
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homoleptic octahedral CoII complexes as precatalysts for regioselective hydroboration of alkenes with high turnover frequencies†
Guoqi Zhang *a,
Haisu Zengab,
Nora Zadoria,
Camila Marinoa,
Shengping Zhengb and
Michelle C. Nearyb
*a,
Haisu Zengab,
Nora Zadoria,
Camila Marinoa,
Shengping Zhengb and
Michelle C. Nearyb
aDepartment of Sciences, John Jay College and PhD Program in Chemistry, The Graduate Center of the City University of New York, New York, 10019 NY, USA. E-mail: guzhang@jjay.cuny.edu
bDepartment of Chemistry, Hunter College, The City University of New York, New York, 10065 NY, USA
First published on 22nd September 2023
Abstract
Homoleptic complexes adopting octahedral coordination modes are usually less active in catalysis due to the saturated coordination around metal centers that prevents substrate activation in a catalytic event. In this work, we demonstrated that a homoleptic octahedral cobalt complex (1) OF 4′-pyridyl-2,2′;6′,2′′-terpyridine that experienced monoprotonation at the non-coordinating pyridyl moiety upon crystallization could serve as a highly efficient precatalyst for the hydroboration of styrene derivatives with Markovnikov selectivity. The solid-state structure of this precatalyst along with relevant homoleptic CoII and FeII complexes has been characterized by X-ray crystallography. In the solid state, 1 features one-dimensional hydrogen-bonded chains that are further stacked by interchain π⋯π interactions. The newly synthesized complexes (1–3) along with several known analogues (4–6) were examined as precatalysts for the hydroboration of alkenes. The best-performing system, 1/KOtBu was found to promote Markovnikov hydroboration of substituted styrenes with high turnover frequencies (TOFs) up to ∼47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1, comparable to the most efficient polymeric catalyst [Co(pytpy)Cl2]n reported to date. Although some limitations in substrate scope as well as functional group tolerance exist, the catalyst shows good promise for several relevant hydrofunctionaliation reactions.
000 h−1, comparable to the most efficient polymeric catalyst [Co(pytpy)Cl2]n reported to date. Although some limitations in substrate scope as well as functional group tolerance exist, the catalyst shows good promise for several relevant hydrofunctionaliation reactions.
Introduction
Hydrofunctionalization of unsaturated bonds provides a powerful tool for incorporating valuable functional groups into hydrocarbon compounds.1 In particular, alkene hydroboration is one of the most popular and convenient ways to approach alkylboronates that have extensive applications in C–C bond forming processes through cross-coupling reactions.2 Great advances have been made in the past decade toward developing metal-based catalysts utilizing various transition metals from precious Ru, Rh and Ir to earth abundant ones such as Fe, Co, Ni, Cu, and Mn,3–7 with the latter being the focus of recent research as nowadays chemists are seeking lower cost and more environmentally sustainable catalytic methodologies.Among many well-defined earth-abundant metal catalysts for alkene hydroboration that have emerged over the past decade, cobalt catalysts turned out to be most attractive with respect to the ligand versatility, good regioselectivity of products and high TOFs.8–13 Numerous cobalt catalyst systems have been reported for either branched (Markovnikov) or linear (anti-Markovnikov) selectivity by Chirik,9 Lu,10 Thomas,11 Huang,12 Findlater13 and our group,8 respectively. While all cobalt-based discrete molecular catalysts reported thus far realized alkene hydroboration with relatively low TOFs (<300 h−1), we have disclosed that a one-dimensional CoII-coordination polymer assembled from a divergent ligand, 4′-pyridyl-2,2′; 6′,2′′-terpyridine (pytpy), enabled Markovnikov-selective hydroboration of aryl alkenes with high TOFs up to 47520 h−1.8a However, the origin of the extremely high catalytic efficiency of this polymeric cobalt catalyst remained unclear. To better understand whether the polymeric structure of catalyst is vital for the unexpected activity, we decided to explore discrete molecular analogues of the polymeric precatalyst by using the same pytpy ligand and different cobalt salts (Scheme 1).
 | ||
| Scheme 1 The development of Co-catalysed hydroboration of alkenes. | ||
To continue our recent efforts on earth-abundant metal catalysis with tpy ligands,14 herein, we report the synthesis and structural characterization of homoleptic octahedral CoII and FeII complexes of pytpy containing tetrafluoroborate or hexafluorophosphate counterions (Scheme 2), and their surprisingly high catalytic activity for regioselective hydroboration of alkenes with high TOFs. Although homoleptic octahedral metal complexes of tpy derivatives have been well explored for electrochemical and photophysical properties, and some have been utilized as supramolecular synthons, they are considered to be less catalytically active as the coordination environment of the metal centers makes it relatively inaccessible during a catalytic event. Nevertheless, several examples of homoleptic Co, Fe and Ni tpy complexes have been reported to promote electrocatalytic reduction of CO2,15 owing to their rich redox chemistry that can be tuned by varying electronic substituents on the tpy backbone.16
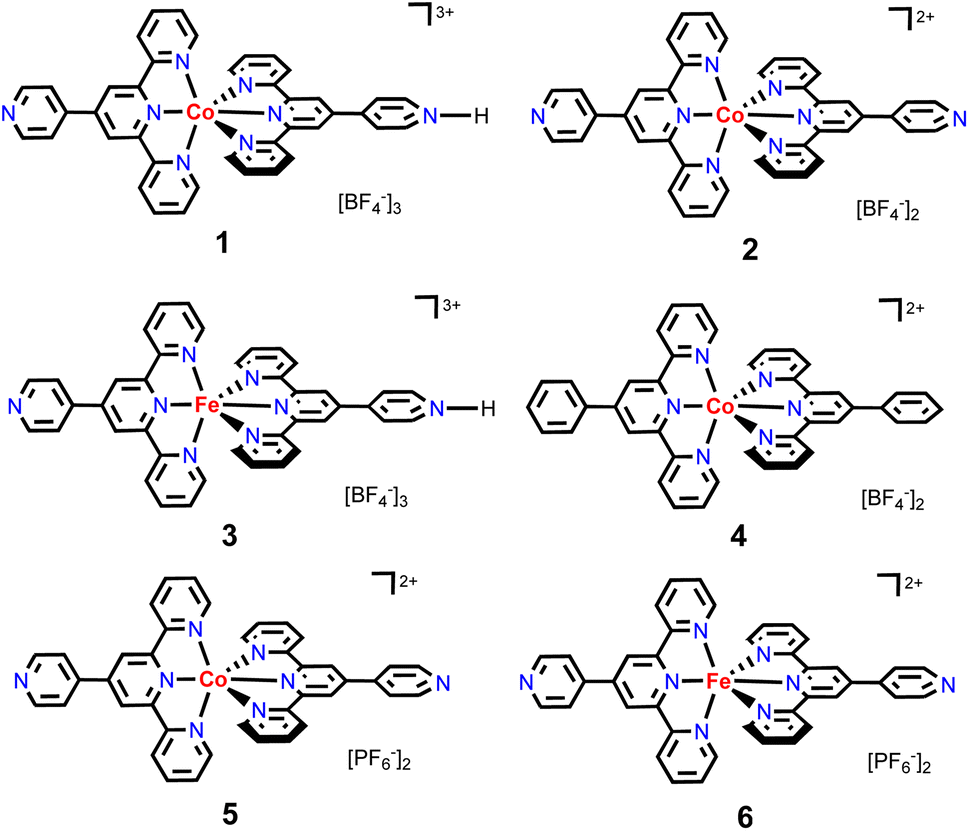 | ||
| Scheme 2 The structures of cobalt and iron complexes 1–6 studied in this work. | ||
Results and discussion
Carefully layering a solution of Co(BF4)2·6H2O in MeOH onto a CH2Cl2–MeOH solution (10 mL, 3:1, v/v) of pytpy over two weeks led to the formation of red block-like crystals of 1 that were suitable for single-crystal X-ray diffraction analysis. A bulk sample of the crystals has been isolated in 92% yield. The solution 1H NMR spectrum of 1 reveals broadened paramagnetic signals that could not be unambiguously assigned. The mass spectrum shows the only peak envelope at 679.1766 that can be assigned to the cation of the complex and the isotope pattern matches with that simulated. The solid-state structure of 1 was confirmed unambiguously by X-ray crystallography as a mono-protonated complex of [Co(pytpy)(H-pytpy)][BF4]3, a product obtained serendipitously during attempts to produce the expected complex [Co(pytpy)2][BF4]2. 1 crystallizes in the monoclinic space group P21/c. The spontaneous partial protonation of pytpy ligand during the complexation with transition metals is reminiscent of known Ru and Fe complexes of the same ligand.17,18 The synthesis of 1 was well reproducible as evidenced from several independent layering experiments as well as the direct reaction between the ligand and Co(BF4)2·6H2O in a CH2Cl2–MeOH solution. In contrast, the originally expected homoleptic complex [Co(pytpy)2][BF4]2 (2) has been synthesized by adopting the standard reaction sequence, i.e. the solution reaction of pytpy and CoCl2·6H2O followed by an anion exchange with excess amount of NaBF4 (see ESI†). X-ray quality single crystals of 2 were obtained by vapor diffusion of diethyl ether into a solution of 2 in acetonitrile over 3 days. The mass spectrum of 2 shows the same peak envelope at 679.1766 as observed in 1. X-ray structural analysis confirmed the structure of 2 as expected and it crystallizes in the monoclinic space group Pc. Co-crystallized solvent molecules of CH2Cl2 and CH3CN were found in each cell of 1 and 2, respectively.
