
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent biological applications of heterocyclic hybrids containing s-triazine scaffold
Muhammad Imran Ali and
Muhammad Moazzam Naseer*
and
Muhammad Moazzam Naseer*
Department of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan. E-mail: moazzam@qau.edu.pk; Fax: +92-5190642241; Tel: +92-5190642129
First published on 17th October 2023
Abstract
s-Triazine possesses an auspicious status in the field of drug discovery and development owing to its presence in many naturally occurring compounds as well as commercially available drugs like enasidenib, gedatolisib, bimiralisib, atrazine, indaziflam, and triaziflam. Easy, cost-effective, and efficient access to its derivatives in addition to their splendid biological activities such as anticancer, anti-inflammatory, antiviral, anticonvulsant, anti-tubercular, antidiabetic, antimicrobial, makes it an attractive heterocyclic nucleus in the field of medicinal chemistry. Other than the direct access of its derivatives from simple commercially available starting materials like amidine, the s-triazine derivatives have also been obtained starting from an inexpensive commercially available 2,4,6-trichloro-1,3,5-triazine (TCT) commonly known as cyanuric chloride. Owing to the high reactivity and the possibility of sequential substitution of TCT, a variety of biologically active heterocyclic scaffolds have been installed on this nucleus in order to have more potent compounds. These s-triazine-based heterocyclic hybrids have been reported to show enhanced biological activities in recent years. Therefore, it is important to summarize and highlight recent examples of these hybrids which is imperative to attract the attention of the drug development community.
1. Introduction
Rapid developments in the field of combinatorial, computational, and experimental chemistry have provided organic chemists access to a library of a large number of bioactive compounds based on heterocycle scaffolds.1,2 Careful analysis suggests that the compounds having two or more bioactive heterocyclic moieties are superior in their biological applications as compared to the compounds having only one heterocyclic moiety.3 Therefore, the hybridization of different bioactive heterocyclic moieties has become an effective approach to afford potent compounds and solve resistance and efficacy problems. Resultantly, the heterocyclic hybrids are envisioned as excellent future candidates for the development of drug candidates.3Triazine is an important heterocyclic scaffold having a diverse biological profile. Depending on the position of nitrogen, there are three isomeric forms of triazine, namely 1,2,3-triazine (1), 1,2,4-triazine (2), and 1,3,5-triazine (3). Out of these three, 1,3,5-triazine (s-triazine) has a unique place in medicinal chemistry4 owing to its symmetric structure and easy access to a variety of its derivatives either directly from simple starting materials (vide infra) or indirectly from one of its commercially available derivatives e.g., cyanuric chloride5 (2,4,6-trichlorotriazine) (4) (Fig. 1). Having three reactive chloro-substituents and the possibility of their sequential substitution at different temperatures has made cyanuric chloride an ideal platform to construct novel drug candidates with optimum physicochemical and biological properties.6 Owing to these reasons, s-triazine is now part of many approved drugs such as altretamine (5) (anti-ovarian cancer),7 tretamine (6) (antineoplastic),8,9 azacitidine (7) (antineoplastic agent),10 almitrine (8) (respiratory stimulant),11 enasidenib (9) (antileukemia),12 gedatolisib (10) (anti-breast cancer),13,14 bimiralisib (11) (anti-breast cancer)15 melarsoprol (12) (anti-trypanosomiasis or sleeping sickness),16 (Fig. 2a). In addition, this heterocyclic nucleus is also part of many commercially available herbicides.17 Most importantly, these herbicides are also found in natural sources, especially in plants which help them in proper growth and protection from epiphytes, climbers, and wild wines.18 Atrazine (13), desethylatrazine (14), desisopropylatrazine (15), simazine (16), terbuthylazine (17), propazine (18), cyanazine (19), cyprazine (20), atraton (21), prebane (22), terbutryn (23), prometon (24), prometryn (25), desmetryn (26), ametryn (27) and dimethametryn (28) are the examples of s-triazine based herbicides17,19,20 (Fig. 2b). Likewise, indiaziflam (29)21 and triaziflam (30)22 are US-FDA-approved insecticides that possess s-triazine as a basic structure (Fig. 2c).
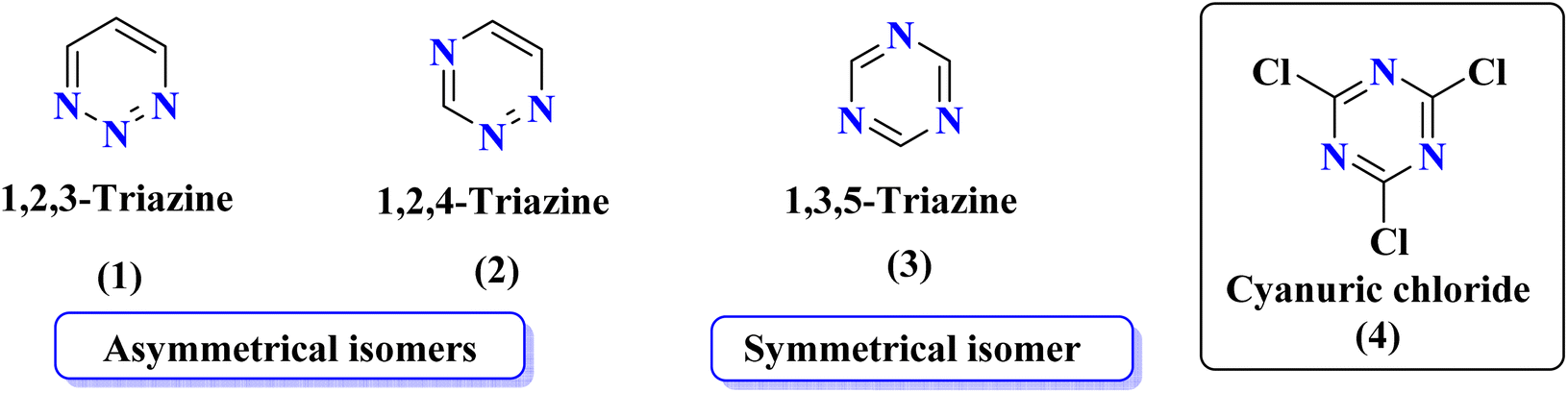 | ||
| Fig. 1 Isomeric forms of triazine (1), (2), and (3), and structure of cyanuric chloride (TCT) (4). | ||
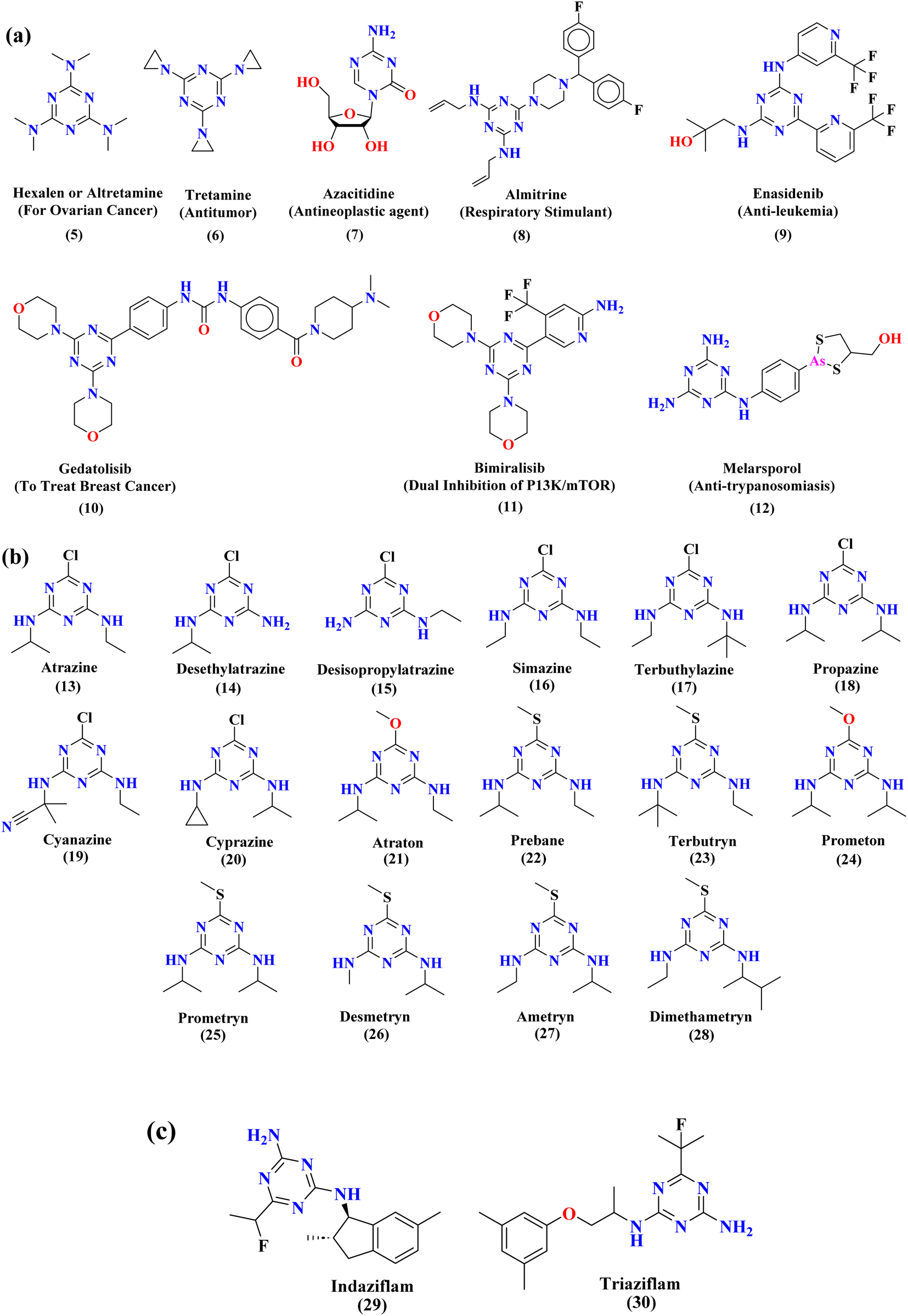 | ||
| Fig. 2 Commercially available drugs (a), commercially available herbicides (b), and commercially available insecticides (c) that possess s-triazine scaffold. | ||
Given the tremendous recent importance of heterocyclic derivatives, scientists are continuously working to generate novel libraries of drug candidates by hybridizing s-triazine nucleus (vide supra) with a wide variety of well-known heterocyclic scaffolds.23 Therefore, it is imperative to summarize such recent studies published in the last five years (2018–2023) to attract not only the attention of the drug development community but also to find the gap in planning novel strategies for the synthesis of pharmacologically more potent novel s-triazine-based hybrid molecules.
2. Summary of synthetic strategies for s-triazine derivatives
A variety of strategies have been used to synthesize s-triazine derivatives from cheap and readily available starting materials (please see Fig. 3). For instance, from zinc dimethyl imidodicarbonimidic (31) by reacting it with activated acid derivatives in a mixture of solvent DCM and pyridine24 (Fig. 3). Derivatives of s-triazine are also reported to be synthesized from amidines (32) by reacting with phosgene gas,25 with tertiary amines via copper catalysis,26 with primary amines under metal-free conditions,26 with alcohols under highly chemo-selective, base-free conditions via aerobic oxidation,27–30 and with diones.31 Similarly, ethyl acetimidate hydrochloride (33) is reacted with two equivalent monosodium cyanamide (NaNHCN), which on further treatment with hydroxylamine hydrochloride yields derivatives of s-triazine.32 Likewise, cycloaddition reactions of DICY (34) with aryl and aliphatic nitriles to yield substituted s-triazines are also reported.33 Cyclization of nitriles (35) under Lewis-acid-catalysis yields substituted symmetrical triazines.34 One-pot synthesis of 1,3,5-triazine can also be carried out via a trimerization reaction of nitriles.35 In the same fashion, reactions of metformin (36) with ester36 or other bile acids are reported to synthesize derivatized s-triazines in good yield.37 The metformin (36) on reaction with primary alcohols under graphene oxide catalysis yields substituted triazines.38 Derivatized 1,3,5-triazines can also be synthesized from rearrangement reactions of benzodiazepinedione (37) via hydrolysis or alcoholysis.39 In industries, derivatives of s-triazine can be synthesized starting from hydrogen cyanide (38).40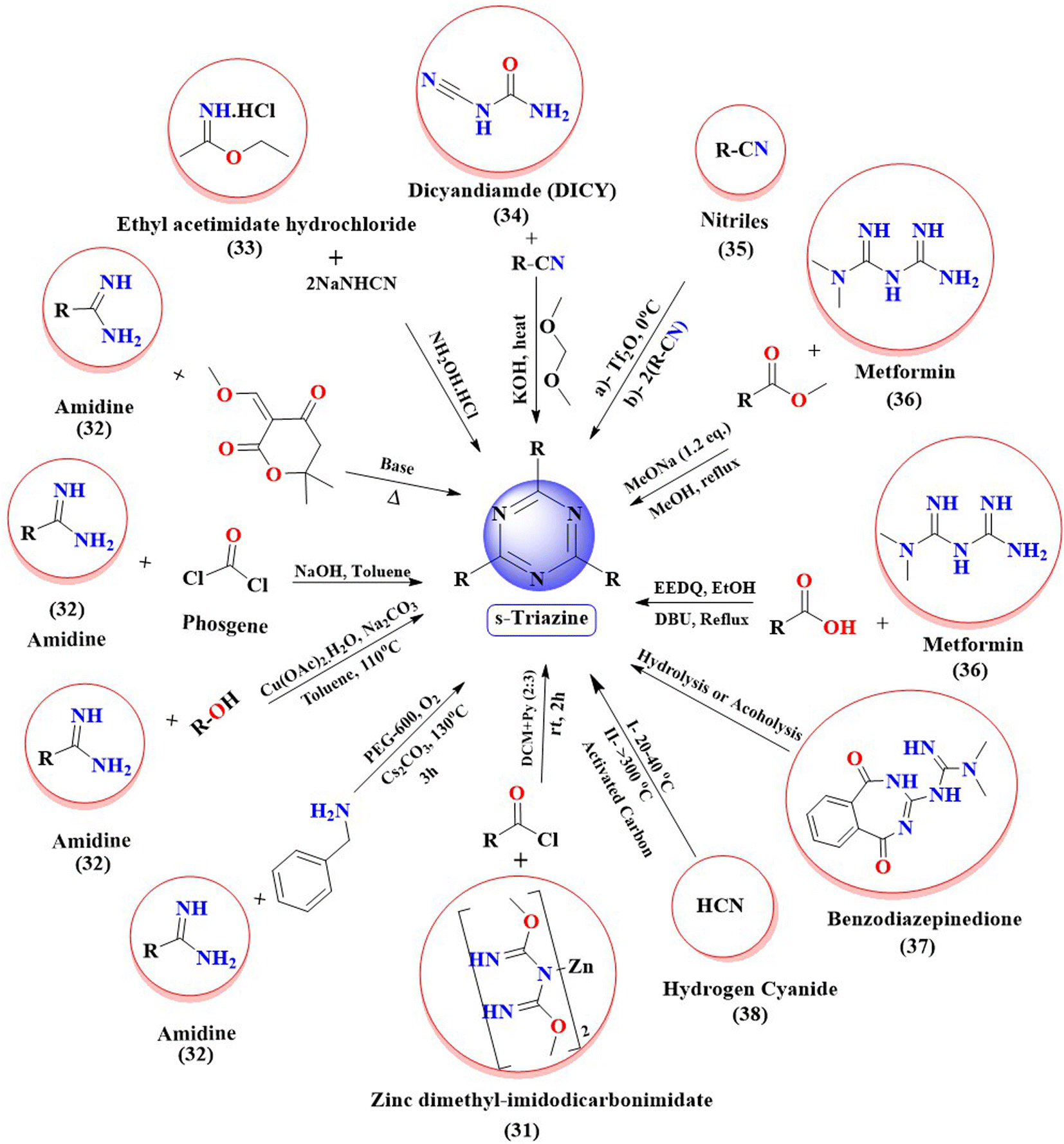 | ||
| Fig. 3 Summary of the various synthetic strategies used for the synthesis of s-triazine derivatives. | ||
Apart from the direct access of triazine derivatives (Fig. 3), they are now easily accessible starting from an inexpensive, commercially available cyanuric chloride or trichlorotriazine (TCT) (4). TCT is a useful and reactive source to synthesize the biologically active leads, different multitopic molecules, and molecular hybrids possessing s-triazine as a main ingredient.5 Any nucleophilic species can be utilized to substitute chlorine atoms from TCT (4) through SNAr mechanism at different temperature conditions (Fig. 4).41
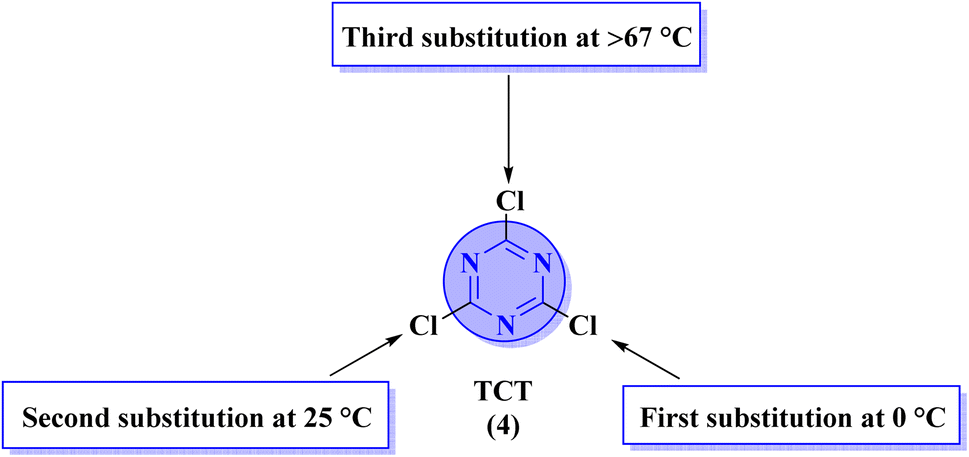 | ||
| Fig. 4 Showing optimum temperature conditions for sequential substitution of three reactive chloro substituents of TCT (4). | ||
3. Biological activities of s-triazine-based heterocyclic hybrids
3.1 Anticancer activity
Cancer is one of the serious health issues globally.42 It is the second major cause of death in the USA.43,44 Cancer is not just caused by genetic factors but also by other carcinogenic environmental factors.45 Some of the recently reported molecules are summarized in Table 1 that have the potential to inhibit the enzymes responsible for cancer and can control angiogenesis.| Compounds | Most potent compound | IC50 of reference drug | Refs. | |
|---|---|---|---|---|
| Substituents | (IC50) | |||
 |
 |
MDA-MB-231 = 23.13 ± 1.29 μM, HeLa = 39.13 ± 0.89 μM, HCT-116 = 29.89 ± 0.87 μM, Huh-7 = 44.01 ± 0.41 μM | Genistein: MDA-MB-231 = 89.11 ± 6.39 μM, HeLa = >100 μM, HCT-116= >100 μMHuh-7 = >100 μM, 5-FU: MDA-MB-231 = 78.04 ± 8.90 μM, HeLa = 67.64 ± 5.85 μM, HCT-116= 15.42 ± 2.88 μM, Huh-7 = 38.37 ± 1.51 μM | 46 |
 |
— | A549 (IC50 41.3 μmol L−1), PA-1 (IC50 10.6 μmol L−1), Huh7 (IC50 19.9 μmol L−1), HeLa (IC50 3.7 μmol L−1), HEK293 (IC50 15.8 μmol L−1) | – | 47 |
 |
 |
MCF-7 = 0.03 ± 0.01μM, Hela = 0.27 ± 0.02 μM, A549 = 0.18 ± 0.01 μM, PI3K = 23.8 nM, mTOR = 10.9 nM | GDC- 0941: MCF-7 = 0.23 ± 0.10 μM, Hela = 0.93 ± 0.06 μM, A549 = 0.59 ± 0.03 μM, PI3K = 6.0 nM, mTOR = 525.0 nM | 48 |
 |
 |
U87-MG = 0.70 ± 0.10 mM, MCF-7 = 0.13 ± 0.02 mM, PC-3 = 0.57 ± 0.15 mM, PI3Kα = 0.32 ± 0.18 nM, PI3Kβ = >100 nM, PI3Kγ = 16.03 ± 3.55 nM, PI3Kδ = >100 nM | ZSTK-474: U87-MG = 0.71 ± 0.01 mM, MCF-7 = 0.13 ± 0.02 mM, PC-3 = 0.58 ± 0.03 mM, PI3Kα = 6.40 ± 0.12 nM, PI3Kβ = 23.89 ± 0.28 nM, PI3Kγ = 3.24 ± 1.84 nM, PI3Kδ = 24.76 ± 3.57 nM | 49 |
 |
 |
BTK = 17.0 ± 1.01 nM, EGFR = >1000 nM, JAK3 = 104.6 ± 0.80 nM | Ibrutinib: BTK = 1.9 ± 1.71 nM, EGFR = 1.7 ± 1.22 nM, JAK3 = 47.9 ± 1.23 nM | 50 |
 |
 |
