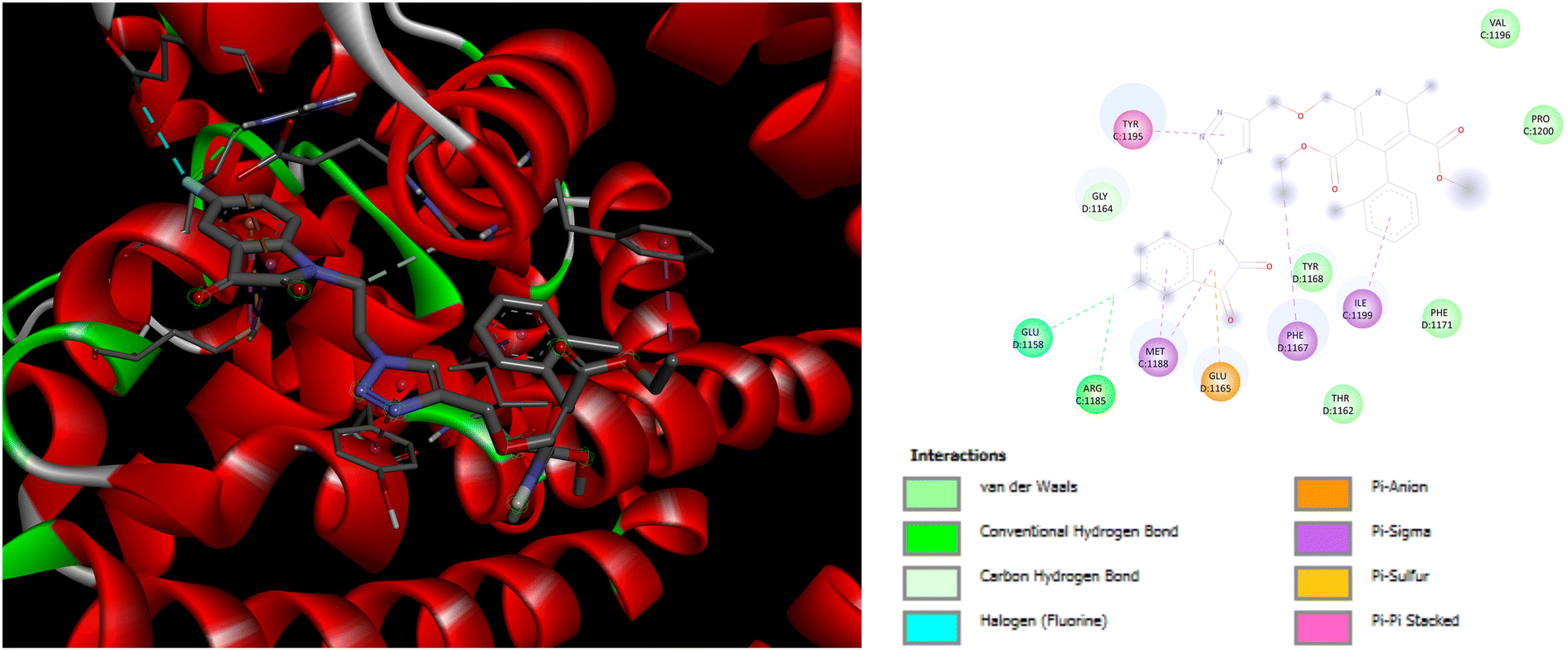
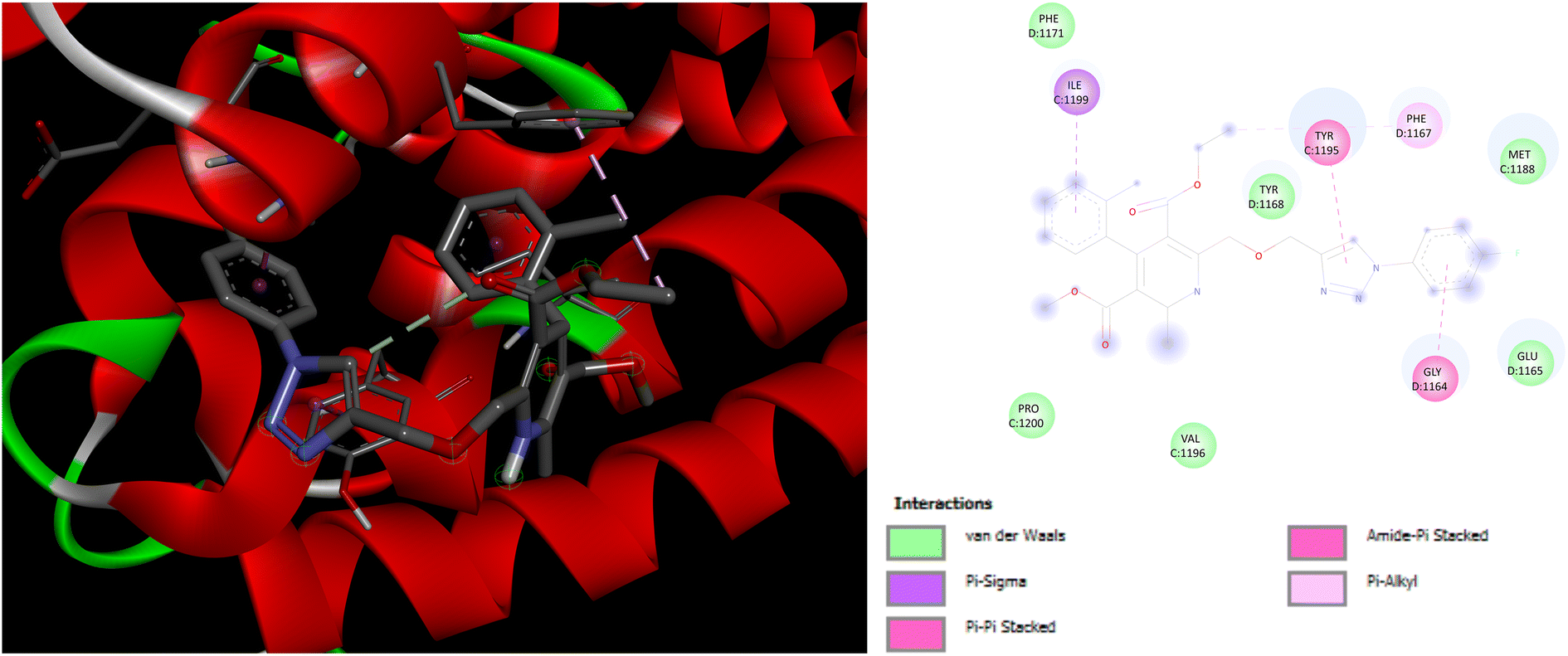
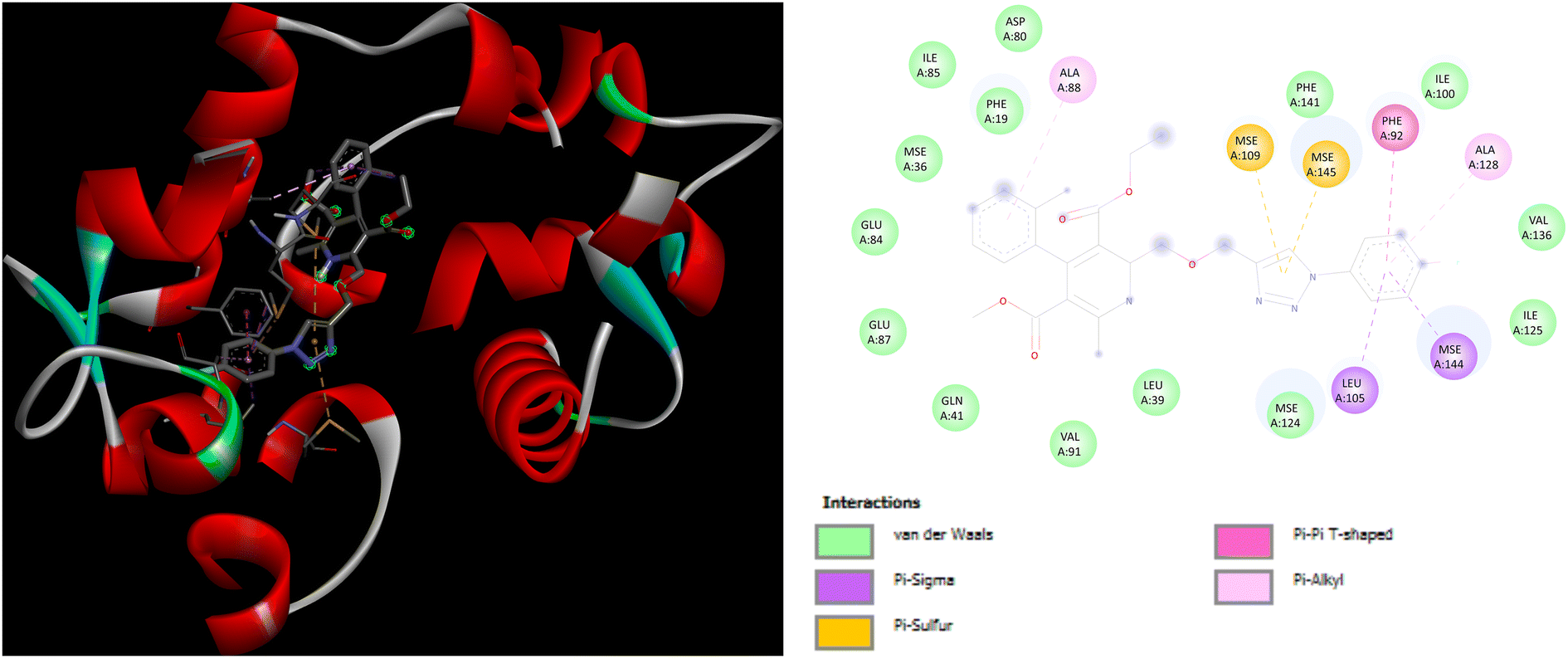
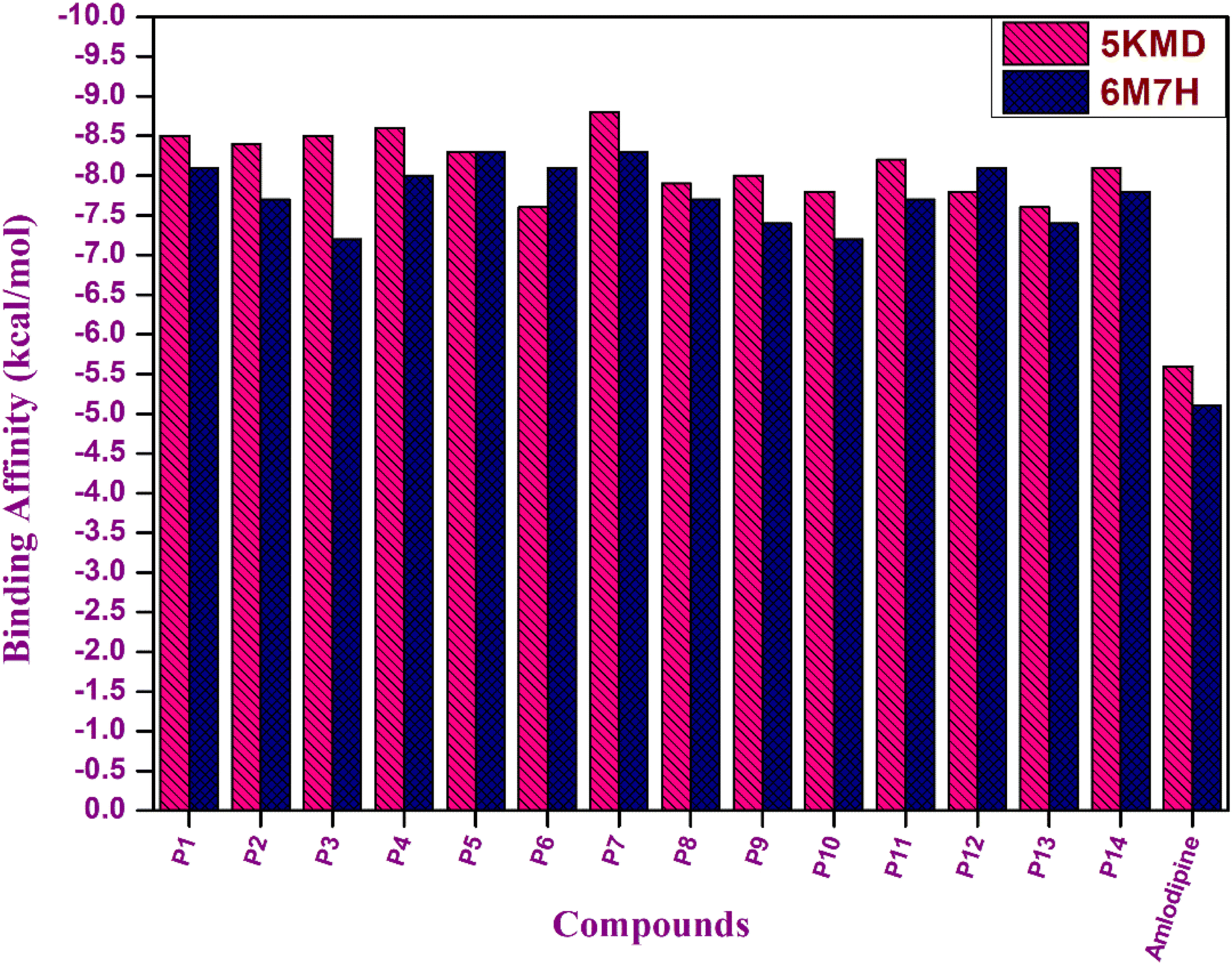
Evaluation of the molecular docking results with unique binding sites on calcium voltage gated (CaV) channels 5KMD demonstrated a remarkable binding affinity of −8.6 kcal mol−1 for P4 containing fluorinated isatin group in case of heteroaryl rings that is considerably higher than that of the standard amlodipine (−5.6 kcal mol−1). However, the bioisoster P7 containing the triazole tethered fluorobenzene ring displayed the highest binding affinity of −8.8 kcal mol−1 for 5KMD. As displayed in Fig. 6, P4 exhibits impressive interactions with a number of amino acids. Significant Pi–Sigma interactions with MET1188, PHE1167, and ILE1199, are exhibited by the isatin ring, –CH3 of –COOEt group and chlorobenzene ring, respectively. Triazole ring is involved in Pi–Pi stacking with TYR1195. Further, –F atom of isatin ring is involved in hydrogen bond interactions with GLU1158 and ARG1185. Pi–Anion interaction is also displayed by the indole ring of isatin with GLU1165. In case of P7, Pi–Sigma interactions are displayed by the chlorobenzene ring with the amino acid residue ILE1199. Amide–Pi Stacked interactions are exhibited by the triazole ring and fluorobenzene ring with TYR1195 and GLY1164. Furthermore, –CH3 of –COOEt group is involved in the Pi–Alkyl interactions with the amino acid residue PHE1167. It is also involved in a number of van der Waals interactions with several amino acid residues (Fig. 7).
