
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vitro anticancer evaluation of Enceleamycin A and its underlying mechanism†
Abujunaid Khanab,
S. Pradeepab and
Syed G. Dastager *ab
*ab
aNCIM-Resource Center, Biochemical Sciences Division, CSIR-National Chemical Laboratory, Pune – 411008, India. E-mail: sg.dastager@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad – 201002, India
First published on 22nd November 2023
Abstract
It has become more crucial than ever to find novel anticancer compounds due to the rise in cancer mortality and resistance to the present chemotherapeutic drugs. Naphthoquinones are regarded as privileged structures for their ability to inhibit various cancers. The current study examined three novel furo-naphthoquinones (Enceleamycins A–C) previously isolated from Amycolatopsis sp. MCC 0218 for their anticancer potential. Enceleamycin A demonstrated considerable cytotoxicity for triple-negative breast cancer (TNBC) MDA-MB-231 cells with an IC50 value of 1.25 μg mL−1 (3.78 μM). It also showed the ability to inhibit MDA-MB-231 cell migration. Enceleamycin A raises intracellular ROS levels in TNBC cells, ultimately leading to apoptotic cell death, as demonstrated by Annexin V/PI staining. The molecular docking and simulation investigation revealed better binding affinity of Enceleamycin A with AKT2, which plays a vital role in breast cancer's invasiveness and chemo-resistance. Enceleamycin A inhibits the AKT2 enzyme in vitro with an IC50 value of 0.736 μg mL−1 (2.22 μM), further validating the docking study. The in silico physicochemical and pharmacokinetics characteristics of Enceleamycin A demonstrated its drug-likeness. Intriguingly, Enceleamycin A is non-hemolytic in nature. Taken together, Enceleamycin A could be a candidate molecule for treating TNBC cells by targeting the AKT2 signaling pathway.
1. Introduction
Cancer is a non-communicable disease and is associated with rapid and uncontrolled cell growth.1 With approximately twenty million new cases and ten million fatalities in the year 2020, it's the second largest cause of mortality globally. With an estimated 2.3 million new cases, breast cancer has surpassed lung cancer as the most frequently detected cancer.2,3 Among breast cancer, 10–15% of all cases are from triple-negative breast cancer (TNBC), which is considered more aggressive with a low survival rate. It does not express the three receptors, namely, progesterone (PR), estrogen (ER), and human epidermal growth factor receptor-2 (HER2).4,5 There are limited options for specific and effective therapy against TNBC due to the absence of validated biomarkers. So chemotherapy is generally used as the standard TNBC treatment, but the prognosis remains poor.6–8 Considering this, developing new anticancer agents with increased potency and high specificity is essential.In several kinds of human cancer, an aberration in the PI3K-AKT-mTOR signaling pathway contributes to tumor development.9 AKT, also known as protein kinase B (PKB), belongs to a serine–threonine kinase family and is essential for the smooth functioning of this pathway.
In several kinds of human cancer, an aberration in the PI3K-AKT-mTOR signaling pathway contributes to tumor development.9 AKT, also known as protein kinase B (PKB), belongs to a serine–threonine kinase family and is essential for the smooth functioning of this pathway. Among the three AKT isomers, AKT2 is closely associated with cancer cell metabolism, angiogenesis, proliferation, metastasis, and drug resistance.10,11
The elevated AKT2 expression is frequently detected in various human tumors, including breast, lung, prostate, pancreatic, colorectal, and ovarian. Overexpression of AKT2 is correlated with cancer aggressiveness and poor survival rates.10,12,13 The AKT2 inhibition in breast cancer effectively reduces the colony formation abilities and invasion of non-cancer stem cells (non-CSC) and CSCs.14 All these functions of AKT2 make this signaling molecule a promising target for cancer therapy.
Naphthoquinones comprise a naphthalene ring system bearing two carbonyl groups, naturally distributed in bacteria, fungi, and plants. They display various biological activities, like anticancer, antibacterial, antifungal, antimalarial, antiviral, antitrypanosomal, and antiinflammatory.15–20 Naphthoquinone like structures as in Fig. 1 have been approved by FDA and are used in various cancer chemotherapy. Mitomycin (a benzoquinone) is used to treat bladder and anal cancer.21,22 Anthraquinones, such as doxorubicin, daunorubicin, idarubicin, mitoxantrone, and epirubicin, are commonly used to treat various solid and hematologic cancers.23 Thus, these molecules continue to attract in developing new drugs for cancer therapy.
 | ||
| Fig. 1 Structure of anticancer drugs with quinone moiety.21–23 | ||
The primary mechanism of the naphthoquinones is suggested to be reactive oxygen species (ROS) formation in the cell. Cancer cells often have a higher concentration of ROS than normal cells because of the higher metabolic demands; producing even higher amounts of ROS by naphthoquinones in the cell can lead to cell death.1,24 Also, the pyrano-naphthoquinone molecules like lactoquinomycin, frenolicin B, and kalafungin were found to have selective inhibition towards the serine–threonine kinase AKT.25 Therefore, the dual-action molecule with the ability to produce ROS and inhibit the AKT2 signaling pathway would result in potent anticancer drug with higher specificity and low resistance.
In our previous study, three novel furo-naphthoquinones, Enceleamycin A–C, were isolated and characterized from rare actinobacteria, Amycolatopsis sp. MCC0218 produced under static incubation.26 As depicted in Fig. 2, Enceleamycin A and B possess an unprecedented pentacyclic oxeto-furo-furo-naphthoquinone structure, with the latter containing a dihydroxylated group, whereas Enceleamycin C has the furo-furo-naphthoquinone skeleton with hydroxy-aldehyde functionalities. The isolated Enceleamycins displayed selective inhibition towards the Gram-positive bacteria.26
 | ||
| Fig. 2 Structure of Enceleamycin A, B and C isolated from Amycolatopsis sp. MCC0218.26 | ||
Based on the anticancer potential of naphthoquinones and the need for novel antineoplastic drug, we have demonstrated the anticancer activity of Enceleamycins against MDA-MB-231, A549, and HeLA cell lines. With the maximum activity, Enceleamycin A was further considered for the ROS formation ability and apoptosis assay in the MDA-MB-231 cells. Moreover, Enceleamycin A displayed binding affinity to the serine–threonine kinase AKT2, determined by the molecular docking and molecular dynamic (MD) simulation, which was further validated in vitro. The molecule's physicochemical and pharmacokinetic characteristics were examined in silico, and its hemolytic effect was also established. The findings from our research can stimulate interest in exploring the microbial naphthoquinones as potential anticancer medication by targeting AKT2 kinase signalling pathway.