Cytotoxic effect (on mouse T-lymphoma cells): PAR = 11.37 ± 0.33 μM, MDR = 16.73 ± 0.35 μM, antiproliferative effects: (on mouse T-lymphoma cells), PAR = 6.71 ± 0.40 μM, MDR = 5.35 ± 0.52 μM | Doxorubicin Cytotoxic effect (on mouse T-lymphoma cells): PAR = 0.7 ± 0.56 μM, MDR = 2.14 ± 0.76 μM, antiproliferative effects: (on mouse T-lymphoma cells), PAR = 0.28 ± 0.06 μM, MDR = 1.75 ± 0.38 μM | 51 |
 |
 |
PI3Kα = 177.41 nM, mTOR = 12.24 nM, MCF-7 = 0.30 ± 0.06 μM, Ovcar-2 = 0.25 ± 0.11 μM, U87MG = 1.25 ± 0.37 μM, A549 = 0.47 ± 0.09 μM, NCI-H460 = 2.31 ± 0.38 μM, H2228 = 0.22 ± 0.03 μM, H1975 = 0.008 ± 0.002 μM, HeLa = 1.04 ± 0.23 μM, HeLa-MDR = 0.37 ± 0.13 μM | GDC-0941: PI3Kα = 6.02 nM, mTOR = 525.03 nM, MCF-7 = 1.20 ± 0.23 μM, Ovcar-2 = 1.25 ± 0.17 μM, U87MG = 3.13 ± 0.31 μM, A549 = 1.03 ± 0.09 μM, NCI-H460 = 2.68 ± 0.37 μM, H2228 = 1.07 ± 0.28 μM, H1975 = 2.42 ± 0.74 μM, HeLa = 3.15 ± 0.17 μM, HeL-MDR = 2.97 ± 0.31 μM | 52 |
 |
 |
PC3 = 0.07 ± 0.0082 μM, A549 = 0.13 ± 0.021 μM, MCF-7 = 0.025 ± 0.0018 μM, A2780 = 0.19 ± 0.073 μM | Etoposide: PC3 = 2.39 ± 1.56 μM, A549 = 3.08 ± 0.135 μM, MCF-7 = 2.11 ± 0.024 μM, A2780 = 1.38 ± 0.56 μM | 53 |
 |
 |
MDA-MB-231 = 97.3 ± 9.3 μM, U-87MG = 10.9 ± 4.1 μM, PANC-1 = 26.7 ± 8.3 μM, A549 = 12.4 ± 6.4 μM, MCF-7 = 8.3 ± 2.1 μM, HDFs = 38.9 ± 8.3 μM, EGFR-PK = 70.3 ± 1.34 nM, PI3K = 6.64 ± 0.15 ng mL−1, AKT = 37.3 ± 0.69 ng mL−1, mToR = 69.3 ± 1.98 ng mL−1 | Tamoxifen: EGFR-PK inhibition, IC50 = 69.1 ± 1.39 nM | 54 |
 |
— | HepG-2 = 29.75 ± 0.03 μM, PC-3 = 17.90 ± 0.03 μM, MCF-7 = 4.65 ± 0.07 μM, A-549 = 7.43 ± 0.04 μM, PBMC = 165.23 ± 18.05 μM, EGFRWT = 0.22 ± 0.05 μM, EGFRT790M = 0.18 ± 0.11 μM | DOX: HepG-2 = 4.51 ± 0.26 μM, PC-3 = 8.11 ± 0.05 μM, MCF-7 = 4.17 ± 0.2 μM, A-549 = 8.20 ± 0.08 μM, PBMC = 250.00 ± 26.56 μM, Erlotinib: HepG-2 = 8.19 ± 0.4 μM, PC-3 = 8.89 ± 0.6 μM, MCF-7 = 4.16 ± 0.2 μM, A-549 = 3.76 ± 0.2 μM, PBMC = 45.75 ± 26.56 μM, EGFRWT = 0.09 ± 0.05 μM, EGFRT790M = 0.55 ± 0.10 μM | 55 |
 |
 |
MCF-7 = 4.53 ± 0.30 μM, HCT-116 = 0.50 ± 0.080 μM, HepG2 = 3.01 ± 0.49 μM, EGFR PK = 61 ± 0.002 nM | Tamoxifen: MCF-7 = 5.12 ± 0.36 μM, HCT-116 = 26.41 ± 4.11 μM, HepG2 = 2.45 ± 0.20 μM, Erlotinib: EGFR PK = 78.65 ± 3.54 nM | 56 |
 |
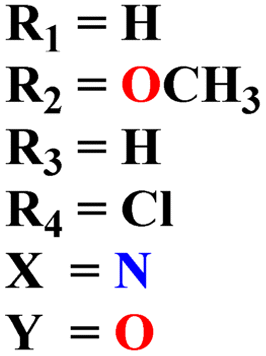 |
Caco-2 = 19.36 ± 3.71 μM, HT-29 = 19.29 ± 2.62 μM, HMVEC-d = 10.86 ± 0.08 μM | — | 57 |
 |
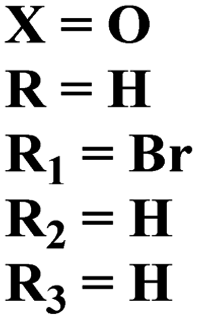 |
Cell growth inhibition (GI%) with 10 μM dose, CCRF-CEM = 79.05, SR = 92.07, NCI-H522 = 66.45, HT29 = 93.39, COLO 205 = 76.23, SF-295 = 70.86, LOX IMVI = 94.77, SK-MEL-2 = 97.36, CAKI-1 = 84.64, MDA-MB-468 = 98.91 | Enasidenib cell growth inhibition (GI%): CCRF-CEM = Not tested, SR = 31.62, HT29 = 19.95, COLO 205 = 15.84, SF-295 = 19.95, LOX IMVI = 15.84, MDA-MB-468 = 19.95 | |
 |
 |
pIC50 = 4.29 | 59 | |
 |
 |
A549 = 2.40 ± 0.64 μM, MCF-7 = 3.28 ± 0.16 μM, HDFs = 3.78 ± 0.55 μM, EGFR = 34.1 ± 1.58 nM, CDK-2 = 108.3 ± 3.12 nM | Doxorubicin (IC50 μM mL−1): A549 = 0.28 ± 0.11, MCF-7 = 0.06 ± 0.02, HDFs = 0.42 ± 0.27, Erlotinib: EGFR = 67.3 ± 2.54 nM, Roscovitine: CDK-2 = 140 ± 3.4 nM | 60 |
 |
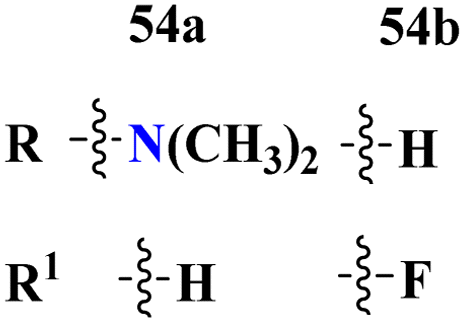 |
54a | Staurosporine | 61 |
| MDA-MB-468 = 3.99 ± 0.21 μM, CCRF-CM = 4.51 ± 0.24 μM, 54b: MDA-MB-468= 1.48 ± 0.08 μM, CCRF-CM = 9.83 ± 0.52 μM | MDA-MB-468 = 6.07 ± 0.03 μM, CCRF-CM = 2.17 ± 0.11 μM | |||
 |
 |
Huh-7 0.4 μM | — | 62 |
 |
 |
Kinase inhibition (IC50 in nM), PI3Kα = 3.41 ± 0.05 nM, mTOR = 8.45 ± 1.28 nM, Anticancer activity (IC50 in μM), MDA-MB321 = 15.83 ± 1.52 μM, MCF-7 = 16.32 ± 0.92 μM, HeLa = 2.21 ± 0.45 μM, HepG2 = 12.21 ± 0.43 μM | Gedatolisib kinase inhibition (IC50 in nM): PI3Kα = 6.04 ± 0.45 nM, mTOR = 2.5 ± 0.23 nM | 63 |
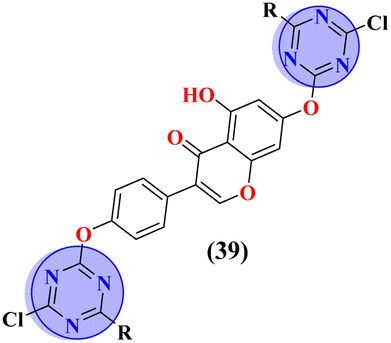
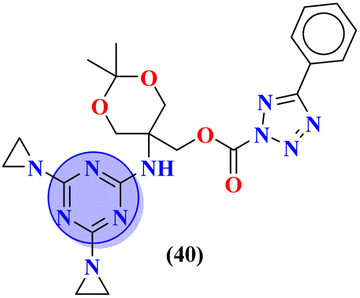
Conjugates (39) represent the molecular hybrids of s-triazine and genistein.46 These hybrids exhibited remarkable action against the cancer cell-lines MDA-MB-231 (breast), HeLa (cervical), HCT-116 (prostate), and Huh-7 (liver). Structure–activity relationship revealed that the hybrid clubbed with 4-methyl piperidine is the most potent and exhibited the highest levels of activity with the least IC50 value in comparison to other substituents for instance N,N-diphenyl amine, N,N-diethyl amine etc. IC50 values of 4-methyl piperidine substituent are HeLa = 39.13 ± 0.89 mM, HCT-116 = 29.89 ± 0.87 mM, and Huh-7 = 44.01 ± 0.41 mM. The most efficient antiproliferative activity is against MDA-MB-231 cells (IC50 = 23.13 mM), which is even better than the standard drug 5-fluorouracil (IC50 = 78.04 mM) (Table 1). Compound (40) is a bioconjugate incorporating the structural component of s-triazine tethered with tetrazole.47 In vitro, analysis of (40) suggested that exhibited good cytotoxicity against human alveolar basal epithelium adenocarcinoma A549 (IC50 = 41.3 mmol L−1), human ovarian teratocarcinoma PA-1 (IC50 = 10.6 mmol L−1), hepatocarcinoma Huh7 (IC50 = 19.9 mmol L−1), cervical cancer HeLa (IC50 = 3.7 mmol L−1), and human embryonic kidney HEK293 (IC50 = 15.8 mmol L−1). After investigation, it was suggested that the possible mechanism of cytotoxicity is through HIF pathway inhibition. This output suggests that these conjugates are remarkable anticancer agents against a wide range of cancerous cell-lines (Table 1).
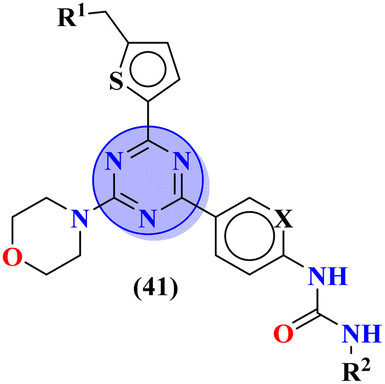
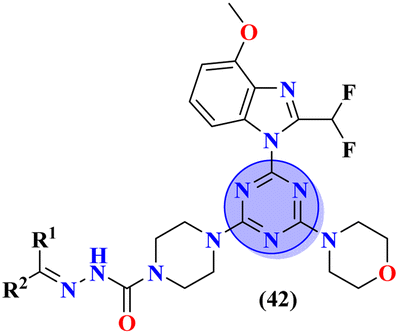
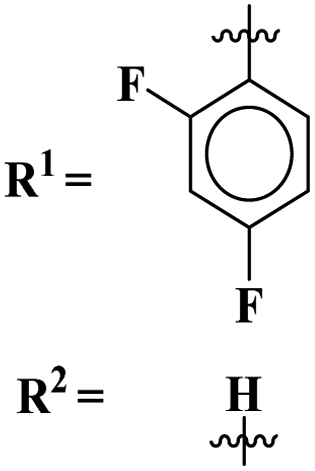
Sun et al., carried out the synthesis of 28 s-triazine-based compounds via coupling of thiophene and aryl urea with s-triazine (41).48 These 2-arylurea-1,3,5-triazine derivatives show prominent inhibition of P13K/mTOR which is an effective anticancer therapy. The cytotoxic activity of these conjugates was evaluated against MCF-7, HeLa, and A549 cancer cell-lines. SAR analysis suggested that cytotoxicity decreases with increasing carbon in the R2 chain linked with arylurea moiety and the most efficient cytotoxicity was observed with the compound having sulfonamide moiety. For this potent compound, IC50 of cytotoxicity was evaluated to be MCF-7 = 0.03 ± 0.01 μM, HeLa = 0.27 ± 0.02 μM, A549 = 0.18 ± 0.01 μM, while for the enzyme inhibition activity, it is noted to be PI3K = 23.8 nM, and mTOR = 10.9 nM (Table 1). To evaluate the anticancer activities of s-triazine, its three distinct series in conjunction with benzimidazole (42) were prepared and its inhibitor effect against P13Kα–β–γ–δ, and three cancer cell-lines, namely, U87-MG, MCF-7, and PC-3 were evaluated.49 SAR showed that compounds with R1 containing 2,4-difluoro substituent and R2 being H showed maximum inhibition effect this may be due to the fact that these two fluorine atoms can show more electron-withdrawing effect on the phenyl ring and make it more susceptible to π–π interactions. Cytotoxicity of these compounds was evaluated to be U87-MG = 0.70 ± 0.10 mM, MCF-7 = 0.13 ± 0.02 mM, PC-3 = 0.57 ± 0.15 mM, PI3Kα = 0.32 ± 0.18 nM, PI3Kβ = >100 nM, PI3Kγ = 16.03 ± 3.55 nM, and PI3Kδ = >100 nM. Moreover, this compound also represented good anticancer activity in, in vivo analysis at 20 mg kg day−1 in comparison to ZSTK-474 administrated at 40 mg kg day−1 (Table 1).
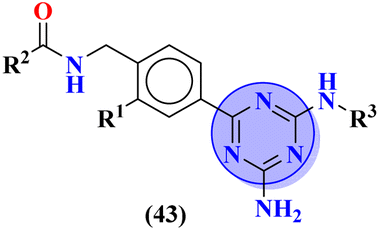
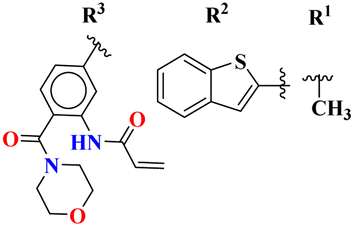
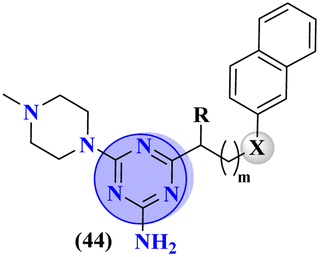
Bruton's tyrosine kinase (BTK) plays a critical and vital role in B-cell development. Its inhibition is proven effective in cancer therapy. A series of 6-amino-s-triazine-based molecular hybrids (43) were synthesized and it's in vitro and in silico inhibitory action was evaluated.50 SAR studies suggested that a compound with R2 benzothiophenyl group proved to be a more potent and irreversible inhibitor against BTK than substituted benzene rings. This SAR was further studied through docking analysis, and it was observed that the benzothiophene moiety can bind more properly into the H3 pocket of BTK to show π–π interaction with Tyr551 which improves the inhibitory action even more than that of the standard drug, ibrutinib. Kinase selectivity of this conjugates is BTK = 17.0 ± 1.01 nM, EGFR = >1000 nM, JAK3 = 104.6 ± 0.80 nM. These findings suggest that the s-triazine and its molecular hybrids can be a promising candidate for blocking the Bruton's tyrosine kinase enzyme irreversibly (Table 1). In order to treat lymphoma, a molecular hybrid of amino-s-triazine tethered piperazine (44) was synthesized and the main motive of the synthesis was to evaluate the difference of activity between thio- and seleno-ether at position X.51 Through all the analyses it was observed that β-naphthyl-thioether derivative proved to be more potent than verapamil by a factor of 11 i.e., 11× times more potent. The cytotoxicity of this lead on mouse T-lymphoma cells was shown to be (sensitive parental) PAR = 11.37 ± 0.33 μM, and (resistant) MDR = 16.73 ± 0.35 μM while, the antiproliferative effects was (sensitive parental) PAR = 6.71 ± 0.40 μM and (resistant) MDR = 5.35 ± 0.52 μM (Table 1).
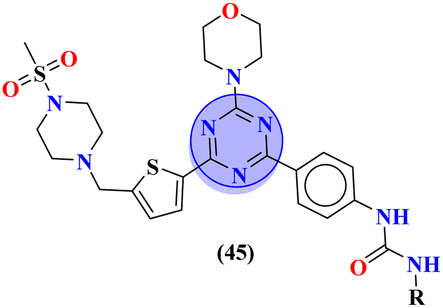
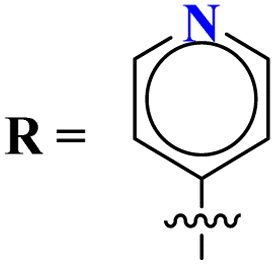
In their studies, S. Xu et al., reported molecular hybrids of s-triazine and thiophene (45). The third scaffold which is pendant with this hybrid is morpholine.52 The anticancer activity of these conjugates was assessed against a panel of 9 different cancer cell-lines and enzymes PI3K/mTOR. Fortunately, 7 compounds among them proved to be more effective than GDC-0941. The inhibitory action of this conjugate was 302.5× better than the lead compound GDC-0941. With the lowest IC50 value being 0.008 ± 0.002 μM. The kinase inhibitory value was noted to be 177.41 and 12.24 nM for PI3K and mTOR respectively. SAR showed that the presence of the arylurea group significantly improved the activity and the conjugate with substituent R = pyridinyl ring displayed 98.04% inhibition highlighting its anticancer potency. The cytotoxicity of these compounds are MCF-7 = 0.30 ± 0.06 μM, Ovcar-2 = 0.25 ± 0.11 μM, U87MG = 1.25 ± 0.37 μM, A549 = 0.47 ± 0.09 μM, NCI-H460 = 2.31 ± 0.38 μM, H2228 = 0.22 ± 0.03 μM, H1975 = 0.008 ± 0.002 μM, HeLa = 1.04 ± 0.23 μM and Hela-MDR = 0.37 ± 0.13 μM (Table 1).