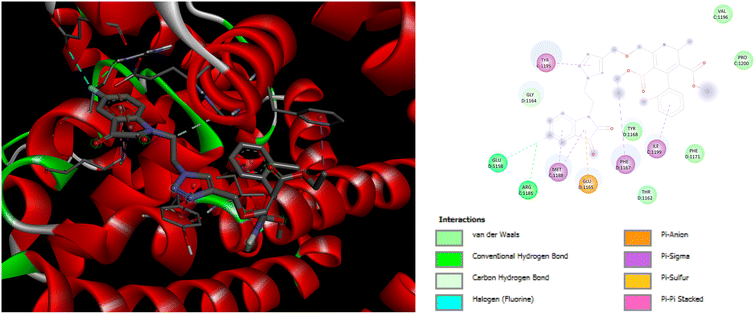 |
| | Fig. 6 3D and 2D representation of binding interactions of P4 with 5KMD. | |
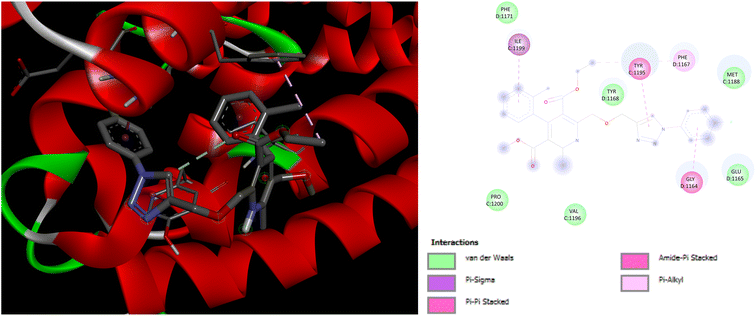 |
| | Fig. 7 3D and 2D representation of binding interactions of P7 with 5KMD. | |
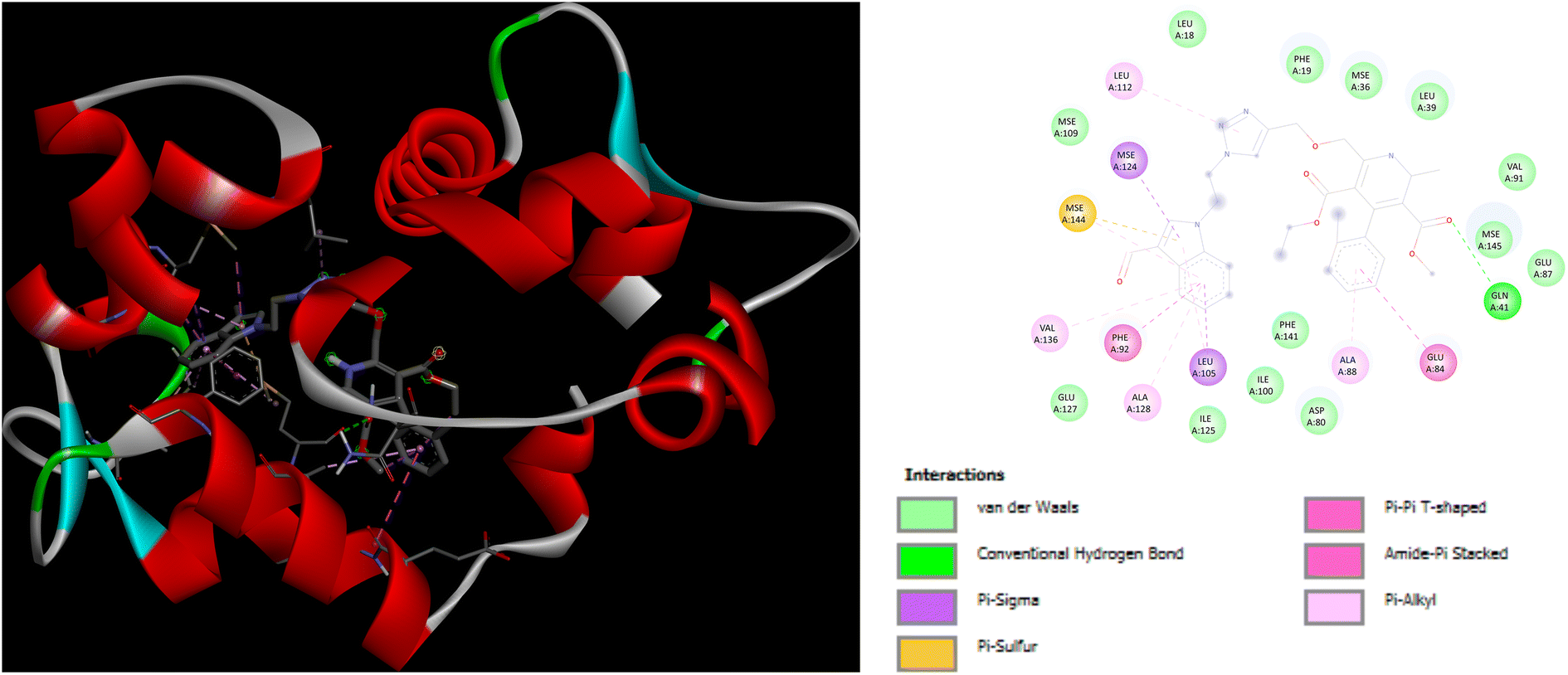
In addition to this, docking studies of these compounds were assessed on the calmodulin (CaM)-dependent protein kinase II (PDB ID: 6M7H). P5 and P7 demonstrated the highest binding affinity of −8.3 kcal mol−1 as compared to amlodipine (−5.1 kcal mol−1). In case of docking interactions of P5 with 6M7H, the indole ring is involved in a number of interactions, namely; Pi–Sigma, Pi–sulfur, Pi–Pi T shaped, and Pi-alkyl interactions with amino acid residues MSE124, LEU105, MSE144, PHE92, VAL136, and ALA128, respectively as depicted in Fig. 8. Further, Pi–Pi T shaped and Pi-alkyl interactions with GLU84 and ALA88, respectively are also exhibited by the chlorobenzene ring of P5 along with the Pi–alkyl interactions of LEU112 with the triazole ring. Fig. 