2. Results and discussion
2.1 Cell viability assay
Previous studies have shown that naphthoquinones exhibit anticancer effects on different types of cancer.15–17 In order to investigate and compare the anticancer activity of novel furo-naphthoquinones, Enceleamycins A–C, the cytotoxicity was evaluated against triple-negative breast cancer cells (MDA-MB-231), lung cancer cells (A549), cervical cancer cells (HeLa) and HFF cells (non-cancer human foreskin fibroblasts) and Vero (non-cancer monkey kidney epithelial cells) by the MTT assay.27 Tables 1 and S2† lists the half maximum inhibitory concentration values whereas, the Fig. 3 and S8† shows the dose–response viability in presence of Enceleamycins and doxorubicin at concentrations varying from 0.1 to 100 μg mL−1.| HFF | MDA-MB-231 | A549 | HeLa | ||||
|---|---|---|---|---|---|---|---|
| CC50a | EC50b | SIc | EC50b | SIc | EC50b | SI | |
| a CC50 is the concentration that achieved 50% cytotoxicity.b EC50 is effective concentration that achieved 50% inhibition.c SI is the selectivity index for the cancer cells with respect to the non-cancer human foreskin fibroblasts (HFF) cells.d A, B, C: Enceleamycin A, B, C, Std: doxorubicin. | |||||||
| A | 3.97 | 1.25 | 3.17 | 1.98 | 2.0 | 1.51 | 2.62 |
| B | 70.95 | 23.49 | 3.02 | 46.68 | 1.51 | 30.53 | 2.32 |
| C | 7.60 | 2.59 | 2.93 | 3.51 | 2.16 | 7.27 | 1.04 |
| Std | 8.96 | 3.28 | 2.73 | 2.68 | 2.83 | 1.69 | 4.49 |
 | ||
| Fig. 3 Viability of MDA-MB-231, A549, HeLa and Vero cells after treatment with Enceleamycin A at different concentration from 0.1 to 100 μg mL−1. Data are presented as mean of ±SEM (standard error of the mean), n = 3. | ||
The Enceleamycins displayed strong to moderate inhibitory activity towards these cell lines. The results indicated that Enceleamycin A has potential activity towards the MDA-MB-231 (TNBC cells) with an EC50 value of 1.25 μg mL−1 (3.78 μM), followed by HeLa and A549 cells with an EC50 value of 1.51 (4.57 μM), and 1.98 μg mL−1 (5.99 μM), respectively. The EC50 value of Enceleamycin A was better than the standard anticancer drug doxorubicin in all the cell lines tested. Furthermore, Enceleamycin A displayed better inhibitory activity towards the cancer cells compared to the non-cancer HFF cells with selectivity index (SI) in the range of 2.0 to 3.17. The selectivity index of Enceleamycin A against cancer cells with respect to non-cancer Vero cells was in the range of 2.35 to 3.73 (Table S2†). Enceleamycin A exhibited strong anticancer activity, followed by Enceleamycin C, whereas Enceleamycin B showed moderate activity. The potential bioactivity of Enceleamycin A and C may be due to the presence of double bond in the second naphthalene ring, which acts as an electron acceptor and results in formation of semiquinone or hydroquinone. The subsequent reduction of Enceleamycin A and C may result in ROS formation and thereby leading to cell death.1,24
With the maximum activity of Enceleamycin A against the MDA-MB-231 cells, the dose–response viability at 4 h, 24 h, and 48 h was determined as shown in Fig. 4. Interestingly, the MDA-MB-231 cell growth was inhibited within 4 hours of treatment by Enceleamycin A with an IC50 value of 7.7 μg mL−1 (23.3 μM). The difference in inhibition after 24 and 48 h of treatment was not much at lower concentrations up to 2 μg mL−1; however, at 4 μg mL−1 and above, maximum inhibition was observed at 48 hours of treatment. The sulforhodamine B (SRB) assay of Enceleamycin A in Table S1 and Fig. S7† demonstrated lethal activity against nine human cancer cells lines originating from breast, liver, colon, oral, cervical, lung, prostate, ovarian, and leukemia. Similar to the results of MTT assay, Enceleamycin A showed better lethal activity than the anticancer drug adriamycin against most cancer cell lines tested. Furthermore, we selected Enceleamycin A for the evaluation of the probable mode of action against MDA-MB-231 cells and its drug-likeness because of its specific potent activity towards the MDA-MB-231 cells, high yield compared to Enceleamycin B and C,26 and lack of treatment options for TNBC cells.5,6
 | ||
| Fig. 4 Dose dependent cell viability of MDA-MB-231 cells at 4, 24 and 48 h treatment with Enceleamycin A. Cell growth inhibition was observed within 4 h treatment with IC50 value of 7.7 μg mL−1 (23.3 μM). Data are presented as mean of ±SEM (standard error of the mean), n = 3. | ||
2.2 Anti-migration assay
Cell migration is crucial during the whole process of cancer development. Cancer cells have the ability to migrate, which is essential to invade surrounding tissues and cause tumor metastasis. The study of cell migration inhibition is appealing as metastatic progression is considered the leading cause of death in cancer patients.28To examine the inhibitory impact of Enceleamycin A on the MDA-MB-231 cell migration, an in vitro anti-migration assay was conducted by a scratch/wound healing assay.29 The images of cell migration in Fig. 5A were recorded in Olympus CKX53 inverted microscope at 0 and 24 hours of treatment and the migration of cells was measured in micrometers using MagVision software. The rate of relative migration for MDA-MB-231 cells treated with Enceleamycin A was 45.25%, 43.01%, 25.55%, and 6.99% at 0 μg mL−1, 0.5 μg mL−1, 1.0 μg mL−1 and 2.0 μg mL−1 respectively. The migration ability of MDA-MB-231 cells was significantly inhibited with 1.0 and 2.0 μg mL−1 concentrations of Enceleamycin A as shown in Fig. 5B. These results demonstrated that Enceleamycin A suppresses the MDA-MB-231 cell's ability to migrate in a concentration-dependent manner.