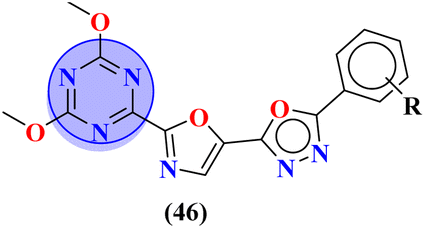
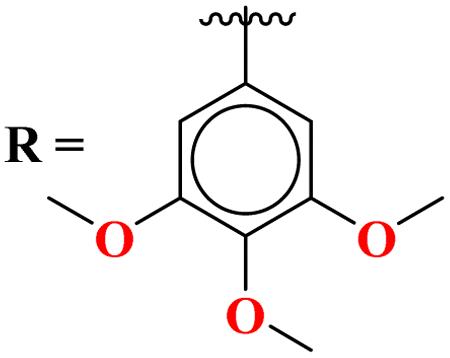
Novel conjugates of oxadiazole tethered with s-triazine (46) were reported by V. Balaraju and coworkers.53 These compounds were assessed as antitumor agents using MTT method against a number of anticancer cell-lines for instance, (PC3) prostate cancer, (A549) lung cancer, (MCF-7) breast cancer, and (A2780) ovarian cancer. Their activity was compared with etoposide, a remarkable standard for antitumor activity. The value of IC50 was in order of PC3 = 0.07 ± 0.0082 μM, A549 = 0.13 ± 0.021 μM, MCF-7 = 0.025 ± 0.0018 μM and A2780 = 0.19 ± 0.073 μM. The SAR studies showed that cytotoxicity is enhanced greatly when substituents are trimethoxy groups on phenyl ring. The reason may lie in the fact that methoxy is a strong electron-donating group and has three strong hydrogen bond acceptor atoms that can bind with the active site of certain enzymes. The data obtained from these studies provide a remarkable insight into the enhanced activity of s-triazine when tethered with oxadiazole (Table 1). Likewise, hybrids of s-triazine with pyrazole and pyrazole-fused cycloalkanone (47) have been synthesized and evaluated as potential anticancer agents54 against triple-negative breast cancer, glioblastoma, lung cancer, pancreatic cancer, human dermal fibroblast via relevant in vitro analysis. Inhibition of enzymes like P13K, AKT, and mToR was also screened. Intriguingly, it was noted that compounds with morpholine moiety proved to be more potent than standard tamoxifen. The most efficient cytotoxicity values of these compounds are U-87 MG = 10.9 ± 4.1 μM, PANC-1 = 26.7 ± 8.3 μM, A549 = 12.4 ± 6.4 μM, MCF-7 = 8.3 ± 2.1 μM, HDFs = 38.9 ± 8.3 μM, EGFR-PK = 70.3 ± 1.34 nM. While the IC50 value for the enzyme inhibitory activity is PI3K = 6.64 ± 0.15 ng mL−1, AKT = 37.3 ± 0.69 ng mL−1, and mToR = 69.3 ± 1.98 ng mL−1 (Table 1).
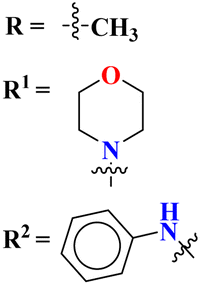
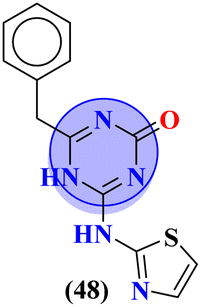
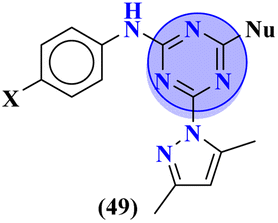
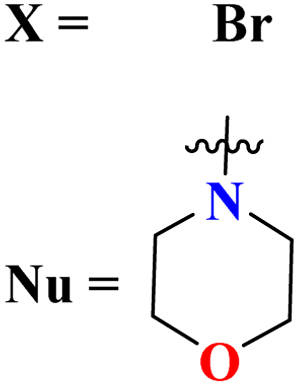
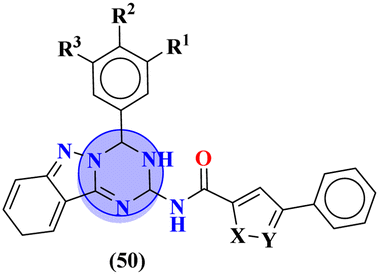
Starting from 2-phenylacetyl isothiocyanate, microwave-assisted synthesis of 1,3,5-triazine tethered thiazole (48) is also reported as a potent anticancer agent.55 This bioconjugate was evaluated against different cancer cell-lines and kinase enzymes epidermal growth factor receptor (EGFR) and results were compared with standard drugs doxorubicin and erlotinib. The notable conjugate was showed the IC50 value as follow, HepG-2 = 29.75 ± 0.03 μM, PC-3 = 17.90 ± 0.03 μM, MCF-7 = 4.65 ± 0.07 μM, A-549 = 7.43 ± 0.04 μM, PBMC = 165.23 ± 18.05 μM, EGFRWT = 0.22 ± 0.05 μM, and EGFRT790M = 0.18 ± 0.11 μM. Through in silico analysis, it was evaluated that the thiazole ring of conjugate showed good binding with methionine and leucine amino acid which supports the enhanced biological activity of conjugate (Table 1). Herein, hybrids of s-triazine and pyrazole are reported (49).56 These hybrids had variations at 4 positions of the phenyl ring and 5 positions of the triazine ring. The biological activities of these compounds were checked for 3 different cancer cell-lines and kinase enzymes. Via SAR studies and in silico inhibitory effect was also noted it was evaluated that conjugate with morpholine moiety and bromine atom at 4 positions of the amino group was noted as potent in comparison to tamoxifen with cytotoxicity MCF-7 = 4.53 ± 0.30 μM, HCT-116 = 0.50 ± 0.080 μM, and HepG2 = 3.01 ± 0.49 μM and IC50 of enzyme inhibitory activity EGFR PK = 61 ± 0.002 nM (Table 1). In search of novel and potential antiproliferation agents, fused ring derivatives (50) of s-triazine and benzimidazole along with an appended oxazole ring linked via an amide bond were synthesized in high yield.57 In vitro analysis against adenocarcinoma cell-lines and endothelial cell-line and IC50 value was quite good for instance, Caco-2 = 19.36 ± 3.71 μM, HT-29 = 19.29 ± 2.62 μM, and HMVEC-d = 10.86 ± 0.08 μM. Further studies were done to insight into the mechanism of action of these drug candidates and it was investigated that the microtubule skeleton was targeted by these conjugates (Table 1).
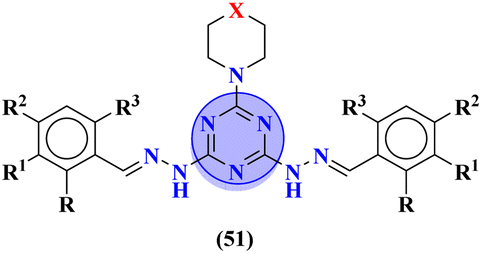
A new series of hydrazones of s-triazine tethered to morpholine (51) were synthesized and their antiproliferation activity was evaluated against 60 different cancer cell-lines and percentage growth inhibition was confirmed which provided useful results of these potent antiproliferation agents. Through comprehensive SAR analysis, it was also noted that the activity of conjugates decreases from a change in the hydrazone of aldehyde to the ketone. Likewise, morpholine substituents (X = O) were more potent than other analogs where X = N or X = CH2. Cell growth inhibition (GI%) with a 10 μM dose can be summed up as, CCRF-CEM = 79.05, SR = 92.07, NCI-H522 = 66.45, HT29 = 93.39, COLO 205 = 76.23, SF-295 = 70.86, LOX IMVI = 94.77, SK, MEL-2 = 97.36, CAKI-1 = 84.64 and MDA-MB-468 = 98.91. Hence, potent antileukemic agents were reported (Table 1).58 Thiophene clubbed s-triazine derivatives (52) were synthesized as anticancer agents and their biological activities were evaluated against human lung adenocarcinoma cells (A549).59 pIC50 of the series ranged from 4.29 to 6.70, enlightening the range of their potency. In addition to in vitro analysis, 3D-QSAR, in silico analysis, and molecular docking were also performed in order to gain more output from compound structure–activity relationships and their binding modes with targeted sites. It was finalized from these findings that conjugates with benzimidazole are more potent than other analogs like substituted benzene ring and aminopyrimidine (Table 1).
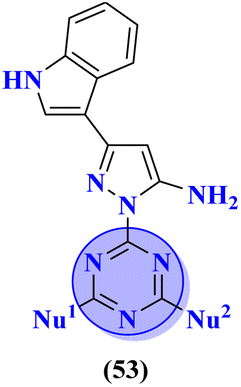
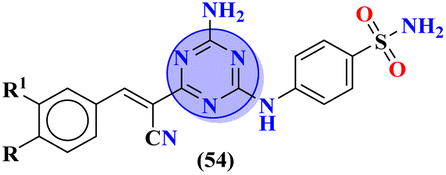
Bioconjugates of pyrazole clubbed s-triazine (53) with an addition motif of indole ring were designed and its antiproliferation activity was evaluated as a kinase inhibitor.60 For this purpose, the inhibitory effect was evaluated against two enzymes, EGFR and CDK. The cytotoxicity of conjugates was also evaluated against three different cancer cell-lines and results were obtained to be A549 = 2.40 ± 0.64 μM, MCF-7 = 3.28 ± 0.16 μM, HDFs = 3.78 ± 0.55 μM, EGFR = 34.1 ± 1.58 nM, and CDK-2 = 108.3 ± 3.12 nM for the most prominent conjugate. Through SAR analysis, it was noted that conjugate having the amalgam of indole, triazine, and pyrazole was proven to be the most effective in controlling cancerous cells via apoptosis. The in silico analysis was also performed in order to analyze the binding affinities (Table 1). Deregulation or overexpression of different isomeric forms of carbonic anhydrase leads to diseases like glaucoma, epilepsy, and cancer. To target and inhibit the overexpression of this enzyme, sulfonamide-based hybrids of s-triazine were reported.61 The biological activity of these compounds was evaluated against breast cancer (MDA-MB-468) and leukemia (CCRF-CM) cell-lines. Two hits were observed in the whole set of compounds where compound 54a when R = N,N-dimethyl and R1 = H with cytotoxicity observed to be MDA-MB-468 = 3.99 ± 0.21 μM, CCRF-CM = 4.51 ± 0.24 μM and the second one is 54b when R = H and R1 = F with cytotoxicity value noted to be MDA-MB-468 = 1.48 ± 0.08 μM and CCRF-CM = 9.83 ± 0.52 μM. The Ki value of 54a and 54b for enzyme hCA-IX is reported to be 190.0 and 38.8 nM respectively. Compound 54b was also able to arrest the G0–G1 and S-phase of the cell cycle (Table 1).
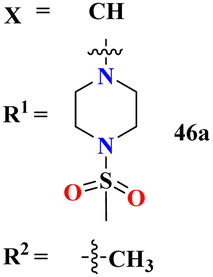
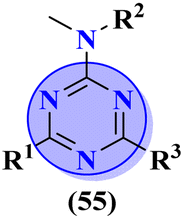
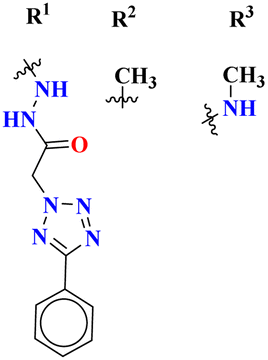
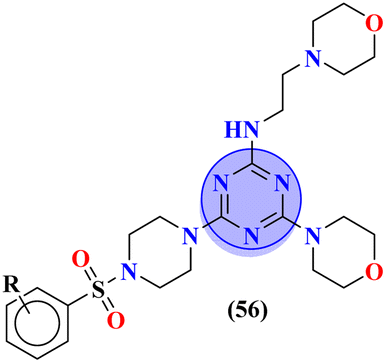
The molecular hybrid of tetrazole and s-triazine (55)62 was prepared in good yield and their biological activities against cancer cell-lines Huh-7 were checked by MTT analysis which was obtained to be Huh-7 0.4 μM. The compound possessing a 5-phenyltetrazol-2-ylacetohydrazide moiety showed good cytotoxic activity. These compounds did not show any cytotoxic activity against normal cells which depicts that these compounds are safe to be used in further clinical trials (Table 1). To the same degree, the molecular hybrids (56) of s-triazine, morpholine, and piperazine were synthesized with good yield. In addition to these three pharmacophores, sulfonamide moiety is also present in these novel molecular hybrids.63 After successful synthesis, these molecular hybrids were evaluated kinase inhibitory action against enzymes i.e., PI3K/mTOR, and four different cancer cell-lines namely, triple-negative breast cancer cells, human breast cancer cells, human cervical cancer cells, and human liver cancer cells and the most efficient potency was found against the human cervical cancer cells (HeLa) with cytotoxicity HeLa = 2.21 ± 0.45 μM. Evaluating SAR it was noted that a compound with 4-OH substituent was able to arrest the G1 phase of HeLa cell-line due to strong electron-donating effect of the hydroxyl group at position 4. The IC50 of this compound is PI3Kα = 3.41 ± 0.05 nM and mTOR = 8.45 ± 1.28 nM. This compound was able to induce apoptosis in cancer cell-lines eventually leading to a reduction in migration and invasion toward normal cells (Table 1).
3.2 Antibacterial activity
The prodigious phenomenon shown by bacteria to resist most of the available antibiotics has made a hot issue for scientists.64 Multidrug resistance (MDR) is known as a global crisis. Although, as with the passage of time resistance is increasing, the clinical trials for new drugs are decreasing. The development of novel antimicrobial agents with minimal financial investment has always been a viable solution to deal with this situation.65 Numerous hybrid analogs of s-triazine combined with various other pharmacophores as part of this aim have been developed with potent antimicrobial activities. Some of the latest antibacterial drug candidates have been reported in Table 2.| Compounds | Most potent compound | MIC/ZOI of ref. drug | Refs. | |
|---|---|---|---|---|
| Substituents | MIC/zone of inhibition | |||
 |
 |
MIC (μM) S. aureus: LATCC 33591 = 3.9, MRSA BRS3 = 0.95, MRSA C1 = 1.95, MRSA C7 = 3.9, MRSA C14 = 3.9, MRSA G1 = 7.8 | MIC (μM), AMK (Amikacin) S. aureus: ATCC 33591= 31.3, MRSA BRS3= 15.6, MRSA C1 = 3.9, MRSA C7 = 7.8, MRSA C14 = 7.8, MRSA G1 = 15.6 | 66 |
 |
 |
Zone of inhibition in (mm), Gram positive: S. aureus = 14.84 ± 0.3, S. faecalis = 14.84 ± 0.4, B. substilis = 14.4 ± 0.3, P. vulgaris = 15.64 ± 0.8, B. pumilus = 16.64 ± 0.4, Gram negative: E. coli = 14.04 ± 0.44, K. penumoniae = 11.04 ± 0.43 | Zone of inhibition in (mm), Norfloxacin Gram positive:S. aureus = 17.44 ± 0.3, S. faecalis = 17.45 ± 0.5, B. substilis = 16.64 ± 0.3, P. vulgaris = 17.64 ± 0.4, B. pumilus = 18.45 ± 0.4, Ciprofloxacin Gram negative: E. coli = 17.45 ± 0.36, K. penumoniae = 17.64 ± 0.65 | 67 |
 |
 |
(MIC) μg mL−1: E. coli = 256, S. aureus = 256 | (MIC) μg mL−1 Chloramphenicol: E. coli = 128, S. aureus = 256 | 68 |
 |
— | ZOI: 60a: E. coli = 30 ± 0.42 mm, B. subtillis = 28 ± 0.22 mm, S. phyogenes = 19 ± 0.56 mm, P. aregeunosa, 60b: E. coli = 35 ± 0.12 mm, B. subtillis = 18 ± 0.32 mm, S. phyogenes = 20 ± 0.56 mm | ZOI: Sulfadiazine: E. coli = 26 ± 0.83 mm, B. subtillis = —, Streptomycin, S. phyogenes = 10 ± 0.66 mm | 69 |
 |
 |
MIC (μg μL−1): E. coli = 1.25, S. enteritidis = 1.25, S. aureus = 1.25, B. subtilis = 1.25 | MIC (μg μL−1): Penicillin, E. coli = 6, S. enteritidis = 6, S. aureus = 1.5, B. subtilis = 1.5 | 70 |
 |
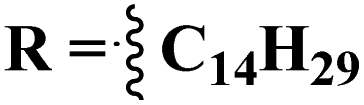 |
MIC (mg L−1): S. aureus = 3.90, E. faecalis = 62.5, E. coli = 62.5, P. aeruginosa = 250, IC50 [mg L−1] = 2.4 | — | 71 |
 |
 |
MIC (μg mL−1): S. aureus = 6.25, P. aeruginosa = 12.5, B. cereus = 100, K. pneumoniae = 100 | MIC (μg mL−1): Ciprofloxacin: S. aureus = 3.125, P. aeruginosa = 3.125, B. cereus = 3.125, K. pneumoniae = 3.125 | 72 |
 |
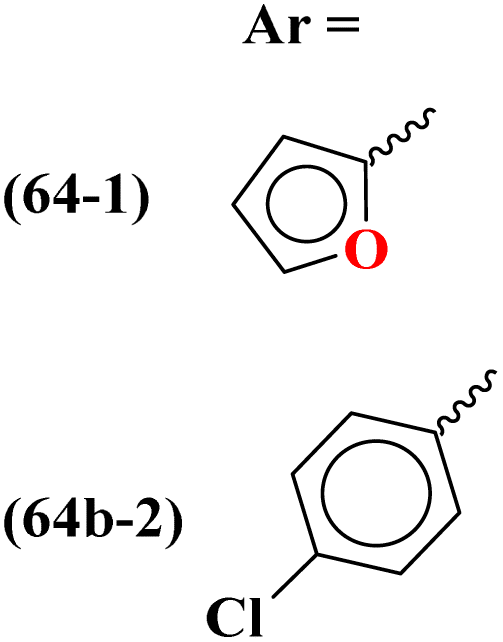 |
MIC (μM): 64b-1: DS = 2.49, MDR = 9.91, XDR = 39.72, 64b-2: DS = 2.28, MDR = 18.14, XDR = 36.31, IC50 (μM) for InhA: 64b-1: 3.90 ± 0.09, 64b-2: 2.47 ± 0.11 | MIC (μM): Isoniazid: DS = 0.88, MDR = Resistant, XDR = resistant, IC50 (μM) for InhA: triclosan = 1.22 ± 0.13 | 73 |
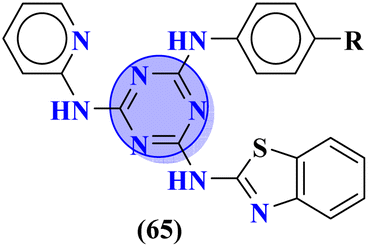 |
R = –CF3 | MIC (μg mL−1): Staphylococcus aureus = 25, Bacillus subtilis = 62.5, Clostridium tetani = 100, Salmonella typhi = 100, Escherichia coli = 12.5, Vibrio cholerae = 250 | Norfloxacin MIC (μg mL−1): Staphylococcus aureus = 100, Bacillus subtilis = 10, Clostridium tetani = 50, Salmonella typhi = 10, Escherichia coli = 10, Vibrio cholerae = 10 | 74 |
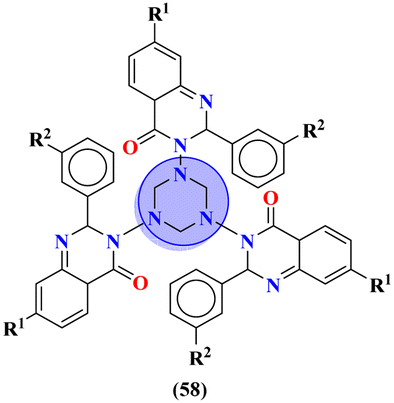
To cope with developed resistance in bacterial strains, molecular hybrids of s-triazine and pyrrole (57) were synthesized and screened for their biological activities against 27 different kinds of S. aureus.66 The biological screening came out with results indicating the conjugates were potent inhibitors. Through the structure–activity relationship, it was noted that the presence of a single imidazole ring in their structure enhanced the activities. The minium inhibitory concentration of this prominent conjugate with pyrazole ring against S. aureus is outed to be ATCC 33591 = 3.9, MRSA BRS3 = 0.95, MRSA C1 = 1.95, MRSA C7 = 3.9, MRSA C14 = 3.9, and MRSA G1 = 7.8 μM (Table 2). New molecular hybrids of s-triazine and quinazoline were synthesized starting from anthranilic acid. After complete synthesis and characterization, these molecular hybrids (58)67 were screened for their biological activity again 5 Gram-positive and 2 Gram-negative strains using Saboraud's medium and Nutrient broth method, and ZOI (zone of inhibition) was noted. After a complete screening and activity SAR analysis, it was confirmed that the compound with R1 = H and R2 = H is the most potent. Zone of inhibition in (mm) can be summarized as Gram positive strains S. aureus = 14.84 ± 0.3, S. faecalis = 14.84 ± 0.4, B. substilis = 14.4 ± 0.3, P. vulgaris = 15.64 ± 0.8, and B. pumilus = 16.64 ± 0.4 while for Gram negative, E. coli = 14.04 ± 0.44, and K. pneumoniae = 11.04 ± 0.43 (Table 2).