9 demonstrates the binding interactions of P7 with 6M7H, showing the Pi–alkyl interactions of fluorobenzene and chlorobenzene ring with ALA128, and ALA88, respectively. Fluorobenzene ring is also associated with the Pi–Pi T-shaped interactions with PHE92, along with Pi–Sigma interactions with MSE144 and LEU105. Residues MSE109 and MSE145 are involved in Pi–sulfur interactions with the triazole ring of P7.
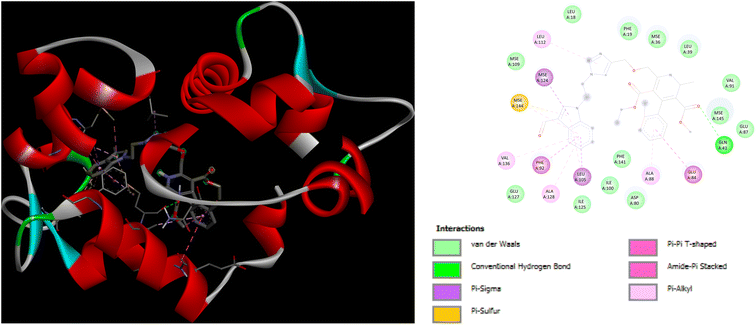 |
| | Fig. 8 3D and 2D representation of binding interactions of P5 with 6M7H. | |
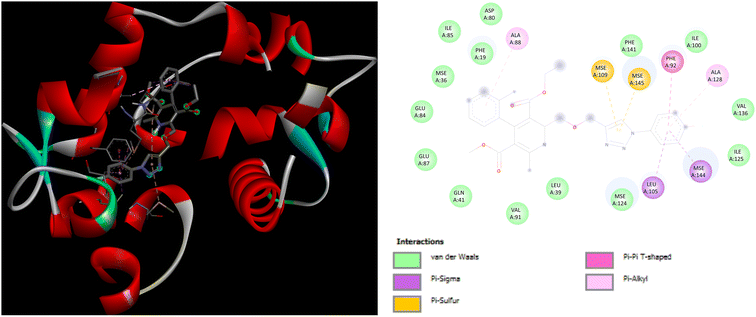 |
| | Fig. 9 3D and 2D representation of binding interactions of P7 with 6M7H. | |
The docking investigations for all the synthesized compounds P1–P14 demonstrated superior binding affinities when compared to the standard amlodipine in case of both the proteins, particularly, P7 exhibited the highest binding affinities with 5KMD and 6M7H, underscoring the significance of these bioisosteres as potential leads for effective anti-hypertensive activity (Fig. 10).