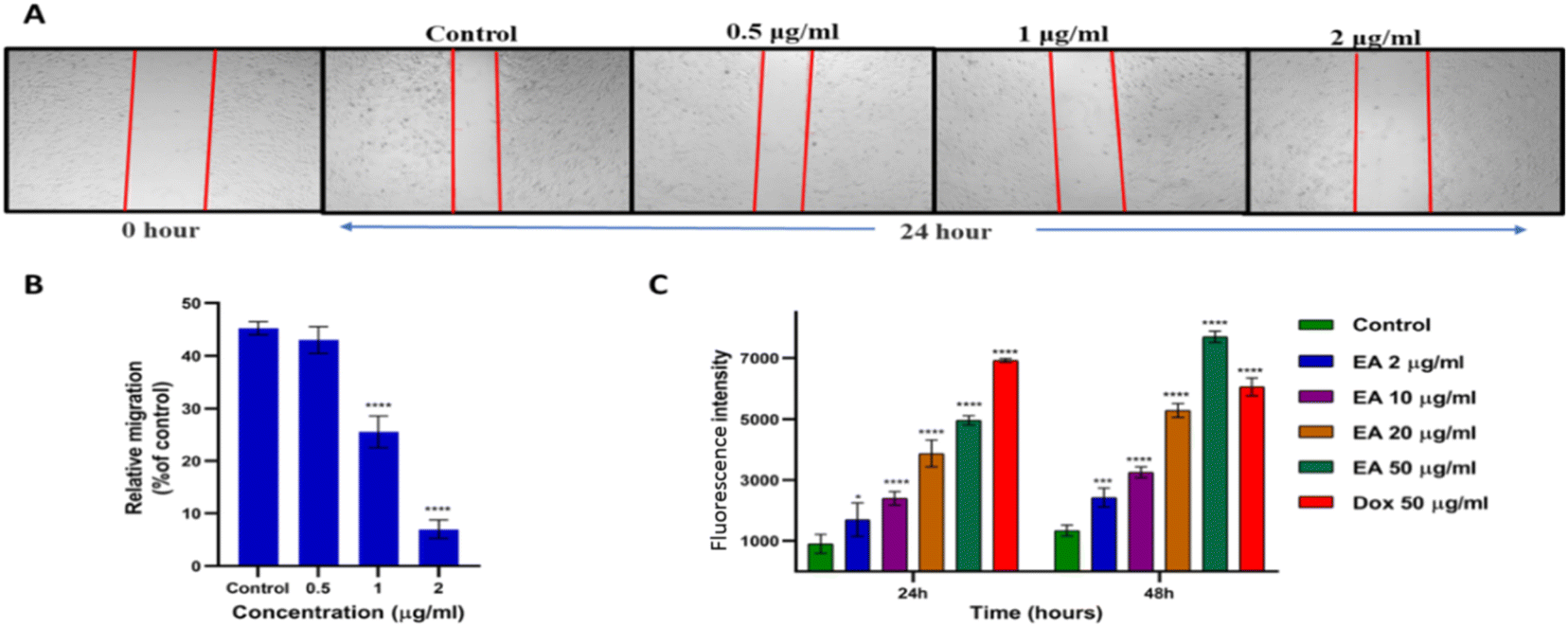 | ||
| Fig. 5 Enceleamycin A effect on the migration and ROS formation ability in MDA-MB-231 cells. (A) Migration of MDA-MB-231 cells in presence of Enceleamycin A at 0, 0.5, 1 and 2 μg mL−1 after 24 hours. (B) Statistical data analyses for MDA-MB-231 cells mobility in presence of Enceleamycin A at 0 (control), 0.5, 1 and 2 μg mL−1 after 24 hours. One-way ANOVA Dunnett test was used to determine statistical significance; ****p < 0.0001. Data are presented as mean of ±SEM (standard error of the mean), n = 3. (C) Statistical data analyses of ROS formation by MDA-MB-231 cells after treatment with Enceleamycin A (EA) at 24 and 48 hours. Two-way ANOVA Dunnett test was used to determine statistical significance; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are presented as mean of ±SEM (standard error of the mean), n = 3. | ||
2.3 Measurements of intracellular reactive oxygen species (ROS)
Since naphthoquinones are known for producing intracellular ROS, we quantified the ROS production after treatment with Enceleamycin A by 2′,7′-dichlorofluorescein diacetate (H2-DCFDA). Cellular esterases deacetylate the cell-permeable dye to a non-fluorescent molecule, which upon oxidation by ROS, transforms into highly fluorescent 2′,7′-dichlorofluorescein (DCF).30 These ROS at lower concentration plays a vital role in the homeostasis of cells; however, when in excess, it leads to the death of cells through apoptosis.31,32 In the ROS assay, the MDA-MB-231 cells in the presence of Enceleamycin A demonstrated a relative increase in fluorescence intensity at 24 and 48 hours as the concentration was raised from 2 to 50 μg mL−1 compared to the untreated control. The graph in Fig. 5C displayed MDA-MB-231 cells after treatment with Enceleamycin A resulted in concentration and time-dependent increase in intracellular levels of ROS. It is known that excessive ROS formation within the cell can induce apoptosis and results in cell death.2.4 Apoptosis detection in MDA-MB-231 cells
To determine whether the ROS production by Enceleamycin A in MDA-MB-231 cells results in apoptotic cell death, we perform the apoptosis assay using flow cytometry. The apoptotic analysis in cancer cells is based on the movement of phosphatidylserine from the inner membrane to the outer membrane, which eventually recognizes by Annexin V. In Fig. 6A, the scatter plot for control and treated cells was demonstrated with four distinct populations of unstained for viable cells, Annexin V-Alexa flour for early apoptotic cells, and Annexin V/propidium iodide dual stained for late apoptotic cells.33 The early/late apoptosis ratio shown in Fig. 6B was 25.8%/2.45%, 49.7%/10.7%, 58.4%/14.85, and 60.65%/19.2% at the concentration 0, 2, 4, and 8 μg mL−1 respectively, after 48 hours of treatment with Enceleamycin A. These observations demonstrated the concentration-dependent apoptosis in the MDA-MB-231 cells by Enceleamycin A. | ||
| Fig. 6 Apoptosis detection in MDA-MB-231 cells after treatment with Enceleamycin A. (A) Enceleamycin A effect on apoptosis of MDA-MB-231 cells based on Annexin V-Alexa flour-488 and PI staining. The cell stages were given as viable-Q3, early apoptotic-Q4 and late apoptotic-Q2. (B) Percentage of viable, early and late apoptotic cells in the population based on flow cytometry data analysis. Data are presented as mean of ±SEM (standard error of the mean), n = 3. | ||
2.5 Molecular docking
The inhibitory activity of the novel ligand Enceleamycin A against the intended protein target of the PI3K-AKT-mTOR pathway was examined using molecular docking. The protein target considered for molecular docking were PI3Kα (4TV3), AKT1 (3O96), AKT2 (1O6L), mTOR (1E7U), and S6K1 (3A62). The top rank from the cluster of a receptor with the lowest binding energy and 0 RMSD was considered appropriate for visualizing the interactions between the protein and ligand complex. The details of the best molecular fit conformations were displayed by Autodock 4.2.6 (ref. 34 and 35) and the best molecular fit pose was visualized by Biovia discovery studio v20.1.0.19295.36,37 Enceleamycin A displayed better binding affinity with AKT2 compared to the other proteins of PI3K-AKT pathway shown in Tables S3, S4 and Fig. S10.† The binding energy calculated was −7.14 kcal mol−1 for the docked complex. The non-bond interactions gave us information about active sites, the nature of bonding, the distance between the native ligand and the protein, and the donor and acceptor atoms. The active sites of the interacting Enceleamycin A and AKT2 displayed in Fig. 7C and Table S5,† were Lys 181, Glu 200, Gly 295, Asp 293, Glu 193, Phe 163, Leu 183, Lys 191, which signifies that the novel ligand is interacting with the A polypeptide chain of AKT2 protein. The interaction distance of active site Lys 181 and Glu 200 was of 2.2 and 2.0 Å, respectively in Fig. 7D by conventional hydrogen bonding. The other interactions include the carbon hydrogen bond for Gly 295 and Asp 293, electrostatic pi–anion for Glu 193, hydrophobic pi–pi stacked for Phe 163, hydrophobic alkyl for Leu 183 and hydrophobic pi–alkyl for Lys 191. Fig. 7E and Table S5† displays the hydrogen bond donor and acceptor details of the interacting complex of Enceleamycin A and AKT2 in which Lys 181 acts as a H-bond donor and Glu 200 as an H-bond acceptor. | ||