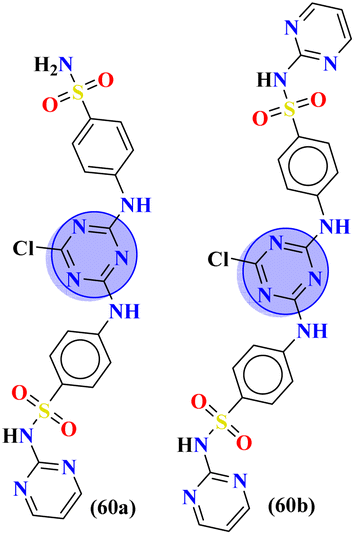
Herein, fused rings molecular hybrids (59) of pyrimidine and triazine are reported.68 Starting from 4-oxopyrimidin-2-yl-thioureas and respective aryl aldehydes, these conjugates were synthesized in good yield and their biological activities were evaluated against E. coli and S. aureus, and their minimum inhibition concentration was noted as (MIC) μg mL−1, E. coli = 256, and S. aureus = 256 (Table 2). Being a versatile scaffold, s-triazine is found to be a very potent antibacterial agent. To evaluate this antibacterial activity, molecular hybrids [(60a) and (60b)] of s-triazine and pyrimidine with additional sulfonamide moiety69 were synthesized in good yield and characterized theoretically and experimentally. Through SAR analysis, it was observed that when s-triazine is tethered to the pyrimidine ring its activities are enhanced meanwhile, when the sulfonamide group is attached activity is also increased since it has a synergic effect on the s-triazine ring. Bioassays were performed, zone of inhibition was calculated and following results were obtained for 60a: E. coli = 30 ± 0.42 mm, B. subtillis = 28 ± 0.22 mm, S. pyogenes = 19 ± 0.56 mm, P. aregenosa, and for 60b: E. coli = 35 ± 0.12 mm, B. subtillis = 18 ± 0.32 mm, and S. pyogenes = 20 ± 0.56 mm (Table 2).
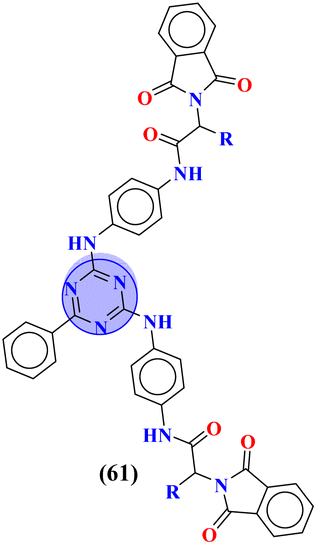
In order to develop new potent antibacterial agents, Asadi and coworkers in 2023, synthesized a series (61) bioconjugates of s-triazine clubbed phthalimide via N-α-amino acids spacer.70 These conjugates were synthesized in moderate to good yield and characterized through spectroscopic techniques. The biological activities of these compounds against 4 strains of bacteria (2 Gram-positive and 2 Gram-negative) were evaluated. Through SAR studies, it was concluded that a compound possessing a hydrophobic iso-butyl chain is the most potent and efficient among the series. The MIC values for this compound were noted which are MIC (μg μL−1): E. coli = 1.25, S. enteritidis = 1.25, S. aureus = 1.25, and B. subtilis = 1.25. In silico docking analyses were also performed with DNA gyrase enzyme to check interactions between ligand and DNA gyrase and the presence of imide, amide, and nitrogen atoms within the ring of s-triazine was highly appreciated for forming hydrogen bonding interactions (Table 2).
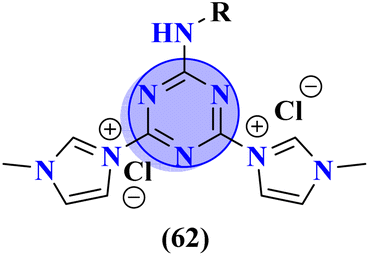
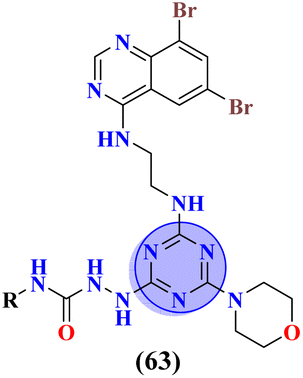
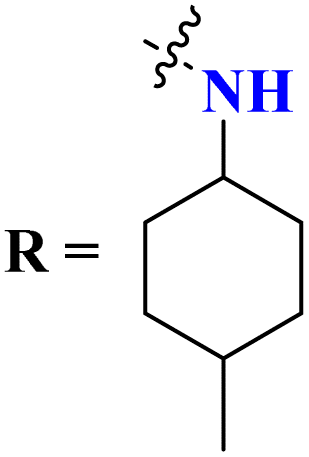
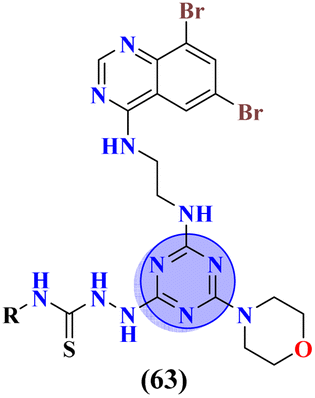
Likewise, molecular hybrids of s-triazine and imidazole (62) are also reported as quaternary ammonium salts as potent antibacterial agents.71 Using the sustainable methodology, 2,4-dichloro-6-alkylamine-s-triazine was reacted with methyl imidazole to synthesize the targeted products in a very good yield of ∼90% in lesser time of 1.5 hours involving haloalkanes as the reactant. The chain length of the alkyl group ranged from C10–C18. During bioassay, the compound with a carbon chain of 14 carbon atoms (tetradecane) came out to be the potent one. These compounds were then evaluated against Gram-positive and Gram-negative bacterial cultures and MIC in (mg L−1) was evaluated to be S. aureus = 3.90, E. faecalis = 62.5, E. coli = 62.5, and P. aeruginosa = 250 (Table 2). Patel and coworkers reported the synthesis of molecular hybrids of quinazoline (63) and s-triazine and additional semi-carbazide moiety.72 After complete synthesis and characterization through different spectroscopic techniques. In SAR analysis, it was noted that the compound possessing 4-methyl cyclohexylamine substituent is the most efficient in comparison to its contrasting scaffold-like aromatic and aliphatic amine. These compounds were evaluated for their biological activities as antibacterial agents against Gram-positive as well as negative strains, and the minimum inhibition concentration (MIC) of the most highlighted compound was noted to be (MIC in μg mL−1) S. aureus = 6.25, P. aeruginosa = 12.5, B. cereus = 100, and K. pneumoniae = 100 which are quite comparable with that of standard drug ciprofloxacin (Table 2). Analogs of these compounds i.e., thiosemicarbazides were also synthesized and evaluated in the same way. It was concluded that thiosemicarbazides exhibited more anti-HIV properties than semicarbazides (Table 3).
| Compounds | Most potent compound | MIC/ZOI of ref. drug | Refs. | |
|---|---|---|---|---|
| Substituents | MIC/zone of inhibition | |||
 |
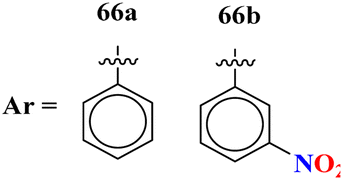 |
The MIC (μg mL−1) against Candida albicans: 66a = 1.475 × 10−8, 66b = 2.1851 × 10−4, (IC50 μg mL−1) for CYP51 inhibition: 66a = 7.451 ± 0.404, 66b = 7.451 ± 0.183 | The MIC (μg mL−1) against Candida albicans: fluconazole = 0.857, (IC50 μg mL−1) for CYP51 inhibition, fluconazole = 1.614 ± 0.087 | 77 |
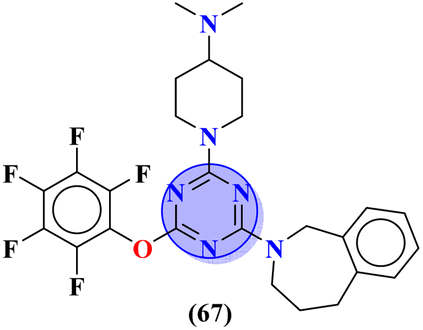 |
— | MIC (μg mL−1): C. albicans SC5314 = 16.08, C. glabrata = 4, C. tropicalis = 8, C. parapsilosis = 8, C. dubliniensis = 8, S. cerevisiae BY4741 = 4 | — | 58 and 78 |
 |
X = –CH2– | MIC (mg mL−1): C. albicans = 3.191 | – | 79 |
 |
R = –CF3 | MIC (μg mL−1): Candida albicans = 250 | Griseofulvin MIC (μg mL−1): Candida albicans = 500 | 74 |
 |
R = 2-OH | IC50 = 12.1 μM, CC50 > 400μM | Remdesivir: IC50 = 0.07 ± 0.04 μM, CC50 > 94.9 μM | 80 |
 |
 |
IC50 (μg mL−1): HIV-1 (IIIB) = 49.13, HIV-2 (ROD) = 49.13, CC50 (μg mL−1): HIV-1 (IIIB) = 49.13, HIV-2 (ROD) = 49.13 | Azidothymidine IC50 (μg mL−1) : HIV-1 (IIIB) = 0.0049, HIV-2 (ROD) = 0.0061, CC50 (μg mL−1), HIV-1 (IIIB) = 0.0074, HIV-2 (ROD) = 0.010 | 72 |
 |
 |
EC50 (μM): human cytomegalovirus > 1.20, herpes simplex virus 1 > 1.20, Varicella-zoster virus > 1.20 | EC50 (μM) Ganciclovir: human cytomegalovirus = 4.65, acyclovir: herpes simplex virus 1 = 0.78, varicella-zoster virus = 3.28 | 81 |
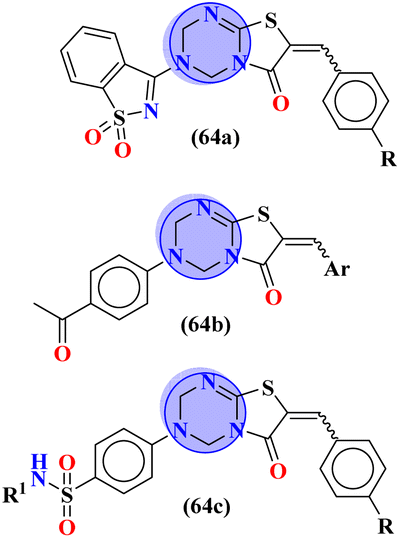
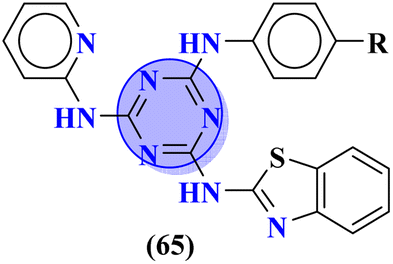
To combat infectious diseases like tuberculosis, 3 new series of s-triazine pendant thiazolidin-4-one were synthesized in good yield starting from thioureas. This thiazolidine-4-one was then fused with triazine to form thiazolo[3,2-a][1,3,5]triazine. This fused ring was then further clubbed with other heterocycles and sulfonamide moiety to enhance the biological activity.73 These compounds were then evaluated for their activity against Mycobacterium tuberculosis (Mtb) drug-sensitive (DS), extensive drug-resistant (XDR), and multi-drug resistant (MDR) strains. In SAR, it was noted compounds with an additional heterocyclic ring furan or an electron-withdrawing group at position 4 of the benzene ring are proven to enhance the biological activity of the compound while any electron-donating group decreases the activities. The MIC in μM is found to be DS = 2.49, MDR = 9.91, and XDR = 39.72 for 64b-1 while for the 64b-2 DS = 2.28, MDR = 18.14, and XDR = 36.3. In addition to tuberculosis, these compounds were also screened for their activity against bronchitis-causing bacteria. Compounds 64b-1 and 64-2 were also screened as inhibiting agents for M. tuberculosis enoyl-acyl carrier protein reductase (InhA) and IC50 (μM) for InhA, 64b-1: 3.90 ± 0.09 and for 64b-2: 2.47 ± 0.11 is calculated (Table 2). In the same manner, two novel series of molecular hybrids (65) of s-triazine with benzothiazole and s-triazine with coumarin were synthesized.74 After complete characterization through FTIR, NMR, and mass spectrometry these novel bioconjugates were further analyzed for their biological activities as a potent medicinal conjugate. From obtained data and in vitro screening it was noted that these series exhibit a remarkable antibacterial activity with MIC in the range of 12.5–62.5 μM and antifungal activity with MIC in the range of 100–200 μM. With the evaluation of SAR analyses, it was noted that hybrid of s-triazine and benzothiazole with electron-withdrawing substituent –CF3 denoted the most promising activities. MIC (μg mL−1) for this compound is calculated to be Staphylococcus aureus = 25, Bacillus subtilis = 62.5, Clostridium tetani = 100, Salmonella typhi = 100, Escherichia coli = 12.5, and Vibrio cholerae = 250 (Table 2). These compounds were also evaluated for their anti-fungal activities MIC against C. albicans came to be in the range of 100–200 μM (Table 3).
3.3 Antifungal and antiviral activities
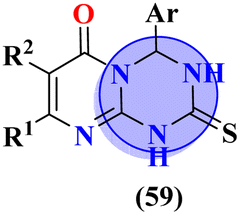
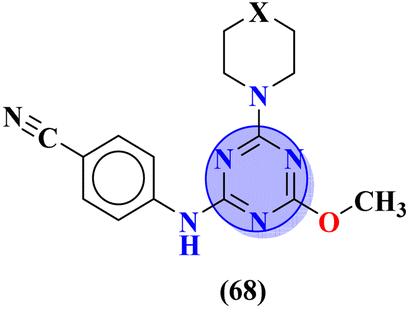
Another major issue in the public health problem is increased due to the ubiquity nature of many fungal species. The developed antifungal drug resistance is the sequel of uncontrolled use of antifungal agents.75 Likewise, CoV-2 is a severe acute respiratory syndrome coronavirus type-2 which was discovered in late 2019. This virus is the cause of the pandemic and global crisis.76 s-Triazine based drug-candidates are also used as an antiviral agent for this virus. Herein, in Table 3 some notable hybrids of s-triazine are tabulated as potent antifungal and antiviral agents.Novel compounds (66) of s-triazine conjugated with tetrazole were synthesized from microwave-assisted one-pot synthesis using amino-tetrazole, aromatic/heteroaromatic aldehydes, and cyanamide in acetic acid as a solvent in moderate to good yield.77 The greenly synthesized new conjugates were screened against C. albicans in comparison to fluconazole as an antifungal agent. While evaluating the SAR, it was noted that the compound with the nitro group present at the meta-position appeared to be the most potent among the set, their MIC was noted in (μg mL−1) 66a = 1.475 × 10−8 for 66b = 2.1851 × 10−4. IC50 in μg mL−1 for enzyme CYP51 inhibition was also calculated 66a = 7.451 ± 0.404 and for 66b = 7.451 ± 0.183. In addition to this, in vitro analysis against lanosterol 14-α-demethylase (CYP51 protein) braced antifungal analysis (Table 3). Compound (67) Me156 (ref. 78) is an s-triazine-based synthetic compound that was screened for its biological activities against human pathogenic fungus specie namely, C. albicans, several nonpathogenic non-albicans Candida strains, and S. cerevisiae and minimum inhibitory concentration (MIC) was noted as C. albicans SC5314 = 16.08, C. glabrata = 4, C. tropicalis = 8, C. parapsilosis = 8, C. dubliniensis = 8, and S. cerevisiae BY4741 = 4 (μg mL−1). It was concluded that (67) inhibits yeast–hyphae transition at significant MIC (Table 3). Molecular hybrids of s-triazine (68) with different heterocycles of 6-membered rings were reported by Dabade and his coworkers.79 In their research work, the concoction of genetic algorithm multiple-linear-regression was used to investigate QSAR and molecular docking of these s-triazine molecular hybrids which braced their motive of potent inhibitors of C. albicans fungal strains. SAR analysis suggested that when the attached 6-memebered heterocyclic ring is piperidine the activity is enhanced remarkably in comparison to other analogs like piperazine. The MIC of this piperidine tethered s-triazine ring is for C. albicans = 3.191 mg mL−1 (Table 3).
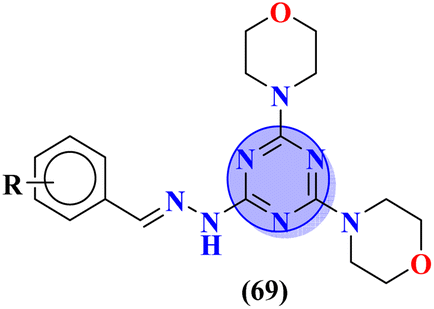
To cure severe acute respiratory syndromes 3 new series (69) of the s-triazine pendant morpholine were synthesized in good yield and its biological activity was screened against SAR-CoV-2.80 SAR suggested one derivative from the series with a strong electron-donating substituent like 2-OH group came out to be potent with the least inhibitory concentration and less toxicity against Caco-2 cells. In vitro ADME analysis and inhibition against helicase enzyme were also studied and IC50 = 12.1 μM and CC50 > 400 μM were obtained (Table 3). Fused ring molecular hybrids of s-triazine and pyrazole (70) are reported as antiviral conjugates.81 Starting from N-(2,2-dichloro-1-cyanoethenyl)-carboxamides, these 5 different substituted molecular hybrids of this series were synthesized in good yield and their biological activity was evaluation against human cytomegalovirus, herpes simplex virus-I, and Varicella-zoster virus. SAR suggested that compounds with simple phenyl rings are more potent than other compounds. EC50 values of these compounds came out to be human cytomegalovirus >1.20 μM, herpes simplex virus 1 > 1.20 μM, and Varicella-zoster virus >1.20 μM (Table 3).