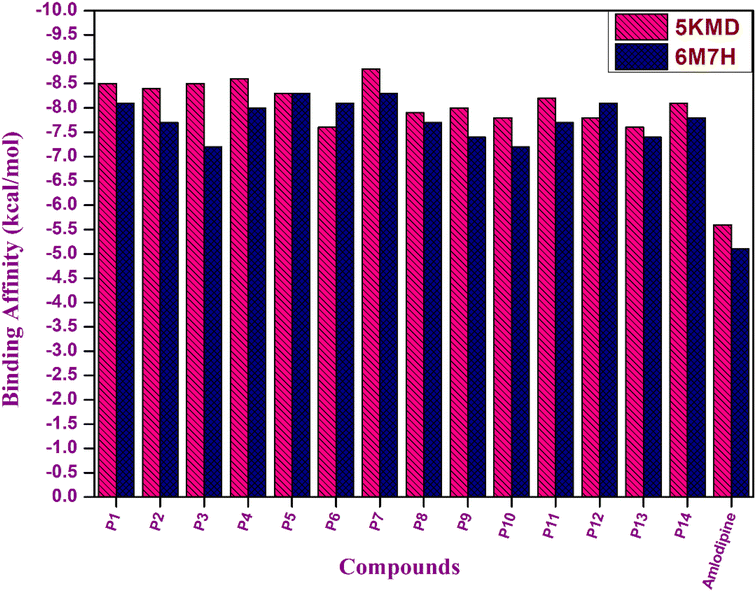 |
| | Fig. 10 Graphical representation of binding affinity of hybrids P1–P14 with target proteins 5KMD and 6M7H. | |
3.3 In silico structure activity relationship (SAR) assessment
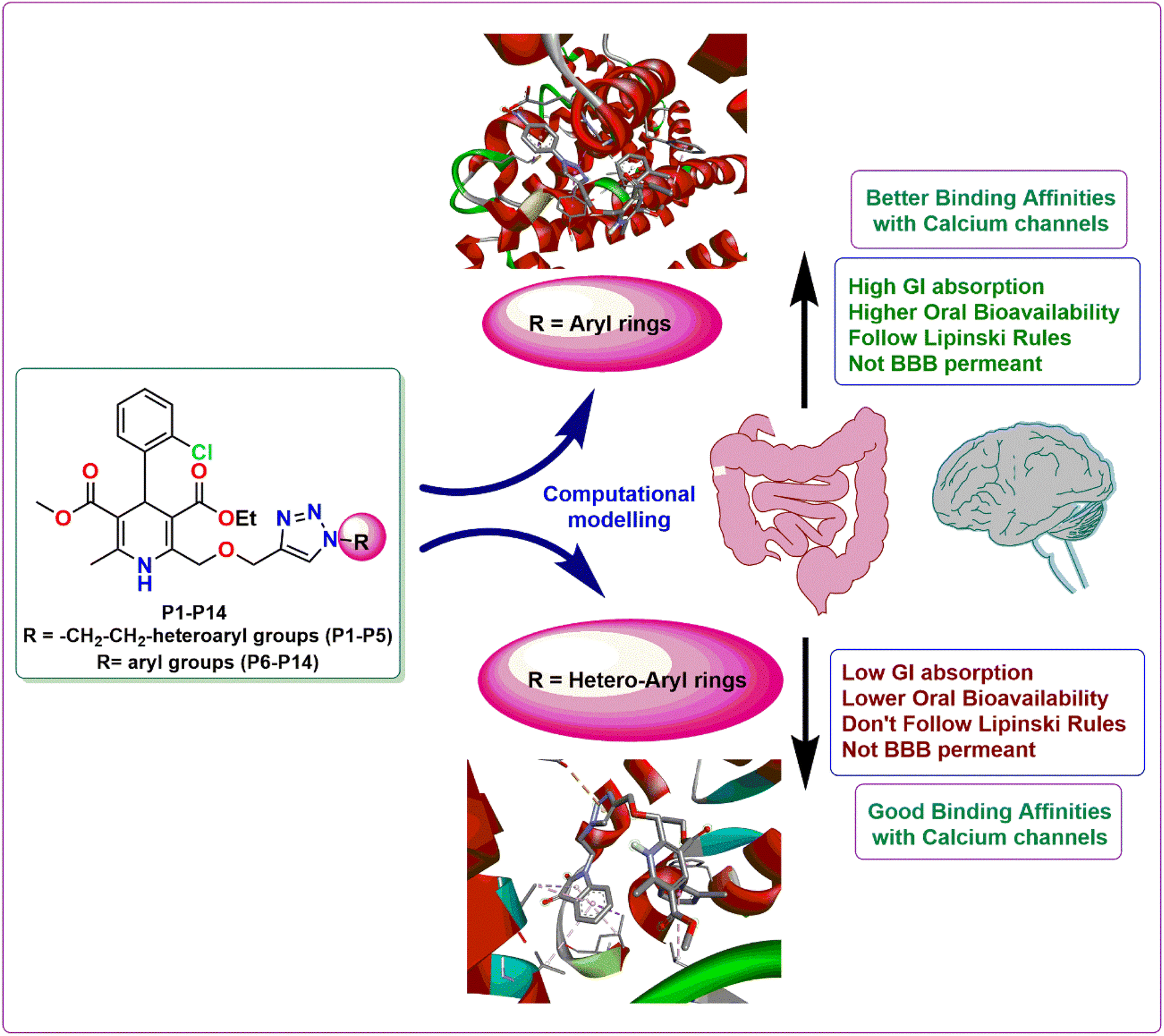
The structure activity relationship is a fundamental concept in drug design that is used to comprehend the relationship between the chemical structure of a compound and its pharmacological activity. Its ultimate goal is to optimize the chemical structure of a compound in order to maximize its intended pharmacological activity while minimizing undesired side effects.22 As evident from the computational evaluation of the synthesized amlodipine bioisosteres P1–P14, hybrids containing heteroaryl rings tethered via triazole linkage (P1–P5) exhibited excellent binding affinity and remarkable interactions with the amino acid residues in case of both the proteins 5KMD and 6M7H as compared to the hybrids possessing the aryl linkage (P6–P14). However, aryl linked hybrids were found to be in accordance with the Lipinski rule and have shown high GI absorption. In contrast, P1–P5 heteroaryl ring containing hybrids displayed two violations in Lipinski rule with low GI absorption (Fig. 11). Hence, it can be inferred that despite of better binding affinity of P1–P5, hybrids P6–P14 possess drug-like attributes and can serve as lead for future investigations. Better computational results than the standard drug amlodipine suggest that the existence of triazole tethered aryl/heteroaryl ring is essential for the manifestation of pharmacological attributes associated with anti-hypertensive activity.
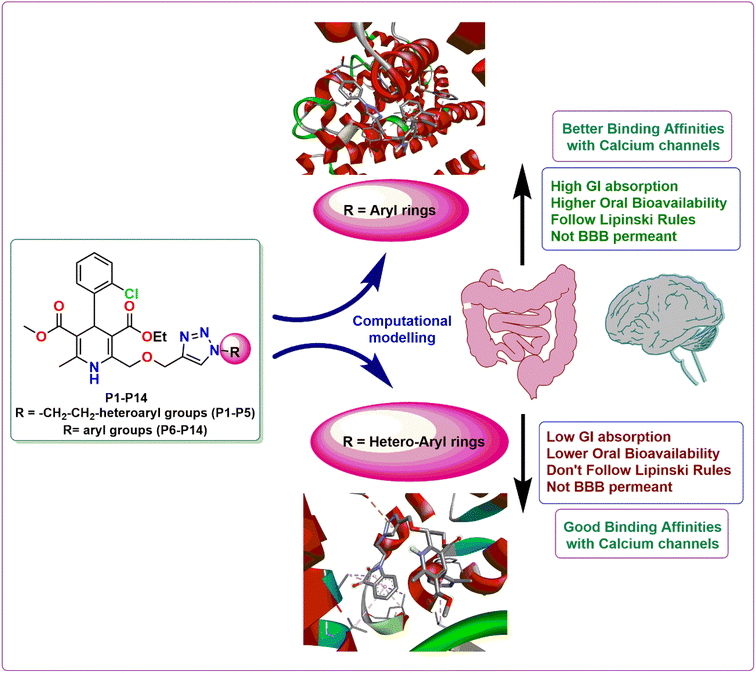 |
| | Fig. 11 Demonstration of the structure activity relationship of the synthesized 1,4-dihydropyridine based amlodipine bioisosteres P1–P14. | |