| Fig. 7 Molecular docking of Enceleamycin A with AKT2 protein and docking validation. (A) Enceleamycin A in protein databank (pdb) format where red indicates oxygen atom, light grey represents hydrogen atom and dark grey represents carbon atom, (B) AKT2 (1O6L) receptor, where α-helices are denoted in red, β-sheets in yellow, and turns & loops in green. (C) Two-dimensional diagram of the docked complex (AKT2 and Enceleamycin A) showing the active sites and the type of interactions. (D) Three-dimensional diagram displaying the interaction of active site Lys 181 and Glu 200 with distance of 2.2 and 2.0 Å, respectively. (E) Three-dimensional diagram with hydrogen bond display of the interacting complex, where the pink shade represents the H-bond donor and green shade represents the H-bond acceptor. (F) Docking validation of native ligand AMP-PNP to AKT2 (1O6L) and displaying the super-imposition of the redocked native ligand protein complex. | ||
To further validate the docking method, the AKT2 (1O6L) protein was re-docked with the native ligand AMP-PNP (phosphoaminophosphonic acid-adenylate ester). Both re-docked, and native ligands interacted with the protein AKT2 with significant overlap and resemblance as shown in Fig. 7F. The RMSD (root mean square deviation) value of the interactions was computed by the DockRMSD and found to be 1.262 Å, considered a reliable docking procedure since the value was <2.0 Å.38 The RMSD value calculated for the superimposed ligand conformation using LS-align was 0.684 Å, which is <1 Å denotes strongly aligned atom pairs.39
2.6 MD simulation analysis
Molecular dynamics (MD) simulation is a computational tool for drug development. It is extensively used to investigate how the molecules shift their form and interact with other molecular species in a variety of environments.40 To confirm the structural stability of the docked complex of Enceleamycin A and AKT2, MD simulations were run for 100 ns using GROMACS package. Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA), a technique for determining binding free energy of protein–ligand complexes, has become more popular. The MM-PBSA technique was implemented via the g_mmpbsa tool41 by which the binding energy of −10.481 ± 3.398 kcal mol−1 was observed as shown in Table S6† during the run of 100 ns demonstrating the potential affinity between the Enceleamycin A and AKT2. For the evaluation of stability of the simulated systems, the trajectories produced after the simulations were analyzed to determine RMSD for backbone atom, Root mean square fluctuation (RMSF) for C-alpha, radius of gyration (Rg) and interacting hydrogen bonds.RMSD calculates the variation between the protein's starting location and final structure. The RMSD values displayed low fluctuation throughout the 100 ns period, and were almost constant from around 70 to 100 ns demonstrating the structural stability of the protein–ligand complex in Fig. 8A. The protein–ligand interactions appear energetically advantageous and contribute to the complexes' stability based on the low RMSD values. RMSF, which evaluates the flexibility of each residue over time, may be used to determine the variations in the protein residues. The RMSF score estimates the stability of protein–ligand complexes, with larger values suggesting less stability and greater flexibility. The major fluctuation was observed only in the designated A (150–170) and B (305–325) region of protein loop which is conserved structurally and functionally for the serine and threonine kinase family42 shown in Fig. 8B. This major fluctuation might be due to the inhibition of AKT2 by Enceleamycin A affecting the phosphorylation responsible for protein functionality.
 | ||
| Fig. 8 Plots displaying the MD simulation data of simulated complex (Enceleamycin A with AKT2 protein) for the 100 ns simulation. (A) Plot displaying the RMSD values of simulated complex for 100 ns. (B) Plot displaying the RMSF value of simulated complex for 100 ns where labeling in the residue index region ranging from 150 to 170 and 305 to 325 correlates with the designated A and B loop in pink colour. These loops contain serine and threonine residues responsible for phosphorylation that shows the higher fluctuation. (C) Plot displaying the radius of gyration (Rg) of simulated complex for 100 ns. (D) Plot displaying the number of hydrogen bonds interacting within the simulated complex for 100 ns. | ||
The understanding of protein's radius of gyration (Rg) is essential to comprehend the effect of inhibitors on the compactness of protein. In our simulation, the radius of gyration was found to be 2.17 and displayed lesser deviation throughout the 100 ns run as depicted in Fig. 8C. The intermolecular H-bonds between interacting atom pairs impacts the stability and molecular recognition process of protein–ligand complex's. To ascertain the dynamic stability of complex, the number of H-bonds interacting with the ligand and receptor protein was measured over the 100 ns run. In Fig. 8D, the hydrogen bond interaction was observed throughout the 100 ns run and the interaction was increased from 70 ns onwards. Hydrogen bond occupancy was similar to the results inferred from molecular docking, with Lys 181 occupying 16.08%, followed by Glu 200 occupying 10.79% during the MD run. Thus, it appears that the predicted system is stable.