3.4 Anti-alzheimer and antioxidant activity
Alzheimer's disease (AD) is a multifactorial neurodegenerative condition that is defined by loss of memory and other perceptive abilities.82 Acetylcholinesterase or butyrylcholinesterase can lead to hydrolysis of acetylcholine and ultimately to AD.83 Likewise, some patients suffering from Alzheimer's are reported to have a high level of free radicals and oxidating agents which promote oxidative stress in the brain.84 s-Triazine-based molecular hybrids are multi-targeted ligands that can act as an antioxidant as well as anti-acetylcholinesterase agents (Table 4).| Compounds | Most potent compound | IC50 ref. drug | Refs. | |
|---|---|---|---|---|
| Substituents | IC50 | |||
 |
 |
(IC50 μM): BuChE = 1.0 ± 0.1 | Donepezil (IC50 μM): BuChE = 2.9 ± 0.5 | 85 |
 |
 |
IC50 = 0.044 μM | Donepezil: IC50= 0.052 μM | 86 |
 |
 |
IC50 (μM) hAChE = 10.82 ± 0.98, hBuChE = 0.046 ± 0.002 | Donepezil: hAChE = 0.027 ± 0.004, hBuChE = 3.17 ± 0.06 | 87 |
 |
 |
Ki = 11 nM | — | 88 |
 |
 |
Ki = 8 nM | — | 89 |
 |
 |
IC50 (μM) EeAChE = 8.49 ± 0.01, EqBuChE = 24.79 ± 0.28, DPPH radical scavenger = 16.72 ± 1.39 | IC50 (μM) Galantamine: EeAChE = 5.0 ± 0.295, EqBuChE = 20.96 ± 0.45, ascorbic acid: DPPH radical scavenger = 10.74 ± 0.36 | 90 |
 |
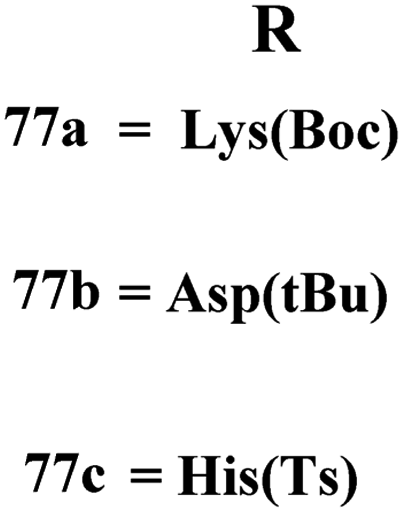 |
IC50 (mM) AChE: 77a = 0.055 ± 0.001, 77b = 0.065 ± 0.002, 77c = 0.067 ± 0.003, BACE1: 77a = 11.09 ± 2.29, 77b = 33.82 ± 3.91, 77c = 14.25 ± 3.45 | IC50 (mM) AChE: Tacrine = 0.046 ± 0.013, donepezil = 0.274 ± 0.08, BACE1: quercetin = 4.89 ± 2.31 | 91 |
 |
 |
IC50 (μg mL−1): DPPH = 25.2 ± 3.1 | IC50 (μg mL) butylated hydroxytoluene: DPPH = 25.2 ± 3.1 | 92 |
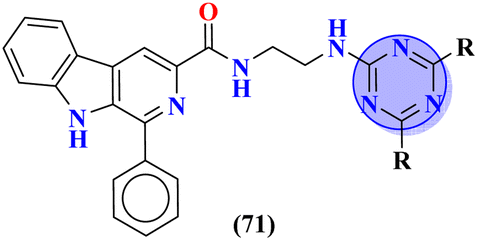
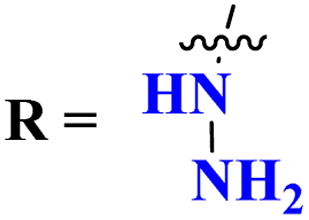
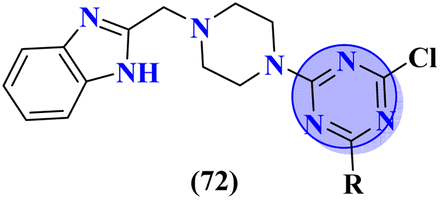
Carboline has three fused ring structures with nitrogen as a heteroatom. The molecular hybrids of s-triazine and carboline (71)85 were prepared through a linker approach in good yield and screened in vitro against cholinesterase enzymes (acetyl and butryl). The IC50 value of these compounds ranged between 1.0 and 18.8 μM with the lowest value of 1.0 ± 0.1 μM which is more efficient than the standard drug Donepezil, IC50 = 2.9 ± 0.5 μM. Compound with a free amino group (R = NHNH2) appeared to be the most potent among the series and virtual docking analysis also supported the bio-screening showing this compound binds in the same site with efficient binding where butyrylthiocholine can bind (Table 4). Benzimidazole is a highly biologically active moiety. This potent moiety was clubbed with the magical scaffold s-triazine to synthesize a new series (72).86 A total of 24 compounds were synthesized and characterized via spectroscopic methods. This series was designed as multi-target ligands to treat Alzheimer's disease. Using colorimetric Ellman's method, synthesized compounds were screened for their cholinesterase inhibition activity. It was evaluated through SAR that the presence of cyclohexyl amine makes the inhibitory action even more potent than the standard drug i.e., IC50 for this compound is 0.044 μM while Donepezil, IC50 = 0.052 μM. In silico ADMET, docking, and MD stimulation suggested these compounds as efficient anti-Alzheimer drug candidates (Table 4).
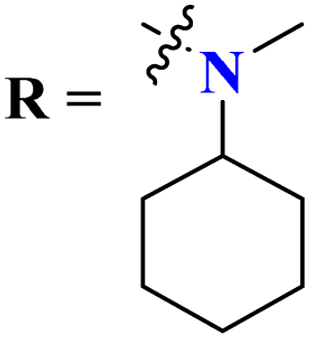
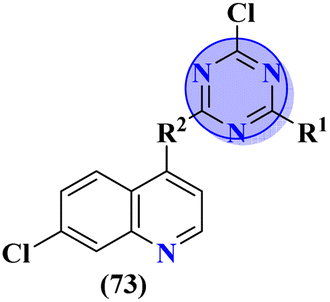
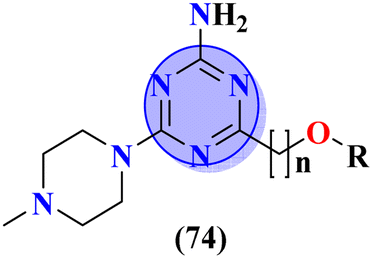
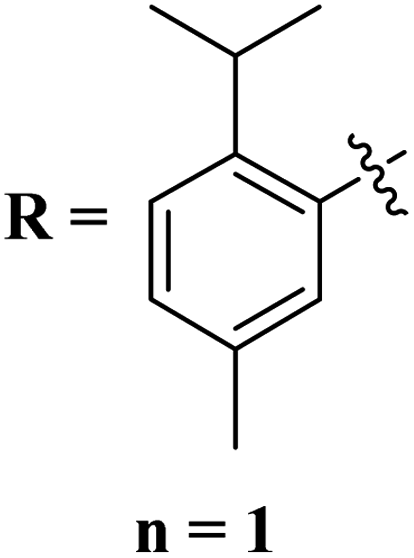
Quinoline is a fused bicyclic, nitrogen-containing heterocyclic compound. This heterocyclic compound was linked with s-triazine to form 16 new s-triazine-quinoline molecular hybrids (73).87 Using colorimetric Ellman's method, synthesized compounds were screened for their cholinesterase inhibition activity, SAR suggested that when R1 is pyrrolidine and R2 the linker between s-triazine and quinoline is piperidin-3-ylmethanamine, the anti-Alzheimer potency is remarkably enhanced than any other amino group present as the linker and the IC50 value was noted to be hAChE = 10.82 ± 0.98 μM and hBuChE = 0.046 ± 0.002 μM. Docking analysis suggested that this compound can interact with and block the active site of butyrylcholinesterase (Table 4). 5-HT6 receptor is abundant in CNS and maintains the presence of neurotransmitters like GABA, dopamine, and acetylcholine which are notable in AD. A decrease in the density of this enzyme improves the mental health of Alzheimer's patients. For this purpose, s-triazine-based compounds (74) were synthesized with N-methylpiperazine scaffold with additional ether linkages.88 In vitro analysis of these conjugates was performed against the 5-HT6 receptor, and it was noted that when alkyl chains are as short as only one carbon atom and the aromatic ring contains a bulky group like iso-propyl the inhibitory action is enhanced to a prominent level the inhibitor constant (Ki) was calculated to be 11 nM (Table 4).
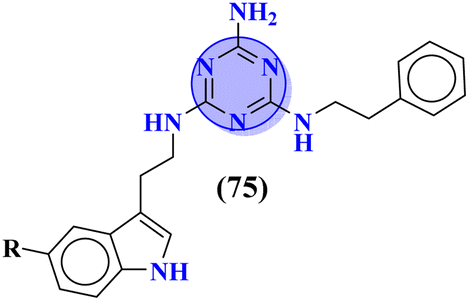
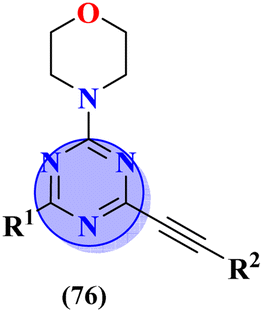
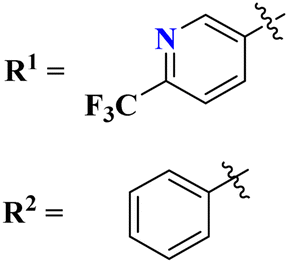
Likewise, keeping in view the psychiatric function of the 5-HT7 receptor, novel s-triazine conjugated benzo-pyrrole compounds (75) were synthesized in good yield and their biological activity was screened in vitro against 5-HT7 receptor and inhibitor constant was noted. From SAR it was evaluated that fluoro-substituent and benzo-pyrrole ring enhances the inhibitory actions of ligand.89 Ki of this compound was evaluated to be 8 nM (Table 4). Reddy and his coworkers reported the molecular hybrids of morpholine and s-triazine (76) to target Alzheimer's disease.90 These molecular hybrids were synthesized in good yield and their inhibitory effect was evaluated against enzymes (acetylcholinesterase and butyrylcholinesterase). Moreover, these compounds were also tested for their antioxidant properties and DPPH free radical scavenger. The most potent compounds in the series exhibited the IC50 for AChE and antioxidants in the range of 7.23 to 10.35 μM and 14.80–27.22 μM respectively. Excellent in vitro and SAR analysis results indicated that the compound with substituted pyridine and R2 being simple phenyl ring are far more potent than other similar compounds with IC50 EeAChE = 8.49 ± 0.01 μM, EqBuChE = 24.79 ± 0.28 μM, and DPPH radical scavenger = 16.72 ± 1.39 μM (Table 4).
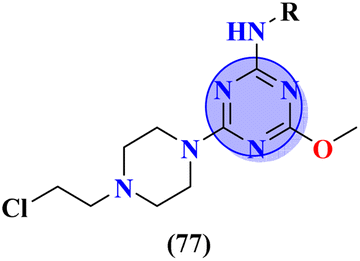
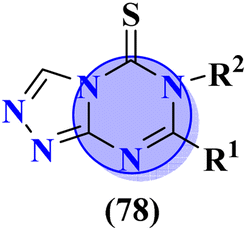
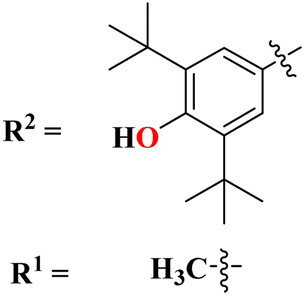
A new series containing molecular hybrids of s-triazine and piperazine was synthesized (77).91 The attributes of this series are that it contains nitrogen mustard and dipeptide residue to enhance the biological activities as anti-Alzheimer. These compounds were tested for their biological activities against enzymes related to Alzheimer i.e., acetylcholinesterase and β-secretase. Compounds with amino acids lysine, aspartic acid, and histidine exhibited highly efficient potency in comparison to standard drugs with IC50 (mM) AChE: 77a = 0.055 ± 0.001, 77b = 0.065 ± 0.002, 77c = 0.067 ± 0.003, BACE1: 77a = 11.09 ± 2.29, 77b = 33.82 ± 3.91, and 77c = 14.25 ± 3.45 (Table 4). Compounds (78) are the molecular hybrids of s-triazine and 1,3,5-triazine which are fused with each other giving rise to novel pharmacophores.92 These bioconjugates were synthesized in good yield via the conventional method and their biological activity was examined as DPPH free-radical scavenger, that is to say, antioxidant. After complete examination and evaluation, and through SAR analysis it was confirmed that compounds possessing hydroxyl group in their structures depicted more efficiency as a free radical scavenger in in vitro analysis than other compounds in the same series due to electron-donating effect. IC50 of this compound was noted to be 25.2 ± 3.1 μg mL−1 and compared with standard antioxidant drugs i.e., BHT 25.2 ± 3.1 μg mL−1 (Table 4).
3.5 Antimalarial activity
According to the World Report Malaria-2021 by WHO, in 2020 a major increment in malarial death by 12% was observed estimating 627![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths globally.93 The species which is the main cause of death in Africa and Southeast Asia is Plasmodium falciparum (Pf). P. falciparum is the deadliest plasmodium species that causes malaria in humans.94 Plasmodium has developed a great resistance against formerly available drugs and a struggle of decades in going all in vain.95 In Table 5 some recently designed antimalarial drug candidates of s-triazine are tabulated which can cope with these resistant plasmodium species and save mankind.
000 deaths globally.93 The species which is the main cause of death in Africa and Southeast Asia is Plasmodium falciparum (Pf). P. falciparum is the deadliest plasmodium species that causes malaria in humans.94 Plasmodium has developed a great resistance against formerly available drugs and a struggle of decades in going all in vain.95 In Table 5 some recently designed antimalarial drug candidates of s-triazine are tabulated which can cope with these resistant plasmodium species and save mankind.
| Compounds | Most potent compound | IC50 ref. drug | Refs. | |
|---|---|---|---|---|
| Substituents | IC50 | |||
 |
 |
IC50 = 9.54 μg mL−1 | — | 96 |
 |
 |
IC50 (μM) 80a: 3D7 = 13.25 ± 0.82, Dd2 = 16.30 ± 0.93, 80b: 3D7 = 88.74 ± 2.14, Dd2 = 14.72 ± 0.82 | Chloroquine: 3D7 = 2.18 ± 0.12, Dd2 = 3.70 ± 0.13 | 97 |
 |
 |
Dead rings trophozoites IC50 (μg mL−1) = 53.85 ± 0.42, Vero cells: CC50 (μg mL−1) = 550.26, SI index = 10.21 | Chloroquine: Dead rings trophozoites: IC50 (μg mL−1) = 0.7 ± 0.004, Vero cells: CC50 (μg mL−1) = 560.29, SI index = 801.28 | 98 |
 |
 |
IC50 (obs.) = 1.36, logIC50 (obs.) = 0.134 |
— | 99 |
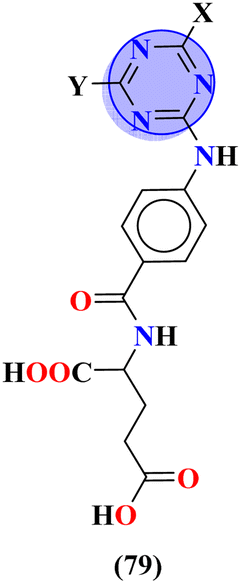
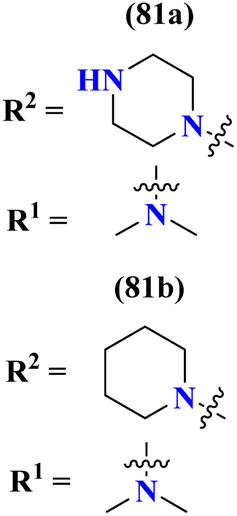
To handle the resistant plasmodium, s-triazine-based novel compounds (79) were designed by Adhikari and coworkers.96 They designed 120 different compounds, and their biological activity was screened for the Dd2 strain of plasmodium which is a chloroquine-resistant strain and IC50 was calculated. SAR suggested that compounds possessing an additional heterocyclic ring of piperidine and a free amino-group on s-triazine have the lowest IC50 value i.e., 9.54 μg mL−1. To support these evaluations, virtual screening like molecular docking against Pf-DHFR-TS protein was performed and interactions were observed (Table 5). Likewise, a new series of 120 compounds of N-(4-aminobenzoyl)-L-glutamic acid pendant s-triazine (80) was successfully synthesized via microwave-assisted synthesis techniques in good yield.97 Before in vitro analysis, these compounds were evaluated through in silico modeling and only 10 compounds out of 120 were selected for further screening. Through SAR analysis, it was observed that this series possessed two main hits, one with piperidine and one with piperazine moiety. These six-membered heterocyclic moieties are the reasons for such splendid antimalarial activities. IC50 of these compounds is obtained as 80a: 3D7 = 13.25 ± 0.82 μM, Dd2 = 16.30 ± 0.93 μM 81b: 3D7 = 88.74 ± 2.14 μM, and Dd2 = 14.72 ± 0.82 μM (Table 5). Gogoi and his coworkers synthesized a novel series of molecular hybrids of pyrazole-tethered s-triazine (81).98 Completing their wet-lab experiments and characterizations, they moved to virtual analysis of their synthesized molecular hybrids and picked the top-three compounds for in vitro analyses and antimalarial agents. In virtual analyses, it was observed that hydrogen bonding has a major contribution with residual amino acids Asp54, Ile164, Arg122, and Tyr170. In vitro analysis showed that these top-three compounds have IC50 in the range of P. falciparum in the range of 53.85 to 100 μg mL−1. SAR suggested that the least IC50 53.85 μg mL−1 value is given by the compound with substituent 4-chloroaniline. This may be due to alkyl-halogen and hydrogen-bond interactions of chlorine and amino group as visualized by molecular docking results (Table 5).
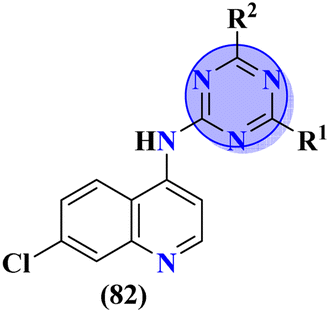
To treat malaria, the major target for medicinal chemists is Plasmodium falciparum dihydrofolate reductase (Pf-DHFR). Mutation in this enzyme leads to resistance to malaria-causing plasmodium. In this regard, molecular hybrids (82) of s-triazine and 4-anilinoquinoline99 were synthesized and their biological profiling and SAR suggested that compounds with an addition morpholine ring are highly potent and show remarkable antimalarial activities with IC50 = 1.36 μg mL−1 and logIC50 = 0.134 μg mL−1 (Table 5).