The ORTEP representations of cations of 1 and 2 are shown in Fig. 1, respectively. In the cations of 1 and 2, Co–N bond lengths around the cobalt coordination centers are within 1.8675(17)–2.1519(19) Å for 1 and 1.875(4)–2.182(4) Å for 2 (see caption of Fig. 1), which are unexceptional compared to the known crystal structures of Co(pytpy)2(PF6)2 complexes.19 The ligand conformations in 1 and 2 are not the same. In both structures, the non-coordinated pyridine ring is twisted with respect to the tpy domain to which it is attached. For 1, the angles between the least squares planes of the rings containing atoms N2 and N4, and N6 and N8 are 33.32° and 34.84°, respectively. For 2, the relevant twist angles are 40.36° and 34.71°. In addition, the deviation away from linearity of the N4⋯Co⋯N8 angle is notable for the cation of 1. The angle of N4⋯Co1⋯N8 is 167.73(2)° for the cation of 1, but it is closer to linear in the cation of 2 (177.44(5)°). This is likely due to the ligand protonation in 1 and the resulting formation of one-dimensional (1-D) hydrogen-bonded chains as well as the major interchain π⋯π stacking between ‘side-arm’ pyridine rings observed in 1 (Fig. 2). A similar situation has been reported in relevant Fe and Ru complexes.17,18 As seen in Fig. 2, the 1-D chain is assembled through N4–H4⋯N8(i) hydrogen bonds (symmetry code i = x − 1, y, z − 1, N4–H4 = 0.84(9) Å, H4⋯N8 = 1.82(9) Å, N4–H4⋯N8(i) = 172(7)°) and the chains are packed by π⋯π interaction of ‘side-arm’ pyridine rings (the closest C⋯C contact is 3.446(4) Å). Other types of π-stacking patterns are also observed in the 3-D packing framework of 1. The intermolecular packing in the cation of 2 is dominated by π⋯π stacking between ‘side-arm’ pyridine rings (the closest C⋯C contact is 3.604(7) Å), similar to that of 1.
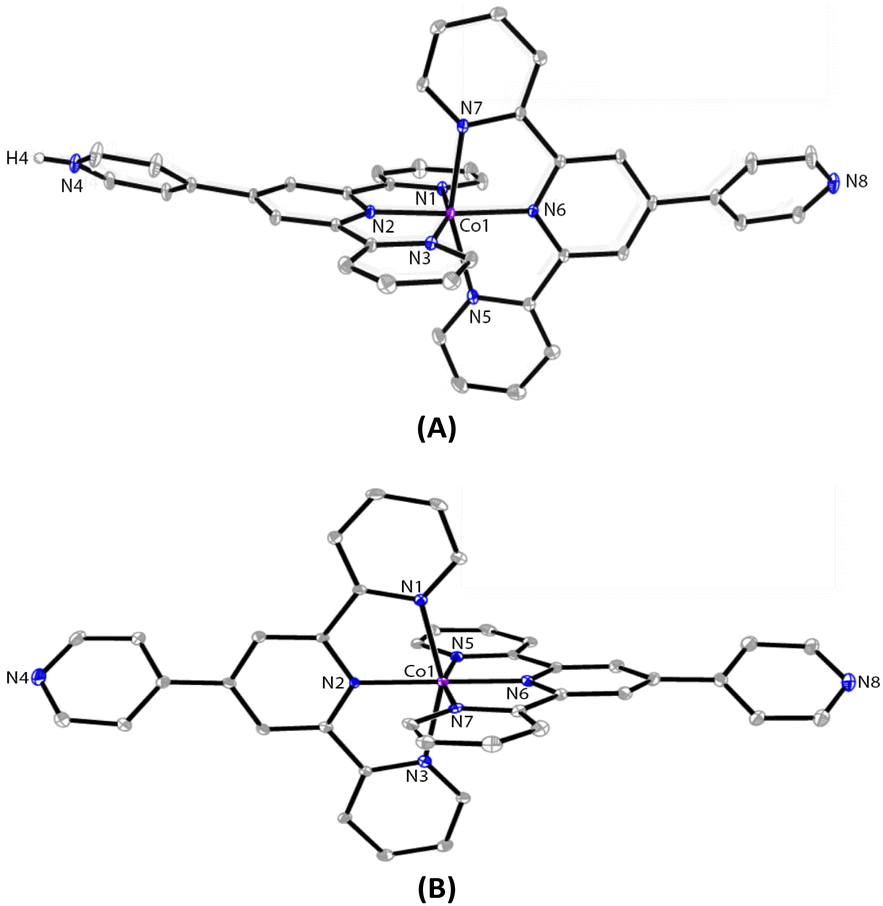 | ||
| Fig. 1 The ORTEP structures of 1 (A) and 2 (B) with thermal ellipsoids drawn at the 30% probability level. BF4− counterions and H atoms bound to C are omitted for clarity. Selected bond parameters for 1: Co1–N1 = 1.998(2), Co1–N2 = 1.8675(17), Co1–N3 = 1.992(2), Co1–N5 = 2.150(2), Co1–N6 = 1.9270(18), Co1–N7 = 2.1519(19), N4–H4 = 0.84(9)Å, N1–Co1–N2 = 81.20(7), N2–Co1–N3 = 81.24(8), N5–Co1–N6 = 78.47(7), N6–Co1–N7 = 79.16(7)°; for 2: Co1–N1 = 2.154(4), Co1–N2 = 1.943(4), Co1–N3 = 2.182(4), Co1–N5 = 1.989(4), Co1–N6 = 1.875(4), Co1–N7 = 1.987(4)Å, N1–Co1–N2 = 78.62(16), N2–Co1–N3 = 77.99(16), N5–Co1–N6 = 81.33(16), N6–Co1–N7 = 80.78(17)°. | ||
 | ||
| Fig. 2 The 1-D hydrogen-bonded chains found in 1 and the π-stacking between the chains (A) and the intermolecular π-stacking in 2 (B). BF4− counterions and co-crystallized solvents are omitted for clarity. | ||