2.7 ADP-Glo kinase assay
An in vitro assay using the AKT2 kinase enzyme system was conducted to confirm further the in silico binding affinity and stability of the docked complex of Enceleamycin A and AKT2.43 The ADP generated during the reaction is measured by the ADP-GloTM Kinase. In the subsequent luciferase reaction, the newly produced ADP is transformed into ATP and generates light. The light generated correlates with the kinase activity. A concentration of less than 1 μg mL−1 of Enceleamycin A proved efficient in inhibiting the AKT2 enzyme. Enceleamycin A displayed an IC50 value of 0.736 μg mL−1 (2.22 μM) for AKT2 as shown in Fig. 9, consistent with a prior study showing that pyrano-naphthoquinones specifically inhibit the AKT kinase enzyme. However, the potential enzyme inhibition activity was comparatively less compared to pyrano-naphthoquinones.25 With further in-depth research, Enceleamycin A can be used as a dual-action anticancer molecule with ROS formation and AKT2 inhibition to reduce the metastasis and invasiveness of cancer. | ||
| Fig. 9 AKT2 kinase inhibition by Enceleamycin A with IC50 value of 0.736 μg mL−1 (2.22 μM). Data are presented as mean of ±SEM (standard error of the mean), n = 3. | ||
2.8 Pharmacokinetics and physicochemical properties
The anticancer candidate molecule should have an approving biopharmaceutical property since it will interact with various cells and macromolecules inside the body. A molecule's acceptable ADMET (absorption, distribution, metabolism, excretion, and toxicity) characteristics are necessary for its consideration as a lead molecule for therapeutic application. The physicochemical and ADMET of Enceleamycin A was evaluated by the SwissADME44 and pkCSM45 webtool. The bioavailability radar in Fig. 10A, displays that the molecule falls under the optimum physicochemical properties (lipophilicity, size, polarity, insolubility, saturation, and flexibility). A molecule should follow Lipinski's46 and Veber's47 rules for drug-like properties. Enceleamycin A comes under all the designated Lipinski's rule of five (mol. wt < 500 Daltons, no. of hydrogen H-bond acceptors < 10, no. of H-bond donors < 5, octanol–water partition coefficients log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P < 5 and molar refractivity < 140) and Veber's rule (topological polar surface area < 140 Å and no. of rotatable bonds < 10) as displayed in Table 2, which signifies its drug likeliness property. The pharmacokinetics displayed in the boiled-egg ADME profile in Fig. 10B and Table 3 shows that the gastrointestinal tract effectively absorbed the molecule and did not penetrate the blood–brain barrier (BBB). The non-permeability of BBB signifies the molecule theoretically does not have side effects on the central nervous system.48
P < 5 and molar refractivity < 140) and Veber's rule (topological polar surface area < 140 Å and no. of rotatable bonds < 10) as displayed in Table 2, which signifies its drug likeliness property. The pharmacokinetics displayed in the boiled-egg ADME profile in Fig. 10B and Table 3 shows that the gastrointestinal tract effectively absorbed the molecule and did not penetrate the blood–brain barrier (BBB). The non-permeability of BBB signifies the molecule theoretically does not have side effects on the central nervous system.48
 | ||
| Fig. 10 ADME property and hemolysis of Enceleamycin A. (A) Bioavailability radar of Enceleamycin A. (B) Boiled-egg ADME profile where white egg part shows the gastro-intestinal absorption and yellow yolk part shows the blood–brain barrier permeability, whereas the horizontal axis displays TPSA value and vertical axis shows the logP value. The blue dot represents Enceleamycin A. (C) Hemolysis assay of Enceleamycin A demonstrating the intact pellet of red blood cells. (D) Percentage of hemolysis of RBCs by Enceleamycin A displaying less than 2% hemolysis at highest tested concentration of 512 μg mL−1 (1550.15 μM). Data are presented as mean of ±SEM (standard error of the mean), n = 3. | ||
| Physicochemical properties | |
|---|---|
| Formula | C17H1407 |
| Molecular weight | 330.29 g mol−1 |
| Number of heavy atoms | 24 |
| Number of aromatic heavy atoms | 6 |
| Number of rotatable bonds | 0 |
| Number H-bond acceptors | 7 |
| Number of H-bond donors | 2 |
| logP |
2.05 |
| Molar refractivity | 78.07 |
| TPSA | 102.29 Å |
| Pharmacokinetic properties | |
|---|---|
| GI absorption | Yes |
| BBB permeation | No |
| P-Glycoprotein substrate | Yes |
| P-Glycoprotein inhibitor | No |
| CYP-450 class enzyme inhibitor | No |
| hERG I and II inhibitor | No |
| logKp (skin permeation) |
−7.79 cm s−1 |
| AMES toxicity | No |
| Hepatotoxicity | No |
| Skin sensitisation | No |
Furthermore, the molecule did not inhibit the P-glycoprotein and CYP-450 class of enzymes, thus having a lower chance of corresponding drug interaction.48 The toxicity of molecules can damage organs and fail in late-stage drug development. So the toxicity of Enceleamycin A was evaluated by the pkCSM web tool, where the compound was negative to the Ames test, which assesses the carcinogenic effect. The molecule was non-hepatotoxic, not sensitive to skin, and a non-inhibitor of hERG I and II.
In addition to Enceleamycin A, the physicochemical and pharmacokinetics of Enceleamycin B and C was also evaluated as shown in Fig. S11, S12 and Tables S7–S14.† The Enceleamycin A displayed most acceptable physicochemical properties and lowest toxicity followed by Enceleamycin C and B. Based on these in silico findings, Enceleamycin A displayed desirable drug-likeness properties for a potential therapeutic candidate.