3.6 Anticonvulsant activity
An abrupt, forceful, erratic movement of the body brought on by the uncontrollable contraction of muscles, particularly linked to brain illnesses like epilepsy is called convulsion.100,101 Despite the accessibility to advanced anti-epileptic medications, still there are numerous problems for chemists, for instance, cytotoxicity, fatal adverse drug reactions, and patient resistance to well-established drugs.102 In an attempt to encounter these issues, s-triazine-based conjugates have been represented in Table 6.| Compounds | Most potent compound | IC50 ref. drug | Refs. | |
|---|---|---|---|---|
| Substituents | % Protection | |||
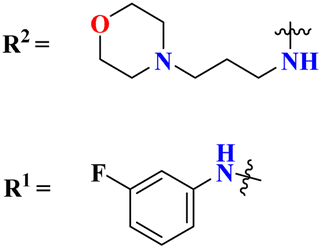
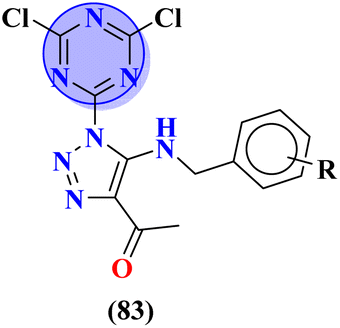 |
 |
% Protection MES: 83a = 100, 83b = 100, 83c = 100, 83d = 100 | % Protection MES: valproate = 100, methaqualone = 100 | 103 |
| PTZ: roscovitine = 100, 83b = 100, 83c = 100, 83d = 100 | PTZ: valproate = 100, methaqualone = 100 | |||
| Therapeutic index: 83a = 3.35, 83b = 2.76, 83c = 5.61, 83d = 4.41 | Therapeutic index: valproate = 2.59, methaqualone = 1.40 | |||
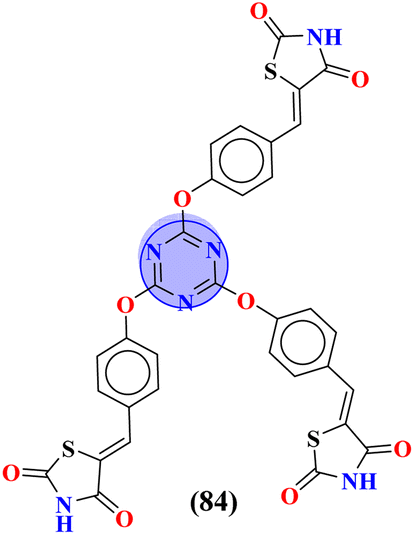 |
— | Docking score = −10.79, Interactions = N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (VAL-86), O (ether)⋯H–O (THR87) C (VAL-86), O (ether)⋯H–O (THR87) |
— | 104 |
To grapple with the health problems due to epilepsy, a new series (83) of s-triazine tethered 1,2,3-triazole was synthesized using click chemistry approach,103 and their biological activity as anticonvulsant agents was evaluated via maximum electroshock seizure (MES), and pentylenetetrazole (PTZ) induced seizure. In the second phase of analysis, compounds with fluoro (83a), chloro (83b), trifluoro-carbon (83c), and nitro (83d) substituents appeared to be more potent than the standard drug methaqualone and valproate due to electron-withdrawing effect on the benzene ring. Percentage protection against MES and PTZ was evaluated to be 100% while the therapeutic index for these 4 hits was calculated as 83a = 3.35, 83b = 2.76, 83c = 5.61, and 83d = 4.41. Gamma amino butyric acid (GABA) was chosen as the target protein for the molecular docking study of these conjugates because convulsions are usually aroused when there is a low concentration of GABA (Table 6). Through the optimization, multicomponent reactions Knoevenagel condensation (method A) and Bucherer–Bergs (method B), a novel series of compounds (84) with two or three biologically active nuclei of imidazolidine-2,4-dione and thiazolidine-2,4-dione (TZD) were prepared and characterize via FT-IR, 1HNMR, 13CNMR, and elemental analysis.104 All the compounds were put through molecular docking experiments for anticonvulsant drug binding (ADB) to the VGCIP i.e., voltage-gated sodium channel inner pore to assess the anticonvulsant activity of these conjugates. According to the outcomes of the in silico molecular interaction conjugate containing the s-triazine scaffold appeared to be the most efficient. The docking score was calculated to be −10.79 kcal mol−1. Major interactions observed are N–H⋯OC (VAL-86), O (ether)⋯H–O (THR87) (Table 6).
3.7 Antidiabetic and anti-obesity activity
Diabetes mellitus is a well-known complex and chronic health issue that greatly adds to high mortality rates by leading to severe health issues, particularly those related to cardiovascular illnesses, renal damage, and paralysis.105 Inhibiting the enzyme glucosidase and dipeptidyl peptidase-4, which catalyzes the hydrolysis of starch in the gut, is an efficient treatment method for managing the glucose brought on by type-2 diabetes.106 However, the currently FDA-approved medications and inhibitors, including acarbose and voglibose, have a number of unfavorable stomach adverse effects that limit their use. Therefore, an intriguing field of study continues to be the hunt for new and more potent inhibitors with fewer adverse effects and lower costs.107 In this regard, s-triazine-based molecular hybrids have been synthesized as potent antidiabetic agents (Table 7).| Compounds | Most potent compound | IC50 ref. drug | Refs. | |
|---|---|---|---|---|
| Substituents | IC50 | |||
 |
 |
IC50 = 44. 72 nM, Ki = 41.74 ± 8.08 nM | Acarbose = 118.0 nM, Ki = 132.74 ± 14.76 nM | 108 |
 |
R = 4-F | IC50 (nM): DPP-4 = 2.32, DPP-8 = >25 000, DPP-9 = >25 000, hERG = 505.21 μM | IC50 (nM): alogliptin, DPP-4 = 3.56, hERG = >34 μM | 109 |
 |
— | Binding energies (DG): 87 = −9.0 to 11.2 kcal mol−1, 88 = −9.2 kcal mol−1, 89 = −10.1 kcal mol−1 | — | 110 |
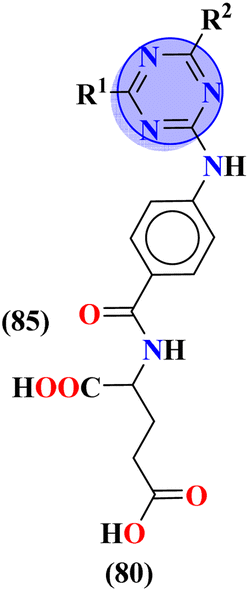
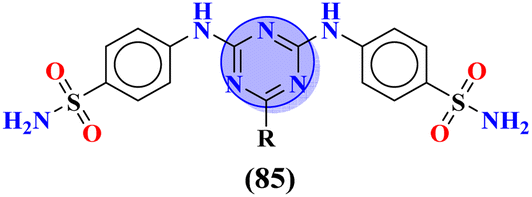
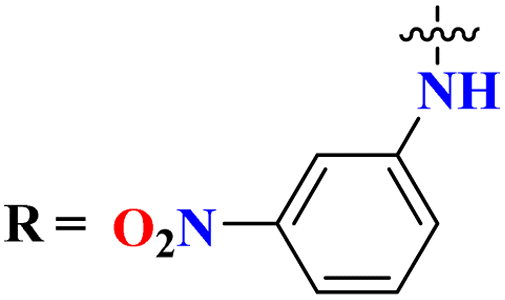
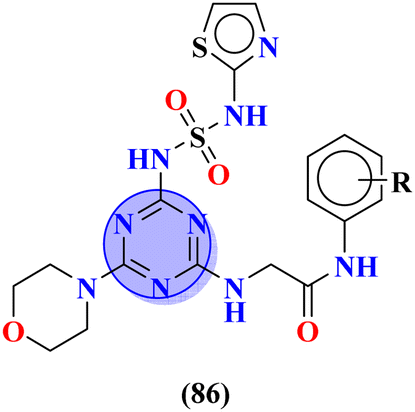
Sulfonamides possess a noble place in the field of medicinal chemistry. This well-potent moiety was clubbed with s-triazine (85) to synthesize molecular hybrids that can serve as multitargeted ligands and cure multiple diseases at the same time.108 Conjugates from this series were evaluated for their inhibitor effect on various enzymes like carbonic anhydrase, acetylcholinesterase, and a-glucosidase and it was concluded form SAR studies that conjugates having an 3-nitro group on linked phenyl ring exhibit more activity than any other analogs due to high electron-withdrawing effect on nitro group. Ki for this conjugate is calculated to be 51.67 ± 4.76 for hCA-I, 40.35 ± 5.74 nM for hCA-II, 41.74 ± 8.08 nM forα-GLY and 335.76 ± 46.91 nM for AChE (Table 7). Gao et al. reported the molecular hybrids of s-triazine with morpholine and thiazole (86). The bioactive thiazole ring was clubbed with s-triazine via a sulfonamide linker.109 These novel molecular hybrids of sulfonamide-thiazole-s-triazine were used as dipeptidyl peptidase-4 (DPP-4) inhibitors to treat diabetes mellitus type II. The in vitro analysis showed that the compound with substituent 4-fluoro has a maximum inhibitory action on DPP-4 with IC50 2.32 nM which is even lower than that of the standard drug, alogliptin. From in silico analysis, it was observed that this compound also showed prominent interactions with the binding site of DPP-4 with hotspot amino acids Ser630, Asp708, and His740. In vivo analyses of blood glucose lower effect were also evaluated (Table 7).
Obesity is brought on by a confluence of excessive food consumption, inactivity, and hereditary vulnerability. To combat obesity and its consequences Badrey and coworkers synthesized molecular hybrids of the s-triazine pendant with thiazole (87, 88) and thiazine (89).110 After the successful synthesis of these conjugates, these conjugates were evaluated through virtual screening against fat mass and obesity-associated (FTO) proteins to analyze the superimposition of these hybrids with the active site. The results of these molecular docking were given in terms of free binding energies (ΔG), 87 = −9.0 to 11.2 kcal mol−1, 88 = −9.2 kcal mol−1, and 89 = −10.1 kcal mol−1. Moreover, through computer-based studies, ADMET, pharmacokinetic studies, and drug–likeness profiles were also evaluated (Table 7).
3.8 Anti-inflammatory activities
Whenever infectious and pathogenic microbes reside in a specific part of tissues or move around the body via the circulatory system they cause inflammation.111,112 Other causes of inflammation may include rupturing or damage of tissues, apoptosis or sudden cell death, ischemia, degeneration, or cancer.113,114 Both innate as well as adaptive immune systems respond against inflammations.114–116 Chronic inflammation is marked by simultaneous sabotage and curing of the damaged and ruptured tissue.117 To encounter the inflammation, molecular hybrids s-triazine have been synthesized with prominent biological properties (Table 8).| Compounds | Most potent compound | % Inhibition of ref. drug | Refs. | |
|---|---|---|---|---|
| Substituents | % Inhibition | |||
 |
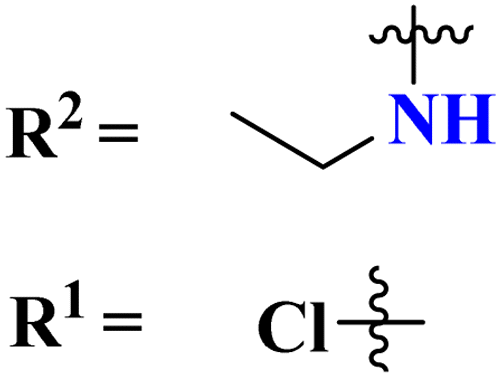 |
% Inhibition at 100 mg kg−1, 93% inhibition (P < 0.001) | % Inhibition at of indomethacin (10 mg kg−1), 90% inhibition | 118 |
 |
 |
% Inhibition at 10 μM, TNF-α = 72%, IL-6 = 79% | % Inhibition of dexamethasone (1 μM), TNF-α = 75%, IL-6 = 81% | 119 |
 |
R = –OCH3 | % Change in paw height, 1 h = 0.30 ± 0.02, 2 h = 0.24 ± 0.01, 3 h = 0.28 ± 0.01, % inhibition = 56%, COX1-IC50 = 6.60 μM, COX2-IC50 = 0.080 μM | — | 120 |
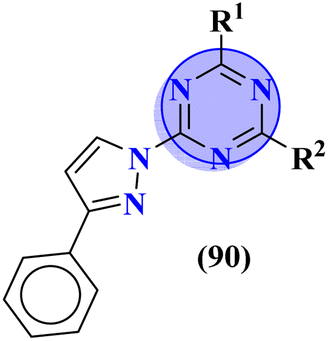
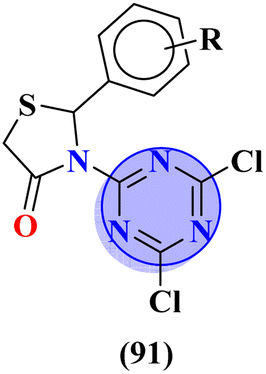
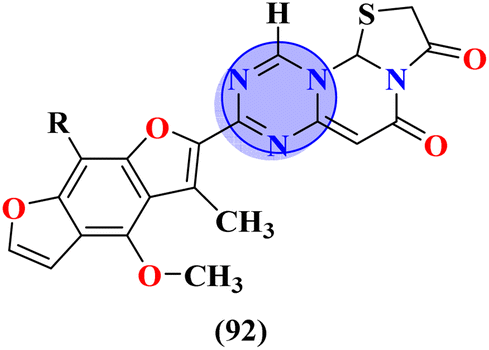
Molecular hybrids of phenylpyrazole and s-triazine (90) were synthesized using conventional methods and characterized by spectroscopic methods.118 These nine molecular hybrids were then evaluated for their biological activities as anti-inflammatory agents against the carrageenan-induced paw edema model in male rats. These analyses were compared with a group of rats with indomethacin as standard. SAR analysis suggested that a hybrid with ethyl amine and free chloro group of s-triazine shows a high percentage of inhibition i.e., 93% at 100 mg kg−1 which is greater than the standard drug (Table 8). In the same manner, to cope with inflammation, molecular hybrids (91) of thiazolidine-4-one and s-triazine were prepared in single step cyclo–condensation reaction of 2-amino-4,6-dichloro-1,3,5-triazine, aromatic aldehydes, and thioglycolic acid.119 These nine molecular hybrids were then evaluated for their biological activities as anti-inflammatory agents against the carrageenan-induced paw edema model in male rats. Through the structure–activity relationship, it was evaluated that the 2,5-difluoro substituent present on the phenyl ring of thiazolidine-4-one enhances the inhibition remarkably. Percentage inhibition at 10 μM is evaluated as TNF-α = 72%, and IL-6 = 79% (Table 8). Furochromones have been reported to possess vast applications in medicinal chemistry. Molecular hybrids (92) include the s-triazine fused with pyrimidine, thiazolidine-4-one, and furochromones.120 This extensive molecular hybrid was then analyzed for its biological activity as anti-inflammatory affects by evaluating the percentage inhibition of edema and cyclooxygenases (COX1 and COX2). SAR suggested that hybrids possessing strong electron-donating groups like methoxy on furochromones are highly potent in controlling inhibition. The percentage inhibition was calculated after every in-cat's paw which can be summarized as, % change in paw height: 1 h = 0.30 ± 0.02, 2 h = 0.24 ± 0.01, 3 h = 0.28 ± 0.01, % inhibition = 56%. While IC50 of enzymes are COX1-IC50 = 6.60 μM, and COX2-IC50 = 0.080 μM (Table 8).
3.9 Anti-Parkinson activities
Parkinson's disease in one of the neurodegenerative diseases which are caused by the loss of dopamine-secreting neurons.121 The currently approved drugs are based on the approach as an alternative to dopamine, but these therapeutic drugs have also been linked to other diseases such as dyskinesia.122 Enzymes like adenosine A2A receptor, GSK-3β, and CK-1δ are hotspot targets to treat neurodegenerative diseases.122 To target these enzymes efficient molecular hybrids of s-triazine have been reported which are included in Table 9.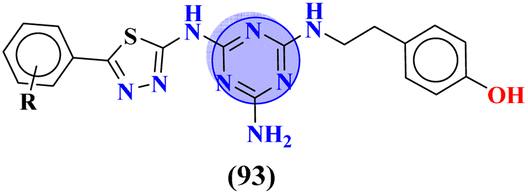
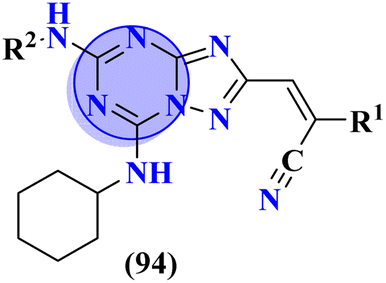
Compounds (93) are molecular hybrids of thiadiazole and s-triazine which are designed, synthesized, and screened as anti-Parkinson's disease.123 After completion of synthesis and characterizations, these conjugates were screened as a radioligand binding assay for adenosine receptors A2AR and A1R. After screening it was concluded that conjugates show selective binding toward A2AR and conjugates with 4-NO2 substituent show the highest efficacy due to the presence of a strong electron-withdrawing group. The Ki for this compound was evaluated to be A2AR = 32.1 nM, A1R = 322.3 nM, and the selectivity index was 10.04 (Table 9). As mentioned before, GSK-3β and CK-1δ are hotspots to cure neurodegenerative disorders. To grapple with these disorders Redenti and coworkers synthesized fused ring molecular hybrids (94) of s-triazine and 1,3,4-triazole and screened these hybrids against GSK-3β and CK-1δ and noted their IC50 values.124 In vitro analysis confirmed that the compound with amide linkage as R2 is non-cytotoxic, and highest efficacy, and can serve as a hit in drug discovery and development. IC50 for this hit is found to be, SK-3β = 0.17 ± 0.02 μM while CK-1δ = 0.68 ± 0.03 μM (Table 9).
3.10 Anti-leishmanial and anti-sleeping sickness activities (antiprotozoal)
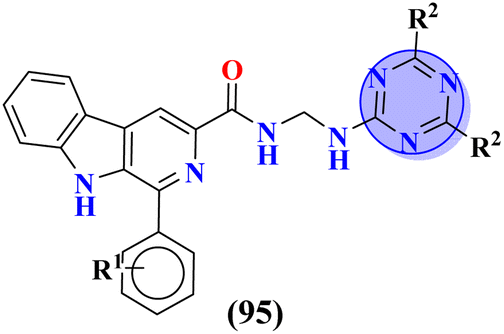
Leishmaniasis is caused by 17 different protozoan species belonging to the genus Leishmania. Many species of Leishmania are resistant to chemotherapy. Moreover, these drugs exhibit high toxicity and other side-effects.125 To cope with this scenario, Barea and co-workers synthesized molecular hybrids (95) of s-triazine and β-carboline alkaloids, and their biological activity was evaluated against promastigote and amastigote forms of Leishmania amazonensis (Table 10). In vitro and SAR analysis showed compound with 4-OMe group at the C1 position of carboline and isopropyl amine linked to s-triazine has the least IC50 value against these forms and low toxicity to J774-A1 cell-lines. IC50 of this compound is Promastigotes = 6.2 ± 1.4 μM, and for Amastigotes = 1.2 ± 0.5 μM, while CC50 is, J774A1 = 145.7 ± 10.1 μM. From further findings, it was confirmed that this compound can alter the cell division cycle which ultimately leads to the death of species126 (Table 10).| Compounds | Most potent compound | IC50 of ref. drug | Refs. | |
|---|---|---|---|---|
| Substituents | IC50 | |||
 |
 |
IC50 (μM): Promastigotes = 6.2 ± 1.4, Amastigotes = 1.2 ± 0.5 | Miltefosine IC50 (μM): promastigotes = 18.5 ± 1.1, amastigotes = 2.4 ± 0.1 | 126 |
| CC50 (μM): J774A1 = 145.7 ± 10.1 | CC50 (μM): J774A1 = 40.5 ± 1.7, Melarsoprol | |||
 |
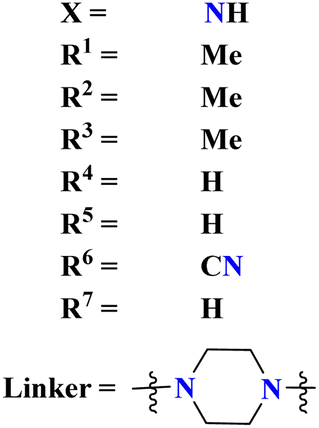 |
IC50 (μM): T. b. bruc. = 0.32, T. b. rhod. = 0.07 | IC50 (μM): T. b. bruc. = 0.03, T. b. rhod. = 0.02 | 128 |
| CC50: MRC-5 = >64 | CC50: MRC-5 = 7.4 | |||
A parasitic ailment termed human African trypanosomiasis also referred to as sleeping sickness, is spread through the bite of the ‘Glossina’ bug, also known as the tsetse fly. It generally results in death if untreated.127 Venkatraj et al., designed, and synthesized dimers (96) of s-triazine linked via piperazine128 as a potent antitrypanosomal agent and screened their biological activity through in vitro and in vivo analyses. It was observed that these dimers show 4–400 times more inhibitory activity as compared to their monomers. IC50 of this dimer is found to be T. b. bruc. = 0.32 μM, T. b. rhod. = 0.07 μM, while CC50 MRC-5 = >64 μM (Table 10).