Next, the reaction of pytpy with Fe(BF4)2·6H2O using the same layering method as for 1 resulted in the isolation of X-ray quality crystals of 3. Complex 3 is isomorphic to 1 and also crystallizes in the monoclinic space group P21/c. A disordered solvent molecule in the asymmetric unit of 3 could not be well modelled and so it was treated as a diffuse contribution using PLATON/SQUEEZE.20 The monoprotonated cation of 3 is shown in Fig. 3, which has been reported previously in compounds [Fe(pytpy)(pytpyH)][Fe(NCS)6]·2H2O and [Fe(pytpy)(pytpyH)][Fe(NCS)6]·MeCN.18 The bending conformation in cation of 3 is very close to that found in 1 (angle N4⋯Fe1⋯N8 is 168.04(6)°), resulting from the intermolecular hydrogen bond N4–H4⋯N8(i) (symmetry code i = x + 1, y, z + 1, N4–H4 = 0.88(2) Å, H4⋯N8 = 1.80(3) Å, N4–H4⋯N8(i) = 167(8)°). Again, a similar 1-D hydrogen-bonded chain was found in 3. As referenced, homoleptic complexes 4–6 (Scheme 2) were synthesized according to the procedure reported previously.21
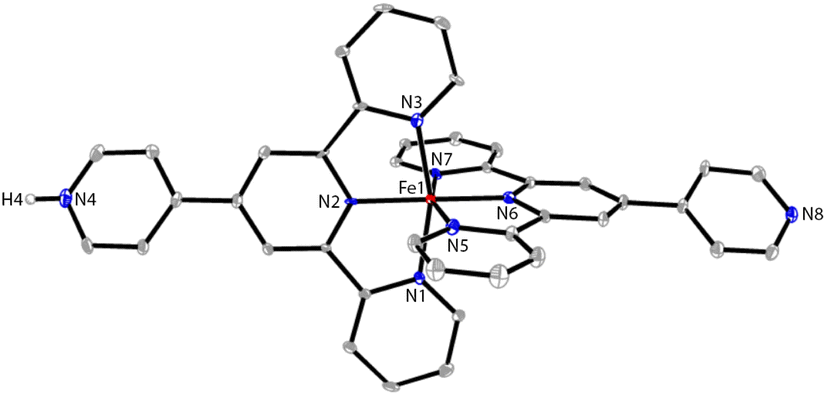 | ||
| Fig. 3 The ORTEP structure of 3 with thermal ellipsoids drawn at the 30% probability level. BF4− counterions and H atoms bound to C are omitted for clarity. Selected bond parameters: Fe1–N1 = 1.982(5), Fe1–N2 = 1.882(5), Fe1–N3 = 1.972(5), Fe1–N5 = 1.972(5), Fe1–N6 = 1.876(5), Fe1–N7 = 1.976(5), N4–H4 = 0.88(2)Å, N1–Fe1–N2 = 81.0(2), N2–Fe1–N3 = 81.2(2), N5–Fe1–N6 = 80.6(2), N6–Fe1–N7 = 81.2(2)°. | ||
In order to evaluate whether new octahedral CoII and FeII complexes could be used as effective precatalysts for alkene hydroboration, we first adopted the optimized conditions as for [Co(pytpy)Cl2]n to perform catalytic hydroboration of styrene with pinacolborane (HBpin). The results of catalytic screening are summarized in Table 1. To our delight, when 1 (0.025 mol%) and KOtBu (1 mol%) were combined in THF, the hydroboration of styrene was realized in 5 min to give the branched (7) and linear alkylboronates (8) in 98% total yield (TOF = ∼47000 h−1 for both regioisomers) and 9:1 regioselectivity (entry 1, Table 1), closely comparable to the results obtained by using polymeric [Co(pytpy)Cl2]n as a precatalyst. This is remarkable and represents the first homoleptic octahedral cobalt(II) complex to enable alkene hydroboration with extremely high efficiency. Likewise, when 2 was used for the reaction under the same conditions, similar regioselectivity was found, while the yield dropped slightly to 80% (TOF = ∼38000 h−1). Interestingly, going from CoII to FeII resulted in a notable loss of catalytic activity, as 3 catalysed the reaction with 17% yield in 5 min (8000 h−1), although the regioselectivity remained (entry 3). In addition, both complexes 4 and 5 are moderately active precatalysts for styrene hydroboration (entries 4 and 5), indicating the importance of both the pytpy ligand and BF4− counterions for high catalytic efficiency. According to these results, it is not surprising to see that complex 6, Fe(pytpy)2(PF6)2, is an inactive precatalyst (entry 6) under standard conditions.
|
|||||
|---|---|---|---|---|---|
| Entry | Precatalyst | Activator | Solvent | Yieldb (%) | Ratioc (b/l) |
| a Conditions: styrene (1.0 mmol), HBpin (1.1 mmol), precatalyst (0.025 mol%), activator (1 mol%) and solvent (0.5 mL), 25 °C, 5 min, N2.b Yield of 7a + 8a, determined by GC analysis with hexamethylbenzene as an internal standard.c Ratio (b/l = 7a: 8a) determined by GC analysis.d Reaction run in the air.e Reaction run using 0.025 mol% of microcrystalline sample of 1. |
|||||
| 1 | 1 | KOtBu | THF | 98 | 9:1 |
| 2 | 2 | KOtBu | THF | 80 | 8:1 |
| 3 | 3 | KOtBu | THF | 17 | 10:1 |
| 4 | 4 | KOtBu | THF | 35 | 8:1 |
| 5 | 5 | KOtBu | THF | 41 | 9:1 |
| 6 | 6 | KOtBu | THF | <2 | — |
| 7 | 1 | KOtBu | Neat | 50 | 3:1 |
| 8 | 1 | KOtBu | Toluene | <1 | — |
| 9 | 1 | KOtBu | CH2Cl2 | 2 | — |
| 10 | 1 | KOtBu | Et2O | 62 | 9:1 |
| 11 | 1 | KOtBu | DMSO | 58 | 5:1 |
| 12 | 1 | NaOtBu | THF | 83 | 6:1 |
| 13 | 1 | LiOtBu | THF | 20 | 3:1 |
| 14 | 1 | KOCH3 | THF | 72 | 5:1 |
| 15 | 1 | K2CO3 | THF | 59 | 8:1 |
| 16 | 1 | LiNTf2 | THF | 2 | — |
| 17 | 1 | NaHBEt3 | THF | 88 | 5:1 |
| 18 | — | KOtBu | THF | 0 | — |
| 19 | 1 | — | THF | 2 | — |
| 20 | Co(BF4)2 | KOtBu | THF | 0 | — |
| 21d | 1 | KOtBu | THF | <5 | — |
| 22e | 1 | KOtBu | THF | 97 | 9:1 |
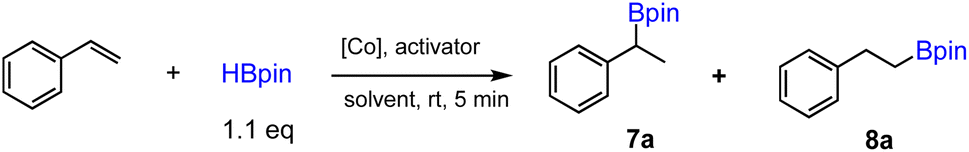
Having established the ability of 1 as the best-performing precatalyst among six homoleptic complexes, we further screened the influence of reaction conditions such as solvents and activators on the catalytic performance. The solvent effect proved to be significant (entries 7–11). Much lower yield and regioselectivity were observed when the reaction was conducted without a solvent. Toluene and dichloromethane were incompatible solvents for this reaction, as only trace amount of product has been detected for reactions in these solvents. Diethyl ether and dimethylsulfoxide are relatively better, yet still inferior to THF in terms of yield and selectivity. Next, we examined the reaction with different activators (entries 12–17). The results suggest the key role played by KOtBu as an activator for both high yield and regioselectivity, while other common activators such as NaOtBu, K2CO3 and NaHBEt3 all led to relatively poor results. Finally, control experiments were carried out to show that both cobalt complex and an activator are required to initiate the reaction (entries 18 and 19). It is also worth noting that when Co(BF4)2 was used instead complex 1 as a precatalyst, no hydroboration of styrene was detected (entry 20). Under the optimal conditions with 1/KOtBu as catalyst, the reaction did not proceed when it was exposed to the air, indicating that a highly air-sensitive reactive intermediate (likely a cobalt hydride species) must have formed and was responsible for the high-efficiency catalysis (entry 21). It is worth mentioning that to achieve the high TOF and regioselectivity, a single crystalline sample was not necessary. Instead, microcrystalline material of 1 could be synthesized in a gram-scale within 1–2 h by simply mixing concentrated, equimolar solutions of pytpy and Co(BF4)2·6H2O in CH2Cl2 and MeOH, respectively (see the ESI†), and the hydroboration of styrene using this microcrystalline sample was found to be equally efficient (entry 22).
To further demonstrate the effectiveness of 1 as a precatalyst for other alkene substrates, we employed the optimized conditions (entry 1, Table 1) to examine substituted and functionalized styrenes. The results for a range of substrates tested are summarized in Scheme 3. First, methyl- and fluoro-substituted styrenes are suitable substrates affording the corresponding alkylboronates with high TOFs and slightly lower regioselectivity. Product 7b was readily isolated from the mixture with 80% yield. However, when 4-chloro- or 2-chlorostyrene was used, the reaction ran slower and moderate yields were obtained in 5 min for both cases. Lower TOF (28000 h−1) was also found in the case of 4- trifluoromethylstyrene as a substrate. Styrene with an electron-donating 4-methoxy group proceeded well with good yield, while the regioselectivity dropped to 5:1 (7g). cis-Stilbene was found to be active substrate for hydroboration to afford 7h with appreciable isolated yield and high TOF. However, 1,1-disubstituted alkene shows poor reactivity under standard conditions (7i). Styrenes containing functional groups such as nitro, amino or pyridyl are inactive substrates, similar to the results reported previously using polymeric [Co(pytpy)Cl2]n as a precatalyst.8a In addition, aliphatic and cyclic alkenes are also reactive substrates, however, poor regioselectivity was obtained for 1-hexene (7n) and anti-Markovnikov selectivity was found for vinylcyclohexane (7o).
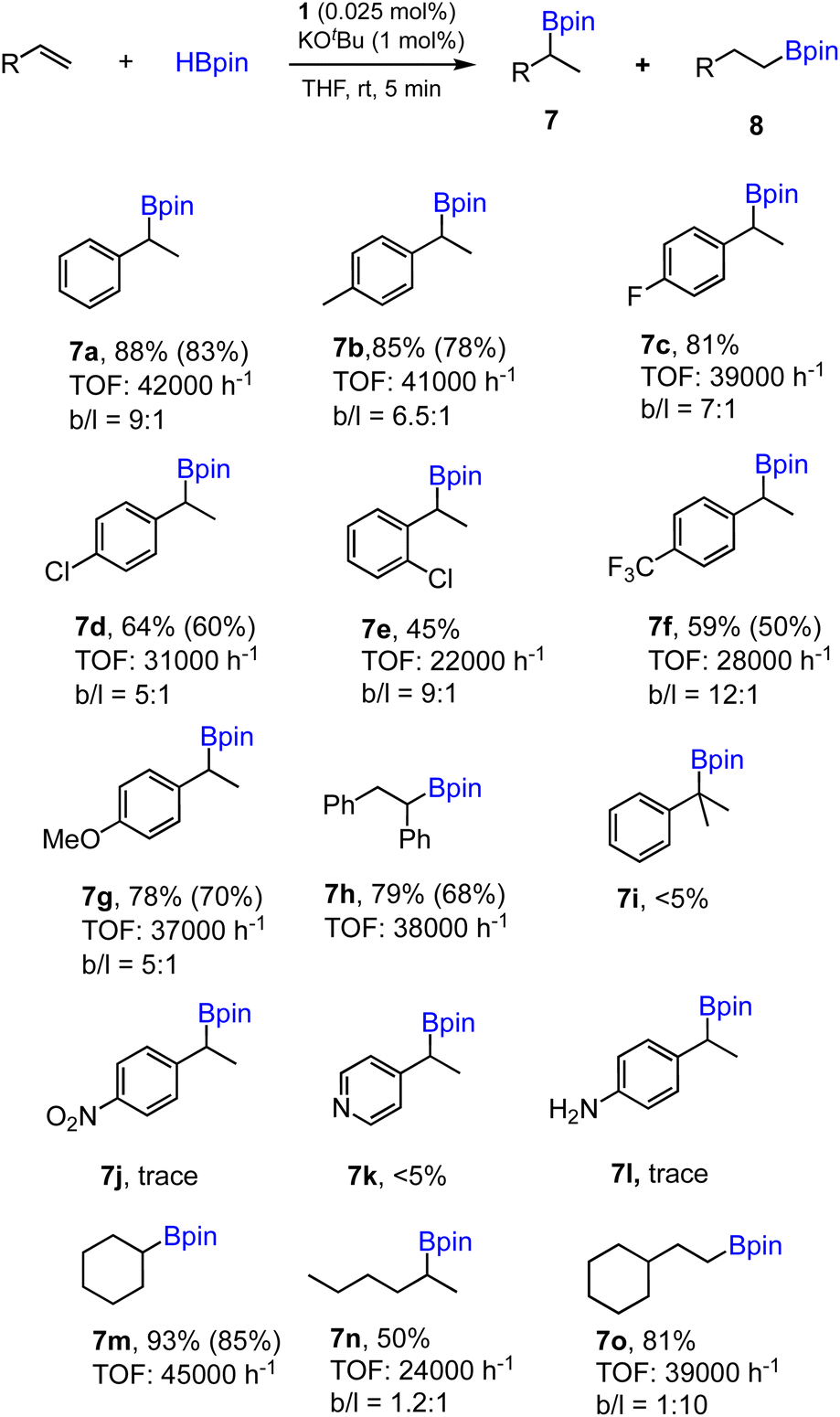 | ||
| Scheme 3 Substrate scope for the hydroboration of alkenes using 1/KOtBu. Conditions: alkene (1.0 mmol), HBpin (1.1 mmol), 1 (0.025 mol%), KOtBu (1 mol%) and THF (0.5 mL), 25 °C, 5 min, N2. Yields of products 7 and the ratio 7/8 (b/l) determined by GC analysis with hexamethylbenzene as an internal standard. TOF determined based on the yield of regioisomer 7 only. Yields of isolated products given in parentheses. | ||
Next, we explored the functional group tolerance of 1/KOtBu system for styrene hydroboration by adding a second reducible substrate, which is so called a fast catalyst robustness screening.22 Thus, styrene was chosen to react with HBpin under standard conditions in the presence of equimolar additives as listed in Table 2. Both ketone and aldehyde showed strongly competing reactions with styrene, as 60% ketone and 95% aldehyde hydroboration was detected as the major reactions, respectively (entries 1 and 2, Table 2). The ester showed little influence on the efficiency of styrene hydroboration with no change on the regioselectivity (entry 3). However, both amide and nitrobenzene have completely suppressed the reactions (entries 4 and 5). Interestingly, butyronitrile is compatible with the reaction, while increasing the regioselectivity to 15:1 (entry 6). Finally, the presence of styrene oxide significantly decreased the yield of 7 albeit the regioselectivity remained. These results indicate somewhat inferior functional group tolerance of homoleptic complex 1, compared to the polymeric precatalyst [Co(pytpy)Cl2]n.8a,14
|
|||
|---|---|---|---|
| Entry | Additive | Yield (7a + 8a)b/% | Ratio (7a/8a)b |
| a Conditions: styrene (0.5 mmol), HBpin (0.5 mmol), 1 (0.025 mol%), KOtBu (1 mol%), other reducible substrate (0.5 mmol) and THF (1 mL), 25 °C, 5 min, N2.b Determined by GC analysis using hexamethylbenzene as an internal standard.c 60% ketone hydroboration was detected.d 95% aldehyde hydroboration was detected. | |||
| 1c | 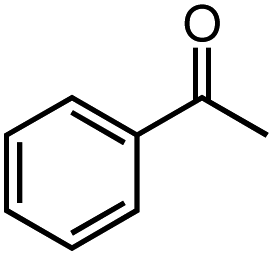 |
40% | 8:1 |
| 2d |  |
4% | — |
| 3 | 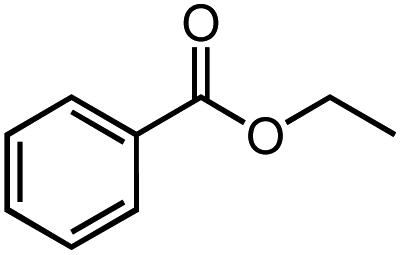 |
72% | 9:1 |
| 4 |  |
6% | — |
| 5 | 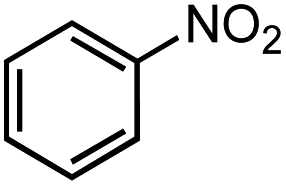 |
3% | — |
| 6 | 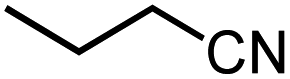 |
90% | 15:1 |
| 7 | 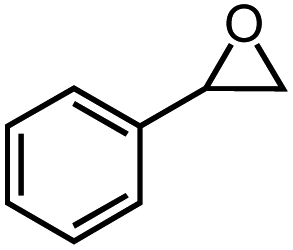 |
35% | 9:1 |
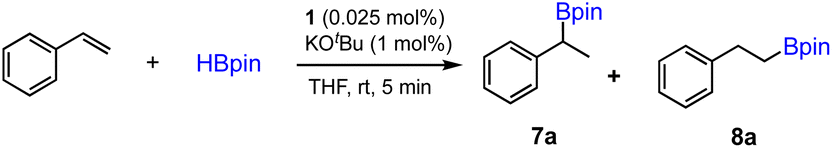
The ability of precatalyst 1 to promote hydroboration and hydrosilylation for several other substrates was further evaluated. The preliminary results are presented in Scheme 4. The hydroboration of ketone was furnished under the standard conditions within 5 min, while phenylacetylene is almost inactive for hydroboration after 1 h. This is in sharp contrast with the results obtained using [Co(pytpy)Cl2]n as precatalyst where very high TOFs could be achieved.14b In addition, hydrosilylation of styrene and phenylacetylene using phenylsilane as a Si–H source has been investigated. It was found that styrene has experienced effective hydrosilylation to afford the anti-Markovnikov product with complete regioselective control. However, hydrosilylation of phenylacetylene was accomplished within 16 h in 95% yield with poor regioselectivity (b/l = 4:5). The results indicate that both reactivity and regioselectivity are highly substrate-dependent when using 1/KOtBu as the catalyst.
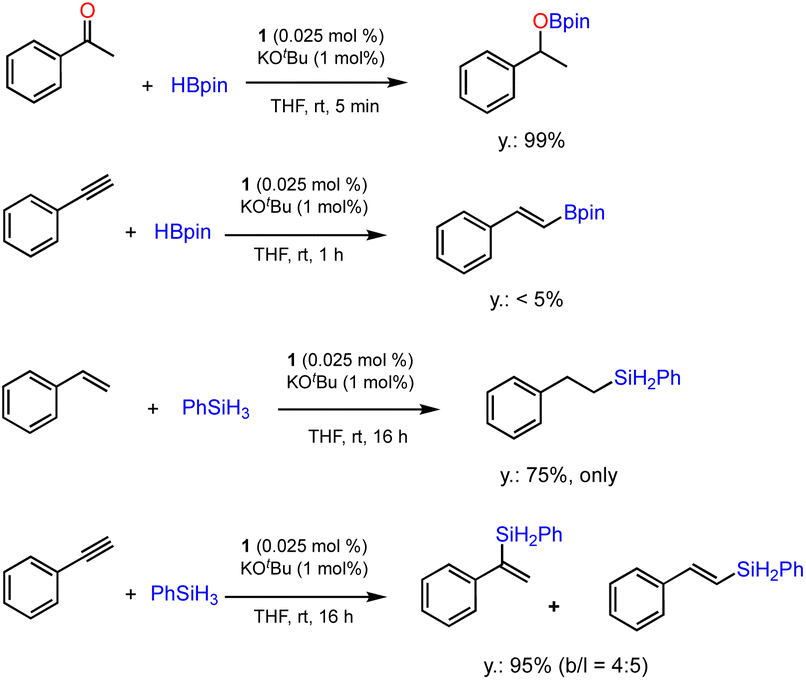 | ||
| Scheme 4 Additional catalytic tests for hydroboration and hydrosilylation reactions using 1. Conditions: substrate (0.5 mmol), HBpin or PhSiH3 (0.55 mmol), 1 (0.025 mol%), KOtBu (1 mol%) and THF (0.5 mL), 25 °C, N2, indicated time. Yields and regioselectivity determined by GC analysis using hexamethylbenzene as an internal standard. | ||
Conclusions
In summary, we have synthesized and characterized homoleptic complexes of cobalt or iron with pytpy ligand. The direct reaction of pytpy and metal tetrafluoroborate resulted in the monoprotonation at the non-coordinating pyridyl moiety, namely [M(pytpy)(H-pytpy)][BF4]3 (M = Co or Fe). X-ray structural analysis reveals the conformational bending of these complexes in the solid state due to the formation of 1-D hydrogen-bonded chains, in comparison with the common complex, Co(pytpy)2(BF4)2. These new complexes (1–3) along with several known analogues (4–6) have been explored as precatalysts for the regioselective hydroboration of alkenes. The best catalytic system 1/KOtBu was found to furnish the hydroboration of styrene with a very high TOF of ∼47000 h−1, comparable to the most efficient precatalyst [Co(pytpy)Cl2]n reported thus far. The method can be applied to a range of styrene derivatives for the regioselective synthesis of branched alkylboronates. However, some limitation of substrates has been disclosed. Catalyst robustness screening experiments provide further insights into the functional group tolerance of current catalyst. Preliminary experiments on relevant substrates for hydroboration and hydrosilylation catalysis were also conducted to extend its applicability for other conversions.
Experimental
General
Unless specified otherwise, all reactions were carried out under a dry N2 atmosphere using standard glovebox and Schlenk techniques. Anhydrous grade solvents and reagents used were obtained from Aldrich or Fisher Scientific and stored over 4 Å molecular sieves. All chemicals of analytical grade including the alkene substrates and additives are used as received from Alfa Aesar, Acros, TCI America or Fisher Scientific without further purification. FT-IR spectra were recorded on a Shimadzu 8400S instrument with solid samples under N2 using a Golden Gate ATR accessory. Elemental analyses were performed by Midwest Microlab LLC in Indianapolis in the US. 1H NMR and 13C NMR spectra were obtained at room temperature on a Bruker AV 400, 500 or 600 MHz NMR spectrometer, with chemical shifts (δ) referenced to the residual solvent signal. HR-MS data were obtained on an Agilent 6550 QToF coupled to an Agilent 1290 Infinity LC system. GC-MS analysis was obtained using a Shimadzu GCMS-QP2010S gas chromatograph mass spectrometer (column: SHRX1-5MS, thickness: 0.25 m, diameter: 0.25 mm, length: 30.0 m; conditions: 30–200 °C, 10 °C min−1, injection temperature: 100 °C; solvent cutoff: 3 min).Synthesis of 1
A solution of pytpy (31.0 mg, 0.100 mmol) in MeOH/CH2Cl2 (10 mL, 1:3, v/v) was placed in a test tube. A blank solution of MeOH/CH2Cl2 (4 mL, 1:1, v/v) was layered on the top of the ligand solution, followed by a solution of Co(BF4)2·6H2O (34.1 mg, 0.100 mmol) in MeOH (8 mL). The tube was sealed and allowed to stand at room temperature for about four weeks, during which time X-ray quality yellow blocks grew at the bottom of the tube. The crystals were collected by decanting the solvent and washed with MeOH and then dried in vacuo. Yield: 43.2 mg (92% based on pytpy). FT-IR (solid, cm−1): 3078m, 1620m, 1597s, 1538s, 1470s, 1428s, 1405s, 1247s, 1031br, 822s, 786s, 733m. 1H NMR (500 MHz, DMSO) δ 83.31 (bs), 48.22 (bs), 31.38 (bs), 30.84 (bs), 12.82 (bs), 10.49 (bs), 9.87 (bs) ppm. HR-MS (ESI positive): 679.1766 ([M-3(BF4−)-H+], Cald. 679.1769). Anal. Calcd. for C40H29B3CoF12N8: C 51.05, H 3.11, N 11.91%. Found C 50.82, H 3.01, N 11.69. In a separate experiment, in a 100 mL flask, pytpy (0.62 g, 2.00 mmol) was dissolved in MeOH/CH2Cl2 (40 mL, 1:3, v/v), to which a solution of Co(BF4)2·6H2O (0.68 g, 2.00 mmol) in MeOH (20 mL) was added dropwise in 3 min. The reaction mixture was allowed to stay for an additional 1.5 h, red microcrystals had formed and were filtered to give bulk sample of 1 (yield: 0.83 g, 88%). Catalytic experiment confirmed its activity and efficiency for styrene hydroboration (entry 22, Table 1).
Synthesis of 2
pytpy (31.0 mg, 0.100 mmol) was dissolved in MeOH/CH2Cl2 (8 mL, 1:3, v/v) in a 20 mL vial, to which was added a solution of CoCl2·6H2O (11.9 mg, 0.050 mmol) in MeOH (3 mL). The mixture was stirred for 15 min at ambient temperature and then a solution of NaBF4 (66.0 mg, 0.600 mmol) in MeOH (2 mL) was added, the resulting precipitate was filtered, washed with MeOH and dried in vacuo. X-ray quality crystals were obtained by slow diffusion of diethyl ether into a concentrated solution of 2 in acetonitrile. Yield: 34.5 mg (81%). FT-IR (solid, cm−1): 3057m, 1619m, 1597s, 1538s, 1470s, 1407s, 1245s, 1053br, 896m, 823m, 789s, 732m. 1H NMR (400 MHz, DMSO) δ 83.42 (bs), 48.26 (bs), 31.69 (bs), 30.80 (bs), 12.82 (bs), 10.43 (bs), 9.92 (bs) ppm. HR-MS (ESI positive): 679.1766 ([M-2(BF4−)], Cald. 679.1769). Anal. Calcd. for C40H28B2CoF8N8: C 56.31, H 3.31, N 13.13%. Found C 56.05, H 3.19, N 13.04.