3. Conclusions
The present study investigated the anticancer potential of three novel furo-naphthoquinones, Enceleamycin A, B and C previously isolated by our group from Amycolatopsis sp. Of the three, Enceleamycin A showed prominent anticancer properties, particularly against the MDA-MB-231 (TNBC) cells with an IC50 value of 1.25 μg mL−1 (3.78 μM). It also inhibits the cell migration ability of TNBC cells. The treatment of MDA-MB-231 cells by Enceleamycin A increases intracellular ROS formation, leading to apoptotic cell death, demonstrated by flow cytometry analysis. Molecular docking and MD simulation showed that Enceleamycin A can be a potential AKT2 inhibitor candidate. The in silico investigation was validated by the in vitro AKT2 enzyme assay, in which Enceleamycin A exhibited IC50 value of 0.73 μg mL−1 (2.22 μM). According to in silico physicochemical and ADMET studies, Enceleamycin A showed good bioavailability and did not violate Lipinski's rule of five, making it a candidate drug-like molecule. The compound showed no prominent hemolytic effect on human RBCs. Altogether, this study presents essential insights about Enceleamycin A possessing therapeutic potential for cancer treatment, particularly triple-negative breast cancer cells.4. Experimental
4.1 Materials
Enceleamycin A (C17H14O7), B (C17H16O9) and C (C17H14O7), used in the experiments were isolated in our previous study from Amycolatopsis sp. MCC 0218.26 Heat-inactivated FBS (Fetal bovine serum) and DMEM (Dulbecco's modified eagle's medium) were obtained from GIBCO. Trypsin EDTA and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Himedia. Standard doxorubicin and H2-DCFDA dye was purchased from Sigma. MDA-MB-231, A549, HeLa, HFF and Vero cells were obtained from NCCS (National Center for Cell Science), Pune.4.2 Cell viability assay
The MTT assay, which measures cellular metabolic activity, was used to examine cell viability.27 The cell lines used for the study were MDA-MB-231, A549, HeLa, HFF and Vero. DMEM with 10% FBS was the media used for the growth and assay of all the cells. The seeding of 10000 cells per well was done of these cells in 96-well plate and incubated at 37 °C with 5% CO2 for 20 hours. The stock solution of 20 mg mL−1 was made of compounds Enceleamycin A, B and C in dimethyl sulfoxide (Hi-Media, Mumbai). The stock solution of the compounds were diluted to 1 mg mL−1 in complete medium (DMEM + 10% FBS) and was further diluted to 100 μg mL−1 and 10 μg mL−1 working solution in DMEM media. The final DMSO concentration at the highest concentration tested at 100 μg mL−1 was 0.5% i.e. below toxicity level and was used as a negative control. The stock solution of 20 mg mL−1 of the standard drug doxorubicin was made in MQ water, filter sterilized and further diluted to working solution in complete media similar to the Enceleamycins. The cells were treated with different concentrations of compound in the range of 0.1 to 100 μg mL−1 for 48 hours. 100 μL of MTT solution (0.5 mg mL−1) was added after the media was removed, and the solution was incubated for 4 h at 37 °C in the dark. Subsequently, the MTT solution was decanted, and the solubilization of formazan crystals was done by adding 100 μl DMSO. Biotek synergy H1 microplate reader was used to capture the reading at 570 nm. In addition, inhibition of MDA-MB-231 cells was determined for Enceleamycin A at 4, 24, and 48 h for the time point study.
4.3 Anti-migration assay
To estimate the anti-migration impact of Enceleamycin A on MDA-MB-231 cells, a scratch/wound healing assay was conducted.28,29 In a 24-well plate, 1.5 × 105 cells per well were seeded in DMEM (10% FBS). After the confluency, linear gaps were scratched at the bottom of the plate by a sterile micropipette tip (200 μL), and the detached cells were subsequently eliminated with 1× phosphate buffer saline (PBS). The adherent cells on the plate were incubated with Enceleamycin A (0.5, 1, and 2 μg mL−1) in serum-free DMEM media for 24 hours at 37 °C with 5% CO2 to eliminate the interference of cell proliferation. The wound images were obtained using an inverted microscope at 0 and 24 h, respectively, and the % of cell migration was calculated with reference to scratch width (SW) at 0 h and 24 h: (SW at 0 h − SW at 24 h/SW at 0 h) × 100.4.4 Measurement of intracellular ROS
The intracellular ROS production level was assessed by the 2′,7′-dichlorofluorescein diacetate (H2-DCFDA) dye.30 In brief, seeding 1 × 104 cells per well of MDA-MB-231 cells was done in 96-well plate and kept for 24 hours. After being washed with DPBS, the cells were incubated at 37 °C in the dark for 30 minutes with H2-DCFDA (20 μM) in DMEM complete media. The culture was treated with a varying concentration of Enceleamycin A (2, 10, 20, and 50 μg mL−1) in DMEM complete media for 24 and 48 h. After incubation, RFU was measured at 485/535 (excitation/emission) in a micro-plate reader.4.5 Apoptosis detection in MDA-MB-231 cell line
The apoptotic-like features in MDA-MB-231 cells after treatment with Enceleamycin A were estimated by Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit (ThermoFisher Scientific).33 In brief, seeding of 3.5 × 105 cells per well of MDA-MB-231 cells was done in a 6-well plate and enable to grow overnight. The culture was treated with Enceleamycin A (2, 4, and 8 μg mL−1) for 48 h. After being washed with binding buffer (1×), the cells were stained for 30 min by Alexa Flour Annexin V and propidium iodide at 37 °C in the dark. Stained cells were assessed for the percentage of apoptotic cells relative to untreated and unstained cells by flow cytometry.4.6 Docking analysis
Canonical SMILES of the novel ligand, Enceleamycin A, were converted to protein data bank format using Open Babel software. The 3-dimensional structure of the PI3K-AKT-mTOR pathway receptors was retrieved from the Protein Data Bank. Water molecules, native ligands, and other heteroatoms from the protein were removed using PyMOL (v.1.74) before docking. Molecular docking was performed using Autodock 4.2.6.34,35 The final DLG file contained important details, viz., top ten conformations for every run consisting of rank, free binding energy (kcal mol−1), mean RMSD, and inhibitory constant (μM). The docking parameter was analyzed based on the lowest binding energy of the ligand–protein complex. Autodock tools 4.2.6, Discover Studio v20.1.0.19295 from Biovia, and PyMOL v.1.74 were used in molecular docking to visualize and study the two-dimensional, three-dimensional and surface annotation of Enceleamycin A interaction with the protein.36,37 The docked complex was validated by redocking of AKT2 with native ligand, and the RMSD value was calculated from the DockRMSD38 and LS-align web tool.394.7 Molecular dynamics simulations