4. Conclusion and future perspectives
A library of chemically diverse s-triazine-based heterocyclic hybrid molecules has been highlighted as promising drug development candidates. The majority of the summarized hybrid molecules have been reported to show more potent in vitro biological activities when compared to the standard drugs. Importantly, the effect of different substituents installed on the heterocyclic hybrid core has also been highlighted. This review having a chemically diverse collection of s-triazine-based hybrid molecules may serve as an ideal platform to identify potential leads and therefore can be an important avenue for the development of novel drugs with high efficacy and low toxicity for the treatment of various challenging diseases.Based on the gilt-edged properties of s-triazine and its presence in a number of already approved commercially available drugs, it can be regarded as the magic nucleus to be incorporated in the design of novel drug candidates. Especially the design of multitarget directed ligands (MTDLs) i.e., the molecules having the capability to act on several targets at a time is a novel approach that is getting increased attention from the scientific community. In this context, the hybrids molecules having multiple heterocyclic moieties including s-triazine may be the ideal MTDLs for future drug design and development.
Data availability
No data was used for the research described in the article.Author contributions
Muhammad Imran Ali, conceptualization, writing - original draft. Muhammad Moazzam Naseer, conceptualization, supervision, final editing of manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors are grateful to the Department of Chemistry, Quaid-i-Azam University for providing necessary research facilities and funding through its URF program.References
- R. Liu, X. Li and K. Lam, Combinatorial chemistry in drug discovery, Curr. Opin. Chem. Biol., 2017, 38, 117–126 CrossRef CAS PubMed.
- X. Xiao, J.-L. Min, W.-Z. Lin, Z. Liu, X. Cheng and K.-C. Chou, iDrug-target: predicting the interactions between drug compounds and target proteins in cellular networking via benchmark dataset optimization approach, J. Biomol. Struct., 2015, 33, 2221–2233 CrossRef CAS PubMed.
- C. Viegas-Junior, A. Danuello, V. da Silva Bolzani, E. J. Barreiro and C. A. M. Fraga, Molecular hybridization: a useful tool in the design of new drug prototypes, Curr. Med. Chem., 2007, 14, 1829–1852 CrossRef CAS PubMed.
- A. Sharma, R. Sheyi, B. G. de la Torre, A. El-Faham and F. Albericio, s-Triazine: a privileged structure for drug discovery and bioconjugation, Molecules, 2021, 26, 864 CrossRef CAS PubMed.
- P. Singla, V. Luxami and K. Paul, Triazine as a promising scaffold for its versatile biological behavior, Eur. J. Med. Chem., 2015, 102, 39–57 CrossRef CAS PubMed.
- G. Blotny, Recent applications of 2, 4, 6-trichloro-1, 3, 5-triazine and its derivatives in organic synthesis, Tetrahedron, 2006, 62, 9507–9522 CrossRef CAS.
- L. A. Hansen and T. E. Hughes, Altretamine, DICP, 1991, 25, 146–152 CrossRef CAS PubMed.
- I. Todd, Drug therapy of cancer, Br. J. Cancer, 1973, 28, 102 CrossRef , PMCID: PMC2009072.
- B. A. Chabner, P. C. Amrein, B. Druker, M. D. Michaelson, C. S. Mitsiades, P. E. Goss, D. P. Ryan, S. Ramachandra, P. G. Richardson and J. G. Supko, Antineoplastic Agents, The Pharmacological Basis of Therapeutics, Mcgraw-Hill (Tx), New York, 9th edn, 2006 Search PubMed.
- M. A. A. Siddiqui and L. Scott, Azacitidine: in myelodysplastic syndromes, Drugs, 2005, 65, 1781–1789 CrossRef CAS PubMed.
- F. J. Golder, M. M. Hewitt and J. F. McLeod, Respiratory stimulant drugs in the post-operative setting, Respir. Physiol. Neurobiol., 2013, 189, 395–402 CrossRef CAS PubMed.
- M. I. Del Principe, G. Paterno, R. Palmieri, L. Maurillo, F. Buccisano and A. Venditti, An evaluation of enasidenib for the treatment of acute myeloid leukemia, Expert Opin. Pharmacother., 2019, 20, 1935–1942 CrossRef CAS PubMed.
- E. S. Kim, Enasidenib: first global approval, Drugs, 2017, 77, 1705–1711 CrossRef CAS PubMed.
- R. E. Shor, J. Dai, S. Y. Lee, L. Pisarsky, I. Matei, S. Lucotti, D. Lyden, M. J. Bissell and C. M. Ghajar, The PI3K/mTOR inhibitor Gedatolisib eliminates dormant breast cancer cells in organotypic culture, but fails to prevent metastasis in preclinical settings, Mol. Oncol., 2022, 16, 130–147 CrossRef CAS PubMed.
- Q. Dai, Q. Sun, X. Ouyang, J. Liu, L. Jin, A. Liu, B. He, T. Fan and Y. Jiang, Antitumor activity of s-triazine derivatives: a systematic review, Molecules, 2023, 28, 4278, DOI:10.3390/molecules28114278.
- C. Burri and J. Keiser, Pharmacokinetic investigations in patients from northern Angola refractory to melarsoprol treatment, Trop. Med. Int. Health, 2001, 6, 412–420 CrossRef CAS PubMed.
- H. M. LeBaron, The Triazine Herbicides, Elsevier, 2011 Search PubMed.
- H. He, Y. Liu, S. You, J. Liu, H. Xiao and Z. Tu, A review on recent treatment technology for herbicide atrazine in contaminated environment, Int. J. Environ. Res. Public Health, 2019, 16, 5129, DOI:10.3390/ijerph16245129.
- C. B. Breckenridge, J. C. Eldridge, J. T. Stevens and J. W. Simpkins, Symmetrical Triazine Herbicides: Hayes' Handbook of Pesticide Toxicology, Elsevier/AP, 2010 Search PubMed.
- E. Knuesli, in The s-Triazine Herbicides, ACS Publications, 1977, pp. 76–92 Search PubMed.
- H. Ahrens, in Indaziflam: An Innovative Broad Spectrum Herbicide, Discovery and Synthesis of Crop Protection Products, ACS Publications, 2015, pp. 233–245 Search PubMed.
- K. Grossmann, S. Tresch and P. Plath, Triaziflam and diaminotriazine derivatives affect enantioselectively multiple herbicide target sites, Z. Naturforsch., C, 2001, 56, 559–569 CrossRef CAS PubMed.
- A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Modern advances in heterocyclic chemistry in drug discovery, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC.
- S. Oudir, B. Rigo, J.-P. Hénichart and P. Gautret, A convenient method for the conversion of a carboxy group into a 4, 6-dimethoxy-1, 3, 5-triazine group: application to N-benzylpyroglutamic acids, Synthesis, 2006, 2845–2848 CAS.
- A. Pinner, Ueber diphenyloxykyanidin, Ber. Dtsch. Chem. Ges., 1890, 23, 2919–2922 CrossRef.
- A. R. Tiwari and B. M. Bhanage, Polythene glycol (PEG) as a reusable solvent system for the synthesis of 1,3,5-triazines via aerobic oxidative tandem cyclization of benzylamines and N-substituted benzylamines with amidines under transition metal-free conditions, Green Chem., 2016, 18, 144–149 RSC.
- J. Shen and X. Meng, Base-free synthesis of 1,3,5-triazines via aerobic oxidation of alcohols and benzamidine over a recyclable OMS-2 catalyst, Catal. Commun., 2019, 127, 58–63 CrossRef CAS.
- Q. You, F. Wang, C. Wu, T. Shi, D. Min, H. Chen and W. Zhang, Synthesis of 1,3,5-triazines via Cu (OAc) 2-catalyzed aerobic oxidative coupling of alcohols and amidine hydrochlorides, Org. Biomol. Chem., 2015, 13, 6723–6727 RSC.
- M. Zeng, T. Wang, D.-M. Cui and C. Zhang, Ruthenium-catalyzed synthesis of tri-substituted 1,3,5-triazines from alcohols and biguanides, New J. Chem., 2016, 40, 8225–8228 RSC.
- F. Xie, M. Chen, X. Wang, H. Jiang and M. Zhang, An efficient ruthenium-catalyzed dehydrogenative synthesis of 2,4,6-triaryl-1,3,5-triazines from aryl methanols and amidines, Org. Biomol. Chem., 2014, 12, 2761–2768 RSC.
- P. Wessig and J. Schwarz, A simple synthesis of 2,4-diaryl-1,3,5-triazines, Monatsh. Chem., 1995, 126, 99–102 CrossRef CAS.
- R. Kumar, N. Kumar, R. K. Roy and A. Singh, Triazines—A comprehensive review of their synthesis and diverse biological importance, Curr. Med. Drug. Res., 2017, 1, 173 Search PubMed.
- Y. Ogata, A. Kawasaki and K. Nakagawa, Kinetics of the formation of benzoguanamine from dicyandiamide and benzonitrile, Tetrahedron, 1964, 20, 2755–2761 CrossRef CAS.
- A. Díaz-Ortiz, A. de la Hoz, A. Moreno, A. Sánchez-Migallón and G. Valiente, Synthesis of 1,3,5-triazines in solvent-free conditions catalysed by silica-supported lewis acids, Green Chem., 2002, 4, 339–343 RSC.
- A. Herrera, A. Riano, R. Moreno, B. Caso, Z. D. Pardo, I. Fernandez, E. Saez, D. Molero, A. Sanchez-Vazquez and R. Martinez-Alvarez, One-pot synthesis of 1, 3, 5-triazine derivatives via controlled cross-cyclotrimerization of nitriles: a mechanism approach, J. Org. Chem., 2014, 79, 7012–7024 CrossRef CAS PubMed.
- S. Ikonen, S. Takala and E. Kolehmainen, Facile synthesis of 5β-cholane-sym-triazine conjugates starting from metformin and bile acid methyl esters: Liquid and solid state NMR characterization and single crystal structure of lithocholyl triazine, J. Mol. Struct., 2009, 936, 270–276 CrossRef CAS.
- M. Vallejo, M. A. Castro, M. Medarde, R. I. Macias, M. R. Romero, M. Y. El-Mir, M. J. Monte, O. Briz, M. A. Serrano and J. J. Marin, Novel bile acid derivatives (BANBs) with cytostatic activity obtained by conjugation of their side chain with nitrogenated bases, Biochem. Pharmacol., 2007, 73, 1394–1404 CrossRef CAS PubMed.
- S. R. Chaurasia, R. Dange and B. M. Bhanage, Graphene oxide as a carbo-catalyst for the synthesis of tri-substituted 1,3,5-triazines using biguanides and alcohols, Catal. Commun., 2020, 137, 105933, DOI:10.1016/j.catcom.2020.105933.
- R. Bardovskyi, O. Grytsai, C. Ronco and R. Benhida, Synthesis and characterization of new heterocycles related to aryl-[e][1,3]-diazepinediones. rearrangement to 2,4-diamino-1,3,5-triazine derivatives, New J. Chem., 2020, 44, 8171–8175 RSC.
- K. Huthmacher and D. Most, Cyanuric acid and cyanuric chloride, Ullmann’s Encycl. Ind. Chem., 2000, 11 DOI:10.1002/14356007.a08_191.
- Ö. Karataş, Y. Ceylan and Z. E. Koç, 2,4,6-Tris (p-aminoanilino)-1,3,5-triazine: synthesis and electron paramagnetic resonance (EPR) analysis, Sakarya Univ. J. Sci., 2022, 26, 1170–1179 CrossRef.
- C. Golias, A. Charalabopoulos and K. Charalabopoulos, Cell proliferation and cell cycle control: a mini review, Int. J. Clin. Pract., 2004, 58, 1134–1141 CrossRef CAS PubMed.
- Z. Abbas and S. Rehman, An overview of cancer treatment modalities, Neoplasm, 2018, 1, 139–157 Search PubMed.
- Centers for Disease Control and Prevention, https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm, accessed 21 September, 2023.
- R. Thilagavathi, S. Priyankha, M. Kannan, M. Prakash and C. Selvam, Compounds from diverse natural origin against triple-negative breast cancer: a comprehensive review, Chem. Biol. Drug Des., 2023, 101, 218–243 CrossRef CAS PubMed.
- J.-P. Zou, Z. Zhang, J.-Y. Lv, X.-Q. Zhang, Z.-Y. Zhang, S.-T. Han, Y.-W. Liu, W.-W. Liu, J. Ji and D.-H. Shi, Design, synthesis and anti-cancer evaluation of genistein-1,3,5-triazine derivatives, Tetrahedron, 2023, 133293, DOI:10.1016/j.tet.2023.133293.
- O. V. Mikolaichuk, E. A. Popova, A. V. Protas, O. S. Shemchuk, L. V. Vasina, Y. N. Pavlyukova, A. A. Potanin, 004F. E. Molchanov, D. N. Maistrenko and K. N. Semenov, Study of biocompatibility, cytotoxic activity in vitro of a tetrazole-containing derivative of 2-amino-4, 6-di (aziridin-1-yl)-1,3,5-triazine, Biochem. Biophys. Res. Commun., 2022, 629, 176–182 CrossRef CAS PubMed.
- X. Sun, B. Zhang, L. Luo, Y. Yang, B. He, Q. Zhang, L. Wang, S. Xu, P. Zheng and W. Zhu, Design, synthesis and pharmacological evaluation of 2-arylurea-1,3,5-triazine derivative (XIN-9): a novel potent dual PI3K/mTOR inhibitor for cancer therapy, Bioorg. Chem., 2022, 129, 106157, DOI:10.1016/j.bioorg.2022.106157.
- Y. Wang, Y. Liu, T. Ge, J. Tang, S. Wang, Z. Gao, J. Chen, J. Xu, P. Gong and Y. Zhao, Based on 2-(difluoromethyl)-1-[4,6-di (4-morpholinyl)-1,3,5-triazin-2-yl]-1H-benzimidazole (ZSTK474), design, synthesis and biological evaluation of novel PI3Kα selective inhibitors, Bioorg. Chem., 2023, 130, 106211, DOI:10.1016/j.bioorg.2022.106211.
- M. Xiao, M. Zhu, S. Wu, L. Ma, L. Qi, S. Ha, S. Xiong, M. Chen, D. Chen and G. Luo, Novel 6-amino-1,3,5-triazine derivatives as potent BTK inhibitors: structure-activity relationship (SAR) analysis and preliminary mechanism investigation, Bioorg. Chem., 2023, 130, 106263, DOI:10.1016/j.bioorg.2022.106263.
- W. Ali, S. Garbo, A. Kincses, M. Nové, G. Spengler, E. Di Bello, E. Honkisz-Orzechowska, T. Karcz, E. Szymańska and E. Żesławska, Seleno-vs. thioether triazine derivatives in search for new anticancer agents overcoming multidrug resistance in lymphoma, Eur. J. Med. Chem., 2022, 243, 114761, DOI:10.1016/j.ejmech.2022.114761.
- S. Xu, L. Luo, X. Sun, Y. Yang, Q. Guo, Z. Jiang and Y. Wu, Design, synthesis and antitumor activity of novel thiophene-triazine derivatives bearing arylurea unit as potent PI3K/mTOR inhibitorss, Bioorg. Med. Chem., 2023, 78, 117133, DOI:10.1016/j.bmc.2022.117133.
- V. Balaraju, S. Kalyani, G. Sridhar and E. Laxminarayana, Design, synthesis and biological assessment of 1,3,4-oxadiazole incorporated oxazole-triazole derivatives as anticancer agents, Chem. Data Collect., 2021, 33, 100695, DOI:10.1016/j.cdc.2021.100695.
- I. Shawish, A. Barakat, A. Aldalbahi, W. Alshaer, F. Daoud, D. A. Alqudah, M. Al Zoubi, M. m. M. Hatmal, M. S. Nafie and M. Haukka, Acetic acid mediated for one-pot synthesis of novel pyrazolyl s-triazine derivatives for the targeted therapy of triple-negative breast tumor cells (MDA-MB-231) via EGFR/PI3K/AKT/mTOR signaling cascades, Pharmaceutics, 2022, 14, 1558, DOI:10.1016/j.cdc.2021.100695.
- H. E. Hashem, A. E.-G. E. Amr, E. S. Nossier, M. M. Anwar and E. M. Azmy, New benzimidazole-, 1,2,4-triazole-, and 1,3,5-triazine-based derivatives as potential EGFRWT and EGFRT790M inhibitors: microwave-assisted synthesis, anticancer evaluation, and molecular docking study, ACS Omega, 2022, 7, 7155–7171 CrossRef CAS.
- I. Shawish, A. Barakat, A. Aldalbahi, A. M. Malebari, M. S. Nafie, A. A. Bekhit, A. Albohy, A. Khan, Z. Ul-Haq and M. Haukka, Synthesis and antiproliferative activity of a new series of mono-and bis (dimethylpyrazolyl)-s-triazine derivatives targeting EGFR/PI3K/AKT/mTOR signaling cascades, ACS Omega, 2022, 7, 24858–24870 CrossRef CAS PubMed.
- M. Robello, S. Salerno, E. Barresi, P. Orlandi, F. Vaglini, M. Banchi, F. Simorini, E. Baglini, V. Poggetti and S. Taliani, New antiproliferative agents derived from tricyclic 3,4-dihydrobenzo[4,5] imidazo[1,2-a][1,3,5] triazine scaffold: synthesis and pharmacological effects, Arch. Pharm., 2022, 355, 2200295, DOI:10.1016/j.cdc.2021.100695.
- A. F. Khalil, T. F. El-Moselhy, E. A. El-Bastawissy, R. Abdelhady, N. S. Younis and M. H. El-Hamamsy, Discovery of novel enasidenib analogues targeting inhibition of mutant isocitrate dehydrogenase 2 as antileukaemic agents, J. Enzyme Inhib. Med. Chem., 2023, 38, 2157411, DOI:10.1080/14756366.2022.2157411.
- R. El-Mernissi, K. El Khatabi, A. Khaldan, L. El Mchichi, M. A. Ajana, T. Lakhlifi and M. Bouachrine, 3D-QSAR, ADMET, and molecular docking studies for designing new 1,3,5-triazine derivatives as anticancer agents, Egpt. J. Chem., 2022, 65, 9–18 Search PubMed.
- I. Shawish, A. Aldalbahi, H. H. Al-Rasheed, M. Ali, W. Alshaer, M. Al Zoubi, S. Al Ayoubi, B. G. De La Torre, F. Albericio and A. El-Faham, Pyrazolyl-s-triazine with indole motif as a novel of epidermal growth factor receptor/cyclin-dependent kinase 2 dual inhibitors, Front. Chem., 2022, 10, 1078163, DOI:10.3389/fchem.2022.1078163.
- A. I. Zain-Alabdeen, T. F. El-Moselhy, N. Sharafeldin, A. Angeli, C. T. Supuran and M. H. El-Hamamsy, Synthesis and anticancer activity of new benzensulfonamides incorporating s-triazines as cyclic linkers for inhibition of carbonic anhydrase IX, Sci. Rep., 2022, 12, 16756, DOI:10.1038/s41598-022-21024-7.
- O. Mikolaichuk, A. Protas, E. Popova, O. Molchanov, D. Maistrenko, V. Ostrovskii, Y. N. Pavlyukova, V. Sharoyko and K. Semenov, Synthesis and in vitro study of cytotoxic activity of new tetrazole-containing 2,4-diamino-1,3,5-triazine derivatives, Russ. J. Gen. Chem., 2022, 92, 1621–1628 CrossRef CAS.
- J. Hu, Y. Zhang, N. Tang, Y. Lu, P. Guo and Z. Huang, Discovery of novel 1,3,5-triazine derivatives as potent inhibitor of cervical cancer via dual inhibition of PI3K/mTOR, Bioorg. Med. Chem., 2021, 32, 115997, DOI:10.1016/j.bmc.2021.115997.