Synthesis of 3
The procedure is similar to that for 1, except that Fe(BF4)2·6H2O (33.8 mg, 0.100 mmol) was used. Brown plate-like crystals of 3 were collected in 85% yield (40 mg). FT-IR (solid, cm−1): 3603w, 3540w, 3075m, 1595s, 1537s, 1482m, 1467m, 1408s, 1286m, 1246m, 1053br, 895m, 821s, 788s, 755s, 733m. HR-MS (ESI positive): 676.1774 ([M-3(BF4−)-H+], Cald. 676.1780).General procedure for 1-catalysed alkene hydroboration
In a glovebox under N2 atmosphere, cobalt catalyst 1 (0.23 mg, 0.25 μmol, 0.025 mol%) and KOtBu (1.12 mg, 1 mol%) was dissolved in THF (0.5 mL) in a 3.8 mL glass vial equipped with a small stir bar. The mixture was stirred for 1 min. Alkenes (1.0 mmol) and pinacolborane (141 mg, 1.1 mmol) were then added. The reaction mixture was allowed to stir at room temperature for 5 min and then the reaction was quenched by exposing the reaction solution to air and adding CH2Cl2 (1 mL) to the solution. The crude reaction mixture was first analyzed by GC-MS to determine the total yields of desired alkylboronates and the ratio of the regioisomeric products by comparing the GC traces with those of authentic samples.8a The reaction mixture for several selected products was then evaporated under reduced pressure and the product was purified through a SiO2 column chromatography using ethyl acetate/hexane as an eluent. The pure alkylboronates of major products were characterized by 1H and 13C NMR spectroscopies.Catalyst robustness screening
In a glovebox under N2 atmosphere, cobalt precatalyst 1 (0.12 mg, 0.025 mol%) and KOtBu (1.12 mg, 1 mol%) was dissolved in THF (0.5 mL) in a 3.8 mL glass vial equipped with a stir bar. The mixture was stirred for 1 min. Styrene (52 mg, 0.5 mmol), additive (0.5 mmol) and pinacolborane (64 mg, 0.5 mmol) were then added sequentially. Hexamethylbenezene (25 mg) was added as an internal standard for GC analysis. The reaction mixture was allowed to stir at room temperature for 5 min. The reaction was quenched by exposing the reaction solution to air and adding CH2Cl2 (1 mL) to the solution. The crude product was analyzed by GC-MS to determine the GC yield and ratio of the regioisomeric products from styrene hydroboration. In each case, the identification of the corresponding boronate esters have been made by comparing their GC retention time and MS data with the authentic samples.8aX-ray crystallography
X-ray diffraction data were collected on a Bruker X8 Kappa Apex II diffractometer using Mo Kα radiation (for 1) or on a Bruker D8 VENTURE diffractometer using Cu Kα radiation (for 2 and 3). Crystal data, data collection and refinement parameters are summarized in Table S1 (ESI†). The structures were solved using a dual-space method and standard difference map techniques and were refined by full-matrix least-squares procedures on F2 with SHELXTL.23 All hydrogen atoms bound to carbon were placed in calculated positions and refined with a riding model [Uiso(H) = 1.2 − 1.5Ueq(C)]. The hydrogen atom bound to nitrogen was located on the difference map and refined freely or with an N–H distance restraint (for 3) [Uiso(H) = 1.2Ueq(N)].Author contributions
G. Z. and S. Z. conceived and supervised the project. H. Z., N. Z. and C. M. performed experimental studies. M. C. N. conducted the X-ray crystallographic analysis. All authors analysed the data and wrote the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the financial support from the National Science Foundation (CHE #1900500 and #2247728) for this work. We also acknowledge the PSC-CUNY awards (65203-0053 and 66076-0054), the Seed grant from the Office for Advancement of Research and the PRISM program at John Jay College, the City University of New York. The X-ray diffractomer used for 2 and 3 was proved by the Air Force Office of Scientific Research under award number FA9550-20-1-0158.Notes and references
- (a) B. T. Cho, Chem. Soc. Rev., 2009, 38, 443–452 RSC; (b) A. Togni and H. Grützmacher, Catalytic Heterofunctionalization, Wiley-VCH, Weinheim, 2001 CrossRef; (c) M. B. Smith and J. March, March's Advanced Organic Chemistry, Wiley-Interscience, Hoboken, NJ, 6th edn, 2007, pp. 1703–1869 Search PubMed; (d) Hydrofunctionalization, Topics in Organometallic Chemistry, ed. V. P. Ananikov and M. Tanaka, Springer–Verlag, Berlin, Heidelberg, 2013, p. 43 Search PubMed.
- A. Singh, S. Shafiei-Haghighi, C. R. Smith, D. K. Unruh and M. Findlater, Asian J. Org. Chem., 2020, 9, 416–420 CrossRef CAS.
- (a) K. Burgess and M. J. Ohlmeyer, Transition-Metal Promoted Hydroborations of Alkenes, Emerging Methodology for Organic Transformations, Chem. Rev., 1991, 91, 1179–1191 CrossRef CAS; (b) C. M. Crudden and D. Edwards, Catalytic Asymmetric Hydroboration: Recent Advances and Applications in Carbon–Carbon Bond-Forming Reactions, Eur. J. Org Chem., 2003, 2003, 4695–4712 CrossRef; (c) Y. Yamamoto, R. Fujikawa, T. Umemoto and N. Miyaura, Iridium-Catalyzed Hydroboration of Alkenes with Pinacolborane, Tetrahedron, 2004, 60, 10695–10700 CrossRef CAS; (d) A. M. Carroll, T. P. O'Sullivan and P. J. Guiry, The Development of Enantioselective Rhodium-Catalysed Hydroboration of Olefins, Adv. Synth. Catal., 2005, 347, 609–631 CrossRef CAS; (e) C. M. Vogels and S. W. Westcott, Recent Advances in Organic Synthesis Using Transition Metal-Catalyzed Hydroborations, Curr. Org. Chem., 2005, 9, 687–699 CrossRef CAS; (f) D. R. Edwards, Y. B. Hleba, C. J. Lata, L. A. Calhoun and C. M. Crudden, Regioselectivity of the Rhodium-Catalyzed Hydroboration of Vinyl Arenes: Electronic Twists and Mechanistic Shifts, Angew. Chem., Int. Ed., 2007, 46, 7799–7802 CrossRef CAS PubMed; (g) S. Kisan, V. Krishnakumar and C. Gunanathan, Ruthenium-Catalyzed Anti-Markovnikov Selective Hydroboration of Olefins, ACS Catal., 2017, 7, 5950–5954 CrossRef CAS; (h) G. Wang, X. Liang, L. Chen, Q. Gao, J. G. Wang, P. Zhang, Q. Peng and S. Xu, Iridium-Catalyzed Distal Hydroboration of Aliphatic Internal Alkenes, Angew. Chem., Int. Ed., 2019, 58, 8187–8191 CrossRef CAS PubMed.
- (a) J. V. Obligacion and P. J. Chirik, Earth-Abundant Transition Metal Catalysts for Alkene Hydrosilylation and Hydroboration: Opportunities and Assessments, Nat. Rev. Chem, 2018, 2, 15–34 CrossRef CAS PubMed; (b) M. D. Greenhalgh, A. S. Jones and S. P. Thomas, Iron-Catalysed Hydrofunctionalisation of Alkenes and Alkynes, ChemCatChem, 2015, 7, 190–222 CrossRef CAS; (c) H. Wen, G. Liu and Z. Huang, Recent advances in tridentate iron and cobalt complexes for alkene and alkyne hydrofunctionalizations, Coord. Chem. Rev., 2019, 386, 138–153 CrossRef CAS; (d) W. Fan, L. Li and G. Zhang, Branched-selective alkene hydroboration catalyzed by earth-abundant metals, J. Org. Chem., 2019, 84, 5987–5996 CrossRef CAS PubMed; (e) S. R. Tamang and M. Findlater, Emergence and Applications of Base Metals (Fe, Co, and Ni) in Hydroboration and Hydrosilylation, Molecules, 2021, 24, 319 Search PubMed; (f) J. Guo, Z. Cheng, J. Chen, X. Chen and Z. Lu, Iron-and cobalt-catalyzed asymmetric hydrofunctionalization of alkenes and alkynes, Acc. Chem. Res., 2021, 54, 2701–2716 CrossRef CAS PubMed.
- C. Li, S. Song, Y. Li, C. Xu, Q. Luo, Y. Guo and X. Wang, Selective hydroboration of unsaturated bonds by an easily accessible heterotopic cobalt catalyst, Nat. Commun., 2021, 12, 3813 CrossRef CAS PubMed.
- M. Zhong, Y. Gagné, T. O. Hope, X. Pannecoucke, M. Frenette, P. Jubault and T. Poisson, Copper-Photocatalyzed Hydroboration of Alkynes and Alkenes, Angew. Chem., Int. Ed., 2021, 133, 14619–14624 CrossRef.
- G. Zhang, H. Zeng, J. Wu, Z. Yin, S. Zheng and J. C. Fettinger, Highly Selective Hydroboration of Alkenes, Ketones and Aldehydes Catalyzed by a Well-Defined Manganese Complex, Angew. Chem., Int. Ed., 2016, 55, 14369–14372 CrossRef CAS PubMed.