4. Conclusion
In conclusion, this study represents a significant advancement in the quest for innovative antihypertensive medications. A promising series of 1,4-dihydropyridine based scaffolds were developed as prospective replacements to amlodipine through a meticulous approach encompassing strategic design, synthesis and extensive in silico evaluation. This comprehensive study involved the use of crystallography, molecular docking, ADMET profiling, and analysis of Structure Activity Relationships (SAR). The examination of crystal structure provided valuable structural insights, laying the foundation for subsequent computational evaluations. The existence of triazole appended aryl/heteroaryl linkage was substantiated by the better molecular docking results with the concerned calcium channels as compared to amlodipine. ADMET and SAR evaluation uncovered pivotal insights for the future refinement of antihypertensive activity. Furthermore, excellent binding affinity, high GI absorption, no BBB permeance, and the druglikeliness nature of the synthesized bioisosteres, especially P6–P10, P12 and P14 as established from the in silico ADMET assessments, suggest their suitability for further development. In particular, P7 showcased extraordinary in silico profiling and its crystal structure manifests the molecular interactions with biological systems. This collective effort establishes a robust foundation for the progression and potential clinical applications of these candidates, signalling a promising avenue in antihypertensive drug discovery. In essence, this research significantly contributes to the growing panorama of innovative therapeutics by offering prospective solutions for hypertension management and underscoring the importance of exploring novel bioisosteres in drug design and development.