MD simulation of the docking complex was carried out with reading biomolecular coordinates, solvation of desired protein, setting periodic boundary conditions, and generating the input files for equilibration and production using the Gromacs v 2020.6 and visualization and analysis using VMD v 1.9.3 and PyMOL v 1.7.4.5 software. The ligand Protein interaction, having the lowest binding energy, from molecular docking is considered for MD simulation. The novel ligand bound to the AKT2 receptor was analyzed using the GROMOS 54A7 force field of the GROMACS simulation software. With the help of MODELLER9 v 14,51 the missing residues from the crystal structure's 1O6L (Ile450, Thr451, Pro452, Pro453, Asp454, Arg455, Tyr456, Asp457, Ser458, Leu459, Gly460, Leu461, Leu462, Glu463, Leu464, Asp464, Gln464, Arg464, Glu465, Glu466) were put back together. By using the Automated Topology Builder (ATB) repository, the force fields of the ligand was created.52 Hydrogen was added to the heavy atoms using the GROMACS module pdb2gmx. The structures were then solvated within cubic periodic box with water stretching 2 Å on all sides outside the protein by applying the simple point charge (SPCE) water model.53 After that, systems were maintained with an appropriate salt concentration of 0.15 M by introducing suitable amount of Na+ and Cl− ions to neutralize the system. The steepest descent method was subsequently employed to minimize the energy of all systems in the solvated state over a time frame of 2000 steps. To perform equilibration in the NVT (no. of atoms, volume, and temperature) ensemble, the systems were then gradually heated to a temperature of 310 K by a V-rescale thermostat with a 0.1 ps coupling constant.54 Further to achieve equilibration in the NPT (no. of atoms, pressure, and temperature) ensemble, the Parrinello–Rahman barostat was implemented to keep solvent density at 1 bar and 310 K with 0.1 ps coupling constant. To check the stability of protein and ligand complex, the resultant each structure generated from NPT equilibration phase was utilized for final NPT ensemble production run for 100 ns simulation.554.8 ADP-Glo kinase assay
The AKT2 kinase activity was measured in the presence of Enceleamycin A by employing ADP-Glo kinase assay and the AKT2 Kinase enzyme system (Promega). The assay is based on quantifying ADP produced during the kinase reaction. The reaction was done in 96-well plates in which 10 μL of AKT2 (10 ng), 5 μL of modified AKT substrate (1 μg), 5 μL of ATP (50 μM), and 5 μL of inhibitor was added followed by an hour-long incubation at ambient temperature. To terminate the kinase reaction, 5 μL of the ADP-Glo reagent was applied for 40 min. At last, 10 μL of kinase detection reagent was added and left for 30 min. Biotek synergy H1 microplate reader detected the luminescence. The relative activity (%) of kinase was calculated compared to the control without an inhibitor.434.9 Physicochemical and pharmacokinetics analysis
In silico physicochemical and pharmacokinetic properties were detected for Enceleamycins using the SwissADME online program.44 The SMILES notations generated from ChemDraw 20.0 was submitted as an input file to the SwissADME web tool, which provides a reliable prediction of physicochemical (TPSA, number of hydrogen bond acceptor and donor, and solubility), pharmacokinetics (GI absorption, BBB permeability, and CYP inhibitor) and drug-likeness (Lipinski's rule of five and Veber's rule) properties. In addition, the toxicity profile, like the Ames test, skin sensitivity, and hepatotoxicity, was generated by the pkCSM web tool.454.10 Hemolysis assay
Hemolytic activity of Enceleamycin A was performed on the RBCs of human blood sample. The blood sample of 5 mL was decanted after being centrifuged at 5000 rpm for 10 min. The RBCs were suspended in Phosphate buffer saline (PBS) and washed three times by centrifugation for 10 min at 5000 rpm, and the pelleted RBCs were resuspended in 25 mL of PBS. The compound was twofold diluted in PBS and added to RBCs suspension to achieve concentrations varying from 1 to 512 μg mL−1. Triton X-100 (1% v/v) and DMSO (0.5%) in PBS were considered positive and negative controls, respectively. The treated suspensions were kept for 60 min at 37 °C and centrifuged at 5000 rpm for 10 minutes. The resultant supernatant was added to the 96-well plate with a flat bottom. At 570 nm, the absorbance was measured, and the relative percentage of hemolysis was estimated compared to the suspension treated with 1% Triton X-100. The following equation calculates the percent of hemolysis: % hemolysis = [absorbance of the sample (treated with Enceleamycin A)] ÷ [absorbance of the positive control (treated with 1% Triton X-100)] × 100.49,50Author contributions
AK: conceptualization, investigation, methodology, and writing – original draft. SP: investigation and methodology. SGD: supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AK thanks UGC, India, for the Senior Research Fellowship (SRF) award. Authors thank Anticancer Drug screening facility (ACDSF) at ACTREC, Tata Memorial Centre, Navi Mumbai for sulforhodamine B assay.Notes and references
- K. W. Wellington, RSC Adv., 2015, 5, 20309–20338 RSC.
- H. ur Rashid, Y. Xu, Y. Muhammad, L. Wang and J. Jiang, Eur. J. Med. Chem., 2019, 161, 205–238 CrossRef PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- D. Karakaş, R. O. Akar, Z. Gökmen, N. G. Deniz and E. Ulukaya, Turk. J. Biol., 2019, 43, 256–263 CrossRef PubMed.
- L. Yin, J. J. Duan, X. W. Bian and S. C. Yu, Breast Cancer Res., 2020, 22, 1–13 CrossRef PubMed.
- H. Yao, G. He, S. Yan, C. Chen, L. Song, T. J. Rosol and X. Deng, Oncotarget, 2017, 8, 1913 CrossRef PubMed.
- X. T. Le, J. Lee, N. T. Nguyen, W. T. Lee, E. S. Lee, K. T. Oh, H. G. Choi, B. S. Shin and Y. S. Youn, Biomater. Sci., 2022, 10, 7117–7132 RSC.
- C. D. Mock, B. C. Jordan and C. Selvam, RSC Adv., 2015, 92, 75575–75588 RSC.
- J. R. Testa and A. Bellacosa, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10983–10985 CrossRef CAS PubMed.
- P. G. Rychahou, J. Kang, P. Gulhati, H. Q. Doan, L. A. Chen, S. Y. Xiao, D. H. Chung and B. M. Evers, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20315–20320 CrossRef CAS PubMed.
- T. Liu, J. Zhu, W. Du, W. Ning, Y. Zhang, Y. Zeng, Z. Liu and J. A. Huang, Respir. Res., 2020, 21, 1–15 CrossRef PubMed.
- M. Riggio, M. C. Perrone, M. L. Polo, M. J. Rodriguez, M. May, M. Abba, C. Lanari and V. Novaro, Sci. Rep., 2017, 7, 44244 CrossRef CAS PubMed.
- A. Bellacosa, D. De Feo, A. K. Godwin, D. W. Bell, J. Q. Cheng, D. A. Altomare, M. Wan, L. Dubeau, G. Scambia, V. Masciullo and G. Ferrandina, Int. J. Cancer, 1995, 64, 280–285 CrossRef CAS PubMed.
- P. Gener, D. Rafael, J. Seras-Franzoso, A. Perez, L. Alamo Pindado, G. Casas, D. Arango, Y. Fernández, Z. V. Díaz-Riascos, I. Abasolo and S. Schwartz Jr, Cancers, 2019, 11, 1058 CrossRef CAS PubMed.
- R. Inagaki, M. Ninomiya, K. Tanaka and M. Koketsu, ChemMedChem, 2015, 10, 1413–1423 CrossRef CAS PubMed.