- (a) G. Zhang, J. Wu, S. Li, S. Cass and S. Zheng, Markovnikov-Selective Hydroboration of Vinylarenes Catalyzed by a Cobalt (II) Coordination Polymer, Org. lett., 2018, 20, 7893–7897 CrossRef CAS PubMed; (b) G. Zhang, J. Wu, M. Wang, H. Zeng, J. Cheng, M. C. Neary and S. Zheng, Cobalt-Catalyzed Regioselective Hydroboration of Terminal Alkenes, Eur. J. Org Chem., 2017, 2017, 5814–5818 CrossRef CAS.
- W. N. Palmer, T. Diao, I. Pappas and P. J. Chirik, High-Activity Cobalt Catalysts for Alkene Hydroboration with Electronically Responsive Terpyridine and α-Diimine Ligands, ACS Catal., 2015, 5, 622–626 CrossRef CAS.
- X. Chen, Z. Cheng and Z. Lu, Cobalt-Catalyzed Asymmetric Markovnikov Hydroboration of Styrenes, ACS Catal., 2019, 9, 4025–4029 CrossRef CAS.
- J. Peng, J. H. Docherty, A. P. Dominey and S. P. Thomas, Cobalt-Catalysed Markovnikov Selective Hydroboration of Vinylarenes, Chem. Commun., 2017, 53, 4726–4729 RSC.
- J. Liu, J. Du, F. Yu, L. Gan, G. Liu and Z. Huang, Thermally Robust and Highly Active PCP Cobalt (II) Catalyst for Linear-Selective Hydroboration of Terminal and Internal Olefins, ACS Catal., 2023, 13, 7995–8003 CrossRef CAS.
- S. R. Tamang, D. Bedi, S. Shafiei-Haghighi, C. R. Smith, C. Crawford and M. Findlater, Cobalt-catalyzed hydroboration of alkenes, aldehydes, and ketones, Org. Lett., 2018, 20, 6695–6700 CrossRef CAS PubMed.
- (a) J. Wu, H. S. Zeng, J. Cheng, S. P. Zheng, J. A. Golen, D. R. Manke and G. Zhang, J. Org. Chem., 2018, 83, 9442–9448 CrossRef CAS PubMed; (b) G. Zhang, S. Li, J. Wu, H. Zeng, Z. Mo, K. Davis and S. Zheng, Org. Chem. Front., 2019, 6, 3228–3233 RSC; (c) G. Zhang, J. Wu, H. Zeng, M. C. Neary, M. Devany, S. Zheng and P. A. Dub, ACS Catal., 2019, 9, 874–884 CrossRef CAS; (d) G. Zhang, J. Wu, S. Zheng, M. C. Neary, J. Mao, M. Flores, R. J. Trovitch and P. A. Dub, J. Am. Chem. Soc., 2019, 141, 15230–15239 CrossRef CAS PubMed; (e) G. Zhang, J. Cheng, K. Davis, M. G. Bonifacio and C. Zajaczkowski, Green Chem., 2019, 21, 1114–1121 RSC; (f) G. Zhang, H. Zeng, S. Zheng, M. C. Neary and P. A. Dub, Vanadium-Catalyzed Stereo-and Regioselective Hydroboration of Alkynes to Vinyl Boronates, ACS Catal., 2022, 12, 5425–5429 CrossRef CAS; (g) G. Zhang, S. Zheng and M. C. Neary, An ionic Fe-based metal–organic-framework with 4′-pyridyl-2,2′:6′,2′′-terpyridine for catalytic hydroboration of alkynes, RSC Adv., 2023, 13, 2225–2232 RSC.
- (a) N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Terpyridine complexes of first row transition metals and electrochemical reduction of CO2 to CO, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC; (b) S. Suhr, N. Schröter, M. Kleoff, N. Neuman, D. Hunger, R. Walter, C. Lücke, F. Stein, S. Demeshko, H. Liu and H. U. Reissig, Spin State in Homoleptic Iron (II) Terpyridine Complexes Influences Mixed Valency and Electrocatalytic CO2 Reduction, Inorg. Chem., 2023, 62, 6375–6386 CrossRef CAS PubMed.
- J. C. Dickenson, M. E. Haley, J. T. Hyde, Z. M. Reid, T. J. Tarring, D. A. Iovan and D. P. Harrison, Fine-tuning metal and ligand-centered redox potentials of homoleptic bis-terpyridine complexes with 4′-aryl substituents, Inorg. Chem., 2021, 60, 9956–9969 CrossRef CAS PubMed.
- J. E. Beves, D. J. Bray, J. K. Clegg, E. C. Constable, C. E. Housecroft, K. A. Jolliffe, C. J. Kepert, L. F. Lindoy, M. Neuburger, D. J. Price and S. Schaffner, Expanding the 4, 4′-bipyridine ligand: Structural variation in {M(pytpy)2}2+ complexes (pytpy= 4′-(4-pyridyl)-2,2′:6′,2′′-terpyridine, M= Fe, Ni, Ru) and assembly of the hydrogen-bonded, one-dimensional polymer {[Ru(pytpy)(Hpytpy)]}n3n+, Inorg. Chim. Acta, 2008, 361, 2582–2590 CrossRef CAS.
- J. E. Beves, E. C. Constable, C. E. Housecroft, C. J. Kepert, M. Neuburger, D. J. Price and S. Schaffner, The conjugate acid of bis {4′-(4-pyridyl)-2,2′:6′,2′′-terpyridine} iron (II) as a self-complementary hydrogen-bonded building block, CrystEngComm, 2007, 9, 1073–1077 RSC.
- Y. Guo, X. L. Yang, R. J. Wei, L. S. Zheng and J. Tao, Spin transition and structural transformation in a mononuclear Cobalt (II) complex, Inorg. Chem., 2015, 54, 7670–7672 CrossRef CAS PubMed.
- (a) A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2005 Search PubMed; (b) A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- (a) E. C. Constable, J. Lewis, M. C. Liptrot and P. R. Raithby, The coordination chemistry of 4′-phenyl-2, 2′: 6′, 2 ′′-terpyridine; the synthesis, crystal and molecular structures of 4′-phenyl-2,2′:6′,2′′-terpyridine and bis (4′-phenyl-2,2′:6′,2′′-terpyridine) nickel (II) chloride decahydrate, Inorg. Chim. Acta, 1990, 178, 47–54 CrossRef CAS; (b) J. E. Beves, E. L. Dunphy, E. C. Constable, C. E. Housecroft, C. J. Kepert, M. Neuburger, D. J. Price and S. Schaffner, Vectorial property dependence in bis {4′-(n-pyridyl)-2,2′:6′,2 ′′-terpyridine} iron (ii) and ruthenium (ii) complexes with n= 2, 3 and 4, Dalton Trans., 2008, 3, 386–396 RSC; (c) P. C. Mondal, V. Singh and B. Shankar, Fe-terpyridyl complex based multiple switches for application in molecular logic gates and circuits, New J. Chem., 2014, 38, 2679–2685 RSC.
- K. D. Collins and F. A. Glorius, Robustness Screen for the Rapid Assessment of Chemical Reactions, Nat. Chem., 2013, 5, 597–601 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, SHELXTL, An Integrated System for Solving, Refining, and Displaying Crystal Structures from Diffraction Data, University of Göttingen, Göttingen, Federal Republic of Germany, 1981 Search PubMed; (b) G. M. Sheldrick, SHELXT-Integrated Space-Group and Crystal-Structure Determination, Acta Crystallogr., 2015, A71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2284208–2284210. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra06113b |
| This journal is © The Royal Society of Chemistry 2023 |