Accession codes
CCDC 2294665 contains the supplementary crystallographic data for ligand S1 for this paper.
Conflicts of interest
The authors declare no conflicts of interests.
Acknowledgements
P. T. is grateful to Council of Scientific and Industrial Research (CSIR) for the financial support as Senior Research Fellowship. All the authors acknowledge Institute of Eminence (IoE), Council of Scientific and Industrial Research (CSIR), Department of Chemistry, University of Delhi, and University Science Instrumentation Centre (USIC), University of Delhi for providing research and characterization facilities.
References
- K. T. Mills, A. Stefanescu and J. He, Nat. Rev. Nephrol., 2020, 16, 223–237 CrossRef CAS PubMed.
- M. Burnier and B. M. Egan, Circ. Res., 2019, 124, 1124–1140 CrossRef CAS PubMed.
- W. B. Kannel, J. Cardiovasc. Pharmacol., 1989, 13, S4–S10 CrossRef PubMed.
- H. Scholz, Cardiovasc. Drugs Ther., 1997, 10, 869–872 CrossRef PubMed.
- K. G. Bulsara and M. Cassagnol, in StatPearls [Internet], StatPearls Publishing, 2023 Search PubMed.
- U. Hostalek-Gottwald and Z. Gaciong, Curr. Med. Res. Opin., 2022, 38, 1047–1053 CrossRef CAS PubMed.
- S. Mostafa, H. Shabana, F. Khalil, I. M. El Mancy, H. A.-M. Zedan, A. Elmoursi, I. G. Ramadan, S. E. D. Mohamed, A. Kassem and I. S. Kamel, High Blood Pressure Cardiovasc. Prev., 2022, 29, 565–576 CrossRef CAS PubMed.
- T. Iida, J. Kanazawa, T. Matsunaga, K. Miyamoto, K. Hirano and M. Uchiyama, J. Am. Chem. Soc., 2022, 144, 21848–21852 CrossRef CAS PubMed.
- B. S. Jayashree, P. S. Nikhil and S. Paul, Med. Chem., 2022, 18, 915–925 CrossRef CAS PubMed.
- J. Striessnig and N. J. Ortner, in Encyclopedia of Molecular Pharmacology, Springer, 2022, pp. 375–383 Search PubMed.
- R. Bansal, P. Jain, G. Narang, A. Kaur, C. Calle and R. Carron, Lett. Drug Des. Discovery, 2023, 20, 1632–1644 CrossRef CAS.
- H. J. Little, Pharmacol. Rev., 2021, 73, 1298–1325 CrossRef PubMed.
- L. Tang, T. M. Gamal El-Din, T. M. Swanson, D. C. Pryde, T. Scheuer, N. Zheng and W. A. Catterall, Nature, 2016, 537, 117–121 CrossRef CAS PubMed.
- C. N. Johnson, R. Pattanayek, F. Potet, R. T. Rebbeck, D. J. Blackwell, R. Nikolaienko, V. Sequeira, R. Le Meur, P. B. Radwański, J. P. Davis, A. V Zima, R. L. Cornea, S. M. Damo, S. Györke, A. L. George and B. C. Knollmann, Cell Calcium, 2019, 82, 102063 CrossRef CAS PubMed.
- B. N. Sağlık, B. K. Çavuşoğlu, D. Osmaniye, S. Levent, U. A. Çevik, S. Ilgın, Y. Özkay, Z. A. Kaplancıklı and Y. Öztürk, Bioorg. Chem., 2019, 85, 97–108 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V Dolomanov, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. V. D. Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- A. Daina, O. Michielin and V. Zoete, J. Chem. Inf. Model., 2014, 54, 3284–3301 CrossRef CAS PubMed.
- R. Guha, J. Comput.-Aided Mol. Des., 2008, 22, 857–871 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Balaram Pani
b,
Balaram Pani