- M. M. Rahman, M. R. Islam, S. Akash, S. Shohag, L. Ahmed, F. A. Supti, A. Rauf, A. M. Aljohani, W. Al Abdulmonem, A. A. Khalil and R. Sharma, Chem.-Biol. Interact., 2022, 368, 110198 CrossRef CAS PubMed.
- E. Leyva, L. I. López, R. F. G. de la Cruz and C. G. Espinosa-González, Res. Chem. Intermed., 2017, 43, 1813–1827 CrossRef CAS.
- V. S. de Sena Pereira, F. da Silva Emery, L. Lobo, F. Nogueira, J. I. Oliveira, U. L. Fulco, E. L. Albuquerque, A. M. Katzin and V. F. de Andrade-Neto, Malar. J., 2018, 17, 1–11 CrossRef PubMed.
- X. Li and Y. Song, Eur. J. Med. Chem., 2023, 260, 115772 CrossRef CAS PubMed.
- M. H. Khraiwesh, C. M. Lee, Y. Brandy, E. S. Akinboye, S. Berhe, G. Gittens, M. M. Abbas, F. R. Ampy, M. Ashraf and O. Bakare, Arch. Pharm. Res., 2012, 35, 27–33 CrossRef CAS PubMed.
- P. Milla, C. Fiorito, F. Soria, S. Arpicco, L. Cattel and P. Gontero, Cancer Chemother. Pharmacol., 2014, 73, 503–509 CrossRef CAS PubMed.
- A. Vinayan and R. Glynne-Jones, Best Pract. Res. Clin. Gastroenterol., 2016, 30, 641–653 CrossRef CAS PubMed.
- G. N. Hortobagyi, Drugs, 1997, 54, 1–7 CrossRef CAS PubMed.
- Y. Kumagai, Y. Tsurutani, M. Shinyashiki, S. Homma-Takeda, Y. Nakai, T. Yoshikawa and N. Shimojo, Environ. Toxicol. Pharmacol., 1997, 3, 245–250 CrossRef CAS PubMed.
- E. J. Salaski, G. Krishnamurthy, W. D. Ding, K. Yu, S. S. Insaf, C. Eid, J. Shim, J. I. Levin, K. Tabei, L. Toral-Barza and W. G. Zhang, J. Med. Chem., 2009, 52, 2181–2184 CrossRef CAS PubMed.
- A. Khan, M. S. Said, B. R. Borade, R. Gonnade, V. Barvkar, R. Kontham and S. G. Dastager, J. Nat. Prod., 2022, 85, 1267–1273 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- C. R. Justus, N. Leffler, M. Ruiz-Echevarria and L. V. Yang, J. Vis. Exp., 2014, 88, e51046 Search PubMed.
- C. Luo, Y. Wang, C. Wei, Y. Chen and Z. Ji, Exp. Ther. Med., 2020, 19, 273–279 CAS.
- N. S. Ng and L. Ooi, Bio-Protoc., 2021, 11, e3877 CAS.
- L. Sun, C. Luo and J. Liu, Food Funct., 2014, 5, 1909–1914 RSC.
- M. Redza-Dutordoir and D. A. Averill-Bates, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2977–2992 CrossRef CAS PubMed.
- C. M. Worsley, R. B. Veale and E. S. Mayne, PLoS One, 2022, 17, e0270599 CrossRef CAS PubMed.
- S. S. Azam and S. W. Abbasi, Theor. Biol. Med. Model., 2013, 10, 1–16 CrossRef PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- A. K. Umar, J. H. Zothantluanga, K. Aswin, S. Maulana, M. S. Zubair, H. Lalhlenmawia, M. Rudrapal and D. Chetia, Struct. Chem., 2022, 33, 1445–1465 CrossRef CAS PubMed.
- Y. D. Gao and J. F. Huang, Dongwuxue Yanjiu, 2011, 32, 262–266 CAS.
- E. W. Bell and Y. Zhang, J. Cheminf., 2019, 11, 40 Search PubMed.
- J. Hu, Z. Liu, D. J. Yu and Y. Zhang, Bioinformatics, 2018, 34, 2209–2218 CrossRef CAS PubMed.
- H. M. De Vivo, M. Masetti, G. Bottegoni and A. Cavalli, J. Med. Chem., 2016, 59, 4035–4061 CrossRef PubMed.
- R. Kumari, R. Kumar, Open Source Drug Discovery Consortium and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- J. Yang, P. Cron, V. M. Good, V. Thompson, B. A. Hemmings and D. Barford, Nat. Struct. Mol. Biol., 2002, 9, 940–944 CrossRef CAS PubMed.
- H. Zegzouti, M. Zdanovskaia, K. Hsiao and S. A. Goueli, Assay Drug Dev. Technol., 2009, 7, 560–572 CrossRef CAS PubMed.
- A. Daina and V. Zoete, Nat. Sci. Rep., 2017, 7, 42717 Search PubMed.
- D. E. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Deliv. Rev., 2012, 64, 4–17 CrossRef.
- D. F. Veber, S. R. Johnson, H. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- Z. Ates-Alagoz, M. M. Kisla, F. Z. Karadayi, S. Baran, T. S. Doğan and P. Mutlu, New J. Chem., 2021, 45, 18025–18038 RSC.
- I. Ali, W. A. Wani, K. Saleem and M. F. Hsieh, RSC Adv., 2014, 4, 29629–29641 RSC.
- I. P. Sæbø, M. Bjørås, H. Franzyk, E. Helgesen and J. A. Booth, Int. J. Mol. Sci., 2023, 24, 2914 CrossRef PubMed.
- B. Webb and A. Sali, Curr. Protoc. Bioinf., 2016, 54, 5–6 CrossRef PubMed.
- A. K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P. C. Nair, C. Oostenbrink and A. E. Mark, J. Chem. Theory Comput., 2011, 7, 4026–4037 CrossRef CAS PubMed.
- M. N. Clifford and J. Wight, J. Sci. Food Agric., 1976, 27, 73–84 CrossRef CAS PubMed.
- B. S. Gangadharappa, R. Sharath, P. D. Revanasiddappa, V. Chandramohan, M. Balasubramaniam and T. P. Vardhineni, J. Biomol. Struct. Dyn., 2020, 38, 3757–3771 CrossRef CAS PubMed.
- R. Chandan, M. S. Chaithanya, M. Aditya and K. S. Kiran, J. Biomol. Struct. Dyn., 2022, 1–9 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplemental data figures. See DOI: https://doi.org/10.1039/d3ra06204j |
| This journal is © The Royal Society of Chemistry 2023 |