- B. Spellberg, J. H. Powers, E. P. Brass, L. G. Miller and J. E. Edwards Jr, Trends in antimicrobial drug development: implications for the future, Clin. Infect. Dis., 2004, 38, 1279–1286 CrossRef CAS PubMed.
- J. Conly and B. Johnston, Where are all the new antibiotics? The new antibiotic paradox, Can. J. Infect. Dis. Med. Microbiol., 2005, 16, 159–160 CrossRef PubMed.
- K. D. Green, A. H. Pang, N. Thamban Chandrika, A. Garzan, A. D. Baughn, O. V. Tsodikov and S. Garneau-Tsodikova, Discovery and optimization of 6-(1-substituted pyrrole-2-yl)-s-triazine containing compounds as antibacterial agents, ACS Infect. Dis., 2022, 8, 757–767 CrossRef CAS PubMed.
- P. S. Yaduwanshi, B. K. Tiwari and N. Sharma, Study the effect of antimicrobial activity of some synthesize noval qunazolino-2,4,6-tri-substituted-s-triazine derivatives, J. Pharm. Negat. Results, 2022, 1597–1607 CAS.
- D. Van Quy, N. Van Hung, N. Stolpovskaya, A. Kruzhilin, S. S. Olshannikova, M. Holyavka, V. Sulimov and K. Shikhaliev, An efficient synthesis of novel 4-aryl-2-thioxo-3,4-dihydro-1H-pyrimido[1,2-a][1,3,5]triazin-6-(2H)-ones and their antibacterial activity, Molbank, 2022, 2022, M1417, DOI:10.3390/M1417.
- S. Noureen, S. Ali, J. Iqbal, M. A. Zia and T. Hussain, Synthesis, comparative theoretical and experimental characterization of some new 1,3, triazine based heterocyclic compounds and in vitro evaluation as promising biologically active agents, J. Mol. Struct., 2022, 1268, 133622, DOI:10.1016/j.molstruc.2022.133622.
- P. Asadi, E. Khodamoradi, G. Khodarahmi, A. Jahanian-Najafabadi, H. Marvi and S. Dehghan Khalili, Novel N-α-amino acid spacer-conjugated phthalimide–triazine derivatives: synthesis, antimicrobial and molecular docking studies, J. Amino Acids, 2023, 1–12 Search PubMed.
- A. Morandini, E. Spadati, B. Leonetti, R. Sole, V. Gatto, F. Rizzolio and V. Beghetto, Sustainable triazine-derived quaternary ammonium salts as antimicrobial agents, RSC Adv., 2021, 11, 28092–28096 RSC.
- J. J. Patel, R. P. Modh, M. Asamdi and K. H. Chikhalia, Comparative biological study between quinazolinyl–triazinyl semicarbazide and thiosemicarbazide hybrid derivatives, Mol. Divers., 2021, 25, 2271–2287 CrossRef CAS PubMed.
- M. H. Younis, E. R. Mohammed, A. R. Mohamed, M. M. Abdel-Aziz, H. H. Georgey and N. M. A. Gawad, Design, synthesis and anti-Mycobacterium tuberculosis evaluation of new thiazolidin-4-one and thiazolo [3,2-a][1,3,5] triazine derivatives, Bioorg. Chem., 2022, 124, 105807, DOI:10.1016/j.bioorg.2022.105807.
- A. R. Zala, D. Kumar, U. Razakhan, D. P. Rajani, I. Ahmad, H. Patel and P. Kumari, Molecular modeling and biological investigation of novel s-triazine linked benzothiazole and coumarin hybrids as antimicrobial and antimycobacterial agents, J. Biomol. Struct. Dyn., 2023, 1–12, DOI:10.1080/07391102.2023.2216293.
- D. S. Perlin, R. Rautemaa-Richardson and A. Alastruey-Izquierdo, The global problem of antifungal resistance: prevalence, mechanisms, and management, Lancet Infect. Dis., 2017, 17, e383–e392 CrossRef PubMed.
- Y. Shi, G. Wang, X.-p. Cai, J.-w. Deng, L. Zheng, H.-H. Zhu, M. Zheng, B. Yang and Z. Chen, An overview of COVID-19, J. Zhejiang Univ., Sci., B, 2020, 21, 343, DOI:10.1631/jzus.B2000083.
- R. A. Mekheimer, G. E.-D. A. Abuo-Rahma, M. Abd-Elmonem, R. Yahia, M. Hisham, A. M. Hayallah, S. M. Mostafa, F. A. Abo-Elsoud and K. U. Sadek, New s-triazine/tetrazole conjugates as potent antifungal and antibacterial agents: design, molecular docking and mechanistic study, J. Mol. Struct., 2022, 1267, 133615, DOI:10.1016/j.molstruc.2022.133615.
- K. A. Conrad, H. Kim, M. Qasim, A. Djehal, A. D. Hernday, L. Désaubry and J. M. Rauceo, Triazine-based small molecules: a potential new class of compounds in the antifungal toolbox, Pathogens, 2023, 12, 126, DOI:10.3390/pathogens12010126.
- S. J. Dabade, D. Mandloi, A. V. Bajaj and H. Atre, QSAR and docking studies of triazine derivatives as NMT inhibitors of Candida albicans, Int. J. Pharm. Sci. Drug Res., 2021, 13(2), 140–146 CAS.
- S. Cesarini, I. Vicenti, F. Poggialini, M. Secchi, F. Giammarino, I. Varasi, C. Lodola, M. Zazzi, E. Dreassi and G. Maga, Privileged scaffold decoration for the identification of the first trisubstituted triazine with anti-SARS-CoV-2 Activity, Molecules, 2022, 27, 8829, DOI:10.3390/molecules27248829.
- Y. S. Velihina, S. Pil’o, V. Zyabrev and V. Brovarets, Synthesis and evaluation of the antiviral activity of 2-(dichloromethyl) pyrazolo| 1,5-a|| 1,3,5| triazines, J. Rep. Natl. Acad. Sci. Ukr., 2019, 7, 75–80, DOI:10.15407/dopovidi2019.07.075.
- P. Scheltens, B. De Strooper, M. Kivipelto, H. Holstege, G. Chételat, C. E. Teunissen, J. Cummings and W. M. van der Flier, Alzheimer's disease, Lancet, 2021, 397, 1577–1590 CrossRef CAS PubMed.
- R. M. Lane, S. G. Potkin and A. Enz, Targeting acetylcholinesterase and butyrylcholinesterase in dementia, Int. J. Neuropsychopharmacol., 2006, 9, 101–124 CrossRef CAS PubMed.
- Y. Christen, Oxidative stress and Alzheimer disease, Am. J. Clin. Nutr., 2000, 71, 621S–629S CrossRef CAS PubMed.
- P. Baréa, V. A. Barbosa, D. A. d. S. Yamazaki, C. M. B. Gomes, C. R. Novello, W. F. d. Costa, G. d. F. Gauze and M. H. Sarragiotto, Anticholinesterase activity of β-carboline-1, 3, 5-triazine hybrids, Braz. J. Pharm. Sci., 2022, 58 DOI:10.1590/s2175-97902022e19958.
- W.-L. Wu, Z.-Y. Wen, J.-J. Qian, J.-P. Zou, S.-M. Liu, S. Yang, T. Qin, Q. Yang, Y.-H. Liu and W.-W. Liu, Design, synthesis, characterization and evaluation of 1,3,5-triazine-benzimidazole hybrids as multifunctional acetylcholinesterases inhibitors, J. Mol. Struct., 2022, 1257, 132498, DOI:10.1016/j.molstruc.2022.132498.
- J.-b. Su, W.-l. Wu, C.-E. Dong, S. Yang, Y.-y. Feng, T. Qin, K.-q. Chen, J.-j. Qian, J.-p. Zou and Y.-H. Liu, Synthesis, characterization, crystal structure and biological evaluation of 1, 3, 5-triazine-quinoline derivatives as butyrylcholinesterase inhibitors, J. Mol. Struct., 2023, 1274, 134391, DOI:10.1016/j.molstruc.2022.134391.
- D. Łażewska, M. Więcek, G. Satała, P. Chałupnik, E. Żesławska, E. Honkisz-Orzechowska, M. Tarasek, G. Latacz, W. Nitek and E. Szymańska, New triazine derivatives as serotonin 5-HT6 receptor ligands, Molecules, 2023, 28, 1108, DOI:10.3390/molecules28031108.
- D. Kułaga, A. K. Drabczyk, G. Satała, G. Latacz, A. Boguszewska-Czubara, D. Plażuk and J. Jaśkowska, Design, synthesis and biological evaluation of novel 1, 3, 5-triazines: effect of aromatic ring decoration on affinity to 5-HT7 receptor, Int. J. Mol. Sci., 2022, 23, 13308, DOI:10.3390/ijms232113308.
- M. Reddy, K. Rao, G. Anusha, G. Kumar, A. Damu, K. R. Reddy, N. P. Shetti, T. M. Aminabhavi and P. V. G. Reddy, In-vitro evaluation of antioxidant and anticholinesterase activities of novel pyridine, quinoxaline and s-triazine derivatives, Environ. Res., 2021, 199, 111320, DOI:10.1016/j.envres.2021.111320.
- D. Maliszewski, A. Wróbel, B. Kolesińska, J. Frączyk and D. Drozdowska, 1, 3, 5-Triazine nitrogen mustards with different peptide group as innovative candidates for AChE and BACE1 inhibitors, Molecules, 2021, 26, 3942, DOI:10.3390/molecules26133942.
- A. Hajri, D. Alimi, K. Rtibi and H. Sebai, Novel 6-aryl-7-alkyl/aryl-[1,2,4] triazolo [4,3-a][1,3,5] triazine-5 (6H)-thiones, processes for their preparation, characterization and evaluation of their in vitro antioxidant activity, Bull. Chem. Soc. Ethiop., 2021, 35, 565–572 CrossRef CAS.
- World Malaria Report 2021, https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2021, accessed 21 September, 2023.
- A. G. Maier, K. Matuschewski, M. Zhang and M. Rug, Plasmodium falciparum, Trends Parasitol., 2019, 35, 481–482 CrossRef PubMed.
- W. H. Wernsdorfer and D. Payne, The dynamics of drug resistance in Plasmodium falciparum, J. Pharmacol. Exp. Ther., 1991, 50, 95–121 CrossRef CAS PubMed.
- N. Adhikari, A. A. K. Choudhury, A. Shakya, S. K. Ghosh, S. J. Patgiri, U. P. Singh and H. R. Bhat, Molecular docking and antimalarial evaluation of novel N-(4-aminobenzoyl)-l-glutamic acid conjugated 1,3,5-triazine derivatives as Pf-DHFR inhibitors, 3 Biotech, 2022, 12, 347, DOI:10.1007/s13205-022-03400-2.
- N. Adhikari, A. A. Choudhury, A. Shakya, S. K. Ghosh, S. J. Patgiri, U. P. Singh and H. R. Bhat, Design and development of novel N-(4-aminobenzoyl)-l-glutamic acid conjugated 1,3,5-triazine derivatives as Pf-DHFR inhibitor: an in-silico and in-vitro study, J. Biochem. Mol. Toxicol., 2022, e23290, DOI:10.1002/jbt.23290.
- P. Gogoi, A. Shakya, S. K. Ghosh, N. Gogoi, P. Gahtori, N. Singh, D. R. Bhattacharyya, U. P. Singh and H. R. Bhat, In silico study, synthesis, and evaluation of the antimalarial activity of hybrid dimethoxy pyrazole 1, 3, 5-triazine derivatives, J. Biochem. Mol. Toxicol., 2021, 35, e22682, DOI:10.1002/jbt.22682.
- H. Hadni and M. Elhallaoui, Molecular docking and QSAR studies for modeling the antimalarial activity of hybrids 4-anilinoquinoline-triazines derivatives with the wild-type and mutant receptor Pf-DHFR, Heliyon, 2019, 5, e02357, DOI:10.1016/j.heliyon.2019.e02357.
- A. McTague, T. Martland and R. Appleton, Drug management for acute tonic-clonic convulsions including convulsive status epilepticus in children, Cochrane Database Syst. Rev., 2018, 1 DOI:10.1002/14651858.CD001905.pub3.
- H. E. Scharfman, The neurobiology of epilepsy, Curr. Neurol. Neurosci. Rep., 2007, 7, 348–354 CrossRef CAS PubMed.
- L. Santulli, A. Coppola, S. Balestrini and S. Striano, The challenges of treating epilepsy with 25 antiepileptic drugs, J. Pharm. Res., 2016, 107, 211–219 CrossRef CAS PubMed.
- A. G. Alhamzani, T. A. Yousef, M. M. Abou-Krisha, M. S. Raghu, K. Y. Kumar, M. K. Prashanth and B.-H. Jeon, Design, synthesis, molecular docking and pharmacological evaluation of novel triazine-based triazole derivatives as potential anticonvulsant agents, Bioorg. Med. Chem. Lett., 2022, 77, 129042, DOI:10.1016/j.bmcl.2022.129042.
- H. Yazdani Nyaki, N. O. Mahmoodi, H. Taherpour Nahzomi and E. Panahi Kokhdan, Two-and three-directional synthesis by 3–7CRs of novel (imidazolidine/thiazolidine)-2, 4-diones: preparation, antibacterial, anticonvulsant, and molecular docking investigation, Res. Chem. Intermed., 2023, 1–27 Search PubMed.
- K. Kaul, J. M. Tarr, S. I. Ahmad, E. M. Kohner and R. Chibber, in Diabetes: An Old Disease, A New Insight, Springer Science and Bussiness Media, 1st edn., 2013, vol. 771, pp. 1–11 Search PubMed.
- B. Ahrén, Emerging dipeptidyl peptidase-4 inhibitors for the treatment of diabetes, Expert Opin. Emerg. Drugs, 2008, 13, 593–607 CrossRef PubMed.
- A. Mushtaq, U. Azam, S. Mehreen and M. M. Naseer, Synthetic α-glucosidase inhibitors as promising anti-diabetic agents: recent developments and future challenges, J. Med. Chem., 2023, 115119, DOI:10.1016/j.ejmech.2023.115119.
- N. Lolak, S. Akocak, C. Türkeş, P. Taslimi, M. Işık, Ş. Beydemir, İ. Gülçin and M. Durgun, Synthesis, characterization, inhibition effects, and molecular docking studies as acetylcholinesterase, α-glycosidase, and carbonic anhydrase inhibitors of novel benzenesulfonamides incorporating 1,3,5-triazine structural motifs, Bioorg. Chem., 2020, 100, 103897, DOI:10.1016/j.bioorg.2020.103897.
- H.-D. Gao, P. Liu, Y. Yang and F. Gao, Sulfonamide-1, 3, 5-triazine–thiazoles: discovery of a novel class of antidiabetic agents via inhibition of DPP-4, RSC Adv., 2016, 6, 83438–83447 RSC.
- M. G. Badrey, S. M. Gomha, A. H. Abdelmonsef and A. A. El-Reedy, Syntheses and molecular docking analysis of some new thiazole and thiazine derivatives as three armed molecules with a triazine ring as a core component: a search for anti-obesity agents, Polycyclic Aromat. Compd., 2023, 1–19 CrossRef.
- D. Artis and H. Spits, The biology of innate lymphoid cells, Nature, 2015, 517, 293–301 CrossRef CAS PubMed.
- G. Pedraza-Alva, L. Pérez-Martínez, L. Valdez-Hernández, K. F. Meza-Sosa and M. Ando-Kuri, Negative regulation of the inflammasome: keeping inflammation under control, Immunol. Rev., 2015, 265, 231–257 CrossRef CAS PubMed.
- S. M. Lucas, N. J. Rothwell and R. M. Gibson, The role of inflammation in CNS injury and disease, Br. J. Pharmacol., 2006, 147, S232–S240 CrossRef CAS PubMed.
- K. L. Rock, J. J. Lai and H. Kono, Innate and adaptive immune responses to cell death, Immunol. Rev., 2011, 243, 191–205 CrossRef CAS PubMed.
- A. Waisman, R. S. Liblau and B. Becher, Innate and adaptive immune responses in the CNS, Lancet Neurol., 2015, 14, 945–955 CrossRef CAS PubMed.
- A. Azab, A. Nassar and A. N. Azab, Anti-inflammatory activity of natural products, Molecules, 2016, 21, 1321, DOI:10.3390/molecules21101321.
- R. de Cássia da Silveira e Sá, L. N. Andrade and D. P. de Sousa, A review on anti-inflammatory activity of monoterpenes, Molecules, 2013, 18, 1227–1254 CrossRef PubMed.
- P. Asadi, M. Alvani, V. Hajhashemi, M. Rostami and G. Khodarahmi, Design, synthesis, biological evaluation, and molecular docking study on triazine based derivatives as anti-inflammatory agents, J. Mol. Struct., 2021, 1243, 130760, DOI:10.1016/j.molstruc.2021.130760.
- R. Shinde, V. Mas and M. Patil, Antiinflammatory activity of triazine thiazolidinone derivatives: molecular docking and pharmacophore modeling, Indian J. Pharm. Sci., 2019, 81, 851–858 CAS.
- A. A. Abu-Hashem, S. A. Al-Hussain and M. E. Zaki, Synthesis of novel benzodifuranyl; 1,3,5-triazines; 1,3,5-oxadiazepines; and thiazolopyrimidines derived from visnaginone and khellinone as anti-inflammatory and analgesic agents, Molecules, 2020, 25, 220, DOI:10.3390/molecules25010220.
- A. E. Lang and A. M. Lozano, Parkinson's disease, N. Engl. J. Med., 1998, 339, 1130–1143 CrossRef CAS PubMed.
- M. J. Armstrong and M. S. Okun, Diagnosis and treatment of Parkinson disease: a review, JAMA, 2020, 323, 548–560 CrossRef PubMed.
- A. Masih, S. Singh, A. K. Agnihotri, S. Giri, J. K. Shrivastava, N. Pandey, H. R. Bhat and U. P. Singh, Design and development of 1,3,5-triazine-thiadiazole hybrids as potent adenosine A2A receptor (A2AR) antagonist for benefit in Parkinson's disease, Neurosci. Lett., 2020, 735, 135222, DOI:10.1016/j.neulet.2020.135222.
- S. Redenti, I. Marcovich, T. De Vita, C. Pérez, R. De Zorzi, N. Demitri, D. I. Perez, G. Bottegoni, P. Bisignano and M. Bissaro, A triazolotriazine-based dual GSK-3β/CK-1δ ligand as a potential neuroprotective agent presenting two different mechanisms of enzymatic inhibition, ChemMedChem, 2019, 14, 310–314 CrossRef CAS PubMed.
- S. L. Croft and G. H. Coombs, Leishmaniasis–current chemotherapy and recent advances in the search for novel drugs, Trends Parasitol., 2003, 19, 502–508 CrossRef CAS PubMed.
- P. Baréa, V. A. Barbosa, D. L. Bidóia, J. C. de Paula, T. F. Stefanello, W. F. da Costa, C. V. Nakamura and M. H. Sarragiotto, Synthesis, antileishmanial activity and mechanism of action studies of novel β-carboline-1,3,5-triazine hybrids, Eur. J. Med. Chem., 2018, 150, 579–590 CrossRef PubMed.
- A. Cavalli and M. L. Bolognesi, Neglected tropical diseases: multi-target-directed ligands in the search for novel lead candidates against Trypanosoma and Leishmania, J. Med. Chem., 2009, 52, 7339–7359 CrossRef CAS PubMed.
- M. Venkatraj, I. G. Salado, J. Heeres, J. Joossens, P. J. Lewi, G. Caljon, L. Maes, P. Van der Veken and K. Augustyns, Novel triazine dimers with potent antitrypanosomal activity, Eur. J. Med. Chem., 2018, 143, 306–319 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |